

Abstract
Mitochondria provide numerous essential functions for cells and their dysfunction leads to a variety of diseases. Thus, obtaining a complete mitochondrial proteome should be a crucial step toward understanding the roles of mitochondria. Many mitochondrial proteins have been identified experimentally but a complete list is not yet available. To fill this gap, methods to computationally predict mitochondrial proteins from amino acid sequence have been developed and are widely used, but unfortunately, their accuracy is far from perfect. Here we describe MitoFates, an improved prediction method for cleavable N-terminal mitochondrial targeting signals (presequences) and their cleavage sites. MitoFates introduces novel sequence features including positively charged amphiphilicity, presequence motifs, and position weight matrices modeling the presequence cleavage sites. These features are combined with classical ones such as amino acid composition and physico-chemical properties as input to a standard support vector machine classifier. On independent test data, MitoFates attains better performance than existing predictors in both detection of presequences and in predicting their cleavage sites. We used MitoFates to look for undiscovered mitochondrial proteins from 42,217 human proteins (including isoforms such as alternative splicing or translation initiation variants). MitoFates predicts 1167 genes to have at least one isoform with a presequence. Five-hundred and eighty of these genes were not annotated as mitochondrial in either UniProt or Gene Ontology. Interestingly, these include candidate regulators of parkin translocation to damaged mitochondria, and also many genes with known disease mutations, suggesting that careful investigation of MitoFates predictions may be helpful in elucidating the role of mitochondria in health and disease. MitoFates is open source with a convenient web server publicly available.
Mitochondria not only function as the provider of ATP but also play crucial roles in the metabolism of amino acids and lipids, the biosynthesis of iron-sulfur clusters, cell signaling pathways, and apoptosis in eukaryotic cells. Moreover, mitochondrial dysfunction has been implicated in a wide variety of medical conditions such as muscle and neurodegenerative disease, cardiovascular disease, diabetes, and cancer (1).
Obtaining the complete proteome of mitochondria is an essential step toward fully understanding its role in health and disease. To this end, ∼900 (in yeast) and 1100 (in mouse) mitochondrial proteins have been identified by large-scale proteomics analyses (2, 3); and compiled with other relevant mitochondrial proteomics data in useful databases such as MitoCarta (3) and MitoMiner (4). However, these lists are probably not yet complete, and indeed fungi and animal mitochondria have been estimated to host ∼1000 and ∼1500 distinct proteins, respectively (5). Thus, many mitochondrial proteins seem to remain undiscovered even in model organisms. If high accuracy can be achieved, prediction of mitochondrial proteins from primary sequence can save time and effort by identifying promising novel candidate mitochondrial proteins.
The vast majority of mitochondrial proteins are encoded in the nuclear genome and imported by translocator complexes in the mitochondrial membranes. These mitochondrial proteins can be classified into two groups based on the type of targeting signal they contain: an N-terminal cleavable targeting signal (presequence); or a noncleavable, internal targeting signal (6). A recent proteomic analysis of yeast estimated that ∼70% of mitochondrial proteins possess a presequence (7). Thus, improved prediction of presequences should contribute to detecting undiscovered mitochondrial proteins.
Presequences reside in the first 10–90 N-terminal residues, exhibit a high composition of arginine and near absence of negatively charged residues (8, 9). Proteins containing such presequences are translocated by the TOM and TIM protein complexes in the outer and the inner membranes, respectively (6, 10, 11). Tom20 and Tom22 in the TOM complex are reported to initiate import of these proteins by recognizing presequence segments capable of forming a local amphiphilic α-helical structure with hydrophobic residues on one face and positively charged residues on the opposite face (6, 12, 13). Widely used prediction tools such as MitoProt, TargetP, Predotar, and TPpred2 were developed with these properties of presequences in mind (14–17).
The cleavage of mitochondrial protein presequences is an important event implicated in efficient protein import (18) and disease (19). Upon import into mitochondria, most presequences are cleaved off by the heterodimer mitochondrial processing peptidase (MPP)1 in the matrix, and some of them subsequently further cleaved by intermediate peptidases such as Oct1 (20) and the recently discovered Icp55 (7). Although methods exist to predict these cleavage sites, their accuracy leaves much room for improvement (7, 21). Because the correct primary sequence of mature proteins is a prerequisite for precise structural modeling, improving the accuracy of cleavage site prediction should be useful for planning protein crystallography experiments or other structural studies of mitochondrial proteins. Also, accurate in silico prediction of the mature N-termini of mitochondrial proteins could in principle be used to improve the identification of N-terminal peptides in shotgun proteomics.
In this study, we describe MitoFates, a novel method for mitochondrial presequence and cleavage site prediction. MitoFates formulates presequence prediction as a binary classification problem, employing a standard support vector machine (SVM) classifier. Our contribution is the preparation of an updated data set incorporating some recent proteomic data; and the selection of classical and novel sequence features such as amino acid composition, physicochemical properties, a novel positive amphiphilicity score, novel presequence motifs, and refined position weight matrices (PWMs) modeling peptidase cleavage sites. On the task of discriminating between presequences and nonpresequences, MitoFates achieves a true positive rate of 76% at a false positive rate of only 1.7%, improving significantly on previous methods. Moreover, MitoFates predicts the position of cleavage sites with an error rate of only ∼29% versus ∼47% for the best previous method.
To investigate the potential of MitoFates to reveal interesting candidate mitochondrial proteins, we looked for undiscovered mitochondrial proteins among 42,217 human proteins (including isoforms such as alternative splicing or translation initiation variants), and obtained 580 candidate undiscovered mitochondrial proteins. Open source software downloads and a convenient MitoFates web server is available at http://mitf.cbrc.jp/MitoFates/.
MATERIALS AND METHODS
Training and Test Data Set
Presequence Prediction
We prepared a data set of 759 presequence containing mitochondrial proteins by combining the data sets of TargetP and Predotar (containing proteins from various eukaryotes) with presequences identified via recent mitochondrial N-terminal proteome measurements on S. cerevisiae (7), and on A.thaliana and O.sativa (22). Based on an initial inspection of the data, when developing MitoFates we decided to discard any putative mature N-termini from these studies that cannot be explained as the product of cleavage by MPP with an arginine at the −2 position (possibly followed by secondary cleavage by Icp55 or Oct1). We made this decision because for the rest of the data we failed to discern any overall pattern in either the local sequence surrounding the putative cleavage sites or the distance from the original N-termini. Presumably, this non-R-2 site data includes proteins processed by proteases such as IMP and m-AAA, possibly some noncanonical MPP cleavage and probably some nonspecific degradation products. Although we did not include these sites when developing MitoFates itself, we did include them in an exploratory clustering experiment described below. Note that we did include plant mature N-termini with an arginine at the −3 position as they could plausibly be explained as the product of canonical MPP cleavage followed by an additional cleavage of one N-terminal residue by a plant counterpart to yeast Icp55. For negative examples, we used 6310 nonmitochondrial proteins with clear UniProt annotation of subcellular localization and 108 noncleaved yeast mitochondrial proteins (7). These sequences (taken from UniProt (23) ver. 2012 10) were selected such that no pair shared more than 80% mutual sequence identity within the positive or negative data sets. To compare the prediction performance of MitoFates with previous methods, we prepared an independent test data set consisting of 78 mitochondrial proteins possessing a presequence and 8934 nonmitochondrial proteins; in such a way that the sequence identity between training and test data sets and within the positive and negative data sets is less than 25%.
Cleavage Site Prediction
We extracted cleavage sites from the proteomic analysis experiments on S. cerevisiae (7) and A.thaliana and O.sativa (22), excluding N-termini inconsistent with canonical MPP cleavage as described in the previous section. As was done for the TargetP (15) data set, we extracted sequences from their original N-terminus to three residues after their cleavage site, and applied redundancy reduction at 40% identity in each taxonomic group. Although the original proteomic data for yeast shows multiple cleavage sites for some proteins, we chose to use only the most frequently observed site in each protein. Although a few proteins that do not contain arginine at −2, are annotated as cleaved by MPP and other intermediate proteases, we excluded them. For our metazoan data set, we extracted presequences as annotated in UniProt. Similar to the yeast and plant cases, we extracted presequences with an arginine at the −2, −3, or −10 positions. In this way, we obtained 104, 76, and 161 MPP cleavage sites in yeast, plant, and animal, respectively, after applying sequence redundancy reduction at a 40% identity level. As an additional human test data set we also extracted presequences with canonical R-2 MPP cleavage sites from the recently developed DegraBase database (24), again applying sequence redundancy reduction at a rate of 40%. We prepared the negative data (sites not cleaved by MPP) by extracting sequences from noncleaved R-2 sites in the positive data set (i.e. we simplify the task to discrimination between arginines belonging to MPP cleavage sites versus other arginines).
Prediction Features
Amino Acid Composition
It has been observed that presequences exhibit biased amino acid composition, with a high frequency of arginine and few negatively charged residues (8, 9). Thus, for presequence prediction we include the frequency of each of the 20 standard amino acids in the first 30 N-terminal residues in our feature set. Moreover, we include the 400 possible dipeptides and the 400 possible skip-two dipeptides (A1xxA2, where x is any residue). This was motivated by the suggestion that the formation of specific secondary structure motifs is important to presequence recognition (25) and the fact that secondary structure is known to correlate with dipeptide and skip-two dipeptide frequency. When predicting cleavage sites, the feature set is computed on the part of the sequence up to the candidate cleavage site, which can be quite short and therefore without adjustment, the composition features would be very sparse. For example, the shortest presequence in our data set is only seven amino acids long, so when computing amino acid composition on this candidate presequence, at least 13 of out 20 possible amino acids would have zero frequency. For cleavage site prediction, we alleviated this problem by smoothing the amino acid frequencies with a 20-component Dirichlet mixture model prior (26) and not using dipeptide features.
Local Sequence Models (Position Weight Matrices) of MPP, Icp55, and Oct1 Cleavage Sites
A large majority of presequences are cleaved by MPP, and many of those by secondary proteases as well. MPP cleavage sites display local sequence tendencies (20), the most conspicuous one being the presence of arginine in the −2 position in nearly all cases, consistent with electrostatic interaction between this arginine and negatively charged residues in MPP (27). After cleavage by MPP, the secondary proteases Oct1 and Icp55 further cleave some presequences, removing eight residues or a single residue, respectively (7). It is reasonable to hope that explicit modeling of this two-step process might improve the prediction of those presequences. Thus, we generated a consensus Position Weight Matrix (PWM) based on the frequencies of amino acids between the −4 position and the +5 position of training set sequences aligned by cleavage site. As with the amino acid composition values described above, we smoothed the observed frequencies in each column of the PWM with a 20-component Dirichlet mixture model (26). The PWM score is calculated as the log-odds ratio between those smoothed frequencies and a background composition based on the mature region of cleaved mitochondrial proteins. To predict if putative MPP cleavage sites are further cleaved by Oct1 or Icp55, we employed PWMs based on the cleavage sites of those peptidases in the training data. By inspection of the training data, we chose the range of positions covered by the PWMs to be [+1, +4] (length 4) and [+1, +2] (length 2) for MPP+Oct1 and MPP+Icp55, respectively (Fig. 1A). Because plant data was rather limited and PWMs require a large number of parameters (19 per column), we chose to use PWMs trained on the more abundant yeast data, even when making predictions for plant proteins (however, we did retrain the length distribution as described below).
Fig. 1.
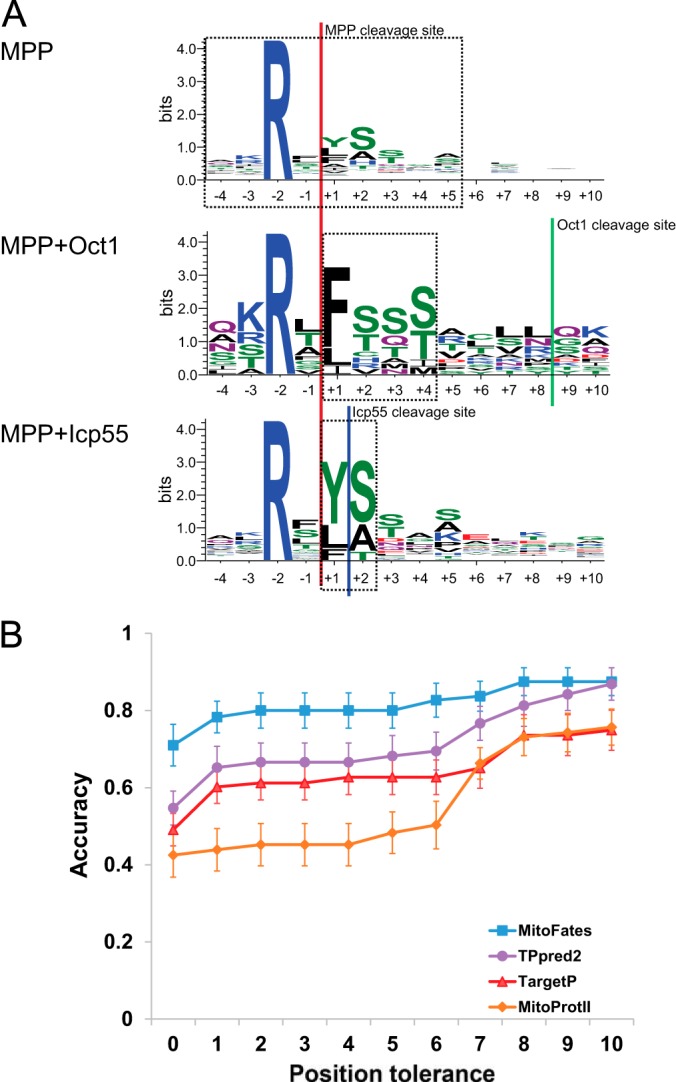
Local sequences and prediction performance of cleavage sites. A, Sequence logo of MPP cleavage sites partitioned into three classes (MPP only, MPP+Icp55, MPP+Oct1) based on recent proteomics data. The dashed line boxes show the range of positions covered by the PWMs for MPP, Oct1, and Icp55. B, Cleavage site accuracy comparison on the yeast data set. Error bars show the standard error of mean estimation based on 10-fold cross validation (only MitoFates is retrained, the other tools are used as distributed without retraining but their prediction accuracy still varies between test folds).
Length Distribution of Presequences
Presequence length is variable, but usually falls within a certain range. To utilize this information we weighted scores at each position according to the probability of finding a cleavage site at that distance from the N-terminus. We implemented this by modeling the presequence length distribution with a mixture model of Gamma distributions, estimating parameters using an Expectation-Maximization learning algorithm (28) on the training data (for metazoan presequence length we prepared a training set based on UniProt annotation). Because yeast, plant, and metazoan presequences each showed distinct length distributions (supplemental Fig. S1A), we chose to learn three separate mixture models. Based on the results of a Kolmogorov-Smirnov goodness of fit test, we chose a 1-component model for yeast and 3-component models for plant and metazoa.
Positively Charged Amphiphilicity Score for Presequence Detection
We defined a simple scoring function that assigns high scores to positions that could form a local α-helical secondary structure with high hydrophobicity on one face and positively charged residues on the opposite face. The scoring scheme is a weighted sum of the standard hydrophobicity moment (29) and a positive charge moment, as expressed in this formula:
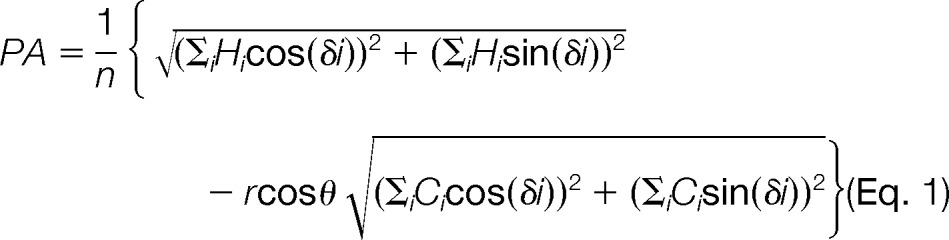 |
where r is a scaling parameter to balance between hydrophobic moment and charge moment, and θ is the angle between the hydrophobic and charge moment vectors (defined so that its effect is maximized when the two moments point in exactly the opposite direction). Hi indicates the hydrophobicity of the ith residue by the Aboderin hydrophobicity scale (30) and similarly Ci indicates the charge (RKH:1, DE: −1, otherwise: 0) of the ith residue. The PA score is normalized by window length n. We used the training data set to optimize r and the helix angle parameter δ empirically, obtaining values of 8.5 and 96° respectively. To compute a single feature score from an amino acid sequence, standard hydrophobicity moments are computed for all possible window sizes of 10 to 20 on the N-terminal 30 residues, and then the PA score is calculated for the window with the maximum hydrophobic moment.
Frequently Observed Hexamers in Presequences
We looked for short sequence motifs that are enriched in the N-terminal region of presequences and hopefully relate to presequence recognition. Using a subset of the training data, we experimented with parameters such as the length of the candidate motifs and the range in which to look for them. After preliminary analysis with motif lengths of five to seven and different N-terminal region lengths, we chose a motif length of six and the first N-terminal 90 residues as the search range. The training subset we used consists of 317 mitochondrial proteins with presequences and 3897 nonmitochondrial proteins sharing no more than 25% sequence identify in their N-terminal regions. To reduce the size of the motif model space (and therefore potentially gain statistical power) we grouped the standard 20 amino acids into five characters based on their physicochemical properties: hydrophobic ϕ (L, F, I, V, W, Y, M, C, A); basic β (R, K, H); acidic α (E, D); polar σ (S, T, N, Q); and secondary structure breaker γ (P, G). We partitioned the N-terminal 90 residues into three blocks of 30 residues each and simply counted the number of sequences with an exact match to each of the 56 possible hexamer motifs in each block of the mitochondrial and nonmitochondrial proteins. We used a Fisher's exact test to compute p values, correcting for multiple testing with LAMP (31), a more sensitive (but still rigorous) method to compute the Bonferroni correction factor than simply multiplying uncorrected p values by 56. We selected motifs with p value < 10−5, yielding a total of 14 hexamers that were all found in the first N-terminal 30 residue block. Thus, we defined 14 binary features taking a value of 1 or 0 based on the presence or absence of the given motif in the first 30 residues. Additionally we defined a combined motif score, defined as the sum of −log10 (p value) for each motif hit. For example, if two motifs were found in the first 30 residues of a query sequence, one of which had a LAMP corrected p value of 10−5 and another of which had a LAMP corrected p value of 10−7 within the training data, the value of the combined motif score feature for that query sequence would be 12.
Physicochemical Propensities
Proteins bound for the endoplasmic reticulum (usually) and peroxisome (often) possess predictable sorting signals in their N- and C-terminals, respectively. To distinguish between mitochondrial presequences and such signal sequences, we partition the N- and C-terminal 90 residues into six blocks of 15 residues, and then compute the average Aboderin hydrophobicity (30), α-helical and β-strand periodicity scores (32, 33), and the density of basic (K, R, H), acidic (D, E), small polar (S, T), aromatic (W, Y, F), and secondary structure breaker (P, G) residues for each block. We also include those compositions computed over the entire sequence in the feature set. Finally, we include four physicochemical propensity based features designed for signal peptide detection as we described in a previous study (33).
For cleavage site prediction, we defined four physicochemical features: average net charge, average hydrophobicity measured by Aboderin scale, number of [KR] residues, and number of [DE] residues. For each potential cleavage site, these features are computed for the N-terminal region up to that site.
Prediction Method
We adopted the Support Vector Machine (SVM) classifier implemented in LIBSVM 3.0 with RBF-kernel (34) for both presequence (presence versus absence) and cleavage site prediction. For cleavage site prediction, we did not use the hexamer motifs or the PA score used in presequence prediction. Given the way we defined positively charged amphiphilicity and trained the hexamer motifs, these features largely reflect presequence recognition rather than cleavage site selection. Because both the hexamer motifs and positively charged amphiphilicity are defined in terms of local sequence and it is conceivable that presequence recognition is somehow coupled to proteolytic cleavage, it might be interesting to see if they can be used to improve cleavage site prediction but we did not pursue that possibility in this study. When predicting cleavage sites, MitoFates computes the prediction score of each position and considers the maximum scoring position to be a MPP cleavage site. MitoFates then uses the PWMs for Oct1 and Icp55 cleavage to predict if the site is further cleaved by one of them. For plants, MitoFates does not consider the possibility of Oct1 cleavage.
Performance Measures
We quantify prediction performance in several ways, including Precision-Recall curve, Matthews correlation coefficient (MCC), and ROC AUC. We used the independent test data to measure presequence prediction. For evaluation of cleavage site prediction, we used 10-fold cross-validation on the yeast and plant data sets, and performance on the independent DegraBase data set for human proteins. Following standard definitions: precision is the fraction of actual presequences among predicted presequences and recall is the fraction of presequences that are successfully predicted. The Matthews correlation coefficient (MCC) (35) is the Pearson's correlation coefficient of the binary vector of the true labels (1 for presequences, 0 for nonpresequences) and the predicted labels. The Area Under the Curve (AUC) of a Receiver Operating Characteristics (ROC) graph is a widely used metric to evaluate binary classification accuracy (36). ROC AUC estimates the probability that a randomly chosen presequence attains a higher prediction score than a randomly chosen nonpresequence. ROC AUC ranges from 0 to 1.0, with perfect prediction yielding 1.0, uninformative prediction 0.5, and perfectly wrong prediction 0.0. We generated the ROC plots in the usual way by sorting instances according to their prediction scores, and then plotting true positive rate (y axis) versus false positive rate (x axis).
Clustering of Yeast Presequences
To explore the trends in our data set, we conducted a clustering analysis of 243 yeast presequences, obtained by applying 40% identity reduction to recent proteomics data (7). Unlike our cleavage site prediction data set, for clustering we included presequences regardless of consistency with canonical R-2 MPP cleavage, although to reduce noise we did remove putative presequences from proteins annotated with a nonmitochondrial localization site. The features we used for clustering are: length, average net-charge, positively charged amphiphilicity score (PA), MPP cleavage site score (PWM score without weighting by the length distribution), the compositions of four charged residues (Arg, Lys, Asp, and Glu), and an ortholog multiple alignment based measure of the degree of evolutionary sequence conservation shown to be predictive of presequences in our earlier work (37) but this time adopting the Jensen-Shannon divergence (38) as the function applied to each column of the multiple alignments. We computed PWM scores for all possible cases: MPP only, MPP+Oct1, and MPP+Icp55; and treated the maximum score as the MPP cleavage score. Because evolutionary sequence conservation scores fluctuate greatly from column to column, we summarized these as two features averaged over the first 18 and 36 N-terminal residues, respectively (36 was chosen as the average yeast presequence length). We clustered this data by application of a Gaussian mixture model (39), and model parameter estimation by the EM algorithm (28) as implemented in Weka (40).
To determine the number of clusters we followed the default Weka criteria. In this empirical wrapper procedure, the data is randomly split into 10 partitions for 10-fold cross validation and the average log-likelihood of the test partition is computed. Starting with one cluster, the number of clusters is incremented until the test partition average log-likelihood stops increasing.
RESULTS
Prediction of Mitochondrial Presequences
We benchmarked presequence prediction performance between our predictor (MitoFates) and four previously developed predictors: TPpred2, TargetP (ver. 1.1), Predotar (ver. 1.03), and MitoProtII (ver. 1.101) on the independent test data containing 78 presequences described in Methods. Fig. 2A shows the 11 point precision-recall curve (PR-curve) of each predictor averaged over 10 random selections of 500 negative test set proteins. MitoFates achieves an average precision of 84% on the PR-curve, outperforming TPpred2, Predotar, TargetP, and MitoProtII, which obtained an average precision of 81%, 79%, 78%, and 74%, respectively. In particular, MitoFates attains better precision for recall values of 50–80% (in this range the average precision of MitoFates, TPpred2, Predotar, TargetP and MitoProtII is 91%, 81%, 82%, 77%, and 77%, respectively). The ROC AUC of MitoFates is also superior to other predictors (Table I). For MitoFates, we focused on two prediction cutoffs (0.5 and 0.385) based on a 5-fold cross-validation test within the training data set (supplemental Fig. S2); 0.5 is the default cutoff determined by LIBSVM (34) with a precision and recall of 0.83 and 0.73, respectively; and 0.385 corresponds to a precision and recall of 0.79 and 0.80. At both prediction cutoff values, MitoFates' Matthews correlation coefficient (MCC) is better than those of other predictors at their default cutoffs. In addition, the PR-curve and ROC AUC of MitoFates is better than TargetP and Predotar even when MitoFates is trained on their training data set (supplemental Fig. S3), suggesting that our novel features contribute to improved prediction accuracy (the training data set of TPpred2 overlapped to a large extent with our test data so we did not do this experiment on the TPpred2 training data). However, the PR-curve and ROC AUC of MitoFates trained on those data sets is inferior to those of MitoFates trained on its original data set, suggesting that the updated MitoFates training data also contributes to its superior performance.
Fig. 2.

MTS discrimination performance comparison between MitoFates and previous predictors on an independent test data set. A, Comparison by PR-curve. B, True positive rate versus false positive rate. C, Statistical significance (vertical axis) of the true positive rate difference between MitoFates and other predictors plotted against false positive rate. For each input sequence the predictors output both a score (for TPpred2 we extracted the GRHCRF-scores from their software) and a label (mitochondrial, ER, or other); the dashed lines show performance based purely on the scores, and the solid lines always count mislabeled mitochondrial proteins as false negatives and nonmitochondrial proteins with nonmitochondrial labels as true negatives, regardless of score.
Table I. Comparison of ROC AUC and MCC on an independent test data set.
| Predictor | ROC AUC | MCC |
|---|---|---|
| MitoFates (cutoff: 0.5) | 0.954 | 0.465 |
| MitoFates (cutoff: 0.385) | 0.446 | |
| TPpred2 | 0.948 | 0.355 |
| Predotar | 0.939 | 0.304 |
| TargetP | 0.933 | 0.242 |
| MitoProtII | 0.941 | 0.217 |
To more rigorously evaluate MitoFates performance relative to previous methods, we tested the statistical significance of the number of true positives at each false positive rate using McNemar's test for paired data (41). As shown in Fig. 2B, the true positive ratio of MitoFates as always equal or greater than the best competitor and this difference is statistically significant near a false positive rate of 1.7%, where MitoFates achieved 76% precision (Fig. 2C). This suggests MitoFates can be a useful method to identify promising candidate presequences for experimental validation with fewer false positives than previous prediction methods.
MitoFates assigned a very high presequence score (LIBSVM estimated probability > 0.99) to five of the 8934 negative test set proteins. Interestingly, although not annotated in UniProt at the time of data set preparation, a literature search revealed that at least two of them have been reported to have mitochondria localization (human Acyl-coenzyme A thioesterase 11 and NipSnap homolog 3A) (42, 43).
We also evaluated MitoFates presequence prediction performance on an additional data set containing 226 matrix proteins obtained from recent human mitochondrial matrix proteome data (44) as a positive test. MitoFates achieved the best performance as measured by PR-curve, ROC AUC, or MCC, on this data set as well (supplemental Fig. S4), although the difference between MitoFates and the second best predictor TPpred2 is modest.
Discrimination Capability of Individual Prediction Features for Presequence Prediction
To examine the discrimination capabilities of each prediction feature, we calculated F-scores (45 and supplemental Text) and Spearman's rank correlation coefficients within the training data set for each feature. By F-score, the best feature is the score of MPP cleavage site (F-score = 0.250), and the next best four are the composition of Arg in the N-terminal 30 residues (0.217), the total hexamer motif score (0.159), the dipeptide composition of Leu-Arg (0.126), and the positively charged amphiphilicity (PA) score (0.126), respectively. Using Spearman's rank correlation coefficient as a measure of feature importance also produces the same top five features in nearly the same order (PA comes fourth and Leu-Arg fifth). Below we discuss some of these top features in some detail.
Prediction of Cleavage Site Location
As described in Methods, our cleavage site predictor uses cleavage site PWMs (Fig. 1A) and other sequence features. To evaluate the performance of MitoFates and other predictors we conducted 10-fold cross validation on a yeast presequence proteomic data set (7). For TPrepd2, MitoProtII, and TargetP we simply ran them as is, without consideration of possible overlap between their training sets and data in the test folds. To simplify this comparison we only compared proteins for which all of TPpred2, MitoProtII, and TargetP predict cleavage somewhere in the protein. This criterion leads to 70, 56, and 79 cleavage sites for yeast, plant, and human, respectively. As shown in Fig. 1B, MitoFates' prediction accuracy on the yeast data set (71%) was considerably higher than the next best predictor TPpred2 (54.7%); or equivalently, TPpred2 makes 1.56 times as many errors as MitoFates. MitoFates also predicts cleavage sites more accurately in plants and human, correctly predicting 72.2% and 51.9%, respectively, improving on the second best predictor's performance of 55.0% and 43.0% (supplemental Fig. S1B and S1C).
Interestingly, the change in accuracy when predictions are allowed to be off by a given distance offers a glimpse into the ability of each predictor to correctly predict secondary peptidase cleavage events. MitoProtII and TargetP show noticeable leaps in accuracy between 0 and 1, and between 6, 7, and 8. The leap between 0 and 1 often comes from under/over-prediction of Icp55 cleavage, whereas the leaps between 6, 7 and 8 mainly result from under/over-prediction of Oct1 cleavage, sometimes combined with over-prediction of Icp55 cleavage.
Such leaps can also be observed in the plant and human data sets as well (supplemental Fig. S1B and S1C), however, MitoFates prediction shows only a single leap between 0 and 1 in plants. Although plants do have a homolog to yeast Oct1 (At5G51540), it appears to localize to chloroplasts (46), and presequences with an arginine in the −10 position (possibly indicating cleavage by MPP+Oct1) are not prevalent in plants (22). Therefore, MitoFates does not consider Oct1-type cleavage for plants. For plant cleavage site prediction, we do take into account the length distribution of presequences in plants (supplemental Fig. S1A), which differs significantly from yeast. For the DegraBase human data set, large leaps in accuracy between 0, 1 and 6, 7, 8 are observed in all predictors including MitoFates (supplemental Fig. S1C). As with the yeast data, these leaps may largely reflect genuine mispredictions of secondary protease cleavage, but it is possible that some fraction may also be explained as intermediate MPP cleavage sites present in the data set that are actually destined for additional cleavage by human counterparts to Icp55 and Oct1.
Although a homolog or counterpart to Icp55 has not been identified in plant mitochondria, presequences with an arginine in the −3 position (suggesting MPP+Icp55-like cleavage) are prevalent. However, these sites differ from R-3 presequences in yeast. In particular, phenylalanine at the −1 position of R-3 presequences (i.e. directly after the inferred MPP cleavage site) is relatively rare in yeast R-3 presequences (Fig. 1A) because those are usually cleaved by Oct1 (yielding an R-10 presequence), but are common in plant R-3 presequences; also consistent with a lack of Oct1-like cleavage in plant mitochondria. Another difference is that plant R-3 presequences sometimes have a methionine in the −1 position, but MitoFates cannot predict them well as it uses PWM's trained on yeast data and yeast Icp55 cleavage sites to not exhibit methionine in that position. These observations suggest that MitoFates prediction of cleavage sites in plants could potentially be improved further, but because presequence cleavage site training data for plants is currently limited, we leave a careful optimization of the Icp55-like peptidase model for plants as future work.
Modification of the Hydrophobic Moment Score for Presequence Prediction
Although the ability to adopt an amphiphilic α-helix has been proposed to be important for presequence recognition (9, 47), prior to this work, attempts to use this feature for prediction have had limited success (16). To investigate this problem, we compared the distributions of maximum hydrophobic moment score (29) in the first 30, 60, and 90 N-terminal residues of proteins containing or lacking presequences. The distributions differ the most in the N-terminal 30 residues, but still overlap each other to a large extent (Fig. 3A, top). We considered one reason for this poor separation may be that the hydrophobic moment calculation does not distinguish between positive and negative charges on the polar face. Given that Tom20 and Tom22 in the TOM complex most likely recognize an amphiphilic helical local structure consisting of hydrophobic and positively charged faces (6, 12, 13), we conjectured that a score that favors positive charges on the polar face might better characterize presequences. Thus, we defined the PA score (Positively charged Amphiphilicity score) that adds a positive charge moment to the hydrophobicity moment as described under “Experimental Procedures” above. This PA score yields much better discrimination (Fig. 3A, bottom). We also note known Tom20 binding sites in the presequences Su9 of N.crassa (48) and ALDH2 of R.norvegicus (12) exhibit a high PA score (data not shown).
Fig. 3.

Positively charged amphiphilicity (PA) score and presequence specific motifs. A, Histograms of the distribution of maximum hydrophobic moment score (above) and PA score (below) in the N-terminal 30 residues of proteins with and without a presequence. B, The top six presequence specific hexamers are listed with their statistical significance, coverage in the positive, and negative training examples, PA score, and a sequence logo depicting their matches in the positive data. Arrows show the hydrophobic moments. Blue and gray indicate higher or lower PA score than the 90th percentile score over all hexamers in the positive and negative examples, respectively.
Novel Hexamer Motifs in Mitochondrial Presequences
Although no obvious consensus sequence is common to presequences, some attempts have been made to find common sequence motifs. Obita et al. proposed the consensus θφχβφφ (where θ, β, φ, and χ represent a hydrophilic, basic, hydrophobic, and any residue, respectively) based on the results of a peptide library experiment measuring the binding of rat ALDH precursor derived peptides to Tom20 (49). However, they noted that their results did not generalize quantitatively to the precursors of other proteins. Indeed their proposed motif covers only 18% of the yeast proteomic presequences (7) and only 19% of the UniProt annotation derived presequences used in this study, while unfortunately also matching 8% of the N-terminal 30 residue region of nonpresequence containing proteins. In a computational approach, motif finding based on discriminative training of Profile Hidden Markov Models (Profile HMMs) found a few tetramer motifs in yeast mitochondrial sequences (50). However, this study did not search only within presequences, but rather included the entire sequence of mitochondrial proteins with and without presequences. Judging based on visual inspection of the motif sequence logos given in their supplementary material, their top mitochondrial motif is essentially a detector for high lysine content, even though in presequences arginine is generally more enriched than lysine. TargetP (15) uses MEME (51) derived PWM motifs to model peptidase cleavage sites but do not report motifs that may be related to other stages of presequence recognition such as Tom20 binding.
Based on our survey of previous work summarized above, we felt that it would be useful to make a new attempt to find presequence-specific motifs. For simplicity, we choose a simple motif model consisting of a string from the degenerate alphabet: hydrophobic φ (L, F, I, V, W, Y, M, C, A), basic β (R, K, H), acidic α (E, D), polar σ (S, T, N, Q), and secondary structure breaker γ (P, G). We chose a reduced alphabet in order to reduce the size of the hypothesis space, and thus, potentially gain statistical power. Of course condensing the alphabet comes at the cost of possibly throwing away important information. We hope this affect was ameliorated by our particular choice of grouping, motivated by consideration of the physico-chemical properties of amino acids in the light of what hints we currently have regarding the mechanism of presequence recognition. To avoid learning motifs specific to a particular family of proteins we conducted this search on the nonredundant (< 25% sequence identity) training data, using LAMP (31), a recently introduced, highly sensitive multiple hypothesis testing method. As an additional control, we performed the same motif search procedure on three 30-residue blocks in the N-terminal 90 region (1–30, 31–60, and 61–90).
We found 14 statistically significant (p value < 10−5) hexamers (Fig. 3B, supplemental Fig. S5), all in the first 30 residue block, and all enriched in the positive (i.e. presequence) examples, rather than the negative examples. Interestingly, most of these hexamers have a PA score above 2.72 (the 90th percentile score over all hexamers in the positive and negative examples), indicating the potential to form amphiphilic helixes with a positively charged hydrophilic face, and we speculate that they may reflect Tom20 binding. On the other hand βφφσσσ, one of the hexamers with a lower PA score, matches the MPP cleavage sites of presequences subsequently cleaved by Oct1 (Fig. 1A).
Presequences exhibit characteristic amino acid composition biased toward arginine and against negatively charged residues. To clarify how the hexamer motifs found by our procedure are influenced by amino acid composition, we reran the motif finding procedure using scrambled sequences of positive examples as negative examples. In 100 random trials, eight of the fourteen hexamers were observed fewer than five times (supplemental Table S1) and therefore these appear to largely reflect amino acid composition bias. On the other hand, the motifs φφσβφφ, φφβσφφ, and βφφσσσ are observed 100 times, 88 times, and 73 times, respectively, indicating that they are specific presequence motifs, reflecting more than amino acid composition. As mentioned above, βφφσσσ matches the MPP cleavage sites of presequences subsequently cleaved by Oct1. To look for a hint into the nature of the other two hexamers, we investigated their matching positions. Interestingly, the other two hexamers frequently occur exactly at the N-terminus (23 and 15 times, respectively) or at the position directly following the N-terminus (10 and 12 times). We confirmed this trend holds under 25% sequence identity redundancy reduction as well (supplemental Fig. S6). One of the matching proteins, Potato formate dehydrogenase, has a φφσβφφ motif (MAMSRVA) near its N terminus (supplemental Table S2A) and it has been reported that deletion of the first and second residues (Met and Ala) or mutation of the third residue (Met) inhibited mitochondrial targeting of this protein (52). Thus, those N-terminal φφσβφφ sequences might constitute a mitochondrial targeting sequence. We note that N-terminal φφβσφφ matches are also frequently found in fungi mitochondrial proteins (supplemental Table S2B) and it would be interesting to experimentally test if these function as mitochondrial targeting signals.
Because arginine is highly enriched in presequences and has a central role in determining MPP cleavage sites, we also tried our motif finding procedure with arginine separated as a distinct character, i.e. with a character set of: hydrophobic, arginine, other basic β (K, H), acidic, polar, and secondary structure breaker. In general, the results were similar to when R, K, and H were grouped together as one (note that the amino acid matching β usually was R anyway, as evident in the sequence logos in supplemental Fig. S5). Rerunning the sequence scrambling test under this alphabet, φφσRφφ is observed in all 100 trials suggesting that arginine is preferred over other basic residues in this motif and that this preference is not simply a consequence of the generally high overall composition of arginine in presequences.
Recently, a long presequence pSu9 was reported to contain two distinct Tom20-binding elements; a Tom20 binding element in the N-terminal half and an element important for efficient protein import in the C-terminal half (48). Thus, we attempted to find motifs characteristic of long presequences by preparing a data set of 102 mitochondrial proteins with long presequences (more than 40 amino acids) and searching for statistically significant hexamers in their N-terminal 90 residues (divided in three blocks of 30 residues each) with LAMP (31). However, even with a lenient p value threshold of 0.05, we found no significant hexamer motifs in the second and third blocks.
Cluster Analysis for Yeast Mitochondrial Presequences
MitoFates improves the state-of-the-art in presequence prediction, but unfortunately still fails to predict a sizable number of presequences. Visual inspection of these false negatives reveals that they usually have fewer positively charged residues or poor score for MPP cleavage, suggesting they may belong to a different class of presequences than the true positives. To investigate this, we clustered 243 yeast presequences as described in Methods. The results suggests yeast presequences can be grouped into at least three clusters (supplemental Table S3), as visualized by primary component analysis (PCA) in Fig. 4A. The largest cluster (cluster I, blue in Fig. 4A) consists of 144 presequences that are strongly enriched for arginine and contain almost no negatively charged residues, exhibit typically low conservation (i.e. average value for presequences), a relatively well defined length distribution centered at an average of 25 residues, high PA score, and significantly higher MPP cleavage scores than other presequences. These properties are consistent with known features of presequences. However, the two remaining clusters differ in some ways from the classical view of presequences.
Fig. 4.

The three clusters of yeast presequences. A, Presequence feature vectors, colored by cluster, shown as mapped to three dimensions by PCA. B, Box plots of the nine features used for clustering are shown for each cluster.
The second largest cluster (cluster II, red in Fig. 4A) contains 64 presequences. Their level of evolutionary conservation and PA scores are similar to cluster I. However, they are much longer (average length of 60 residues), have lower net charge because of lower arginine and higher D+E composition, and have significantly lower MPP cleavage scores than cluster I (Fig. 4B). The low average MPP cleavage score can be explained by a high proportion of presequences lacking a canonical MPP cleavage site arginine (the fraction of the proteins containing arginine at the −2, −3, or −10 position is 84%, 30%, and 9%, in clusters I, II, and III, respectively). Overall amino acid composition might be expected to be less biased for long presequences, simply because the average is taken over a longer region. Nevertheless, the low MPP cleavage scores for presequences in cluster II suggests that some may be cleaved by other proteases. In fact, cluster II includes the presequences of Ccp1, MrpL32, Cy1, and Gut2; known substrates of the inner membrane proteases m-AAA and Imp (11). Cluster II also contains Imo32, which is cleaved by MPP and Oct1 (53), but at a highly atypical MPP cleavage site with a cysteine substituted for the nearly invariant arginine at the −2 position. Like other cluster II presequences, Imo32 is longish (38 residues) but has relatively few arginines (only two).
The 35 presequences in the third cluster (cluster III, yellow in Fig. 4A) differ the most from other presequences in their high evolutionary sequence conservation. Like cluster II, they exhibit a lower average net-charge than cluster I, and in fact 40% of these presequences have a negative net-charge. We did not use the MitoFates 14 presequence hexamer motifs during clustering, but noticed that cluster III sequences have very few matches to them (56%, 42%, and 14% of precursors match at least one hexamer motif in clusters I, II, and III, respectively). Interestingly, 13 of the 35 presequences in cluster III are derived from the mitochondrial proteins annotated with dual localization or nonmitochondrial localization in UniProt. Low net charge and PA score are consistent with previously reported characteristics of dual-localized mitochondrial proteins (54). Cluster III also includes some presequences with higher average net-charge (enriched in lysine rather than arginine). Most (six) of these are ribosomal protein presequences. However, ribosomal proteins presequences are also found in the other clusters (23 and six of them in Cluster I and II, respectively).
Although not perfect, MitoFates can predict typical presequences, like those found in cluster I, relatively reliably. To further improve in silico prediction of mitochondrial localization from amino acid sequence, it will be necessary to better characterize the remaining presequences. Also, there is a need to develop accurate prediction methods for mitochondrial proteins localized without the use of presequences, via internal or C-terminal targeting signals (55, 56).
Human Proteome Analysis by MitoFates
Analysis of Human Mitochondrial Intermembrane Space Proteome
Most presequence bearing proteins are localize to the matrix, but some localize to the inner membrane via arrest of a hydrophobic segment during translocation. In addition, some proteins are released to the intermembrane space (IMS) after proteolysis by inner membrane proteases (e.g. HtrA2 and Opa1/Mgm1) (10). Conversely, some IMS proteins, such as Tim9 and Tim10 localize to the IMS via alternative pathways (6, 11) and it is unclear what portion of IMS proteins localize via presequence dependent translocation. Fortunately, a recent proteomics advance (57) gave us the opportunity to obtain 127 human proteins in or at least partially accessible from the intermembrane space (IMS) and examine the proportion of them that are predicted by MitoFates to have presequences (supplemental Table S4). MitoFates predicted 43 of the 127 proteins to have presequences, of which 24 are annotated or predicted as single-pass membrane proteins and two as multiple-pass membrane proteins. Of the remaining 17 proteins, eight contain N-terminal hydrophobic segments. Considering the transport pathways to the inner membrane and IMS mentioned above this result seems reasonable.
Candidate Undiscovered Mitochondrial Proteins in the Human Proteome
According to recent estimates (5), animal mitochondria are expected to contain proteins from ∼1500 different genes. In MitoCarta, the most comprehensive animal mitochondria proteomics study to date, Pagliarini et al. (3) identified 1098 mouse mitochondrial proteins with an estimated false positive rate of 10%. Thus, hundreds of mitochondrial proteins may remain undiscovered. Under its default threshold (0.385 corresponding to an estimated 79% precision and 80% recall), MitoFates predicts 1847 human proteins (from 1167 genes) to contain mitochondrial targeting presequences (supplemental Table S5), including 580 predicted presequence genes (851 proteins) without annotation as mitochondrial proteins in either UniProt or Gene Ontology (supplemental Table S6). We hope that this list of predicted mitochondrial presequence proteins (including isoforms) will be a useful resource for prioritizing experimental investigation of novel candidates.
Candidate Mitochondrial Proteins with Differentially Localized Isoforms
In humans and many other eukaryotic organisms, most protein genes produce multiple isoforms via mechanisms such as alternative splicing and alternative translation initiation. Many cases are known in which isoforms of the same gene exhibit differential subcellular localization. For example, aldehyde dehydrogenase 7A1 (ALDH7A1), which plays an important role in protecting cells and tissues from hyperosmotic stress, has both mitochondrial and nonmitochondrial isoforms that are differentially expressed in a tissue-specific manner. Mitochondrial ALDH7A1 is thought to utilize an alternate upstream start codon, resulting in the addition of a presequence (58). Several genes like ALDH7A1, possessing both mitochondrial and nonmitochondrial targeted isoforms have been identified, but a comprehensive list of such proteins is not available. Thus, we applied MitoFates to search for candidate differentially localized isoforms, obtaining 517 candidate genes (supplemental Table S7), a sizable (44%) percentage of predicted mitochondrial genes. In about 90% of these candidates, the maximum score difference between isoforms is larger than 0.4, reflecting major changes in the N-termini of those sequences (supplemental Fig. S7).
Predicted Mitochondrial Proteins Related to Human Disease
Mitochondrial disorder is implicated in a wide range of diseases, including Parkinson's disease, a neurodegenerative disease affected by mitochondrial dysfunction. Recent studies reported that the Parkinson's disease associated genes PINK1 and parkin function in selective degradation of mitochondria (mitophagy) preventing the accumulation of dysfunctional mitochondria. PINK1 is rapidly degraded in healthy mitochondria but accumulates on the surface of membrane potential deficient mitochondria where it recruits parkin to ubiquitylate the damaged mitochondria (59–62). Hasson et al. (63) recently performed a screening experiment for regulators that have an impact on parkin translocation to damaged mitochondria, using genome wide small interfering RNA. In that study, SIAH3 was identified as a novel mitochondrial protein that inhibits PINK1 accumulation after mitochondrial damage by reducing parkin translocation. Encouragingly, MitoFates can predict SIAH3, even though TargetP and Predotar cannot, suggesting that MitoFates may have the ability to find other undiscovered mitochondrial proteins among candidate parkin translocation regulators. Thus, we ran MitoFates on protein sequences from supplemental Table S1 of Hasson et al. (63), yielding 72 novel mitochondrial protein candidates from 42 genes (supplemental Table S8). In addition to those 42 genes, MitoFates predicted one of the isoforms of Rhomboid-related protein 3 (RHBDL3, UniProt AC: Q495Y4) to have a presequence. This is interesting because another rhomboid protease family protein, PARL, regulates PINK1 accumulation by mitochondria membrane potential dependent cleavage of PINK1 (64). This isoform of RHBDL3 shows weak positive regulation of parkin translocation in the Hasson et al. screening study, but it is possible that the protein localizes to mitochondria and mediates parkin translocation by cleaving some proteins in a membrane potential dependent manner.
In addition, we attempted to predict undiscovered mitochondrial proteins having disease mutations by performing MitoFates prediction against the humsavar data (human polymorphisms and disease mutations data available from UniProt; http://www.uniprot.org/docs/humsavar). MitoFates predicts 31 novel candidate genes with 266 mutations related to 40 diseases (supplemental Table S9B). Also 158 genes having 1608 mutations from 164 diseases are predicted that already are annotated as mitochondrial proteins (supplemental Table S9A). The predictions include the recently identified mitochondrial protein F-box/LRR-repeat protein 4 (FBXL4) having a mutation causing mitochondrial encephalopathy (65).
Some mutations in presequences may lead to disease through mislocalization of mitochondrial proteins. For example, mitochondrial pyruvate dehydrogenase E1 component subunit alpha, somatic form (PDHA1) has a mutation R10P (the 10th residue Arg is mutated to Pro) reducing the efficiency of mitochondrial translocation of PDHA1, resulting in pyruvate dehydrogenase E1-alpha deficiency (66). Interestingly, the mutation occurs in a position corresponding to both a φφσβφφ hexamer match and the region with the maximum PA score within the N-terminal 30 residues. The mutation of positively charged Arg to the secondary structure breaker Pro might lead to loss of mitochondria localization ability of PDHA1 by disruption of the amphiphilic helix and/or reduction of net positive charge. In another example, mitochondrial DNA polymerase subunit gamma-1 (POLG) has a disease mutation R3P related to progressive external ophthalmoplegia in its predicted presequence (67). Similar to PDHA1, this mutation is located in the region with the highest PA score, suggesting that our PA score and hexamer motifs might give hints as to the likelihood that disease mutations result in protein mislocalization.
MitoFates Webserver
We developed a MitoFates webserver for easy use, available at http://mitf.cbrc.jp/MitoFates/. The default threshold is set to 0.385, corresponding to an estimated 79% precision and 80% recall. The MitoFates webserver can accept multiple protein sequences at a time. The output shows prediction results with predicted cleavage sites (of MPP and secondary proteases) and presequence hexamer motif hits having a maximum PA score (Fig. 5). More information is available at http://mitf.cbrc.jp/MitoFates/usage.html. The source code for MitoFates is also available at the website.
Fig. 5.

An example of prediction output from the MitoFates web server is shown.
DISCUSSION
Our main result is the MitoFates prediction method, which predicts presequences and their cleavage sites more accurately than previous methods. In particular, MitoFates achieves a sizable gain over previous methods in the accuracy of presequence cleavage site prediction. This improved accuracy should be useful for applications that can benefit from accurate prediction of mature protein N-termini such as crystallography and mass-spectrometry. It is interesting that our method performs well on plants and animals (supplemental Fig. S1) even though it is trained only on the sequence characteristics of yeast cleavage sites with only the length distribution of the cleavage portion customized for plants and animals. Apparently this reflects exceedingly strong functional conservation of MPP across species (20) and is consistent with previous Kohonen network analysis (8), which failed to find species-specific presequence motifs. In addition, the secondary protease Oct1/MIPEP is conserved from yeast to mammals and corresponding R-10 cleavage sites are widely observed in fungal and metazoan species. Icp55, the other secondary protease considered by MitoFates, is experimentally confirmed only in yeast, but R-3 cleavage sites are observed in all of yeast, metazoa, and plant. Therefore it seems likely that a counterpart of yeast Icp55 functions in the mitochondrial matrix of plants and metazoa. One candidate protease is XPNPEP3, a human homolog of yeast Icp55 that was recently confirmed to localize to mitochondria in renal cells (68).
We also applied MitoFates prediction to the human proteome; providing candidate lists of presequence containing proteins, protein isoforms with differential localization, and potentially disease related mitochondrial proteins. We hope these lists will prove helpful in prioritizing experiment work on those topics.
Supplementary Material
Footnotes
Author contributions: Y.F., K.T., P.H., and K.I. designed research; Y.F., J.T., S.F., and K.I. performed research; Y.F., P.H., and K.I. wrote the paper.
* This work was supported by a grant of the Platform for Drug Discovery, Informatics, and Structural Life Science, Grants-in-Aid for Scientific Research on Innovative Areas (“Matryoshka-type evolution”, No. 3308) from Ministry of Education, Culture, Sports, Science and Technology (MEXT), and Grant-in-Aid for JSPS Fellows (No. 12J06550).
 This article contains supplemental Figs. S1 to S7, Tables S1 to S9, and Text.
This article contains supplemental Figs. S1 to S7, Tables S1 to S9, and Text.
1 The abbreviations used are:
- MPP
- mitochondrial processing peptidase
- PWM
- position weight matrices
- SVM
- support vector machine
- MCC
- Matthews correlation coefficient
- AUC
- area under the curve
- ROC
- receiver operating characteristic curve.
REFERENCES
- 1. Duchen M. R., Szabadkai G. (2010) Roles of mitochondria in human disease. Essays Biochem. 47, 115–137 [DOI] [PubMed] [Google Scholar]
- 2. Reinders J., Zahedi R. P., Pfanner N., Meisinger C., Sickmann A. (2006) Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 5, 1543–1554 [DOI] [PubMed] [Google Scholar]
- 3. Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S.-E., Walford G. A., Sugiana C., Boneh A., Chen W. K., Hill D. E., Vidal M., Evans J. G., Thorburn D. R., Carr S. A., Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith A. C., Blackshaw J. A., Robinson A. J. (2012) MitoMiner: a data warehouse for mitochondrial proteomics data. Nucleic Acids Res. 40, D1160–D1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meisinger C., Sickmann A., Pfanner N. (2008) The mitochondrial proteome: from inventory to function. Cell 134, 22–24 [DOI] [PubMed] [Google Scholar]
- 6. Chacinska A., Koehler C. M., Milenkovic D., Lithgow T., Pfanner N. (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vögtle F. N., Wortelkamp S., Zahedi R. P., Becker D., Leidhold C., Gevaert K., Kellermann J., Voos W., Sickmann A., Pfanner N., Meisinger C. (2009) Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 139, 428–439 [DOI] [PubMed] [Google Scholar]
- 8. Schneider G., Sjöling S., Wallin E., Wrede P., Glaser E., von Heijne G. (1998) Feature-extraction from endopeptidase cleavage sites in mitochondrial targeting peptides. Proteins: Struct., Funct., Genet. 30, 49–60 [PubMed] [Google Scholar]
- 9. von Heijne G. (1986) Mitochondrial targeting sequences may form amphiphilic helices. EMBO J. 5, 1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neupert W., Herrmann J. M. (2007) Translocation of proteins into mitochondria. Biochemistry 76, 723–723 [DOI] [PubMed] [Google Scholar]
- 11. Schmidt O., Pfanner N., Meisinger C. (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 11, 655–667 [DOI] [PubMed] [Google Scholar]
- 12. Abe Y., Shodai T., Muto T., Mihara K., Torii H., Nishikawa S., Endo T., Kohda D. (2000) Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100, 551–560 [DOI] [PubMed] [Google Scholar]
- 13. Yamano K., Yatsukawa Y., Esaki M., Hobbs A. E., Jensen R. E., Endo T. (2008) Tom20 and Tom22 share the common signal recognition pathway in mitochondrial protein import. J. Biol. Chem. 283, 3799–3807 [DOI] [PubMed] [Google Scholar]
- 14. Claros M. G. (1995) MitoProt, a Macintosh application for studying mitochondrial proteins. Comput. Appl. Sci. 11, 441–447 [DOI] [PubMed] [Google Scholar]
- 15. Emanuelsson O., Nielsen H., Brunak S., von Heijne G. (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016 [DOI] [PubMed] [Google Scholar]
- 16. Small I., Peeters N., Legeai F., Lurin C. (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4, 1581–1590 [DOI] [PubMed] [Google Scholar]
- 17. Savojardo C., Martelli P. L., Fariselli P., Casadio R. (2014) TPpred2: improving the prediction of mitochondrial targeting peptide cleavage sites by exploiting sequence motifs. Bioinformatics 30, 2973–2974 [DOI] [PubMed] [Google Scholar]
- 18. Greene A. W., Grenier K., Aguileta M. a., Muise S., Farazifard R., Haque M. E., McBride H. M., Park D. S., Fon E. a. (2012) Mitochondrial processing peptidase regulates PINK1 processing, import, and Parkin recruitment. EMBO Rep. 13, 378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Driest S. L., Gakh O., Ommen S. R., Isaya G., Ackerman M. J. (2005) Molecular and functional characterization of a human frataxin mutation found in hypertrophic cardiomyopathy. Mol. Genet. Metab. 85, 280–285 [DOI] [PubMed] [Google Scholar]
- 20. Gakh O. (2002) Mitochondrial processing peptidases. Biochim. Biophys. Acta 1592, 63–77 [DOI] [PubMed] [Google Scholar]
- 21. Candat A., Poupart P., Andrieu J.-P., Chevrollier A., Reynier P., Rogniaux H., Avelange-Macherel M.-H., Macherel D. (2013) Experimental determination of organelle targeting-peptide cleavage sites using transient expression of green fluorescent protein translational fusions. Anal. Biochem. 434, 44–51 [DOI] [PubMed] [Google Scholar]
- 22. Huang S., Taylor N. L., Whelan J., Millar a. H. (2009) Refining the definition of plant mitochondrial presequences through analysis of sorting signals, N-terminal modifications, and cleavage motifs. Plant Physiol. 150, 1272–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bairoch A. (2007) UniProtKB/Swiss-Prot. Methods Mol. Biol. 406, 89–112 [DOI] [PubMed] [Google Scholar]
- 24. Crawford E. D., Seaman J. E., Agard N., Hsu G. W., Julien O., Mahrus S., Nguyen H., Shimbo K., Yoshihara H. A., Zhuang M., Chalkley R. J., Wells J. A. (2013) The DegraBase: a database of proteolysis in healthy and apoptotic human cells. Mol. Cell. Proteomics 12, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saitoh T., Igura M., Obita T., Ose T., Kojima R., Maenaka K., Endo T., Kohda D. (2007) Tom20 recognizes mitochondrial presequences through dynamic equilibrium among multiple bound states. EMBO J. 26, 4777–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hughey R., Krogh A. (1996) Hidden Markov models for sequence analysis: extension and analysis of the basic method. Comput. Appl. Biosci. 12, 95–107 [DOI] [PubMed] [Google Scholar]
- 27. Taylor A. B., Smith B. S., Kitada S., Kojima K., Miyaura H., Otwinowski Z., Ito A., Deisenhofer J. (2001) Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9, 615–625 [DOI] [PubMed] [Google Scholar]
- 28. Dempster A. P., Laird N. M., Rubin D. B. (1977) Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Series B 39, 1–38 [Google Scholar]
- 29. Eisenberg D., Weiss R. M., Terwilliger T. C. (1984) The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. U.S.A. 81, 140–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aboderin A. (1971) An empirical hydrophobicity scale for α-amino-acids and some of its applications. Int. J. Biochem. 2, 537–544 [Google Scholar]
- 31. Terada A., Okada-Hatakeyama M., Tsuda K., Sese J. (2013) Statistical significance of combinatorial regulations. Proc. Natl. Acad. Sci. U.S.A. 110, 12996–13001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imai K., Mitaku S. (2005) Mechanisms of secondary structure breakers in soluble proteins. Biophysics 1, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Imai K., Fujita N., Gromiha M. M., Horton P. (2011) Eukaryote-wide sequence analysis of mitochondrial β-barrel outer membrane proteins. BMC Genomics 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang C.-C., Lin C.-J. (2011) LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2, 27 [Google Scholar]
- 35. Matthews B. W. (1975) Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 405, 442–451 [DOI] [PubMed] [Google Scholar]
- 36. Fawcett T. (2006) An introduction to ROC analysis. Pattern Recogn. Lett. 27, 861–874 [Google Scholar]
- 37. Fukasawa Y., Leung R. K. K., Tsui S. K. W., Horton P. (2014) Plus ça change—evolutionary sequence divergence predicts protein subcellular localization signals. BMC Genomics 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Capra J. A., Singh M. (2007) Predicting functionally important residues from sequence conservation. Bioinformatics 23, 1875–1882 [DOI] [PubMed] [Google Scholar]
- 39. Celeux G., Govaert G. (1992) A classification EM algorithm for clustering and two stochastic versions. Comput. Stat. Data Anal. 14, 315–332 [Google Scholar]
- 40. Hall M., Frank E., Holmes G., Pfahringer B., Reutemann P., Witten I. H. (2009) The WEKA data mining software: an update. ACM SIGKDD Explor. Newsl. 11, 10–18 [Google Scholar]
- 41. Dietterich T. G. (1998) Approximate statistical tests for comparing supervised classification learning algorithms. Neural. Comput. 10, 1895–1923 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Y., Li Y., Niepel M. W., Kawano Y., Han S., Liu S., Marsili A., Larsen P. R., Lee C. H., Cohen D. E. (2012) Targeted deletion of thioesterase superfamily member 1 promotes energy expenditure and protects against obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 5417–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Verhagen A. M., Kratina T. K., Hawkins C. J., Silke J., Ekert P. G., Vaux D. L. (2007) Identification of mammalian mitochondrial proteins that interact with IAPs via N-terminal IAP binding motifs. Cell Death Differ. 14, 348–357 [DOI] [PubMed] [Google Scholar]
- 44. Rhee H. W., Zou P., Udeshi N. D., Martell J. D., Mootha V. K., Carr S. A., Ting A. Y. (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y. W., Lin C. J. (2006) Combining SVMs with various feature selection strategies. Feature Extraction, pp. 315–324, Springer, Berlin Heidelberg [Google Scholar]
- 46. Peltier J.-B., Ytterberg A. J., Sun Q., van Wijk K. J. (2004) New functions of the thylakoid membrane proteome of Arabidopsis thaliana revealed by a simple, fast, and versatile fractionation strategy. J. Biol. Chem. 279, 49367–49383 [DOI] [PubMed] [Google Scholar]
- 47. Heijne G., Steppuhn J., Herrmann R. G. (1989) Domain structure of mitochondrial and chloroplast targeting peptides. Eur. J. Biochem. 180, 535–545 [DOI] [PubMed] [Google Scholar]
- 48. Yamamoto H., Itoh N., Kawano S., Yatsukawa Y. I., Momose T., Makio T., Matsunaga M., Yokota M., Esaki M., Shodai T., Kohda D., Aiken Hobbs A. E., Jensen R. E., Endo T. (2010) Dual role of the receptor Tom20 in specificity and efficiency of protein import into mitochondria. Proc. Natl. Acad. Sci. U.S.A., 108, 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Obita T., Muto T., Endo T., Kohda D. (2003) Peptide library approach with a disulfide tether to refine the Tom20 recognition motif in mitochondrial presequences. J. Mol. Biol. 328, 495–504 [DOI] [PubMed] [Google Scholar]
- 50. Tien-ho Lin R. F. M., Bar-Joseph Z. (2011) Discriminative Motif Finding for Predicting Protein Subcellular Localization. IEEE/ACM Trans. Comput. Biol. Bioinf. 9, 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bailey T. L., Elkan C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Sec. Int. Conf. Intel. Syst. Mol. Biol. 2, 28–36 [PubMed] [Google Scholar]
- 52. Ambard-Bretteville F., Small I., Grandjean O., Colas des Francs-Small C. (2003) Discrete mutations in the presequence of potato formate dehydrogenase inhibit the in vivo targeting of GFP fusions into mitochondria. Biochem. Biophys. Res. Commun. 311, 966–971 [DOI] [PubMed] [Google Scholar]
- 53. Vögtle F. N., Prinz C., Kellermann J., Lottspeich F., Pfanner N., Meisinger C. (2011) Mitochondrial protein turnover: role of the precursor intermediate peptidase Oct1 in protein stabilization. Mol. Biol. Cell 22, 2135–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dinur-Mills M., Tal M., Pines O. (2008) Dual targeted mitochondrial proteins are characterized by lower MTS parameters and total net charge. PLoS One 3, e2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee C. M., Sedman J., Neupert W., Stuart R. A. (1999) The DNA helicase, Hmi1p, is transported into mitochondria by a C-terminal cleavable targeting signal. J. Biol. Chem. 274, 20937–20942 [DOI] [PubMed] [Google Scholar]
- 56. Ieva R., Heisswolf A. K., Gebert M., Vogtle F. N., Wollweber F., Mehnert C. S., Oeljeklaus S., Warscheid B., Meisinger C., van der Laan M., Pfanner N. (2013) Mitochondrial inner membrane protease promotes assembly of presequence translocase by removing a carboxy-terminal targeting sequence. Nat. Commun. 4, 2853. [DOI] [PubMed] [Google Scholar]
- 57. Hung V., Zou P., Rhee H. W., Udeshi N. D., Cracan V., Svinkina T., Carr S. A., Mootha V. K., Ting A. Y. (2014) Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brocker C., Lassen N., Estey T., Pappa A., Cantore M., Orlova V. V., Chavakis T., Kavanagh K. L., Oppermann U., Vasiliou V. (2010) Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress. J. Biol. Chem. 285, 18452–18463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vincow E. S., Merrihew G., Thomas R. E., Shulman N. J., Beyer R. P., MacCoss M. J., Pallanck L. J. (2013) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 6400–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Okatsu K., Oka T., Iguchi M., Imamura K., Kosako H., Tani N., Kimura M., Go E., Koyano F., Funayama M., Shiba-Fukushima K., Sato S., Shimizu H., Fukunaga Y., Taniguchi H., Komatsu M., Hattori N., Mihara K., Tanaka K., Matsuda N. (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., Magrane J., Moore D. J., Dawson V. L., Grailhe R., Dawson T. M., Li C., Tieu K., Przedborski S. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hasson S. A., Kane L. A., Yamano K., Huang C. H., Sliter D. A., Buehler E., Wang C., Heman-Ackah S. M., Hessa T., Guha R., Martin S. E., Youle R. J. (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504, 291–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jin S. M., Lazarou M., Wang C., Kane L. a., Narendra D. P., Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bonnen P. E., Yarham J. W., Besse A., Wu P., Faqeih E. A., Al-Asmari A. M., Saleh M. A., Eyaid W., Hadeel A., He L., Smith F., Yau S., Simcox E. M., Miwa S., Donti T., Abu-Amero K. K., Wong L. J., Craigen W. J., Graham B. H., Scott K. L., McFarland R., Taylor R. W. (2013) Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet. 93, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Takakubo F., Cartwright P., Hoogenraad N., Thorburn D. R., Collins F., Lithgow T., Dahl H. H. (1995) An amino acid substitution in the pyruvate dehydrogenase E1 alpha gene, affecting mitochondrial import of the precursor protein. Am. J. Hum. Genet. 57, 772–780 [PMC free article] [PubMed] [Google Scholar]
- 67. Stewart J. D., Tennant S., Powell H., Pyle A., Blakely E. L., He L., Hudson G., Roberts M., du Plessis D., Gow D., Mewasingh L. D., Hanna M. G., Omer S., Morris A. A., Roxburgh R., Livingston J. H., McFarland R., Turnbull D. M., Chinnery P. F., Taylor R. W. (2009) Novel POLG1 mutations associated with neuromuscular and liver phenotypes in adults and children. J. Med. Genet. 46, 209–214 [DOI] [PubMed] [Google Scholar]
- 68. O'Toole J. F., Liu Y., Davis E. E., Westlake C. J., Attanasio M., Otto E. A., Seelow D., Nurnberg G., Becker C., Nuutinen M., Karppa M., Ignatius J., Uusimaa J., Pakanen S., Jaakkola E., van den Heuvel L. P., Fehrenbach H., Wiggins R., Goyal M., Zhou W., Wolf M. T., Wise E., Helou J., Allen S. J., Murga-Zamalloa C. A., Ashraf S., Chaki M., Heeringa S., Chernin G., Hoskins B. E., Chaib H., Gleeson J., Kusakabe T., Suzuki T., Isaac R. E., Quarmby L. M., Tennant B., Fujioka H., Tuominen H., Hassinen I., Lohi H., van Houten J. L., Rotig A., Sayer J. A., Rolinski B., Freisinger P., Madhavan S. M., Herzer M., Madignier F., Prokisch H., Nurnberg P., Jackson P. K., Khanna H., Katsanis N., Hildebrandt F. (2010) Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J. Clin. Invest. 120, 791–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.