

Abstract
Sufficient light is essential for the growth and physiological functions of photosynthetic organisms, but prolonged exposure to high light (HL) stress can cause cellular damage and ultimately result in the death of these organisms. Synechococcus sp. PCC 7002 (hereafter Synechococcus 7002) is a unicellular cyanobacterium with exceptional tolerance to HL intensities. However, the molecular mechanisms involved in HL response by Synechococcus 7002 are not well understood. Here, an integrated RNA sequencing transcriptomic and quantitative proteomic analysis was performed to investigate the cellular response to HL in Synechococcus 7002. A total of 526 transcripts and 233 proteins were identified to be differentially regulated under HL stress. Data analysis revealed major changes in mRNAs and proteins involved in the photosynthesis pathways, resistance to light-induced damage, DNA replication and repair, and energy metabolism. A set of differentially expressed mRNAs and proteins were validated by quantitative RT-PCR and Western blot, respectively. Twelve genes differentially regulated under HL stress were selected for knockout generation and growth analysis of these mutants led to the identification of key genes involved in the response of HL in Synechococcus 7002. Taken altogether, this study established a model for global response mechanisms to HL in Synechococcus 7002 and may be valuable for further studies addressing HL resistance in photosynthetic organisms.
Cyanobacteria are a large group of prokaryotes that have photosystem II (PS II)1, photosystem I (PS I), and carry out oxygenic photosynthesis. They play important roles in global carbon and nitrogen cycles. Using solar energy, cyanobacteria generate the reducing equivalents for CO2 fixation and synthesis of carbohydrates and other metabolite building blocks. In addition to reducing equivalents, the photosynthetic apparatus generates a proton gradient across the thylakoid membranes for ATP synthesis.
Light is a constantly changing environmental factor and cyanobacteria must have the ability to acclimate to changing light conditions. The acclimation can be divided into short-term and long-term processes (1, 2). Short-term acclimation includes state transitions, protective energy dissipation (3, 4), changes in the energy transfer efficiency from the harvesting complex to PS II (5), and the formation of nonfunctional PS II reaction centers (6, 7). These responses occur rapidly and are usually completed within several minutes. The long-term acclimation is much slower and it may take up to several days to complete the processes that involve changes in the composition, structure, and function of the photosynthetic apparatus as well as other photosynthesis-related components. The process occurs at different stages of gene expression, including mRNA synthesis (transcription); protein biosynthesis (translation); and post-translational modification.
Studies have shown that there is an upper limit of light intensity beyond which a cyanobacterium absorbs more energy than its energy consumption and dissipation, leading to photoinhibition and photodamage (8). The light intensity that causes photoinhibition varies in different cyanobacterial strains. Synechococcus sp. strain PCC 7002 (hereafter Synechococcus 7002), a unicellular, euryhaline cyanobacterium, is the one that can grow in the highest light intensity among the cyanobacteria tested and/or reported (9–11). Although most of the cyanobacteria cannot tolerate a light intensity of 1000 μmol photons/m2/s, Synechococcus 7002 can grow rapidly at a light intensity of 2000 μmol photons/m2/s, making it a very nice organism to study the mechanisms of acclimation to high-light (HL). Synechococcus 7002 has a completely sequenced genome (http://www.ncbi.nlm.nih.gov/) and can be easily genetically transformed (12) with a versatile system (13). It has been used for studies on various aspects of photosynthetic electron transport as well as CO2 fixation and reduction, it is also a model organism for studies in biofuel development (14, 15).
Recently, physiological responses to changing light intensity have been studied (1, 16, 17), and some of the relevant molecular mechanisms have been described (2, 4, 18). However, the mechanism of HL acclimation is still not well understood. To analyze the molecular components and regulatory mechanisms of HL-acclimation networks, especially for genes/proteins involved in long-term acclimation, we studied transcription and translation of Synechococcus 7002 in response to HL conditions with methods coupling RNA sequencing (RNA-Seq) and mass spectrometry. With the Next-Generation Sequencing (NGS) technology, RNA-Seq has become a powerful tool for transcriptomic profiling (19, 20). Tandem mass tags (TMT)-based quantitative proteomics has been widely used and proven to be a reliable method for determining protein expression levels (21–23). Here, we demonstrated that an integrated study coupling NGS-based RNA-Seq transcriptomics and quantitative TMT-LC-MS/MS proteomics has gained a system level understanding of the functional components involved in acclimation to HL. A large number of genes were found to be responsive to HL as demonstrated at levels of both transcripts and proteins. Further gene knockout and comparative growth analysis revealed several important molecular components of the long-term HL acclimation network in Synechococcus 7002. To the best of our knowledge, this work represents the first combined functional transcriptomic and proteomic analysis of HL response mechanisms in cyanobacteria.
EXPERIMENTAL PROCEDURES
Sample Preparation
Synechococcus 7002 cells were grown in culture tubes (Φ40 mm × 200 mm) containing medium A supplemented with 1 mg/ml NaNO3 as nitrogen source (15, 24). The cultures were grown at 38 °C at 250 μmol photons m−2 s−1 and were bubbled with 1% (v/v) CO2 in air. Cell density was measured on a UV-1750 spectrophotometer (Shimadzu). Cells were grown to OD730 nm = 0.7 and were inoculated at an OD730 nm = 0.05. Then the cultures were continuously illuminated at 2000 μmol photons/m2/s for HL treatment, or at 50 μmol photons/m2/s (normal light, NL) as the control. The cultures were grown at 38 °C and were bubbled with 1% (v/v) CO2 in air. Illumination (50 μmol photons/m2/s or 2000 μmol photons/m2/s) was provided by a halogen floodlight and light intensities were measured using a LI-250A light meter (Li-COR, Lincoln, NE). The growth temperature was maintained by using a refrigerated water circulator. One milliliter of culture samples were collected and OD was measured by spectrophotometer every 8 h. For both conditions, two biological replicates were performed independently. Growth experiments were repeated at least three times to confirm the growth patterns. Cells were harvested at the early/mid-exponential phase when OD730 nm = 2.5 by centrifugation at 6600 × g for 10 minutes (min) at 4 °C for RNA-Seq transcriptomic analysis and proteomic analysis, respectively (Fig. 1). For both NL and HL-grown cells, the samples were aliquoted for protein and RNA extraction. A cell suspension from an exponential-phase culture grown at 38 °C in medium A+ at a light intensity of 50 μmol photons/m2/s or 2000 μmol photons/m2/s with OD730 = 1.0 contained (0.88 ± 0.1) × 108 cells or (1.13 ± 0.1) × 108 cells in 1 ml as determined by microscopic count.
Fig. 1.
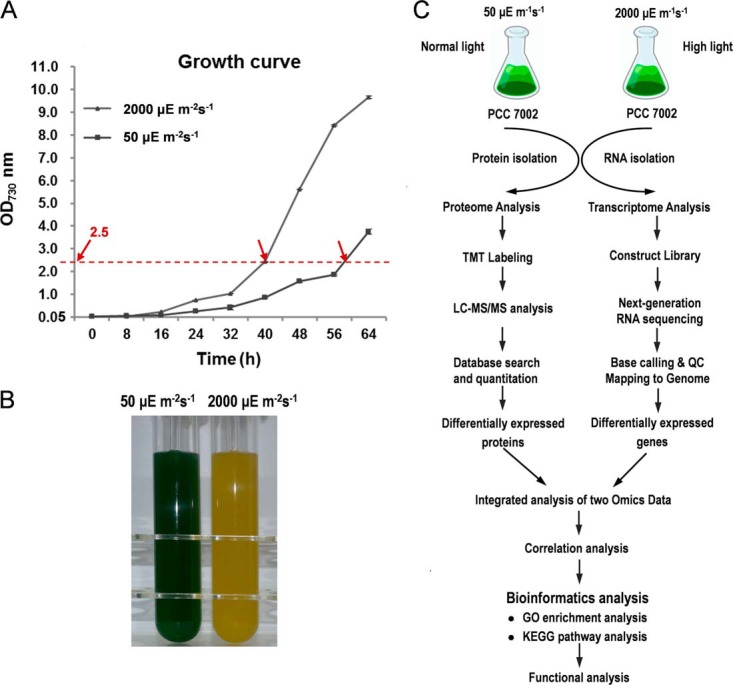
Sampling and scheme for this study. A, Growth curves of Synechococcus 7002 under 50 μEm−2s−1 (NL conditions) and 2000 μEm−2s−1 (HL conditions). Each point represents the mean ± standard deviation (S.D.) of three biological replicates. Arrows indicate time points where samples for RNA-Seq analysis and quantitative proteomics were collected. B, Cultures of Synechococcus 7002 under NL and HL conditions. (C) The overall workflow of this study.
RNA Extraction and Illumina Sequencing
Total RNA was extracted with Trizol reagent (Invitrogen, Gaithersburg, MD) according to the manufacturer's protocols. Briefly, liquid nitrogen grinding was used for cell-disruption and Trizol reagent was added to the grinding powder (100 mg/ml). After centrifugation at 10,000 × g, 4 °C for 10 min to remove the cell debris, phenol/chloroform was applied for the extraction of RNA and isopropyl alcohol for precipitation of RNA at room temperature for 15 min. Then the RNA was washed with 75% ethanol to remove organic pollution and the air-dried RNA was suspended in DEPC-treated H2O. RNase-free DNase I (Fermentas, Hanover, MD) was used to remove the remaining genomic DNA. RNA integrity was examined on a 1% agarose gel and RNA concentration was measured on a Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA). rRNA was subsequently eliminated with RiboMinusTM Transcriptome Isolation Kits for Bacteria (Invitrogen) and Magnetic stand (Invitrogen, Gaithersburg, MD) with some modifications according to the manufacturer's manual. The cDNA libraries were constructed from 0.5 μg RNA sample using a TruSeq Stranded total RNA sample preparation kit (Illumina, San Diego, CA) following the guidelines of the manufacturers. After sequencing libraries were denatured with sodium hydroxide and diluted to 14 pmol/L with hybridization buffer (Illumina), sequencing was performed on an Illumina GAIIx platform at SinoGenoMax Co., Ltd (Beijing, China). A paired-end sequencing strategy was used and the sequencing length was 81 bp. Two biological replicates were sequenced for each condition.
Data Analysis
Quality control was carried out on the Illumina GA reads with a perl script (threshold of Q20) to filter the low quality paired-end reads. The left high quality reads of each replicate obtained from the two conditions were mapped to the complete genome sequence of Synechococcus 7002 individually with a Burrows-Wheeler Aligner (BWA) (25) allowing 3 mismatches on a read. Reads that were not mapped to the reference genome and that were mapped to rRNA-coding regions were eliminated from the alignment results, and ambiguously mapped reads (i.e. those with more than one potential match in the genome) were also removed. As previously described (26, 27), the unambiguously mapped reads were used to compile a coverage profile for each sample which reflects the depth of sequence data at each position in the Synechococcus 7002 genome. For comparative purposes, coverage profiles were normalized based on the total number of unambiguously mapped reads across the genome for each sample. The expression level for a given gene in each sample was measured as the mean coverage depth for all nucleotides in that gene. Genes with coverage <2 were removed.
Protein Extraction and Digestion
The cells were washed twice with PBS buffer, resuspended in lysis buffer containing 20 mm Tris-Cl (pH 7.5), 150 mm NaCl, 1% Triton X-100, 1× protease inhibitor mixture and 1× phosphatase inhibitor mixture (Thermo Fisher Scientific). Samples were sonicated at 135 W for 30 min on ice using an ultrasonic processor (JY92-IIN, Ningbo Scientz Biotechnology Co., Ltd, Ningbo, China). Cellular debris was removed by centrifugation at 12,000 × g for 30 min at 4 °C, and the resulting supernatants were stored in aliquots at −80 °C until further use. Protein concentration was determined using the 2D Quant kit (GE Healthcare Waukesha, WI) according to the manufacturer's protocol. The samples were reduced with 5 mm DTT (Sigma, St. Louis, MO) at 56 °C for 30 min and alkylated with 15 mm iodoacetamide (IAA) (Sigma) for 30 min at room temperature. Samples were then digested overnight at 37 °C with sequencing grade modified trypsin (1:50 w/w) (Promega, Madison, WI).
TMT Labeling
After trypsin digestion, peptides were desalted with a Strata X-C18 SPE column (Phenomenex, Torrance, CA) and vacuum-dried. Peptides from two independent samples were reconstituted in 0.5 m TEAB and processed according to the manufacturer's protocol for the 6-plex TMT kit (the labeling reagent of 128, 129, 130, and 131 was used for 4-plex labeling). Briefly, ten units of TMT reagent (defined as the amount of reagent required to label 1.5 mg of protein) were thawed and reconstituted in 40 μl ACN. The peptide mixtures were then pooled and incubated for 2 h at room temperature, desalted, dried by vacuum centrifugation, and reconstituted in 10% formic acid (FA).
Strong Cation Exchange Fractionation
Peptides were fractionated using strong-cation exchange as described previously (28). In brief, strong-cation exchange was performed using a Zorbax BioSCX-Series II column (0.8 mm×50 mm, 3.5 μm). Solvent A consisted of 0.05% FA in 20% ACN, solvent B consisted of 0.05% formic acid, 0.5 m NaCl in 20% ACN. The following gradient was used: 0–0.01 min (0–2% B); 0.01–8.01 min (2–3% B); 8.01–14.01 min (3–8% B); 14.01–28 min (8–20% B); 28–38 min (20–40% B); 38–48 min (40–90% B); 48–54 min (90% B); 54–60 min (0% B). A total of 14 fractions were collected and dried in vacuo.
LC-MS/MS Analysis
Peptides were dissolved in 0.1% FA, directly loaded onto a reversed-phase column (360 μm O.D. × 75 μm I.D.) packed in-house with 3-μm Reprosil-Pur C18-AQ resin (Dr. Maisch, Ammerbuch, Germany) and eluted with a linear gradient of 5–20% solvent B (0.1% FA in 98% ACN) for 50 min and 20–35% solvent B for 10 min at a constant flow rate of 300 nl/min on an EASY-nLC 1000 UPLC system. The separated peptides were analyzed with a Q ExactiveTM Plus hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific). Intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using 27% normalized collision energy (NCE) with 12% stepped NCE; ion fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 3 × 104 in the MS survey scan with 15.0 s dynamic exclusion. The electrospray voltage applied was 1.8 kV. Automatic gain control (AGC) was used to prevent overfilling of the ion trap; 1 × 105 ions were accumulated for generation of MS/MS spectra. For MS scans, the m/z scan range was 350 to 1600 Da. The fixed first mass was set to 100 m/z for TMT quantification.
Database Search
Raw data files were processed to generate peak list files using Proteome Discoverer software (Thermo Fisher Scientific, v. 1.3.0.339). The filtering parameters used were as follows: (1) Allowed precursor mass range was 350 Da to 5000 Da, (2) Precursor charge state was allowed from 1 to 5, (3) Signal to noise ratio was set as 1.5, (4) For precursors with unrecognized charge state, default charge states of 2 and 3 were allowed. The resulting MS/MS data were processed using MaxQuant software (v.1.4.1.2) with an integrated Andromeda search engine (29, 30). The protein database used for MS/MS searches was downloaded from Cyanobase (http://genome.kazusa.or.jp/cyanobase, 3,186 CDSs, released 2012) for Synechococcus 7002. Trypsin/P was specified as the cleavage enzyme allowing up to 2 missed cleavages. The precursor charge states allowed were from 1 to 5. Mass error was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Carbamidomethylation (C), TMT6plex (K) and TMT6plex (N-term) were set as fixed modifications. Oxidation of methionine (M) was set as a variable modification. Detection of at least two matching peptides per protein was set as a requirement for unambiguous identification. The TMT datasets were quantified using the centroid peak intensity with the ‘reporter ions quantifier’ mode. For all experiments, only unique peptides were considered for protein quantification. The peptide false discovery rate (FDR) was set to 1% and minimum peptide score was set to 13.0. The minimum peptide length was set at 7. All the other parameters in MaxQuant were set to default values.
Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
RNA was reverse transcribed into first-strand cDNA with the high-capacity cDNA reverse transcription kit with RNase inhibitor (Invitrogen). Gene transcription was measured using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) and the LightCycler 480 Real-Time PCR System (Roche Diagnostics Ltd, Mannheim, Germany). The 16S or 23S rRNA gene was used as the endogenous control gene for normalizing expression of the target gene. Triplicate technical replicates were performed for duplicate cultures. ΔCT values were obtained by subtracting the average values of experimental genes from an average of the control gene for each sample. Using a Welch approximation for unequal group variances, a p value was estimated based on the t-distribution that resulted from a between-subjects t test evaluating the control RNA relative to a given experimental RNA. Primers used for qRT-PCR are shown in supplemental Table S1.
Production of Polyclonal Antibodies
Anti-PsaC (PS I subunit VII), CpcG (phycobilisome rod-core linker polypeptide cpcG (l-RC 28.5)), RbcL (ribulose bisphosphate carboxylase large subunit), PsaD (PS I subunit II), ApcA (allophycocyanin alpha subunit) or SYNPCC7002_F0063 (hereafter F0063) polyclonal antibodies were produced and purified via affinity chromatography by ABclonal Co. (Wuhan, Hubei, China). Briefly, polyclonal antibodies of PsaC, CpcG, PsaD, ApcA were generated against the following synthetic peptides: PsaC, CKAGQIASSPRTED; CpcG, EQGEIPFNIKSPR; PsaD, VFPSGETQFLYPLDGVPSEKVNEGR; and ApcA, CDRIKAFVGGAARLR. To produce antibodies against RbcL or F0063, the full-length cDNA of rbcL or F0063 was amplified, PCR products were cloned into the pGEX-4T expression vector (Amersham Pharmacia Biotech, Piscataway, NJ) at the BamHI-XhoI restriction sites, and the resulting plasmid was transformed into E. coli strain BL21 (DE3) for overexpression of RbcL. Cells growing logarithmically were treated with 1 mm isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at 30 °C. The fusion proteins were then purified by His-tag affinity chromatography. Following purification of these antigens, immunization and sampling of the anti-sera from rabbit were performed by ABclonal Co. (Wuhan, China), according to standard operating procedures. The specificity of the generated antibodies was determined by the manufacturer using ELISA and Western blotting.
Western blotting
Equal amounts of proteins (10 μg) from both HL and NL grown cells were prepared as described previously, denatured in SDS sample buffer, and separated by 12% SDS-PAGE. Proteins were stained with Coomassie Brilliant Blue R250 or transferred to polyvinylidene fluoride (PVDF) membranes (GE Healthcare). After blocking with 5% nonfat milk, membranes were incubated overnight with PsaC, CpcG, RbcL, PsaD, ApcA and F0063 protein-specific antibodies (1:1000 dilution), followed by a 1 h incubation with a 1:3000 dilution of peroxidase-conjugated anti-rabbit IgG (KPL, Gaithersburg, MD) at room temperature. Chemiluminescence was detected by using the SuperSignal® West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and the gray-scale of Western blots was recorded using ImageQuant TL (GE Healthcare). Immunoblots were performed in three independent experiments and bands of interest analyzed by ImageJ (http://rsb.info.nih.gov/nih-image/) were expressed as mean ± S.D.
Bioinformatics Analysis
Functional enrichment analysis of differentially expressed transcripts and proteins between HL grown cells and NL grown cells was performed to identify significantly overrepresented GO terms and KEGG pathways using DAVID 6.7 (31). The significance of the enrichment was statistically evaluated with a modified Fisher's exact test (EASE score of p value) (31). For GO term enrichment, the GO FAT annotation available in DAVID was used. GO FAT is a subset of the GO term set created by filtering out the broadest ontology terms in order to not overshadow more specific ones. GO terms with p value <0.05 and fold enrichment >1.5 are considered to be significantly enriched.
Construction and Analysis of Gene Knockout Mutants
F0063 was disrupted by replacing an internal EcoRI fragment with a nonpolar cassette conferring clindamycin resistance, and psbU was disrupted by replacing an internal PstI fragment with a nonpolar cassette conferring kanamycin resistance. The gene-deleted mutants for psaC, psaD, psaF, psbC, psbO, apcF, cpcG (l-RC 28.5), SYNPCC7002_A0568, SYNPCC7002_A1479 or SYNPCC7002_A1480 were constructed by replacement of DNA sequence with a kanamycin cassette.
The resultant plasmid was used to transform motile Synechococcus 7002 wild-type cells and one of the antibiotic-resistant transformants was selected for further study. Complete segregation of the mutation was confirmed by PCR and DNA sequencing. PCR primers for mutant construction and validation are listed in supplemental Table S1 in the Supplementary Material. The knockout mutants of 12 selected gene were comparatively grown under 50 μmol photons/m2/s (NL) and 2000 μmol photons/m2/s (HL) for 72 h, respectively.
RESULTS
Overview of Transcriptomic Analysis
RNA-Seq was performed for four samples at the two light intensities in Synechococcus 7002, and produced 34 million (81bp) uniquely non-rRNA reads. The obtained reads represent an average of ∼130 times Synechococcus 7002 genome lengths and transcripts were detected for nearly all the predicted ORFs. After filtering, a total of 7,549,172 and 12,874,548 effective reads were obtained when growing Synechococcus 7002 under HL and NL conditions, respectively. Further analysis showed that 3180 and 3179 out of the whole 3186 genes in the genome were covered under HL stress and NL, respectively (supplemental Table S2). Candidate genes involved in HL adaption were chosen according to the following criteria: (1) more than 2-fold change after normalization, and (2) statistically significant level p < 0.05. Finally, the transcription of 526 genes was detected to be associated with HL response, including 311 up-regulated genes and 215 down-regulated genes (Table S3).
Overview of Quantitative Proteomics Analysis
Protein samples of HL grown and NL grown Synechococcus 7002 cells were subjected to TMT-based proteomic analysis. The overview of the proteomic results such as protein mass distribution, peptide distribution and length of peptides are presented in supplemental Fig. S1. A total of 1746 proteins and 25,229 unique peptides were quantified in our experiment (supplemental Table S4andsupplemental Table S5), representing 54.8% of the 3186 predicted proteins in the Synechococcus 7002 proteome (http://genome.kazusa.or.jp/cyanobase). Using a cutoff of 1.40-fold change and a p value less than 0.05, we determined that 233 proteins were differentially regulated under HL stress. Among these proteins, 128 were up-regulated and 105 were down-regulated upon HL exposure (supplemental Table S6). All raw data has been deposited in the PeptideAtlas database (http://www.PeptideAtlas.org) with the identifier PASS00642.
Validation of Changes in Gene Expression Using qRT-PCR Analysis
To validate the RNA-Seq results, qRT-PCR was used to quantify changes in the transcript levels of 20 selected genes after HL treatment. These genes have different transcript abundance, length, change tendency (up-regulated, down-regulated or unchanged) and distribution (on chromosome or on plasmid). The RNA-Seq and qRT-PCR results for the 20 tested genes were strongly correlated (R2 = 0.95, slope = 0.9855), and the expected trend in the expression pattern was obtained (Fig. 2A and 2B). These results further proved that our transcriptome data were reliable.
Fig. 2.

Validation of the proteome data and transcriptome data. A, Comparison of log2 expression of 20 selected differentially regulated genes measured by RNA-Seq and qRT-PCR. Positive and negative log2 expression ratios represent up- and down-regulation after HL exposure, respectively. Each data point is calculated from averages of biological triplicates. B, Results derived from RNA-Seq analysis (white bars; n = 2) were compared with those from qRT-PCR (gray bars; n = 3). C, Western blot analysis of protein expression levels of five representative proteins quantified by proteomics after HL exposure. Fold changes of protein levels were determined using Image J software and normalized by protein concentration. D, The mRNA expression level of the validated proteins as detected by qRT-PCR.
Validation of Changes in protein Expression Using Western blot Analysis
To confirm the results from the proteomic study, Western blot analysis were performed to examine the expression status of several of the quantified proteins; PsaC, CpcG, RbcL, PsaD, ApcA and F0063. Results from Western blot and densitometric analysis were consistent with the quantitative proteomic results (Fig. 2C and 2D), confirming the reliability of our proteomic data. The transcript levels of these genes were determined by qRT-PCR, which were also consistent with the RNA-Seq results. The primers for qRT-PCR are listed in supplemental Table S1.
Comparison of Transcriptome and Proteome Data
To estimate the reproducibility of the TMT-based quantitative proteomic and RNA-Seq results, linear regression analysis based on the log2-transformed protein ratios or gene coverage depth was performed for pair-wise comparison of the two experiment replicates. The observed R2 values revealed a relatively strong linear correlation between the two experiment replicates for both quantitative proteomic and RNA-Seq data (supplemental Fig. S2). These findings indicate a high level of reproducibility between replicate data sets.
We conducted a correlation analysis between the quantitative proteomic and RNA-Seq transcriptomic data. Globally, the expression levels of all the quantified proteins and their corresponding mRNAs showed limited correlation (r = 0.2390) (Fig. 3A). However, a higher correlation was observed between the differentially expressed proteins (DEPs) and their corresponding mRNAs (r = 0.4074) (Fig. 3A). The expression ratio of proteins and their corresponding mRNAs with the same or different direction of change (both > 1 or both <1) were also plotted, and higher positive or negative correlation was indicated (Fig. 3B and C).
Fig. 3.

Correlations between protein and mRNA expression. A, Scatterplot of the relationship between genes quantified in both transcriptomic and proteomic data sets. Scatterplots and correlation coefficients between proteins and mRNA expression ratios which have the same (B) or opposite (C) changing tendency. The red plot indicates differentially expressed proteins and black plot indicates non-differentially expressed proteins. D, Scatterplots for protein and mRNA expression classified by nine key clusters of GO categories. All mRNA and protein ratios were log2-transformed.
Within certain clusters of Gene Ontology (GO) groups, the degree of correlation was higher than the global correlation, such as “cell dividing, killing, chaperones, chemotaxis and detoxification” (r = 0.5339), “photosynthesis” (r = 0.4755), “biosynthesis of cofactors, prosthetic groups and carriers” (r = 0.4047), “DNA replication, restriction, modification, recombination and repair” (r = 0.3452), and “cell envelope” (r = 0.3308) (Fig. 3D).
Transcriptomic analysis identified 526 differentially expressed genes (DEGs), and 293 of them have quantitative information on their respective proteins (55.7%) as detected by MS (supplemental Table S3). A total of 64 genes were detected to be regulated at both transcription (≥ 2-fold and p value ≤ 0.05) and translation (≥ 1.4 fold and p value ≤ 0.05) levels, of which 50 genes have the same direction of change and 16 genes have the opposite direction of change in the two levels. The 526 genes differentially expressed at the transcript level and 233 genes differentially expressed at protein level (64 genes were differentially expressed on both mRNA and protein level) under the HL condition were classified into 27 categories according to their GO function (Fig. 4A). We also classified the 233 DEPs in to different groups according to their GO function (Fig. 4B). These genes and proteins were mainly involved in photosynthesis and related pathways, transport and binding, energy metabolism, DNA replication and repair, transcription, and translation.
Fig. 4.

Classification of all DEGs and DEPs based on their GO categories. A, Classification of the 526 DEGs and 233 DEPs (64 genes were differentially expressed on both mRNA and protein level) under HL conditions. Circle nodes indicate DEGs that do not have protein quantitative information. Square nodes indicate genes that only differentially expressed on protein level. Diamond nodes indicate DEGs that have protein quantitative information. Red represents up-regulation, green represents down-regulation and gray represents no change in expression. As for genes have both the mRNA and protein quantitative information, the color represents the expression change on protein level. B, Classification of the 233 DEPs under HL conditions. Red represents up-regulation, green represents down-regulation.
Our transcriptomic and proteomic analyses showed that many hypothetical proteins coding genes were differentially regulated after HL treatment (Fig. 4), 211 of the 526 (40.1%) differentially expressed transcripts were hypothetical genes, and 77 of the 233 (33.0%) DEPs were hypothetical proteins (supplemental Tables S3 and S6). We suggest that these hypothetical genes are indeed expressed and may play important roles in the HL acclimation.
We constructed a heatmap (Fig. 5A) to compare the expression patterns of the 1746 quantified proteins and their corresponding transcripts and the expression patterns of the 233 HL-induced proteins and their corresponding transcripts (Fig. 5B), the heatmap patterns also show the lack of correlation between mRNA levels and proteins.
Fig. 5.

Hierarchical clustering analysis of transcriptomic and proteomic data, based on the expression data. A, Heatmap of all the quantified proteins and their corresponding mRNAs. B, Heatmap of all the DEPs and their corresponding mRNAs. C, Heatmap of the gene and protein expression patterns of key genes/proteins in the six functional groups indicated. The heat map is linked by a dendrogram representing clustering of transcriptomic data or proteomic data (left side). The color code is as follows: yellow indicates up-regulated transcripts or proteins; blue indicates down-regulated transcripts or proteins; black indicates unchanged transcripts or proteins. Each row represents the log2 (HL/NL) of a gene or protein. The color scale of the heatmap ranges from saturated blue (value, −3.0) to saturated yellow (value, 3.0) in the natural logarithmic scale.
The mRNAs/proteins were selected to represent six functional groups, of relevance to photosynthesis, protein production and chlorophyll biosynthesis (Fig. 5C). Genes encoding subunits of photosystem I (PS I and PS II were all significantly down-regulated at the protein level. However, at the mRNA level, genes encoding subunits of PS I were shown to be unchanged or down-regulated whereas genes encoding subunits PS II tended to be up-regulated. Gene and protein expression of subunits of phycobilisomes (PBS) or NADH dehydrogenases tended to change in the same direction, whereas gene and protein expression of enzymes involved in chlorophyll biosynthesis or ribosome subunits tended to change in the opposite direction.
Pathway and GO Term Enrichment Analyses of the HL-Responsive Transcripts and Proteins
As indicated in Fig. 6, The KEGG pathway enrichment analysis showed that three KEGG pathways were differentially regulated at both mRNA and protein level: “Photosynthesis-antenna proteins” (syp00196), “Photosynthesis” (syp00195), and “Oxidative phosphorylation” (syp00190) (supplemental Table S7–1).
Fig. 6.

Enrichment analysis of the HL-responsive genes and proteins. GO terms and KEGG pathways enriched in the DEGs and DEPs under HL stress. p < 0.05 was considered as significant.
GO term enrichment analysis showed that several biological processes, including “photosynthesis”, “oxidative reduction”, “electron transport chain”, “generation of precursor metabolites and energy” and “photosynthesis, light reaction” were significantly enriched in both DEPs and DEGs, suggesting that these processes are very active during the HL treatment (supplemental Table S7–2).
GO cellular component terms, including “thylakoid,” “photosynthetic membrane,” “light-harvesting complex,” and “thylakoid membrane” and GO molecular function terms, including “electron carrier activity” and “oxidoreductase activity, acting on NADH or NAPDH” were all significantly enriched in both DEPs and DEGs (supplemental Table S7–3 and supplemental Table S7–4).
Changes in the Transcript and Protein Abundance of Genes Encoding Thylakoid-Located Complexes
PS II-Related Genes and Proteins
The psb genes, which encode subunits of PS II, were predominantly induced at the transcript level under HL stress. The three psbA genes that encode the D1 protein in cyanobacteria are under strict regulation to guarantee the proper functioning of the PS II (32). The transcription of these genes is modulated in response to changes in light intensity and O2 level (33–35). The transcript abundance of three psbA genes (SYNPCC7002_A1418, SYNPCC7002_A0157, SYNPCC7002_A2164) in Synechococcus 7002 increased about 3- to 5-fold when grown under the HL condition. Similar induction pattern by HL treatment was observed in the psbD (SYNPCC7002_A2199) that encodes the reaction center D2 protein and psbM. Transcripts of genes involved in the oxygen-evolving complex, psbO, psbU and many other genes that encode small subunits of PS II, including psbC, psbD1, psbB, psbP, psbL and psbJ were all slightly induced by the HL treatment. However, all the subunits of PS II that were quantified by mass spectrometry were significantly decreased at the protein level (supplemental Table S8).
PS I-Related Genes and Proteins
In contrast with the case of genes encoding subunits of PS II, the PS I genes generally decreased at the transcript level. The quantitative proteomic analysis revealed that the PS I subunits were all significantly reduced under HL stress. Declining PS I content would be expected to lower the susceptibility of the cells to HL damage particularly under prolonged exposure (9).
Phycobilisome-Related Genes and Protein
Phycobilisomes serve as the main antennae for photosynthesis in cyanobacteria, and they transfer excitation energy to both photosystems (36, 37). There was an overall decrease in transcript levels for phycocyanin and phycocyanin-associated linker proteins (3- to 6.4-fold; supplemental Table S8), whereas the transcript level of allophycocyanin-associated genes decreased to a lesser extent (maximally ∼2-fold reduction; supplemental Table S8). Additionally, the protein expression levels of phycobiliproteins were all significantly decreased.
The ndh Genes and Proteins
The transcript levels of many genes encoding NADH dehydrogenase subunits were increased 1.5- to 4-fold (supplemental Table S8). Transcripts for ndhD2 were 5.7-fold higher in cells exposed to HL than in normal conditions (supplemental Table S8). However, transcripts for ndhD1, encoding a paralogous form of NdhD subunit for the Type-1 NADH dehydrogenase complex remained unchanged in HL grown cells. Almost all of the NADH dehydrogenase subunits were up-regulated (1.4- to 2-fold) at the protein level. The Type-1 NADH dehydrogenase complex is required for cyclic electron flow (38), so we predict that it has a preferential involvement of cyclic electron flow at HL intensity.
Changes in the Transcript and Protein Abundance of the Other Genes
Calvin-Benson-Bassham Cycle (CBB Cycle)
The transcript levels of many genes involved in the CBB cycle were 2- to 3.5-fold higher in cells exposed to HL (supplemental Table S8). The protein expression of the large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO), RbcL was significantly induced. Genes encoding the structural components of carboxysomes (ccmK, ccmL, ccmM, ccmN) were also significantly increased at transcript level (1.5- to 4-fold), although they were only slightly up-regulated at the protein level.
CO2 Uptake Mechanism
Interestingly, transcript levels for the genes encoding the so-called inducible CO2 uptake mechanism (ndhD3, ndhF3, cupA, and cupS) (39) were significantly induced (maximum increase of ∼6-fold); however, the transcript levels for the constitutive CO2-concentrating mechanism (ndhD4, ndhF4, cupB) remained constant, which disagrees with results obtained in previous studies on short-term exposure to HL in Synechocystis sp. PCC 6803 and Synechococcus 7002 (15, 40).
Chlorophyll Biosynthesis
Most genes encoding the enzymes of chlorophyll biosynthesis remained constant at the transcript level when grown under HL-condition, with the exception of the transcripts for chlH (SYNPCC7002_A1000, SYNPCC7002_A1018) and chlL (SYNPCC7002_A2347) genes, encoding magnesium-chelatase and protochlorophyllide reductase, which increased ∼2-fold upon HL treatment. However, six proteins in chlorophyll biosynthesis were found to be significantly decreased about 1.4- to 1.8- fold, which are ChlM, HemF, HemJ, Hox1, PcyA, and Por.
Flavoproteins
Flavoproteins have previously been reported to act as oxygen photoreductases in Synechocystis sp. PCC 6803 (41, 42). HL treatment increased the transcript level of SYNPCC7002_ A1321, a flavoprotein coding gene by ∼2-fold. The transcript level for SYNPCC7002_A1743, another flavoprotein coding gene, remained unchanged. However, the protein expressions of these two genes were both significantly induced. This observation is in accordance with their functions as catalysts in dissipation of excess electrons via the Mehler reaction (42).
Chaperone and ROS Scavenging Enzyme
Despite the increasing energy consumption during HL, the cells could suffer damage from ROS at HL. Thus, genes that have chaperonin-like roles such as heat shock proteins were expected to be up-regulated. Our results show that genes encoding the molecular chaperones increased 2- to 6-fold at the transcript level (supplemental Table S8). However, protein products of these genes only slightly increased or remained constant (1.7-fold at maximum).
Genes that encode scavenging enzymes for ROS were also expected to be up-regulated under HL conditions in which production of ROS may be accelerated (43). However, only SYNPCC7002_A0970, which encodes glutathione peroxidase, was induced at both the transcript and protein level (∼5- and 2-fold, respectively). Transcription of other antioxidant enzymes such as katG (SYNPCC7002_A2422, catalase), sodB (SYNPCC7002_A0242, Mn-superoxide dismutase), and SYNPCC7002_A0117, which encodes another glutathione peroxidase, was not significantly affected. These results suggest that glutathione peroxidase encoding by SYNPCC7002_A0970 may have an important function in resistance to ROS under HL stress.
High-Light-Inducible Polypeptides (HLIPs)
The hli genes, present in cyanobacteria, algae and vascular plants, encode small proteins [high-light-inducible polypeptides (HLIPs)]. In our study, three HLIPs were found to be significantly induced after exposure to HL. The protein expression of SYNPCC7002_A0858 (hliA) was significantly induced (∼2.5-fold), whereas the transcript level remained constant. SYNPCC7002_A0186 was up-regulated at the transcript level (about 3.4-fold), but the protein expression of this gene was not detected by proteomic profiling. The expression of SYNPCC7002_A0602 (hliA) significantly increased at both the mRNA and protein levels.
Characterization of HL-Sensitive Mutants
To confirm the involvement in HL response, 12 genes differentially regulated under HL stress were selected for gene mutant construction, including three subunits of PS I (psaC, psaD, psaF), three subunits of PS II (psbC, psbO, psbU), allophycocyanin beta-18 subunit apcF and phycobilisome rod-core linker polypeptide cpcG (l-RC 28.5). Four genes encoding hypothetical proteins (SYNPCC7002_A0568, SYNPCC7002_A1479, SYNPCC7002_A1480, SYNPCC7002_F0063; hereafter A0568, A1479, A1480, and F0063) were also selected for mutant construction. These selected genes were all significantly down-regulated at the protein level upon HL stress as determined by quantitative proteomics, except for F0063, which was significantly up-regulated (1.65-fold, p < 0.05), and A0568, which was slightly up-regulated (1.33-fold, p < 0.05). Four genes (psaC, psaD, A1479 and A1480) were down-regulated at the transcript level, whereas A0568 was significant induced (about 10-fold, p < 0.05). However, the mRNA expression levels of other genes were not changed.
As shown in Fig. 7, the growth of the knockout mutants ΔA1479, ΔA0568, ΔpsbC and ΔpsaC did not show much difference when compared with wild-type under both NL and HL conditions, suggesting that these genes were not indispensable for the normal growth and HL acclimation. The inactivation of pasD, psaF, psbO, psbU, and F0063 also showed slower growth under NL condition (p < 0.01), and these mutants could not grow at all under HL treatment, suggesting that they were HL-sensitive lethal mutants. These mutants are necessary for the normal growth of Synechococcus 7002 and are indispensable in HL acclimation. Comparative analysis showed that although there was no visible difference in terms of growth patterns between the wild type and the mutants under the NL condition, the ΔcpcG mutant grew slower than the wild type under HL stress (p < 0.01), suggesting that it is more sensitive to HL and may be involved in HL resistance. Interestingly, the growth of ΔapcF and ΔA1480 showed no difference in comparison with wild type under NL, but these two mutants grew faster than wild type under the HL condition (p < 0.01).
Fig. 7.

Time courses of growth of the wild-type and gene knockout mutants. The wild-type and gene knockout mutants were cultivated under NL conditions with (A) or without (C) glycerol in the growth medium; or cultivated under HL conditions with (B) or without (D) glycerol in the growth medium.
DISCUSSION
Synechococcus 7002 is known to be extremely tolerant to HL intensity (9, 10), and sunlight intensity is one of the key environmental factors in natural habitats of cyanobacteria. Global investigation on HL acclimation of cyanobacteria has been conducted at transcript level (15, 40). This transcriptomic data revealed extensive changes in cellular transcript levels in response to HL stress and provided novel insights into the molecular mechanisms of response to HL stress and the genes of potential importance for the adaptation of HL in Synechococcus 7002. Although informative, transcript abundances do not necessarily reflect cellular protein levels because protein expression is influenced by an array of post-transcriptional regulatory mechanisms and the correlation between protein and mRNA levels is generally modest (44–46). It is necessary to analyze the response of Synechococcus 7002 to HL at both the transcriptomic and proteomic levels in an effort to gain systems-level information. Therefore, we employed an integrated quantitative proteomic and transcriptomic approach to investigate the HL acclimation mechanisms of Synechococcus 7002, aiming at identifying novel gene components and regulatory mechanisms in response to HL acclimation.
In this study, the RNA-Seq combined with the quantitative proteomic analyses showed that 526 genes and 233 proteins were differentially regulated and could be related with the response and resistance of Synechococcus 7002 to HL stress. A conceptual model that summarizes the function of the key DEPs was developed to decipher the global molecular mechanisms involved in HL responses in Synechococcus 7002. As shown in Fig. 8, HL stress induced the degradation of phycobiliproteins or the reduction of phycobilisome size. Photosystem content was also reduced to avoid absorption of excess light energy. These changes may originate from the down-regulation of genes that encode enzymes for biosynthesis of photosynthetic pigments (hem and chl genes), structural components of phycobilisome (apc and cpc genes), and subunits of photosystems (psa and psb genes). Although many psb genes did not change or were only slightly induced at transcript levels, psbA genes, which encode D1, were strongly up-regulated. It is likely that the elevated level of psbA transcripts makes the increasing turnover rate of the D1 protein under HL conditions possible (33). In contrast with photochemical reactions down-regulated by HL, CO2 fixation was accelerated. It is not surprising that the rbcL gene was significantly induced at both the transcript and protein levels, because the reaction catalyzed by RuBisCO is the primary rate-limiting factor of the CBB cycle under saturating light intensity. The ccm genes encoding components of the CO2-concentrating mechanism were also induced. Up-regulation of ndh genes, involved in high affinity CO2 uptake (47, 48) may also help to increase the availability of CO2 under HL. Despite the increasing energy consumption during HL, the cells could suffer damage from ROS at HL. Proteins encoding ROS scavenging enzymes (SYNPCC7002_A0970, glutathione peroxidase) and proteins that have chaperonin-like roles, GroES (co-chaperonin GroES) and HtpG (heat shock protein 90), were up-regulated. Accumulation of HLIPs under HL intensities proved to be associated with the pigment alteration (i.e. decrease in light-harvesting pigments, accumulation of the carotenoid myxoxanthophyll and decrease in PS I-associated chlorophylls) and stabilization of PS I trimers to protect cells under HL stress (49–52). In our study, three HLIPs in Synechococcus 7002 were found to be significantly induced after HL exposure. It is likely that the HLIPs in Synechococcus 7002 are critical for survival when absorbing excess excitation energy and may allow the cells to cope more effectively under HL conditions. Enzymes in the carotenoid synthesis pathway were all significantly induced at the transcript level (about 9- fold at maximum), and two enzymes, CruG (carotenoid 2-O-rhamnosyltransferase) and CrtP (phytoene desaturase) were significantly up-regulated at the protein level (1.5-fold). Previous studies have shown that orange carotenoid protein (OCP)-related non-photochemical -quenching mechanism is a very important regulatory mechanism in the quenching and dissipation of excess light energy in some cyanobacteria (53, 54). Therefore, we deduce that this is another protection mechanism against HL-caused damage in Synechococcus 7002.
Fig. 8.

Putative model of the regulation mechanisms involved in HL acclimation in Synechococcus 7002. Proteins colored in yellow were indispensable upon HL stress. CrtU, beta-carotene desaturase; HemF, coproporphyrinogen III oxidase; HemJ, coproporphyrinogen III oxidase; PcyA, phycocyanobilin:ferredoxin oxidoreductase; ChlM, Mg-protoporphyrin IX methyl transferase; Hox1, heme oxygenase (decyclizing); Por, protochlorophyllide oxidoreductase.
Under conditions of HL stress, cells also systematically regulated their transcription, translation, and post-translational modification functions (supplemental Table S8). For example, Protein expression of enzymes in DNA replication, repair and modification increased (For example, ParA, GvrA, and PhrA). Transcriptional regulators (NusB, SigD, SYNPCC7002_A2523), and ribosomal proteins (RpsE, RplB, RplU, and RplT) were also found to be significantly induced at the protein level (supplemental Table S8). We suggest that Synechococcus 7002 bacteria accelerate the key processes in genetic central dogma to cope with the increased demands of DNA damage-repairing and protein synthesis under HL exposure.
Comparative growth analysis of the knockout mutants of HL-responsive genes and the wild type under NL and HL condition revealed several important HL-sensitive mutants and HL-sensitive lethal mutants, which will be discussed below.
ΔcpcG was revealed to be a HL-sensitive mutant, which grew normally under NL, but was inhibited under HL. CpcG1 (homolog of SYNPCC7002_A0811), which connects the rods with the core of major allophycocyanins was reported to be involved in state transitions in Synechocystis 6803 (55). Our results support the previous notion that state transitions are important in acclimating to light intensities (3, 56), and suggested that cpcG may contribute to the HL acclimation of Synechococcus 7002.
In Synechococcus 7002, ApcF plays an important role in energy transfer from PBS to PS II. ΔApcF mutant strain showed a higher tolerance to HL stress than the wild type (Fig. 7D), which is in accordance with a previous study (37). This is possibly because when apcF is inactivated, energy transfer from PBS to PS II is less efficient. The ΔA1480 mutant strain showed the same growth pattern as the ΔApcF, indicating that it may function as a negative regulator in HL acclimation mechanisms (Fig. 7D).
ΔpsbO, ΔpsbU, ΔpsaD, ΔpsaF and ΔF0063 were HL-sensitive lethal mutants. These mutant strains showed slower growth under the NL condition and could not grow at all under HL conditions. PsbO and PsbU are two extrinsic proteins of PS II in cyanobacteria, which stabilize the oxygen-evolving complex and PS II (57–60). Mutation studies of PsbO and PsbU in Synechocystis 6803 revealed that ΔpsbO or ΔpsbU mutant was more susceptible to photodamage when exposed to HL intensity, exhibiting a more rapid degradation of the D1 protein (59–65). The complete growth arrest of ΔpsbO, ΔpsbU observed in our study suggests that PsbO and PsbU in Synechococcus 7002 are important for normal cell growth and are indispensable in HL acclimation process.
The PsaD subunit, as a conserved peripheral protein on the reducing side of PS I, is involved in the docking of ferredoxin to PS I reaction centers and assembly of other peripheral subunits (66). A previous study in Synechocystis 6803 revealed that the photoautotrophic growth of ΔpsaD is much slower than that of wild type cells (67). The PsaF subunit of PS I also has dispensable accessory roles in the function and organization of the complex (68). Therefore, it was not surprising that ΔpsaD and ΔpsaF mutant of Synechococcus 7002 could not grow under HL and showed slower growth rate under NL compared with wild type.
F0063, with its putative role as an acetyltransferase (belongs to GCN5-related N-acetyltransferase (GNAT) superfamily) (supplemental Fig. S4), was significantly induced upon HL treatment. Previous phosphoproteomic analysis of Synechococcus 7002 indicated that post-translational modifications are deeply involved in the photosynthesis process (69). It is well established that lysine acetylation is one of the most common post-translational modifications to proteins in both eukaryotes and prokaryotes and plays important roles in many cellular physiological processes (70, 71). Up-regulation of F0063 under HL conditions suggests that protein acetylation may be an important regulation mechanism in response to HL stress.
In conclusion, the integrated transcriptomic and proteomic analysis revealed multiple levels of regulation in response to HL in Synechococcus 7002, including possible post-translational regulation. Comparative growth analysis of the knockout mutants led to the identification of key genes involved in response to HL in Synechococcus 7002. Our results provide novel insights into the global response mechanisms to HL in Synechococcus 7002 and may be valuable for further studies addressing HL resistance in photosynthetic organisms in general.
Supplementary Material
Footnotes
Author contributions: T.L., F.G., and J.Z. designed research; Q.X., J.F., S.L., G.Z., Z.Q., Z.C., Y.W., H.H., Y.L., and T.L. performed research; Q.X., J.F., S.L., Z.C., T.L., F.G., and J.Z. analyzed data; Q.X., F.G., and J.Z. wrote the paper.
* This work was supported by National Basic Research Program of China (973 Program, 2012CB518700), the National Natural Science Foundation of China (Grant No. 21207153 To Q. X. and Grant No. 31370746 To F. G.), and the Knowledge Innovation Program of the Chinese Academy of Sciences (Grant No. Y35E04-1-501 to Q. X.).
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S8.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S8.
1 The abbreviations used are:
- PS I
- photosystem I
- PS II
- photosystem II
- HL
- high light
- TMT
- tandem mass tags.
REFERENCES
- 1. Anderson J. M. (1986) Photoregulation of the composition, function, and structure of thylakoid membranes. Annu. Rev. Plant. Physiol. 37, 93–136 [Google Scholar]
- 2. Anderson J. M., Chow W. S., Park Y.-I. (1995) The grand design of photosynthesis: acclimation of the photosynthetic apparatus to environmental cues. Photosynth. Res. 46, 129–139 [DOI] [PubMed] [Google Scholar]
- 3. Campbell D., Hurry V., Clarke A. K., Gustafsson P., Oquist G. (1998) Chlorophyll fluorescence analysis of cyanobacterial photosynthesis and acclimation. Microbiol. Mol. Biol. Rev. 62, 667–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niyogi K. K. (1999) Photoprotection revisited: genetic and molecular approaches. Annu. Rev. Plant Physiol. 50, 333–359 [DOI] [PubMed] [Google Scholar]
- 5. Hassidim M., Keren N., Ohad I., Reinhold L., Kaplan A. (1997) Acclimation of Synechococcus strain WH7803 to ambient CO2 concentration and to elevated light intensity. J. Phycol. 33, 811–817 [Google Scholar]
- 6. Chow W. (1994) Advances in molecular and cell biology, pp. 151–196, JAI Press, Greenwich, CT [Google Scholar]
- 7. Anderson J. M., Park Y. I., Chow W. (1997) Photoinactivation and photoprotection of photosystem II in nature. Physiol. Plant 100, 214–223 [Google Scholar]
- 8. Asada K., Foyer C., Mullineaux P. (1994) Causes of photooxidative stress and amelioration of defense systems in plants, pp. 77–104, CRC Press, Boca Raton, FL [Google Scholar]
- 9. Sakamoto T., Bryant D. (2002) Synergistic effect of high-light and low temperature on cell growth of the Δ12 fatty acid desaturase mutant in Synechococcus sp. PCC 7002. Photosynth. Res. 72, 231–242 [DOI] [PubMed] [Google Scholar]
- 10. Nomura C. T., Sakamoto T., Bryant D. A. (2006) Roles for heme-copper oxidases in extreme high-light and oxidative stress response in the cyanobacterium Synechococcus sp. PCC 7002. Arch. Microbiol. 185, 471–479 [DOI] [PubMed] [Google Scholar]
- 11. Batterton J. C., Jr., Van Baalen C. (1971) Growth responses of blue-green algae to sodium chloride concentration. Arch. Mikrobiol. 76, 151–165 [DOI] [PubMed] [Google Scholar]
- 12. Stevens S. E., Porter R. D. (1980) Transformation in Agmenellum quadruplicatum. Proc. Natl. Acad. Sci. U.S.A. 77, 6052–6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Y., Alvey R. M., Byrne P. O., Graham J. E., Shen G., Bryant D. A. (2011) Photosynthesis Research Protocols, pp. 273–293, Humana Press, New York, NY [Google Scholar]
- 14. Ludwig M., Bryant D. A. (2012) Acclimation of the global transcriptome of the cyanobacterium Synechococcus sp. Strain PCC 7002 to nutrient limitations and different nitrogen sources. Front. Microbiol. 3, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bryant D. A., Ludwig M. (2011) Transcription profiling of the model cyanobacterium Synechococcus sp. Strain PCC 7002 by Next-Gen (SOLiD™) sequencing of cDNA. Front. Microbiol. 2, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foy R., Gibson C. (1982) Photosynthetic characteristics of planktonic blue-green algae: changes in photosynthetic capacity and pigmentation of Oscillatoria redekei Van Goor under high and low light. Brit. Phycol. J. 17, 183–193 [Google Scholar]
- 17. Foy R., Gibson C. (1982) Photosynthetic characteristics of planktonic blue-green algae: The response of twenty strains grown under high and low light. Brit. Phycol. J. 17, 169–182 [Google Scholar]
- 18. Hihara Y. (1999) The molecular mechanism for acclimation to high light in cyanobacteria. Curr. Opin. Plant Biol. 1, 37–50 [Google Scholar]
- 19. Pinto A., Melo-Barbosa H., Miyoshi A., Silva A., Azevedo V. (2011) Application of RNA-seq to reveal the transcript profile in bacteria. Genet. Mol. Res. 10, 1707–1718 [DOI] [PubMed] [Google Scholar]
- 20. Wang Z., Gerstein M., Snyder M. (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guerreiro A. C., Benevento M., Lehmann R., van Breukelen B., Post H., Giansanti P., Maarten Altelaar A. F., Axmann I. M., Heck A. J. (2014) Daily rhythms in the cyanobacterium Synechococcus elongatus probed by high-resolution mass spectrometry-based proteomics reveals a small defined set of cyclic proteins. Mol. Cell. Proteomics 13, 2042–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nie S., Lo A., Wu J., Zhu J., Tan Z., Simeone D. M., Anderson M. A., Shedden K. A., Ruffin M. T., Lubman D. M. (2014) Glycoprotein biomarker panel for pancreatic cancer discovered by quantitative proteomics analysis. J. Proteome Res. 13, 1873–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amaral A., Paiva C., Attardo Parrinello C., Estanyol J. M., Ballesca J. L., Ramalho-Santos J., Oliva R. (2014) Identification of proteins involved in human sperm motility using high-throughput differential proteomics. J. Proteome Res. 13, 5670–5684 [DOI] [PubMed] [Google Scholar]
- 24. Ludwig M., Bryant D. A. (2012) Synechococcus sp. strain PCC 7002 transcriptome: acclimation to temperature, salinity, oxidative stress, and mixotrophic growth conditions. Front. Microbiol. 3, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Passalacqua K. D., Varadarajan A., Ondov B. D., Okou D. T., Zwick M. E., Bergman N. H. (2009) Structure and complexity of a bacterial transcriptome. J. bacteriol. 191, 3203–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoder-Himes D. R., Chain P. S., Zhu Y., Wurtzel O., Rubin E. M., Tiedje J. M., Sorek R. (2009) Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc. Natl. Acad. Sci. U.S.A. 106, 3976–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Graaf E. L., Altelaar A. F., van Breukelen B., Mohammed S., Heck A. J. (2011) Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. J. Proteome Res. 10, 4334–4341 [DOI] [PubMed] [Google Scholar]
- 29. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 30. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p. p. b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotech. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 31. Huang D. W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mulo P., Sakurai I., Aro E.-M. (2012) Strategies for psbA gene expression in cyanobacteria, green algae and higher plants: From transcription to PSII repair. BBA-Bioenergetics 1817, 247–257 [DOI] [PubMed] [Google Scholar]
- 33. Mohamed A., Jansson C. (1989) Influence of light on accumulation of photosynthesis-specific transcripts in the cyanobacterium Synechocystis 6803. Plant mol. Biol. 13, 693–700 [DOI] [PubMed] [Google Scholar]
- 34. Summerfield T. C., Toepel J., Sherman L. A. (2008) Low-oxygen induction of normally cryptic psbA genes in cyanobacteria. Biochemistry-US 47, 12939–12941 [DOI] [PubMed] [Google Scholar]
- 35. Sander J., Nowaczyk M., Buchta J., Dau H., Vass I., Deák Z., Dorogi M., Iwai M., Rögner M. (2010) Functional characterization and quantification of the alternative PsbA copies in Thermosynechococcus elongatus and their role in photoprotection. J. Biol. Chem. 285, 29851–29856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ashby M. K., Mullineaux C. W. (1999) Cyanobacterial ycf27 gene products regulate energy transfer from phycobilisomes to photosystems I and II. FEMS Microbiol. Lett. 181, 253–260 [DOI] [PubMed] [Google Scholar]
- 37. Dong C., Tang A., Zhao J., Mullineaux C. W., Shen G., Bryant D. A. (2009) ApcD is necessary for efficient energy transfer from phycobilisomes to photosystem I and helps to prevent photoinhibition in the cyanobacterium Synechococcus sp. PCC 7002. BBA-Bioenergetics 1787, 1122–1128 [DOI] [PubMed] [Google Scholar]
- 38. Mi H., Endo T., Schreiber U., Ogawa T., Asada K. (1992) Electron donation from cyclic and respiratory flows to the photosynthetic intersystem chain is mediated by pyridine nucleotide dehydrogenase in the cyanobacterium Synechocystis PCC 6803. Plant Cell Physiol. 33, 1233–1237 [Google Scholar]
- 39. Ogawa T., Mi H. (2007) Cyanobacterial NADPH dehydrogenase complexes. Photosynth. Res. 93, 69–77 [DOI] [PubMed] [Google Scholar]
- 40. Hihara Y., Kamei A., Kanehisa M., Kaplan A., Ikeuchi M. (2001) DNA microarray analysis of cyanobacterial gene expression during acclimation to high light. Plant Cell 13, 793–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hackenberg C., Engelhardt A., Matthijs H. C., Wittink F., Bauwe H., Kaplan A., Hagemann M. (2009) Photorespiratory 2-phosphoglycolate metabolism and photoreduction of O2 cooperate in high-light acclimation of Synechocystis sp. strain PCC 6803. Planta 230, 625–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Helman Y., Tchernov D., Reinhold L., Shibata M., Ogawa T., Schwarz R., Ohad I., Kaplan A. (2003) Genes Encoding A-Type Flavoproteins Are Essential for Photoreduction of O2 in Cyanobacteria. Curr. Biol. 13, 230–235 [DOI] [PubMed] [Google Scholar]
- 43. Hideg E., Barta C., Kalai T., Vass I., Hideg K., Asada K. (2002) Detection of singlet oxygen and superoxide with fluorescent sensors in leaves under stress by photoinhibition or UV radiation. Plant Cell Physiol. 43, 1154–1164 [DOI] [PubMed] [Google Scholar]
- 44. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 45. de Sousa Abreu R., Penalva L. O., Marcotte E. M., Vogel C. (2009) Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5, 1512–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu L., Candille S. I., Choi Y., Xie D., Jiang L., Li-Pook-Than J., Tang H., Snyder M. (2013) Variation and genetic control of protein abundance in humans. Nature 499, 79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ohkawa H., Price G. D., Badger M. R., Ogawa T. (2000) Mutation of ndh genes leads to inhibition of CO2 uptake rather than HCO3− uptake in Synechocystis sp. strain PCC 6803. J. bacteriol. 182, 2591–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ohkawa H., Sonoda M., Katoh H., Ogawa T. (1998) The use of mutants in the analysis of the CO2-concentrating mechanism in cyanobacteria. Can. J. Bot. 76, 1035–1042 [Google Scholar]
- 49. He Q. F., Dolganov N., Bjorkman O., Grossman A. R. (2001) The high light-inducible polypeptides in Synechocystis PCC6803 - Expression and function in high light. J. Biol. Chem. 276, 306–314 [DOI] [PubMed] [Google Scholar]
- 50. Funk C., Vermaas W. (1999) A cyanobacterial gene family coding for single-helix proteins resembling part of the light-harvesting proteins from higher plants. Biochemistry 38, 9397–9404 [DOI] [PubMed] [Google Scholar]
- 51. Havaux M., Guedeney G., He Q. F., Grossman A. R. (2003) Elimination of high-light-inducible polypeptides related to eukaryotic chlorophyll a/b-binding proteins results in aberrant photoacclimation in Synechocystis PCC6803. BBA-Bioenergetics 1557, 21–33 [DOI] [PubMed] [Google Scholar]
- 52. Wang Q., Jantaro S., Lu B., Majeed W., Bailey M., He Q. (2008) The high light-inducible polypeptides stabilize trimeric photosystem I complex under high light conditions in Synechocystis PCC 6803. Plant Physiol. 147, 1239–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kirilovsky D. (2007) Photoprotection in cyanobacteria: the orange carotenoid protein (OCP)-related non-photochemical-quenching mechanism. Photosynth. Res. 93, 7–16 [DOI] [PubMed] [Google Scholar]
- 54. Kirilovsky D. (2010) The photoactive orange carotenoid protein and photoprotection in cyanobacteria. Advan. Experiment Med. Biol. 675, 139–159 [DOI] [PubMed] [Google Scholar]
- 55. Kondo K., Mullineaux C. W., Ikeuchi M. (2009) Distinct roles of CpcG1-phycobilisome and CpcG2-phycobilisome in state transitions in a cyanobacterium Synechocystis sp. PCC 6803. Photosynth. Res. 99, 217–225 [DOI] [PubMed] [Google Scholar]
- 56. Herbert S. K., Martin R. E., Fork D. C. (1995) Light adaptation of cyclic electron transport through photosystem I in the cyanobacterium Synechococcus sp. PCC 7942. Photosynth. Res. 46, 277–285 [DOI] [PubMed] [Google Scholar]
- 57. Philbrick J., Diner B., Zilinskas B. (1991) Construction and characterization of cyanobacterial mutants lacking the manganese-stabilizing polypeptide of photosystem II. J. Biol. Chem. 266, 13370–13376 [PubMed] [Google Scholar]
- 58. Burnap R. L., Sherman L. A. (1991) Deletion mutagenesis in Synechocystis sp. PCC6803 indicates that the manganese-stabilizing protein of photosystem II is not essential for oxygen evolution. Biochemistry-US 30, 440–446 [DOI] [PubMed] [Google Scholar]
- 59. Veerman J., Bentley F. K., Eaton-Rye J. J., Mullineaux C. W., Vasil'ev S., Bruce D. (2005) The PsbU subunit of photosystem II stabilizes energy transfer and primary photochemistry in the phycobilisome-photosystem II assembly of Synechocystis sp. PCC 6803. Biochemistry-US 44, 16939–16948 [DOI] [PubMed] [Google Scholar]
- 60. Inoue-Kashino N., Kashino Y., Satoh K., Terashima I., Pakrasi H. B. (2005) PsbU provides a stable architecture for the oxygen-evolving system in cyanobacterial photosystem II. Biochemistry-US 44, 12214–12228 [DOI] [PubMed] [Google Scholar]
- 61. Shen J.-R., Ikeuchi M., Inoue Y. (1997) Analysis of the psbU gene encoding the 12-kDa extrinsic protein of photosystem II and studies on its role by deletion mutagenesis in Synechocystis sp. PCC 6803. J. Biol. Chem. 272, 17821–17826 [DOI] [PubMed] [Google Scholar]
- 62. Balint I., Bhattacharya J., Perelman A., Schatz D., Moskovitz Y., Keren N., Schwarz R. (2006) Inactivation of the extrinsic subunit of photosystem II, PsbU, in Synechococcus PCC 7942 results in elevated resistance to oxidative stress. FEBS Lett. 580, 2117–2122 [DOI] [PubMed] [Google Scholar]
- 63. Clarke S. M., Eaton-Rye J. J. (1999) Mutation of Phe-363 in the photosystem II protein CP47 impairs photoautotrophic growth, alters the chloride requirement, and prevents photosynthesis in the absence of either PSII-O or PSII-V in Synechocystis sp. PCC 6803. Biochemistry-US 38, 2707–2715 [DOI] [PubMed] [Google Scholar]
- 64. Kimura A., Eaton-Rye J. J., Morita E. H., Nishiyama Y., Hayashi H. (2002) Protection of the oxygen-evolving machinery by the extrinsic proteins of photosystem II is essential for development of cellular thermotolerance in Synechocystis sp. PCC 6803. Plant cell Physiol. 43, 932–938 [DOI] [PubMed] [Google Scholar]
- 65. De Las Rivas J., Barber J. (2004) Analysis of the structure of the PsbO protein and its implications. Photosynth. Res. 81, 329–343 [DOI] [PubMed] [Google Scholar]
- 66. Chitnis V. P., Jungs Y. S., Albee L., Golbeck J. H., Chitnis P. R. (1996) Mutational analysis of photosystem I polypeptides. Role of PsaD and the lysyl 106 residue in the reductase activity of the photosystem I. J. Biol. Chem. 271, 11772–11780 [DOI] [PubMed] [Google Scholar]
- 67. Chitnis P. R., Reilly P. A., Nelson N. (1989) Insertional inactivation of the gene encoding subunit II of photosystem I from the cyanobacterium Synechocystis sp. PCC 6803. J. Biol. Chem. 264, 18381–18385 [PubMed] [Google Scholar]
- 68. Xu Q., Yu L., Chitnis V. P., Chitnis P. R. (1994) Function and organization of photosystem I in a cyanobacterial mutant strain that lacks PsaF and PsaJ subunits. J. Biol. Chem. 269, 3205–3211 [PubMed] [Google Scholar]
- 69. Yang M. K., Qiao Z. X., Zhang W. Y., Xiong Q., Zhang J., Li T., Ge F., Zhao J. D. (2013) Global phosphoproteomic analysis reveals diverse functions of serine/threonine/tyrosine phosphorylation in the model cyanobacterium Synechococcus sp. strain PCC 7002. J. Proteome Res. 12, 1909–1923 [DOI] [PubMed] [Google Scholar]
- 70. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 71. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.