

Abstract
Objective
Atherosclerosis, the cause of 50% of deaths in westernised societies, is widely regarded as a chronic vascular inflammatory disease. Vascular smooth muscle cell (VSMC) inflammatory activation in response to local pro-inflammatory stimuli contributes to disease progression and is a pervasive feature in developing atherosclerotic plaques. Therefore, it is of considerable therapeutic importance to identify mechanisms that regulate the VSMC inflammatory response.
Approach and Results
We report that myocardin, a powerful myogenic transcriptional coactivator, negatively regulates VSMC inflammatory activation and vascular disease. Myocardin levels are reduced during atherosclerosis, in association with phenotypic switching of smooth muscle cells. Myocardin deficiency accelerates atherogenesis in hypercholesterolemic ApoE−/− mice. Conversely, increased myocardin expression potently abrogates the induction of an array of inflammatory cytokines, chemokines and adhesion molecules in VSMCs. Expression of myocardin in VSMCs reduces lipid uptake, macrophage interaction, chemotaxis and macrophage-endothelial tethering in vitro, and attenuates monocyte accumulation within developing lesions in vivo. These results demonstrate that endogenous levels of myocardin are a critical regulator of vessel inflammation.
Conclusions
We propose myocardin as a guardian of the contractile, non-inflammatory VSMC phenotype, with loss of myocardin representing a critical permissive step in the process of phenotypic transition and inflammatory activation, at the onset of vascular disease.
Keywords: Myocardin, Smooth muscle, Inflammation, Atherosclerosis
Introduction
Cardiovascular diseases are the cause of substantial mortality and morbidity across the Western and, increasingly, the developing worlds.1,2 Chronic inflammation is a pathological process that represents a common, critical hallmark in the development of multiple occlusive vascular diseases including transplant vasculopathy, interventional restenosis and atherosclerosis.3,4 Vascular smooth muscle cells (VSMCs) undergo a unique, coordinated morphological and functional transition between contractile and synthetic states in injury and disease.5 Inflammatory activation of medial VSMCs refers to the attainment of a phenotypic state in which cells secrete a diverse range of pro-inflammatory mediators such as cytokines, chemokines and adhesion molecules, in response to a variety of signals in the surrounding milieu. VSMC inflammatory activation contributes to vessel pathophysiology, vascular disease progression and ultimately, adverse clinical outcome.6,7 Therefore, it is of considerable therapeutic importance to identify mechanisms that regulate the VSMC inflammatory response.
Myocardin (Myocd) is a potent myogenic coactivator, expressed specifically in contractile smooth and cardiac muscle tissues in adulthood, and respective progenitor cells during embryonic development.8,9,10,11 Myocardin associates strongly with serum response factor (SRF) and stabilizes binding at the degenerate CC(A/T-rich)6GG (CArG) cis-elements of all known CArG-dependent SMC marker genes.5,12 Myocardin homozygous-null (Myocd−/−) mice exhibit impaired SMC development and die at embryonic day 10.5,13 although Myocd−/− embryonic stem cells retain some capacity to differentiate into SMC lineages in vitro and in vivo.14,15 However, myocardin-deficient VSMCs show reduced expression of contractile apparatus and exhibit ultrastructural features associated with the synthetic phenotype.16 A growing number of studies report a correlation between vascular injury or disease progression, and reduced myocardin expression17,18,19,20,21 or alternative splicing of myocardin pre-mRNA.22 Furthermore, Lovren and colleagues23 observed that attenuated lesion development following miR-145 administration to VSMCs in hypercholesterolemic ApoE−/− mice coincided with increased myocardin levels. However, despite previous in vitro evidence that myocardin negatively regulates VSMC proliferation,9,11,24 this association is consistently regarded as secondary to disease progression, and myocardin is referred to purely as a marker of SMC contractile gene expression. Recently, we set out to address this shortcoming, and identified myocardin as a critical regulator of the vessel injury response in vivo.25 Re-expression of myocardin suppressed neointima formation in murine carotid arteries, while myocardin-heterozygous-null (Myocd+/−) mice displayed augmented neointima formation with increased VSMC migration and proliferation. Utilising both loss and gain-of-function models, in vitro and in vivo, we now demonstrate that myocardin is a central negative regulator of VSMC inflammatory activation and associated vascular disease.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Myocardin expression is reduced in VSMCs at the onset of vascular disease
Studies in mice and swine have shown that expression of myocardin in the vascular wall is decreased following injury and in atherosclerosis, associated with reduced SMC marker gene expression.17,18,19,20 To examine the reproducibility and temporal dynamics of these observations in an established model of atherosclerosis, we examined myocardin expression following placement of a non-constrictive collar around the common carotid arteries of high fat diet-fed LDLR−/− mice.26,27 In this model, significant neointimal lesions develop by 6 weeks following collar placement. A marked downregulation of myocardin RNA expression was confirmed two weeks post-surgery (P<0.01, Figure 1A), accompanied by concomitant fall in SMC marker gene expression (Figure 1B). These data demonstrate that downregulation of myocardin is an early and consistent feature of vascular disease, as in vascular injury.25 Subsequent analysis of healthy aortic and diseased carotid human artery samples confirmed that myocardin expression is similarly reduced in diseased human vessels compared to healthy controls (P<0.05; Figure 1C). Differential myocardin expression is maintained in their respective human artery VSMC cultures (Figure 1D), indicating that the change is intrinsic to VSMCs, and not attributable to differential VSMC numbers relative to other cell types within healthy and diseased vessel walls.
Figure 1. Loss of myocardin accelerates early atherosclerotic lesion development in a murine model of high-fat diet.


(A–B) Quantification of myocardin (A) and smooth muscle markers, smooth muscle α-actin (Acta2) and smooth muscle myosin heavy chain (Myh11; B) mRNA expression in collar-induced atherosclerosis in carotid arteries of ApoE−/− mice (n=3). (C–D) Myocardin mRNA expression in normal human aorta and diseased carotid vessels (C; n=3) and in SMCs derived from human aorta and diseased carotid plaques (D; n=3). (E) Hematoxylin and eosin staining of aortic root sections from ApoE−/−.Myocd+/+ (wild type, WT) and ApoE−/−.Myocd+/− (Myocd+/−) mice fed on high-fat diet for 8 weeks. Scale bar, 250 μm. (F–G) Quantification of aortic root medial (F) and plaque area (G) from WT (n=15) and Myocd+/− (n=11) mice fed on high-fat diet. (H) Oil red O staining of aortic root sections from WT and Myocd+/− mice fed on high-fat diet for 8 weeks. Scale bar, 250 μm. (I) Quantification of lipid rich regions by oil red O staining in the aortic root in WT (n=5) and Myocd+/− (n=6). Data are presented as mean±s.e.m. *P<0.05 Student’s t-test. ns indicates no statistical significance.
Myocardin deficiency is causal for accelerated vascular disease
Myocd+/− mice have reduced aortic myocardin expression, but similar levels of myocardin-like transcription factors Mkl1 and Mkl2, compared to wild-type littermates (Figure IA of the online only Data Supplement).25 These mice show a novel phenotype of augmented neointima formation following vascular injury, with increased VSMC migration and proliferation.25 To determine whether reduced myocardin expression in VSMCs represents a critical permissive step in early vascular disease progression, Myocd+/− mice were crossed onto an ApoE−/− background.28 Mice were then administered high fat diet for 8 weeks, before analysis of atherosclerotic lesion development in the aortic root. A relatively short period of fat-feeding was selected to visualise early stages of vascular disease, whereby accelerated progression could present most clearly. ApoE−/−. Myocd+/− mice exhibited a significant, close to 50% mean increase in plaque area, compared to ApoE−/−. Myocd+/+ (wild type; WT) littermate controls (P=0.05; Figure 1E–G). Total medial area was unchanged between groups. A complete set of aortic root cross-section images analysed in this study is provided in Figure IIA of the online-only Data Supplement. A second independent study further substantiated these findings, showing a 25% mean increase in oil red O staining of lipid rich regions in the aortic root of ApoE−/−. Myocd+/− mice compared to ApoE−/−. Myocd+/+ controls (P=0.04, Figure 1G-H). Data from these loss-of-function studies provide the first compelling evidence that endogenous levels of myocardin negatively regulate the progress of atherosclerosis in vivo.
Myocardin-deficient SMCs exhibit increased potential for inflammatory activation
VSMC inflammatory activation contributes to the propagation of inflammation in the developing lesion, and to the progression of vascular disease. We hypothesized that myocardin may reduce vascular disease through suppression of VSMC phenotypic transition and inflammatory activation. It was reasoned that reduced VSMC inflammatory activation within a developing lesion would manifest in reduced inflammatory mediator production and reduced leukocyte infiltration. Indeed, increased numbers of Lamp2 (Mac3)-positive macrophage-like cells were observed in lesions of Myocd+/− mice compared with WT littermate controls (P<0.01; Figure 2A and 2B). IgG negative controls confirmed the specificity of Lamp2 immunostaining in the aorta, while immunostaining against an alternative macrophage cell surface marker, CD68, recapitulated the Lamp2 staining results (Figures III and IV of the online-only Data Supplement). While we consider these cells as macrophages of circulating leukocyte origin, we do not exclude the possibility that a proportion are VSMCs that have downregulated smooth muscle marker gene expression, upregulated macrophage markers and assumed macrophage-like morphology.29,30 Consistent with increased leukocyte accumulation, we found significantly greater levels of circulating chemokine monocyte chemotactic protein-1 (CCL2) in Myocd+/− animals compared to WT littermates 4 weeks after initiation of the high fat diet (Figure 2C). Immunostaining revealed similarly increased levels of CCL2 in the aortic lesions of Myocd+/− mice, compared to WT littermate controls (Figure V of the online-only Data Supplement).
Figure 2. Myocardin haploinsufficiency potentiates infiltration of macrophages and inflammation during atherosclerotic lesion development.
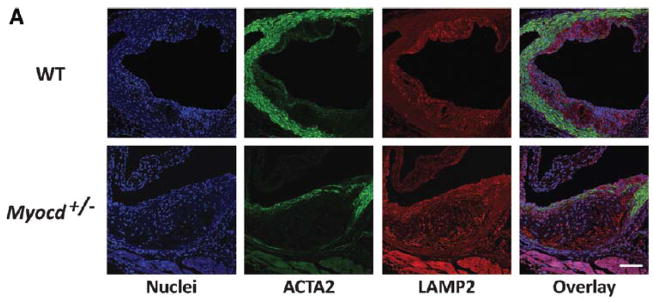


(A) Confocal images of aortic root from high-fat fed ApoE−/− .Myocd+/+ (wild type, WT) and ApoE−/−. Myocd+/− (Myocd+/−) mice labelled with antibodies directed against smooth muscle α-actin (ACTA2; green) and lysosome-associated membrane protein 2 (LAMP2; red). 4′,6-diamidino-2-phenylindole (DAPI; blue) counterstaining indicates nuclei. Scale bar, 100 μm. (B) Quantification of macrophages (cells positive for LAMP2) in the plaque regions of the aortic root from WT (n=5) and Myocd+/− (n=5) mice fed on high-fat diet for 8 weeks. (C) Levels of serum monocyte chemotactic protein-1 (CCL2) in WT and Myocd+/− mice fed on high-fat diet for 4 weeks. Data are presented as a mean fold change relative to week 0. (D) Expression of inflammatory marker mRNA in mouse aortic root, after 1 week high fat diet. (E–H) mRNA expression of interleukin-6 (Il-6; E), Ccl2 (F), Cebpb (G) and Cebpd (H) in VSMCs derived from WT and Myocd+/− aortas after 4 hr stimulation with interleukin-1beta (IL-1B; n=3). Data are presented as mean±s.e.m. *P<0.05, ** P<0.01 and ***P<0.001 Student’s t-test.
Analysis of aortic root tissue harvested after 1-week high fat diet revealed early differences in inflammatory gene expression between Myocd+/− and WT mice (Figure 2D). Levels of the atherogenic cytokine interleukin (Il)-6 were significantly increased in aortic roots of Myocd+/− mice (P<0.05).31 We also found increased expression of pro-inflammatory CCAAT/enhancer-binding protein (C/Ebp) transcription factors Cebpb and Cebpd in Myocd+/− aorta. No significant change in Ccl2 expression was observed, possibly reflecting specific temporal regulation of inflammatory gene expression, or clouding of differential signal from VSMCs by other cell types present in whole aortic root tissue.
To test whether increased inflammatory activation and leukocyte infiltration may indeed be driven specifically by myocardin-deficient VSMCs, medial aortic VSMCs were cultured from Myocd+/− mice and WT littermate controls. These cells were then exposed to varying concentrations of the pro-inflammatory cytokine IL-1β, which has been shown to elicit an inflammatory phenotypic state in VSMCs.32 IL-1β-induced IL-6 release is an established indicator of VSMC inflammatory activation.33 IL-1β induced a marked upregulation of Il-6 and Ccl2 mRNA, and levels were consistently and significantly higher in VSMCs derived from Myocd+/− mice (Figure 2E and 2F). Furthermore, the mRNA expression of Cebpb and Cebpd was similarly increased in myocardin-deficient VSMCs (Figure 2G and 2H). Il-6 and Ccl2 strongly recruit macrophages and monocytes to developing lesions, regulate the local expression of adhesion molecules, cytokines and the pro-coagulant tissue factor, and propagate inflammation in the nascent lesion.7,34,35 CEBPB and CEBPD bind at the promoters of multiple inflammatory response genes, including Il-6 and Ccl2, and synergistically upregulate and sustain gene expression following inflammatory stimulation.36,37,38 Upregulation of Il-6, Ccl2 and Cebp factors in myocardin deficient VSMC is therefore highly suggestive of fundamental transition towards a pro-inflammatory state. Enhanced VSMC inflammatory activation in myocardin-deficient VSMCs thereby provides an endogenous mechanism by which accelerated vascular disease may proceed.
Myocardin deficiency amplifies inflammatory responses in human VSMC
To test the relevance of myocardin deficiency in human disease, human coronary artery VSMC (HCASMC) were grown and transfected with myocardin-specific small interfering RNA (siRNA). A reduction of roughly 50% myocardin expression was observed compared to controls (Figure VI of the online-only Data Supplement). Consistent with observations in mouse VSMCs, reduced myocardin expression in HCASMC caused marked increases in IL-1β-induced IL6 and CCL2 production (P<0.001), both in terms of mRNA expression and protein secretion, as determined by enzyme-linked immunosorbence assay (ELISA) (Figure 3A–3C). Consistent with data from the Myocd+/− aortas, CEBPB and CEBPD expression were also increased in myocardin-deficient HCASMC (P<0.001 and P<0.01, respectively). Importantly, inhibition of CEBPB and CEBPD expression in the presence of myocardin deficiency was able to partially reverse the increase in inflammatory IL-6 and CCL2 marker production (Figure 3B and 3C). This finding is in line with our prior observation that siRNA-mediated inhibition of CebpB and CebpD can suppress IL-1β-induced expression of inflammatory mediators such as Il-6 in rat aortic VSMC (RASMC) (data not shown). These data are thus consistent with our previous findings and support the relevance of myocardin in regulating vascular inflammation in human disease, regulation that may occur at least in part through the inhibition of intermediate inflammatory pathway mediators such as CEBPB and CEBPD.
Figure 3. Myocardin antagonises interleukin-1beta-induced inflammatory pathways in vascular smooth muscle cells.
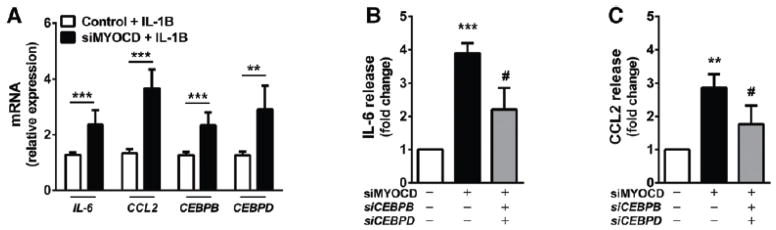


(A) Expression of inflammatory marker mRNA in human coronary SMC (HCASMC) transfected with scrambled control siRNA or siRNA against myocardin (siMYOCD) for 24 hr, before treatment with 20 ng/ml interleukin-1beta (IL-1B). (B–C) Release of interleukin-6 (IL-6; B) and monocyte chemotactic protein-1 (CCL2; C) from HCASMCs transfected with scrambled control siRNA or siRNA against myocardin (siMYOCD), CEBPB (siCEBPB) and CEBPD (siCEBPD), as indicated, for 24 hr, before treatment with 20 ng/ml interleukin-1beta (IL-1B). IL-6 and CCL-2 release into surrounding media was quantified by enzyme-linked immunosorbent assay (ELISA). Data are presented as mean±s.e.m. n=3 independent experiments in biological triplicate. **P<0.01 and ***P<0.001 compared to scrambled siRNA-only control; #P<0.05 compared to siMYOCD-only treated cells. Rat aortic VSMCs were then transduced with adenoviral vectors (Ad) expressing dominant negative myocardin (Ad.Myocd-DN) control, or myocardin (Ad.Myocd). (D) mRNA expression of inflammatory and anti-inflammatory genes determined by real time quantitative PCR and presented in a heat map format, for VSMCs treated with Ad.Myocd-DN control or Ad.Myocd and stimulated for 4 hr with 20 ng/ml IL-1B. Red (high) and blue (low) depict differential gene expression relative to mean expression across all treatments. Source data are available for this figure (Supplementary Fig 1). (E–F) Quantification of IL-6 (E) and CCL2 (F) release after 24 hr IL-1B stimulation. (G-H) Cebpb (G) and Cebpd (H) mRNA expression in rat VSMCs treated with Ad.Myocd-DN control or Ad.Myocd and stimulated for 4 hr with 20 ng/ml IL-1B. Expression of CEBPB and CEBPD in rat VSMCs treated with Ad.LacZ or Ad.Myocd followed by IL-1B stimulation (I). Analysis of Il6 and Ccl2 gene promoters by chromatin immunoprecipitation (ChIP) (J). VSMCs overexpressing Myocd or Myocd-DN constructs were stimulated for 4 hours with IL-1B. Cellular lysates were incubated with anti-CEBPB antibody or IgG antibody (control). Bound Il6 and Ccl2 promoter DNA was then quantified by real-time PCR. Data are presented as mean±s.e.m, n=3 independent experiments in biological triplicate. *P<0.05, ** P<0.01 and ***P<0.001 Student’s t-test. ns indicates no statistical significance.
Myocardin expression suppresses inflammatory activation in cultured VSMC
We next set out to test whether, conversely, augmented myocardin expression in VSMCs protects against the transition towards the inflammatory phenotype. Rat aortic VSMC, which grow reproducibly in culture and express low levels of endogenous myocardin, were treated with adenoviral expression constructs encoding myocardin (Ad.Myocd), or β-galactosidase (Ad.LacZ) and Myocd-DN (Ad.Myocd-DN) negative expression controls. Myocd-DN encodes a truncated form of myocardin that lacks the C-terminal transcription activating domain.8 Efficient expression of relevant constructs was confirmed by Western Blotting (Figure IC–ID of the online-only Data Supplement). Myocardin and contractile SMC marker gene expression was significantly upregulated following Ad.Myocd administration (Figure VII of the online-only Data Supplement). Ad.Myocd-DN exerted no discernible dominant-negative effect39, likely due to low endogenous levels of myocardin in cultured rat VSMC.9 Due to closer molecular similarity to Ad.Myocd, Ad.Myocd-DN was therefore employed as the optimal negative expression control for in vitro studies. Complete datasets containing no-virus and Ad.LacZ controls are shown in Figure VIII of the online-only Data Supplement. The inhibition of lipopolysaccharide (LPS)-induced tumour necrosis factor (TNF-α) and IL-6 production in aortic VSMC by myocardin has been reported previously.24 Given the complexity of the atherogenic inflammatory process,7 we elected to expand on this finding to investigate the effect of myocardin on the IL-1β-stimulated expression of a selection of cytokines, chemokines and adhesion molecules with documented roles in atherosclerosis. Real time RT-PCR expression data is presented in heat-map format for ease of visualisation (Figure 3D) and in full as bar charts (Figure VIII of the online-only Data Supplement). Expression of myocardin was found to strongly and reproducibly suppress the IL-1β-induced expression of a diverse range of pro-inflammatory factors including IL-6, CCL-2, NOS2, CXCL2, CX3CL1, VCAM-1 and CSF-1. Notably, suppression was not universal, with ICAM-1 apparently unaffected, and CSF-3 significantly upregulated in the presence of myocardin. Myocardin also significantly reduced the expression of inflammation-mediating IL-1 receptors IL-1R1 and IL-1R2, the IL-6 receptor IL-6R, the TNFα receptor TNFRSF1A and the oxidized low-density lipoprotein scavenger receptor OLR-1, which is consistent with reduced potential for inflammatory activation. Furthermore, myocardin expression upregulated the putative anti-inflammatory cytokine IL-19,40,41 and two members of the immunosuppressive suppressor of cytokine signalling (SOCS) family, SOCS-2 and SOCS-6.42 Given our previous finding of increased inflammatory activation in HCASMC treated with MYOCD siRNA, we tested whether Ad.Myocd-DN might exhibit a dominant-effect in this setting of potentially higher endogenous myocardin. However, no significant effect was observed, although there was a trend towards decreased expression of the smooth muscle marker gene MYH11 in the presence of Ad.Myocd-DN (Figure IX of the online-only Data Supplement). Speculatively, the truncated mouse Myocd encoded in Ad.Myocd-DN may not compete well with endogenous human Myocd in HCASMC, except possibly at particularly specific, myocardin-sensitive promoters or enhancer elements. Mouse Myocd encoded by Ad.Myocd was, on the other hand, able to potently suppress IL-1β-induced inflammatory marker gene expression and IL-6/CCL2 secretion in HCASMC (Figure X of the online-only Data supplement).
Production of inflammatory factors in VSMCs is controlled pre-translationally, typically at the level of transcription.7,43 Analysis of cell supernatants by ELISA confirmed a marked induction of IL-6 and CCL2 protein secretion from IL-1β-stimulated rat VSMCs, which was robustly abrogated following administration of Ad.Myocd (P<0.001; Figure 3E and 3F). IL-6 release in response to stimulation by TNF-α and LPS was also suppressed by myocardin (Figure XI of the online-only Data Supplement). Increasing Ad.Myocd multiplicity of infection (MOI) resulted in a roughly linear decrease in CCL2 secretion (Figure XIE of the online-only Data Supplement). Consistent with previous results, mRNA expression of Cebpb and Cebpd was induced by inflammatory stimulation but strongly repressed in the presence of myocardin (P<0.05; Figure 3G and 3H). Reduced abundance of both active isoforms of CEBPB, LAP* and LAP,44 and CEBPD, were observed by Western blotting (Figure 3I). Furthermore, analysis of the Il6 promoter by chromatin immunoprecipitation (ChIP) revealed a marked increase in CEBPB protein binding following IL-1β stimulation, which was abrogated in the presence of Myocd expression (Figure 3J). However, no significant changes in binding of CEBPB to the Ccl2 promoter were detected. Binding of CEBPD was also not detected, despite analysis of multiple putative binding regions at the Ccl2 promoter (Figure XII of the online-only data supplement), although this may be attributable to a paucity of ChIP-grade antibodies available to target CEBPD.
Taken together, these data demonstrate that myocardin expression can actively and specifically suppress multiple facets of inflammatory activation in cultured VSMC, including the intracellular inflammatory pathway mediators CEBPB and CEBPD and furthermore, stimulate one or more distinct anti-inflammatory responses.
Myocardin suppresses lipid uptake in human VSMC
To corroborate previous in vitro data, further studies were performed to test the functional relevance of reduced VSMC inflammatory activation in the presence of increased myocardin expression. We previously noted that myocardin expression causes downregulation of the scavenger receptor OLR-1 (Figure 3D). OLR-1 mediates oxidised low-density lipoprotein (oxLDL) uptake in VSMCs, contributing to foam cell formation and progression of vascular disease.45 To examine whether OLR-1 downregulation translated functionally to reduced lipid uptake, HCASMC were incubated with fluorescent dil-labelled oxLDL. A marked, significant reduction in dil-oxLDL uptake in Ad.Myocd-treated VSMCs was measured at both two and three hours post addition, versus controls (P<0.01; Figure 4). Myocardin-induced OLR-1 downregulation in VSMC is thereby consistent with a functional reduction in pathological cellular lipid uptake.
Figure 4. Myocardin reduces the uptake of oxidised lipoproteins.
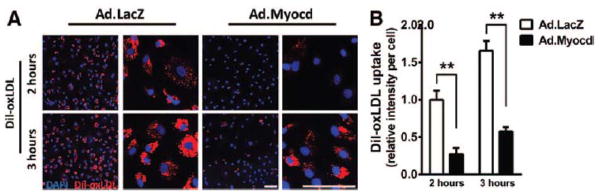
Human coronary smooth muscle cells (HCASMCs) were transduced with adenoviral vectors (Ad) expressing beta-galactosidase (Ad.LacZ; control) or myocardin (Ad.Myocd) then incubated with Dil-ox-LDL (10 mg/ml) for indicated time intervals. (A) Effect of myocardin on the uptake of fluorescent-labelled oxidised lipoprotein (Dil-ox-LDL; red). 4′,6-diamidino-2-phenylindole (DAPI; blue) counterstaining incidates nuclei. Scale bar, 100 μm. (B) Quantification of accumulated Dil-oxLDL in HCASMCs. Data are presented as mean±s.e.m. ** P<0.01 Student’s t-test.
Myocardin expression in VSMC suppresses macrophage chemotaxis, adhesion and lipid uptake in vitro
Activated VSMCs are known to contribute to the recruitment and maturation of circulating leukocytes, and subsequent propagation of lesion inflammation, through the secretion of a diverse assortment of chemokines including CCL2, CSF-1, IL-6 and CX3CL1.7,46,47 We reasoned that myocardin expression in VSMCs would attenuate the leukocyte chemotactic response through downregulation of many such factors. To test this, a trans-well migration assay model was adopted, wherein cultured VSMC were assessed for their ability to induce migration of fluorescently labelled macrophages through a trans-well insert membrane (Figure XIII of the online-only Data Supplement). Pre-stimulation of control rat VSMCs with IL-1β produced a marked induction of macrophage chemotaxis across the trans-well insert membrane, and this migration was significantly attenuated by myocardin expression in the VSMCs (P<0.05; Figure 5A). In a separate experiment, adhesion of macrophages following incubation with a rat VSMC monolayer was similarly quantified. Lesion-dwelling macrophages are known to interact with resident VSMCs, providing synergistic enhancement of pro-inflammatory responses such as IL-6 and CCL2 production.48 Again, macrophage adherence was increased when VSMCs were pre-treated with IL-1β, but this increase was strongly abrogated in VSMCs that had been previously administered with Ad.Myocd (P<0.01; Figure 5B). This is consistent with our observation that myocardin expression reduces the abundance of VSMC surface adhesion molecules such as CX3CL1 and VCAM. Another critical interaction in early atherogenesis is the tethering of circulating monocytes to activated endothelial cells (ECs) at the vessel wall.49 While ECs do not themselves express myocardin, ECs and underlying VSMCs conceivably participate in extensive inflammatory cross-talk. Thus, reduced VSMC inflammatory activation could translate to reduced EC stimulation and reduced EC-leukocyte tethering. Accordingly, conditioned media were collected from pre-treated rat VSMCs, and used to pre-stimulate EC monolayers. Macrophage adhesion to the monolayers was then quantified as previously (Figure XIIB of the online-only Data Supplement). Results demonstrated a small but significant increase in macrophage tethering to ECs treated with conditioned media from IL-1β-stimulated VSMCs. This increase was not seen in ECs treated with conditioned media from Ad.Myocd-treated VSMCs (P<0.05; Figure 5C). In a final in vitro functional assay, conditioned media from pre-treated rat VSMCs was tested for its ability to influence lipid uptake in macrophages, which represents another central process in atherosclerotic lesion progression.4 Macrophages were maintained in the conditioned media and then assayed for uptake of fluorescent Dil-labelled acetylated-LDL. Lipid uptake was significantly increased following exposure to conditioned media from IL-1β-treated VSMCs, but this increase was completely abrogated when the VSMCs had been treated with Ad.Myocd (Figure 5D and 5E). In order to validate the relevance of these findings across species boundaries, macrophage chemotaxis and interaction assays were further repeated with HCASMC in place of rat VSMC, and the same myocardin-mediated suppression of both processes was consistently observed (Figure XIVA and XIVB of the online-only Data Supplement). Therefore, myocardin-induced repression of VSMC inflammatory activation may functionally regulate several key processes in atherogenesis, including EC-leukocyte tethering, leukocyte chemotaxis, VSMC-leukocyte interaction and macrophage lipid uptake.
Figure 5. Myocardin expression in VSMC reduces macrophage chemotaxis, adhesion and uptake of lipids.

(A) Myocardin expression in vascular smooth muscle cells (VSMC) reduces macrophage chemotaxis. Rat VSMCs were transduced with adenoviral vectors (Ad) expressing either dominant negative myocardin (Ad.Myocd-DN) control or myocardin (Ad.Myocd) and pre-stimulated for 6 hr with 20 ng/ml interleukin-1beta (IL-1B). Fluorescent-labelled RAW264.7 macrophages were applied to upper chambers of coated transwell inserts. After 20 hr incubation, un-migrated macrophages on the upper trans-well surface were removed and remaining migrated macrophages were quantified. Quantification of fluorescent-labelled macrophages was performed using fluorescence microscopy and image analysis with ImageJ software. Data are presented as mean±s.e.m., n=3 independent experiments in biological triplicate. *P<0.05 and ** P<0.01 Student’s t-test. (B) Myocardin expression in VSMC reduces macrophage adhesion. Confluent VSMC monolayers were treated with Ad.Myocd-DN control or Ad.Myocd and pre-stimulated for 24 hr with 20 ng/ml IL-1B. VSMC monolayers were incubated for 45 min with fluorescent-labelled RAW264.7 macrophages, before washing and quantification of adhered macrophages, as above. (C) Ad.Myocd-treated VSMCs induce diminished endothelial-macrophage cell interaction. Conditioned media was collected from Ad.Myocd-DN (control) or Ad.Myocd transduced, unstimulated or IL-1B stimulated VSMCs. Human umbilical vein endothelial cell (HUVEC) monolayers were maintained in the conditioned media for 24 hr. The HUVEC monolayers were then incubated for 45 min with fluorescent-labelled RAW264.7 macrophages, before washing and quantification of adhered macrophages, as above. (D) VSMCs transduced with Ad.Myocd reduce the uptake of acetylated low-density lipoprotein (Ac-LDL) by macrophages. Conditioned media was collected from Ad.Myocd-DN (control) or Ad.Myocd transduced, unstimulated or IL-1B stimulated VSMCs. RAW264.7 macrophages were maintained in the conditioned media for 16 hr. The macrophages were then incubated for 3 hours with Dil-labelled Ac-LDL (Dil-Ac-LDL; red). Scale bar, 100 μm. (E) Quantification of Dil-Ac-LDL accumulation in RAW264.7 macrophages. Data are presented as mean±s.e.m., n=3 independent experiments in biological triplicate. *P<0.05, ** P<0.01 and ***P<0.001 Student’s t-test.
Myocardin overexpression reduces neointimal response and macrophage accumulation in vivo
Transluminal wire injury is an established procedure to induce endothelial denudation and vascular injury response in mouse carotid arteries. Performing transluminal wire injury in atherosclerosis-prone ApoE−/−, hypercholesterolemic mice50 is further known to induce the formation of a neointima that shares numerous characteristic features with developing atherosclerotic lesions, including significant macrophage infiltration and even the appearance of cholesterol crystals. This model was therefore adopted as a means to study the functional effects of increased myocardin expression in VSMCs, during a vascular disease-like situation, in vivo. Wire injury of left common carotids was performed and Ad.LacZ control or Ad.Myocd adenoviruses were applied intra-luminally. Efficient transduction and elevated expression of Myocd and SMC marker genes following Ad.Myocd administration is documented in our recent report,25 since to reduce animal usage, both studies utilised sections from the same mice. Ad.LacZ permeation and subsequent beta-galactosidase expression throughout the vessel media was demonstrated and is reproduced here in Figure IB of the online-only Data Supplement. We hypothesised here that administration of Ad.Myocd would reduce macrophage accumulation at the site of carotid injury. Formation of a Lamp2-positive macrophage-rich neointima was evident in both groups, but appeared attenuated in arteries administered with Ad.Myocd. Manual quantification of Lamp2-positive cells in each section confirmed that application of Ad.Myocd to VSMCs at the site of injury had significantly suppressed macrophage accumulation at the site of vascular injury by two-fold (P<0.05; Figure 6A and 6B). This result is consistent with the previous in vitro data. These studies therefore provide strong in vivo evidence to support the conclusion that increased expression of myocardin in VSMC is functionally protective against the onset of inflammatory vascular disease.
Figure 6. Myocardin inhibits accumulation of macrophages in the neointima.

(A) Hematoxylin and eosin (H&E) and confocal images of carotid arteries from mice transduced with adenoviral vectors expressing beta-galactosidase (Ad.LacZ) control or Ad.Myocd. Carotid sections were labelled with antibodies directed against smooth muscle α-actin (ACTA2; green) and lysosome-associated membrane protein 2 (LAMP2; red). 4′,6-diamidino-2-phenylindole (DAPI; blue) counterstaining indicates nuclei. Scale bar, 100 μm. (B) Quantification of macrophages (cells positive for LAMP2) in the neointimal layers. Data are presented as mean±s.e.m. *P<0.05 Student’s t-test. Ad.LacZ (n=12) and Ad.Myocd (n=13). (C) Schematic illustrating a proposed model whereby reduced myocardin expression in contractile medial vascular smooth muscle cells (VSMCs) is a necessary permissive step in VSMC inflammatory activation following exposure to atherogenic stimuli, leading to increased leukocyte infiltration and progression of atherosclerosis. Dashed arrow represents VSMC-independent routes of disease progression.
Discussion
Atherosclerosis is a multi-factorial, chronic inflammatory disease of the vasculature. The development of this condition involves a complex interplay between cells resident in the vascular wall, and infiltrating cells of the innate and adaptive immune system. Contractile, quiescent VSMCs can undergo phenotypic transitions and become proliferative, migratory, and secrete extracellular matrix. VSMCs may additionally undergo a process of inflammatory activation, wherein cells release a diverse range of chemokines and cytokines, take up oxidised lipids, and interact with infiltrating immune cells to help propagate inflammation and disease. An expanding body of literature continues to report correlative associations between pathological VSMC phenotypic modulation, vascular injury or vascular disease progression, and reduced expression of myocardin.17,18,19,20,21 However, no study to date has addressed the question of whether reduced myocardin expression in VSMC is necessary or indeed a potentiating factor in the progression of these clinically important phenomena. We recently identified myocardin as a critical regulator of the arterial response to wire injury.25 We now show for the first time that myocardin is a central negative regulator of VSMC inflammatory activation and pathological vascular disease. Myocardin haploinsufficiency promoted accelerated inflammation and atherosclerosis in hypercholesterolemic mice, compared to myocardin-normal controls. VSMCs cultured from myocardin-heterozygous-null mice retained a phenotype of reduced myocardin levels and increased inflammatory activation potential compared to wild-type controls. Furthermore, knockdown of myocardin expression reproduced the effect of increased inflammatory activation potential in cultured human coronary artery smooth muscle cells. Conversely, raised expression of myocardin in VSMCs potently but specifically suppressed the induction of an array of inflammatory cytokines, chemokines and adhesion molecules, while upregulating one or more anti-inflammatory mediators. Functionally, expression of myocardin in VSMCs reduced oxidised lipid uptake, leukocyte chemotaxis, leukocyte-VSMC interaction, leukocyte-endothelial tethering and leukocyte lipid uptake in vitro, and attenuated macrophage accumulation within a developing murine lesion in vivo.
Myocardin-null mice die during early development,13 and thus cannot be used as a loss-of-function model in atherosclerosis. However, myocardin-heterozygous-null mice are viable, and we recently reported a phenotype of reduced aortic myocardin expression, and augmented neointima formation in response to injury with increased SMC migration and proliferation.25 Our finding that myocardin-heterozygous-null mice exhibit accelerated atherogenesis is consistent with the hypothesis that myocardin depletion is a causal factor in the pathological VSMC phenotypic transition. Together these studies present a compelling demonstration that endogenous levels of myocardin represent a critical regulator of both the vessel injury response, and vascular disease. This is corroborated by the in vitro observation that VSMCs cultured from myocardin-heterozygous-null aortas retained reduced myocardin levels and an increased propensity for inflammatory activation, compared to myocardin wild-type controls. Similarly increased inflammatory activation in human coronary artery smooth muscle cells following siRNA-mediated depletion of myocardin further supports these findings and the relevance of myocardin in regulating VSMC inflammatory activation and vascular disease in humans.
Myocardin potently and specifically reduced the production of a diverse range of pro-inflammatory cytokines, chemokines and adhesion molecules by cultured VSMCs in response to IL-1β stimulation. Myocardin strongly inhibited the expression of the intracellular pro-inflammatory pathway mediators CEBPB and CEBPD, which are known to synergistically upregulate and sustain inflammatory gene expression following SMC stimulation.36,37,38 Importantly, suppression of these mediators was able to partially reverse the increased inflammatory activation in myocardin-deficient VSMCs, suggesting that suppression of CEBP factors is a mechanism through which myocardin regulates inflammatory gene expression. That this reversal was only partial, however, implies the existence of additional levels of myocardin-mediated inflammatory regulation, possibly involving the upregulation of anti-inflammatory mediators IL-19, SOCS-2 and SOCS-6, which provide interesting avenues for future research.40,41,42 Of interesting note, the specific colony stimulating factor (CSF3) found in our study to be upregulated by myocardin in VSMC has been recently linked to decreased plaque area, lipid accumulation and macrophage infiltration into atherosclerotic lesions of ApoE−/− mice. 51
A caveat of these experiments is the difficulty of adequately reducing a complex process such as atherosclerosis to a basic in vitro model. Much current research analyses cellular responses to a single stimulus in culture, whereas cells in vivo are exposed to multiple stimuli simultaneously and elicit integrated responses accordingly. Crosstalk between individual stimuli can significantly affect the SMC inflammatory phenotype.7 Compounding these complexities are species differences, and the fact that cells dispersed from organs rarely behave as in vivo. In an effort to address some of these issues, the present study demonstrates reproducible suppression of a diverse array of VSMC inflammatory responses by myocardin, after IL-1β stimulation and presents consistent results using cells isolated from rat, mouse and human sources.
Basic models of VSMC/macrophage and VSMC/EC/macrophage cellular interaction were employed to simulate events that occur in a developing lesion, and to assess how increased myocardin expression in VSMCs might influence such interactions. Myocardin expression in VSMCs was shown to suppress oxidised lipid uptake, macrophage chemotaxis, endothelial tethering, VSMC-macrophage interaction and macrophage lipid uptake in vitro. Testing of these effects in vivo was performed using a mouse model of lesion-inducing vascular injury.50 Indeed, adenoviral-mediated expression of myocardin in VSMCs significantly reduced macrophage accumulation in developing lesions. Together, these data provide compelling evidence that myocardin is a central regulator of vascular disease, and furthermore, that strategies to prevent or reverse the loss of myocardin expression in VSMCs illustrated in schematic Figure 6C, could protect against inflammation, disease onset and progression. On the strength of presented and previous evidence,25 we propose myocardin as a guardian of the quiescent, differentiated, contractile and non-inflammatory VSMC phenotype, with loss of myocardin representing a critical permissive step in allowing the process of VSMC phenotypic modulation and inflammatory activation to occur at the onset of vascular disease. Methods to maintain or increase myocardin expression may protect aortic and coronary artery VSMCs from entering dysfunctional programs of synthetic and inflammatory activation. 70% of clinical events in patients with coronary artery disease still cannot be prevented with modern drug therapy, including statins, emphasising an urgent need for supplementary treatment strategies.52 Ultimately, myocardin may represent an attractive therapeutic target with multiple anti-proliferative, anti-migratory and potent, specific anti-inflammatory properties. A future challenge may be to target such therapies specifically to aortic and coronary VSMCs, since studies report deleterious effects of increased myocardin expression in cerebral VSMCs53 and cardiomyocytes54. However, successfully tailored myocardin-activating therapies could overcome problems of individual inflammatory pathway redundancy, yet maintain a high degree of VSMC-specificity, thereby reducing off-target, undesirable side effects on the immune system as a whole. Further research into the nature of VSMC inflammation and the specific mechanisms by which myocardin regulates this process will aid in this endeavour, and await exploration.
Supplementary Material
Significance.
Cardiovascular diseases are the cause of substantial global mortality and morbidity, and have a significant inflammatory basis. Vascular wall smooth muscle cells contribute to disease by dividing, migrating into the intimal plaque region, releasing pro-inflammatory mediators, and interacting with infiltrating leukocytes. A substantial degree of clinical events in patients with vascular disease remain non-preventable with modern drug therapy. We recently identified myocardin, a powerful myogenic transcriptional coactivator, as an inhibitor of vascular smooth muscle migration after vessel injury. We now demonstrate that myocardin negatively regulates vascular smooth muscle inflammation and associated vascular disease, thereby revealing the potential of myocardin as an attractive, novel therapeutic target that elicits wide-ranging suppression of multiple inflammatory mediators, in addition to anti-proliferative and anti-migratory effects. We therefore anticipate further important research into the nature of vascular smooth muscle cell inflammation in vascular disease, and the specific mechanisms by which myocardin regulates this process.
Acknowledgments
Sources of funding
This work was supported by Wellcome Trust funding for MAJ (Studentship 086799/Z/08/Z), British Heart Foundation grants (PG/10/007/28184) for AT, and (RG/08/009/25841) for MRB, and SS (FS/13/29/30024), the Cambridge NIHR Biomedical Research Centre and the NIH for JM (NIH HL-117907).
Nonstandard Abbreviations and Acronyms
- Ad
adenoviral
- CCL2
chemokine monocyte chemotactic protein-1
- CEBPB
CCAAT/enhancer-binding protein (C/Ebp) transcription factor beta
- CEBPD
CCAAT/enhancer-binding protein (C/Ebp) transcription factor delta
- EC
endothelial cell
- HCASMC
human coronary artery smooth muscle cell
- LacZ
beta-galactosidase
- Lamp2
lysosomal-associated membrane protein 2
- Myocd
myocardin
- Myocd-DN
myocardin dominant-negative
- oxLDL
oxidised low-density lipoprotein
- RASMC
rat aortic vascular smooth muscle cell
- SOCS
suppressor of cytokine signalling
- (V)SMCs
(vascular) smooth muscle cells
- WT
wild-type
Footnotes
Author contributions: MAJ, AT and SS conceived the study, designed the experiments and prepared the manuscript; MAJ, AT, AS, XL and IB performed the experiments and analysed the data; MRB, NWM and JM provided critical analysis; SS supervised the project and all authors edited the manuscript. We thank Nichola Figg for processing and sectioning the blood vessels and James Harrison, Lauren Baker and Dr Laure Gambardella for skilled technical assistance. The authors would like to thank Prof. Gary Owens for the adenovirus vectors – LacZ, FLAG-tagged myocardin and myocardin dominant negative and Prof. Eric Olson for the myocardin heterozygous-null mice.
Disclosure
None
References
- 1.Dahlöf B. Cardiovascular disease risk factors: epidemiology and risk assessment. Am J Cardiol. 2010;105:3A–9A. doi: 10.1016/j.amjcard.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Celermajer DS, Chow CK, Marijon E, Anstey NM, Woo KS. Cardiovascular disease in the developing world: prevalences, patterns, and the potential of early disease detection. J Am Coll Cardiol. 2012;60:1207–1216. doi: 10.1016/j.jacc.2012.03.074. [DOI] [PubMed] [Google Scholar]
- 3.Cuneo AA, Autieri MV. Expression and function of anti-inflammatory interleukins: the other side of the vascular response to injury. Curr Vasc Pharmacol. 2009;7:267–276. doi: 10.2174/157016109788340721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 6.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2004;47:168–180. doi: 10.1159/000250095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 10.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–2437. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S, Wang DZ, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2003;100:9366–9370. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pipes GC, Sinha S, Qi X, Zhu CH, Gallardo TD, Shelton J, Creemers EE, Sutherland L, Richardson JA, Garry DJ, Wright WE, Owens GK, Olson EN. Stem cells and their derivatives can bypass the requirement of myocardin for smooth muscle gene expression. Dev Biol. 2005;288:502–513. doi: 10.1016/j.ydbio.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Hoofnagle MH, Neppl RL, Berzin EL, Teg Pipes GC, Olson EN, Wamhoff BW, Somlyo AV, Owens GK. Myocardin is differentially required for the development of smooth muscle cells and cardiomyocytes. Am J Physiol Heart Circ Physiol. 2011;300:H1707–H1721. doi: 10.1152/ajpheart.01192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Cheng L, Li J, Chen M, Zhou D, Lu MM, Proweller A, Epstein JA, Parmacek MS. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest. 2008;118:515–525. doi: 10.1172/JCI33304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000;106:1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendrix JA, Wamhoff BR, McDonald OG, Sinha S, Yoshida T, Owens GK. 5′ CArG degeneracy in smooth muscle alpha-actin is required for injury-induced gene suppression in vivo. J Clin Invest. 2005;115:418–427. doi: 10.1172/JCI22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tharp DL, Wamhoff BR, Turk JR, Bowles DK. Upregulation of intermediate-conductance Ca2+-activated K+ channel (IKCa1) mediates phenotypic modulation of coronary smooth muscle. Am J Physiol Heart Circ Physiol. 2006;291:H2493–H2503. doi: 10.1152/ajpheart.01254.2005. [DOI] [PubMed] [Google Scholar]
- 20.Minami T, Kuwahara K, Nakagawa Y, et al. Reciprocal expression of MRTF-A and myocardin is crucial for pathological vascular remodelling in mice. EMBO J. 2012;31:4428–4440. doi: 10.1038/emboj.2012.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starke RM, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez F, Hasan DM, Rosenwasser RH, Owens GK, Koch WJ, Dumont AS. Cigarette smoke modulates vascular smooth muscle phenotype: implications for carotid and cerebrovascular disease. PLoS One. 2013;8:e71954. doi: 10.1371/journal.pone.0071954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Veer E, de Bruin RG, Kraaijeveld A, et al. The RNA-Binding Protein Quaking is a Critical Regulator of Vascular Smooth Muscle Cell Phenotype. Circ Res. 2013;113:1065–1075. doi: 10.1161/CIRCRESAHA.113.301302. [DOI] [PubMed] [Google Scholar]
- 23.Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N, Steer BM, Ingram AJ, Gupta M, Al-Omran M, Teoh H, Marsden PA, Verma S. MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation. 2012;126:S81–90. doi: 10.1161/CIRCULATIONAHA.111.084186. [DOI] [PubMed] [Google Scholar]
- 24.Tang RH, Zheng XL, Callis TE, Stansfield WE, He J, Baldwin AS, Wang DZ, Selzman CH. Myocardin inhibits cellular proliferation by inhibiting NF-kappaB(p65)-dependent cell cycle progression. Proc Natl Acad Sci U S A. 2008;105:3362–3367. doi: 10.1073/pnas.0705842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talasila A, Yu H, Ackers-Johnson M, Bot M, van Berkel T, Bennett MR, Bot I, Sinha S. Myocardin Regulates Vascular Response to Injury Through miR-24/-29a and Platelet-Derived Growth Factor Receptor-β. Arterioscler Thromb Vasc Biol. 2013;33:2355–2365. doi: 10.1161/ATVBAHA.112.301000. [DOI] [PubMed] [Google Scholar]
- 26.von der Thüsen JH, van Berkel TJ, Biessen EA. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation. 2001;103:1164–1170. doi: 10.1161/01.cir.103.8.1164. [DOI] [PubMed] [Google Scholar]
- 27.Bot M, Bot I, Lopez-Vales R, van de Lest CH, Saulnier-Blache JS, Helms JB, David S, van Berkel TJ, Biessen EA. Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am J Pathol. 2010;176:3073–3084. doi: 10.2353/ajpath.2010.090009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 29.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuett H, Oestreich R, Waetzig GH, et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:281–290. doi: 10.1161/ATVBAHA.111.229435. [DOI] [PubMed] [Google Scholar]
- 32.Alexander MR, Murgai M, Moehle CW, Owens GK. Interleukin-1β modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-κB-dependent mechanisms. Physiol Genomics. 2012;44:417–429. doi: 10.1152/physiolgenomics.00160.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feinberg MW, Watanabe M, Lebedeva MA, Depina AS, Hanai J, Mammoto T, Frederick JP, Wang XF, Sukhatme VP, Jain MK. Transforming growth factor-beta1 inhibition of vascular smooth muscle cell activation is mediated via Smad3. J Biol Chem. 2004;279:16388–16393. doi: 10.1074/jbc.M309664200. [DOI] [PubMed] [Google Scholar]
- 34.Niu J, Kolattukudy PE. Role of MCP-1 in cardiovascular disease: molecular mechanisms and clinical implications. Clin Sci (Lond) 2009;117:95–109. doi: 10.1042/CS20080581. [DOI] [PubMed] [Google Scholar]
- 35.Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, Giesen PL, Nemerson Y, Taubman MB. Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J Biol Chem. 1997;272:28568–28573. doi: 10.1074/jbc.272.45.28568. [DOI] [PubMed] [Google Scholar]
- 36.Poli V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- 37.Baccam M, Woo SY, Vinson C, Bishop GA. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: involvement of NF-kappa B, AP-1, and C/EBP. J Immunol. 2003;170:3099–3108. doi: 10.4049/jimmunol.170.6.3099. [DOI] [PubMed] [Google Scholar]
- 38.Sato Y, Nishio Y, Sekine O, Kodama K, Nagai Y, Nakamura T, Maegawa H, Kashiwagi A. Increased expression of CCAAT/enhancer binding protein-beta and -delta and monocyte chemoattractant protein-1 genes in aortas from hyperinsulinaemic rats. Diabetologia. 2007;50:481–489. doi: 10.1007/s00125-006-0480-4. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida T, Sinha S, Dandré F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–864. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 40.Gallagher G, Eskdale J, Jordan W, Peat J, Campbell J, Boniotto M, Lennon GP, Dickensheets H, Donnelly RP. Human interleukin-19 and its receptor: a potential role in the induction of Th2 responses. Int Immunopharmacol. 2004;4:615–626. doi: 10.1016/j.intimp.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 41.Cuneo AA, Herrick D, Autieri MV. Il-19 reduces VSMC activation by regulation of mRNA regulatory factor HuR and reduction of mRNA stability. J Mol Cell Cardiol. 2010;49:647–654. doi: 10.1016/j.yjmcc.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kranzhöfer R, Maraganore JM, Baciu R, Libby P. Systemic thrombin inhibition by Hirulog does not alter medial smooth muscle cell proliferation and inflammatory activation after vascular injury in the rabbit. Cardiovasc Drugs Ther. 1999;13:429–434. doi: 10.1023/a:1007808123953. [DOI] [PubMed] [Google Scholar]
- 44.Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 45.Aoyama T, Chen M, Fujiwara H, Masaki T, Sawamura T. LOX-1 mediates lysophosphatidylcholine-induced oxidized LDL uptake in smooth muscle cells. FEBS Lett. 2000;467:217–220. doi: 10.1016/s0014-5793(00)01154-6. [DOI] [PubMed] [Google Scholar]
- 46.Hastings NE, Feaver RE, Lee MY, Wamhoff BR, Blackman BR. Human IL-8 regulates smooth muscle cell VCAM-1 expression in response to endothelial cells exposed to atheroprone flow. Arterioscler Thromb Vasc Biol. 2009;29:725–731. doi: 10.1161/ATVBAHA.109.184382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spagnoli LG, Bonanno E, Sangiorgi G, Mauriello A. Role of inflammation in atherosclerosis. J Nucl Med. 2007;48:1800–1815. doi: 10.2967/jnumed.107.038661. [DOI] [PubMed] [Google Scholar]
- 48.Chen L, Frister A, Wang S, Ludwig A, Behr H, Pippig S, Li B, Simm A, Hofmann B, Pilowski C, Koch S, Buerke M, Rose-John S, Werdan K, Loppnow H. Interaction of vascular smooth muscle cells and monocytes by soluble factors synergistically enhances interleukin-6 and MCP-1 production. Am J Physiol Heart Circ Physiol. 2009;296:H987–H996. doi: 10.1152/ajpheart.01158.2008. [DOI] [PubMed] [Google Scholar]
- 49.Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18:228–232. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krohn R, Raffetseder U, Bot I, Zernecke A, Shagdarsuren E, Liehn EA, van Santbrink PJ, Nelson PJ, Biessen EA, Mertens PR, Weber C. Y-box binding protein-1 controls CC chemokine ligand-5 (CCL5) expression in smooth muscle cells and contributes to neointima formation in atherosclerosis-prone mice. Circulation. 2007;116:1812–1820. doi: 10.1161/CIRCULATIONAHA.107.708016. [DOI] [PubMed] [Google Scholar]
- 51.Sinha SK, Mishra V, Nagwani S, Rajavashisth TB. Effects of G-CSF on serum cholesterol and development of atherosclerotic plaque in apolipoprotein E-deficient mice. Int J Clin Exp Med. 2014;7:1979–1989. [PMC free article] [PubMed] [Google Scholar]
- 52.Klingenberg R, Hansson GK. Treating inflammation in atherosclerotic cardiovascular disease: emerging therapies. Eur Heart J. 2009;30:2838–2844. doi: 10.1093/eurheartj/ehp477. [DOI] [PubMed] [Google Scholar]
- 53.Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, Streb JW, Guo H, Rubio A, Van Nostrand W, Miano JM, Zlokovic BV. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xing W, Zhang TC, Cao D, Wang Z, Antos CL, Li S, Wang Y, Olson EN, Wang DZ. Myocardin induces cardiomyocyte hypertrophy. Circ Res. 2006;98:1089–1097. doi: 10.1161/01.RES.0000218781.23144.3e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.