

Abstract
The use of extracellular matrix (ECM)1 scaffolds, derived from decellularized tissues for engineered organ generation, holds enormous potential in the field of regenerative medicine. To support organ engineering efforts, we developed a targeted proteomics method to extract and quantify extracellular matrix components from tissues. Our method provides more complete and accurate protein characterization than traditional approaches. This is accomplished through the analysis of both the chaotrope-soluble and -insoluble protein fractions and using recombinantly generated stable isotope labeled peptides for endogenous protein quantification. Using this approach, we have generated 74 peptides, representing 56 proteins to quantify protein in native (nondecellularized) and decellularized lung matrices. We have focused on proteins of the ECM and additional intracellular proteins that are challenging to remove during the decellularization procedure. Results indicate that the acellular lung scaffold is predominantly composed of structural collagens, with the majority of these proteins found in the insoluble ECM, a fraction that is often discarded using widely accepted proteomic methods. The decellularization procedure removes over 98% of intracellular proteins evaluated and retains, to varying degrees, proteoglycans and glycoproteins of the ECM. Accurate characterization of ECM proteins from tissue samples will help advance organ engineering efforts by generating a molecular readout that can be correlated with functional outcome to drive the next generation of engineered organs.
Organ transplantation is an established, lifesaving therapy for patients with chronic end-stage diseases. However, transplantation as a therapeutic option is limited by availability of suitable donor organs (1). Although advancements in surgical techniques, such as successful implementation of bilateral lung transplants and improved immunosuppressant treatments, have led to more successful outcomes in recent years, the percentage of people that die while on the transplant wait list has increased (2, 3). One attractive approach to meet this demand is the in vitro generation of organs using decellularized tissues as scaffolds for recellularization. For complex organs such as the lung, these tissue scaffolds can be derived from a donor organ that would have otherwise been unfit for transplantation. This whole organ scaffold can be recellularized using a patient's own primary or stem-derived cells, thus eliminating many issues related to graft/host incompatibility. This approach was recently used to generate lungs that, when implanted in rat recipients, allowed for gas exchange (4, 5). However, examination of the lung indicated leakage of erythrocytes into the alveolar space, indicating a compromised capillary-endothelial barrier. These exciting results highlighted the potential of the method for organ transplantation but also the need for improved molecular readouts to guide engineering efforts.
Efficient reseeding of decellularized scaffolds has been shown to be dependent on retaining native ECM structural integrity and elasticity (6). Local variations in expression of abundant proteins in the ECM scaffolding (collagens, laminins, fibronectins) have been correlated to variance in cell repopulation and subsequent proliferation (7). It is thought that retaining specific ECM components and architectures may allow cells to be directed back to a tissue-specific niche during reseeding and that small changes in abundance of these molecular cues can drastically affect the recellularization process (8). Current methods used to characterize the protein composition of native and acellular tissues involve antibody- or dye-based staining, hydroxyproline assays assessing collagen content, or relative quantification of proteins by liquid chromatography tandem mass spectrometry (LC-MS/MS) (9, 10). All of these methods either fall short in specificity, accurate quantification, or both. A more complete and accurate method for protein characterization would provide a valuable tool for tissue engineering efforts, while shedding light on the possible molecular mechanisms resulting in cell seeding variability and alterations in mechanical properties of engineered lung tissues.
Current relative quantification strategies (iTRAQ, Spectral Counting, dimethyl labeling, others) (11–15) perform well when the majority of protein in samples does not change, there are approximately equal increases and decreases in protein levels, or in cases where proteins that are known not to change in abundance can be used for normalization. However, normalization steps often employed have the potential to introduce experimental bias (16). The decellularization process differentially removes and enriches proteins in the ECM scaffolding, depleting some proteins with high efficiency while leaving others mostly intact. This makes relative comparisons between native and decellularized lung challenging. Although strategies can be employed in an attempt to normalize data (17), there is a distinct advantage to quantification methods using stable isotope labeled (SIL) peptides in this application. Here, we developed ECM targeted, isotopically labeled peptides using the QconCAT approach first described by Beynon et al. (18). SIL quantification allows for intra- and intersample comparison of heterogeneous tissues, such as native organs and decellularized scaffolds, with high accuracy and precision.
The ECM is largely responsible for defining the biomechanical properties of organs. Maintaining structural rigidity and native microarchitecture through the decellularization process makes an acellular organ a good candidate to serve as a tissue scaffold (19, 20). These same characteristics are a central reason why the ECM is challenging to characterize using common bottom-up proteomics approaches (21). Currently accepted and widely used digestion methods require proteins to be solubilized for bottom-up proteomic analysis (22). Recent papers have reported characterization of the ECM fraction from tissues through the use of strong chaotropes (11, 21, 23–27) or cellular fractionation followed by strong detergent (10, 28, 29). However, in our experience, these protocols invariably yield various sizes of an insoluble protein-containing pellet when applied to a variety of tissue samples (heart, lung, and mammary gland). On one end of the spectrum, methods utilizing deglycosylation and enzymatic digestions for clarification of partial solubilized protein slurries yields good ECM coverage with a high number of spectral matches for collagen alpha-1(I), a highly abundant ECM protein in lung (28). On the other end of the spectrum, methods using only detergents or chaotropes for solubilization result in protein pellets that are generally removed prior to LC-MS/MS analysis. These pellets often contained a majority of fibrillar proteins, resulting in quantitative errors. Consistent with this finding, several of these studies characterizing tissue engineered lungs do not report the identification of collagen alpha-1(I) (8, 10, 30). We believe these observations result from a failure to solubilize and enzymatically digest insoluble ECM proteins. To this end, we explored the use of chemical digestion of the insoluble pellet to improve coverage of the ECM proteome from tissue. This method has been used to quantify protein levels from native and decellularized lungs to determine decellularization specificity and efficiency. The accurate characterization of ECM proteins from lung samples should advance tissue engineering efforts by yielding a readout that can be correlated with functional outcome to drive further development.
MATERIALS AND METHODS
Reagents Section
Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. The pET-21b vector was obtained from Novagen (Merck, Darmstadt, Germany). Molecular biology reagents were obtained from New England Biolabs (Ipswich, MA). Sodium chloride and ammonium persulfate were purchased from Acros Organics (part of Thermo Fisher Scientific) and TEMED from Fisher Scientific (Pittsburgh, PA). Consumables were from Axygen Inc. (Union City, CA). Bis-acrylamide was from Amresco (Solon, OH). Formic acid (FA), trifluoroacetic acid (TFA), and potassium chloride were obtained from Fluka (Buchs, Switzerland), and acetonitrile (ACN) was from Burdick & Jackson (Morristown, NJ). Trypsin (sequencing grade, TPCK treated) was from Promega (Madison, WI).
Organ Procurement and Decellularization
All animal procedures were conducted in accordance with Association for Assessment and Accreditation of Laboratory Animal Care guidelines and were approved by the Yale Institutional Animal Care and Use Committee. Organs were harvested and decellularized for comparative analysis by the proteomic methods described here. For native controls, lungs from two adult Fischer 344 rats were procured. An additional two lungs were procured and decellularized as previously described (31). Briefly, extracted heart, lung, and trachea blocs were cannulated via the pulmonary artery and trachea. The lungs were mounted in a bioreactor and decellularized via perfusion with a hypertonic detergent solution for 2–3 h (500 ml) at 37 °C. After treatment with decellularization solution, benzonase endonuclease (90 U/ml) was administered via the trachea, followed by extensive rinsing with PBS via the pulmonary artery. Fully rinsed lungs were decontaminated to eliminate any remaining bacterial burden with antibiotics/antimycotics for at least 48 h.
QconCAT Design and Purification
Qconcat constructs were designed to quantify structurally and functionally relevant extracellular matrix proteins, and common non-ECM proteins often found in ECM preparations. In general, we sought to cover the majority of readily identified ECM proteins from our global proteomic experiments. In house data, datasets from Naba et al. (28) PeptideAtlas (32), and Global Proteome Machine DataBase (33) were used to select protein specific and quantotypic (34) peptides. The following considerations were used to prioritize peptides for inclusion: (1) We attempted to exclude peptides with M, N-terminal Q or E, poly K, or R termini, commonly sequenced in multiple charge states and known post-translational modification sites; (2) peptides longer than eight residues but shorter than 14 residues were preferred; (3) peptides unique to a single protein or protein family; and (4) when possible, we attempted to select peptides with homology across Homo sapiens (60% of peptides conserved), Mus (79% of peptides conserved across rodent models), and the remainder were specific to Rattus sequences (17%). Using these principles, two QconCAT genes were designed and synthesized (Genewiz Inc., eQ1 & eQ2) covering 56 proteins with 74 peptides (sequences can be found in supplemental material). The gene product was cloned into the NdelI and BamHI sites of the pET-21b vector. The constructs were transformed into an E. coli BL21-DE3 LysA ArgA auxotroph (35), plated on M9 media supplemented with 13C6-Arg and 13C6-Lys in two successive rounds, then expressed and purified as previously described (36). Briefly, this strain was grown in 250 ml M9 media supplemented with 13C6-Arg and 13C6-Lys (Isotec) for 3 h at 37 °C (time to reach an OD600 of 0.7). Following a 4 h induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside, cells were pelleted by centrifugation and lysed with a detergent solution (BugBuster®, Novagen) using high energy sonication, three rounds of 30 s (Sonics® Ultrasonic Processor, Model GE505, Power: 90%). After centrifugation, soluble material was removed and Inclusion bodies were washed with the lysis solution and then solubilized in 6 m guanidine-hydrochloride. Immobilized metal affinity chromatography (IMAC) over nickel-NTA resin (GenScript®) was used to purify the QconCAT polypeptides. Isotopically labeled QconCAT proteins were initially quantified by monitoring the UV signal at A280 and by Bradford assays. Final quantification was determined through a dilution series with quality control Alcohol dehydrogenase (ADH, SwissProt P00330) peptides from the QconCAT and characterized QC standard yeast ADH digests (Michrom Bioresources, Inc.). A time-course analysis of QconCAT digestion in a background of soluble E. coli proteins (1:50 wt:wt) revealed complete digestion of the QconCAT at 6 h and peptide stability for up to 36 h in digestion conditions. Time-course digests were analyzed on a MALDI-TOF (Voyager DE-STR, ABSciex) and results were analyzed by comparing fully tryptic peptide abundances versus miss-cleaved peptides.
Sample Preparation
Approximately 5mg of lyophilized native (i.e. nondecellularized) and decellularized adult Fischer 344 lungs were processed in duplicate. Samples were pulverized in liquid nitrogen using a ceramic mortar and pestle followed by homogenization (Polytron, Model PT 10/35, Brinkman Instruments Inc.) on medium speed in 1.5 ml of a high salt buffer (50 mm Tris-HCl, 0.25%CHAPS, 25 mm EDTA, 3 m NaCl, pH 7.4) supplemented with 10 μl/ml fresh protease inhibitor (Protease Arrest™, G-biosciences). Homogenized tissue was spun at 15,000 rpm (4 °C) for 15 min. The resultant supernatant, referred to as Fraction 1, was removed, and the pellet was further extracted with 0.5 ml high salt buffer two times to yield soluble Fractions 2 and 3. The pellet was then vortexed at room temperature for 30 min in 0.3 ml of freshly prepared urea extraction buffer (8 m Urea, 100 mm ABC, 25 mm TCEP, pH 8.0, passed over mixed ion exchange resin (Amberlite®, MP Biomedicals, LLC)) and spun; the soluble portion was Fraction 4. Finally, the resulting pellet was chemically digested with 100 mm CNBr in 86% TFA overnight in the dark. This final fraction was washed with ddH20 and speed-vacced to dryness three times, then brought up in urea extraction buffer to yield Fraction 5.
Detergent/Chaotrope Removal and Protein Digestion
The 13C6 labeled QconCAT standards were added to each fraction at a ratio of 1:400 wt:wt, a ratio empirically determined in pilot experiments to maximize the number of peptides in the quantifiable range. The protein concentration of each fraction was determined by Bradford assay. Urea and CHAPS were removed from samples through tube gel polymerization as previously described (37). Briefly, 30μgs of sample was added to a solution of ammonium persulfate, bis-acrylamide, and TEMED to polymerize the samples in an Eppendorf tube. Samples were then reduced, alkylated, and digested with trypsin as described (12). Briefly, gel pieces were washed and then reduced with 5 mm DTT for 25 min at 64 °C and alkylated with 20 mm Iodoacetamide in the dark at room temperature for 45 min. Gel pieces were subsequently washed with ddH20, 25 mm ABC/50% acetonitrile and finally 100% acetonitrile and then dried on a speed-vac. Dried gel plugs were then digested with 5 ng/μl sequencing grade trypsin (Promega) by incubating at 4 °C for 30 min (38) and then 37 °C overnight. Tryptic digests were acidified with 1% FA and peptides were extracted with three subsequent washes of 50% ACN and 0.1% FA. Peptides were concentrated and acetonitrile removed on a speed-vac and then brought up to final volume (72μls).
Liquid Chromatography Tandem Mass Spectrometry
Samples were analyzed on both the LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) and the QTRAP®5500 triple quadrupole mass spectrometer (ABSciex) coupled with an Eksigent nanoLC-2D and Agilent 1200 LC system, respectively. On the LTQ Orbitrap Velos/Eksigent system, 8 μl of sample was loaded onto a trapping column (ZORBAX 300SB-C18, 5 × 0.3 mm, 5 μm) and washed with 2% ACN, 0.1% FA at a flow rate of 10 μl/min for 10 min. The trapping column was then switched online with the nanopump at a flow rate of 600 nl/min. Peptides were separated on an in-house-made 100 μm i.d. x 150 mm fused silica capillary packed with Synergi Hydro-RP C18 Resin (Phenomex; Torrance, CA) over a 85 min gradient from 6–40% ACN. The flow rate was adjusted to 350 nl/min after 10 min to increase the effective separation the peptides. MS data acquisition was performed using Xcalibur (version 2.1) software. Collision-induced dissociation was used to produce the fragment ions in the linear ion trap from the precursor ions, which were measured in the Orbitrap mass analyzer. For every MS scan, the 20 most intense ions were selected for fragmentation, and masses selected for fragmentation were then excluded for a duration of 120 s after a repeat count of 3.
A targeted, scheduled selected reaction monitoring (SRM) approach was performed using the QTRAP® 5500. 8 μl of each sample was injected and directly loaded onto an Agilent C18 column (Zorbax SB-C18, 5 μm 150 × 0.5 mm) with 5%ACN, 0.1% FA at 30 μl/min for 3 min. A gradient of 5–28% ACN was run for 61 min to differentially elute QconCAT peptides. The mass spectrometer was run in positive ion mode with the following settings: a source temperature of 200 °C, spray voltage of 5300V, curtain gas of 20 psi, and a source gas of 35psi (nitrogen gas). Transition selection and corresponding elution time, declustering potential, and collision energies were specifically optimized for each peptide of interest using Skyline's (39) stepwise methods set up. Method building and acquisition were performed using the instrument supplied Analyst® Software (Version 1.5.2).
Data Analysis
For global proteomics, peak lists were generated from the .raw files using PAVA (UCSF) and searched using an in house Mascot server (Version 2.3, Matrix Science). Peptide tolerance was set at ± 10 ppm with MS/MS tolerance set at ± 0.6 Da from spectra acquired on the LTQ Orbitrap Velos. Full trypsin specificity was required, and one missed cleavage was allowed; carbamidomethylation on cysteine was defined as a fixed modification; 13C6 Arg and Lys were defined as heavy labels; methionine oxidation and proline hydroxylation were defined as variable modifications for the database searches. Files were searched against an in-house concatenated International Protein Index (IPI) Rat database containing 39,925 proteins (Final Release, 02/2013). Result files from MASCOT were consolidated using Scaffold (Version 3.1, Proteome Software), where peptide spectral match, and total ion current result files were exported for further analysis. This export included 122,158 spectra mapping to peptides at a 99% confidence interval and 653 proteins at a 95% confidence interval with at least two peptides per protein resulting in an false discovery rate (FDR) of 0.1%. The mass spectrometry data have been deposited to the ProteomeXchange Consortium Database (40) via the PRoteomics IDEntifications partner repository, the assigned dataset identifier is PXD001213 and DOI 10.6019/PXD001213.
SRM data obtained on the QTRAP® were directly loaded into a Skyline® file containing all expected precursor ions. Transition quality, peak shape, and peak boundaries were manually validated prior to export of integrated 12C/13C peak areas for each peptide. Data were exported, and the average peptide ratio was determined by taking the average of the 12C/13C ratio of the four transitions selected for identification and quantification of each peptide. Ratios outside the limits of quantification (LOQ, 1 fmol for most peptides) and below the isotope incorporation percentage (98.4–99.8%) for each reporter peptide were thrown out. Limits of detection (LOD), LOQ, linear dynamic range, and digestion efficiency were all controlled for and empirically determined (as reviewed (41)) prior to running biological samples. Briefly, for each peptide, (I) LOD was defined as the lower limit to achieve a signal/noise of at least 3, (II) LOQ was defined as the upper and lower limits that achieve a coefficient of variance (CV) of less than 20%, (III) linear dynamic range was determined by plotting the integrated peak areas of a gradient of the light peptide (12C form) against constant heavy peptide (13C form) and achieving an R value of at least 0.95, and (IV) digestion efficiency was optimized to the method at which the presence of peptides with missed tryptic cleavage sites (screened by MALDI-TOF) accounted for less than 1% of the fully trypsinized peptide probe. Control peptides from yeast alcohol dehydrogenase were included in each QconCAT and a dilution series with a commercial ADH digest (Michrom Bioresources, Inc.) was used to determine the concentration of QconCAT polypeptides. All data were compiled in a spreadsheet for calculating protein abundance, and gene ontology annotations were extracted using the Database for Annotation, Visualization, and Integrated Discovery (42).
RESULTS
Sample Preparation Method Development
To provide molecular characterization of the ECM from native and decellularized lungs, a sample preparation approach that allowed for as complete tissue protein characterization as possible was required. Primary objectives of this work were to identify and quantify the proteins that remain after the decellularization procedure and, of equal importance, characterizing the proteins that were removed. Initial tissue preparation for bottom-up proteomics using published protocols consistently resulted in an insoluble pellet after chaotrope or detergent extraction. Amino acid analysis revealed that this pellet contained protein with a high percentage of glycine and proline compared with the detergent- and chaotrope-soluble fractions. This amino acid profile led us to our first hypothesis; the chaotrope insoluble pellet, which is commonly discarded in proteomic approaches, is primarily composed of fibrillar proteins of the ECM. This class of proteins is known to contain complex crosslinks (43) and therefore is likely to be insoluble in common buffer systems, even those that have strong protein denaturing and solubilizing properties.
Here we developed an extended protein extraction procedure aimed at characterizing both the chaotrope soluble and insoluble protein in tissue samples. The procedure, outlined in Fig. 1, begins with an initial extraction using a high salt, CHAPS buffer with mechanical disruption to remove detergent-soluble proteins. This first step is aimed at removing the bulk of cellular proteins while minimizing the extraction of ECM proteins. Reducing the salinity, varying the detergent, or utilizing deglycosylases and/or nucleases at this step in the procedure reduced the extraction efficiency of cellular proteins or altered the profile of ECM proteins in the remaining pellet as determined by initial nontargeted LC-MS/MS analysis.
Fig. 1.
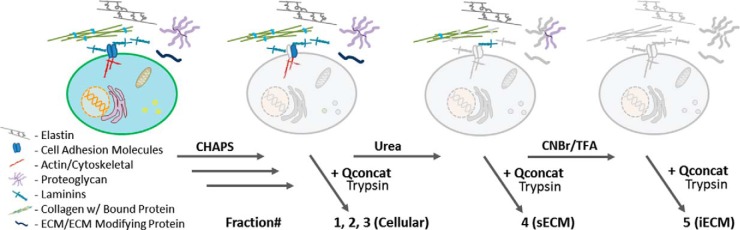
Protein extraction method applied to native and decellularized lungs. Cellular proteins are extracted using a high salt CHAPS buffer and mechanical tissue homogenization, solubleECM (sECM) is extracted using a 6 m urea buffer with vortexing and insolubleECM (iECM) is extracted after chemical digestion. After solubilization, proteins are digested for downstream discovery LC-MS/MS or targeted LC-SRM analysis.
The most widely accepted ECM extraction protocols involve differential tissue fractionation followed by solubilizing the remaining protein in a strong denaturing buffer; radioimmune precipitation assay buffer (Nonidet P-40, sodium deoxycholate and SDS) (10, 29), SDS (23, 24), guanidine-hydrochloride (11, 25), or urea (21, 26–28). Our pilot studies using these protocols resulted in relatively similar ECM protein profiles and uniformly yielded a protein-containing pellet after extraction. Based this observation and our previous amino acid analysis results that identified protein in the chaotrope insoluble pellet, we employed a cyanogen bromide (CNBr) digestion step after chaotrope buffer extraction (Fig. 1). CNBr selectively hydrolyzes the C-terminal peptide bond of methionine, generating protein fragments that can be extracted and subjected to enzymatic digestion for integration into our analytical workflow. Global LC-MS/MS analysis of this fraction from native lung, which we term chaotrope insoluble ECM (iECM), revealed a subset of ECM proteins found in the chaotrope soluble (sECM) fraction with varied relative abundances as determined by label free methods (Fig. 2). The iECM fraction contained a greater percentage of fibrillar ECM molecules than any other fraction. Gene ontology analysis of proteins accounting for 95% of total ion current for each fraction revealed that 67% of the spectral intensity for identifications in this fraction mapped to fibrillar ECM proteins, 18% to basement membrane, and 1% to other ECM proteins, while only 14% mapped to plasma and intracellular proteins (Fig. 2). In contrast, the native lung's cellular fractions (F1–3) resulted in no fibrillar ECM identifications, with 98% mapping to proteins associated with plasma and cellular localization as determined by gene ontology analysis. The sECM fraction was split, with 44% of the total ion current mapping to fibrillar proteins, and 40% to plasma and cellular proteins. Although a unique protein profile exists between the sECM and iECM fraction in terms of peptide spectral matches, few unique proteins were identified in the iECM fraction at this level of analysis.
Fig. 2.
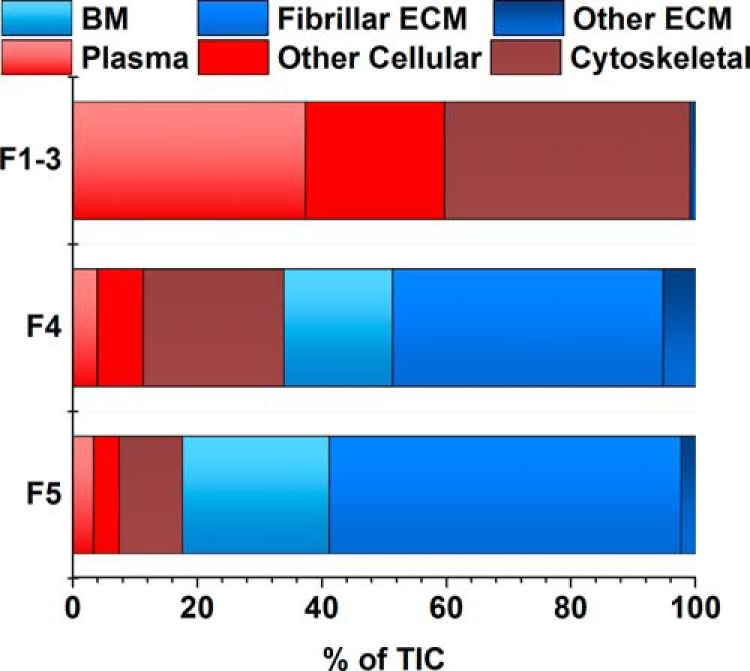
iECM Fraction has unique protein profile. Peptide spectrum matches accounting for 95% of the total ion current from untargeted Semiquantitative analysis of fractions by protein class determined by gene ontology annotations proteomic analysis of native lung (nondecellularized). Peptide identifications in each fractional group (F1–3, cellular; F4, sECM; F5, iECM), separated by protein class as determined by gene ontology analysis. Individual proteins used to generate figure can be found in supplemental Table 1. Totals were averaged from two lungs analyzed in triplicates, n = 6.
Relative quantitative proteomics is widely used to compare protein level differences between samples. We compared peptide spectral matches between native and decellularized lungs to assess both relative protein level differences between samples, as well as the changes in the sECM and iECM protein profile between the two lung types. As shown in a subset of representative proteins in Table I (global proteomics data can be found in supplemental Table S1), the majority of ECM proteins are identified in Fractions 4 and 5 (sECM and iECM, respectively). Interestingly, when we compare the ratio of proteins found in a decellularized lung to that of a native lung between those fractions, a different profile emerges between the two sets. For example, analyzing just the cellular (F1–3) and sECM (F4) fractions of COL6A1, would suggest a nearly threefold decrease in protein abundance after decellularization. However, the complete profile of the lung (F1–5) shows only a 14% decrease due to the iECM fraction containing more than twice the spectral matches in the decellularized lung compared with the native. Additionally, global proteomics suggests efficient removal of prominent structural ECM proteoglycans and glycoproteins such as decorin and fibrillin-1, a surprising finding based on the known interaction of these proteins with those not depleted. While additional fractionation may provide the identification of these proteins, anomalies such as this will remain a problem for proteins near the limit of detection. In addition, the stochastic nature of peptide selection will remain a confounding factor when attempting to quantify proteins in complex tissue proteomes.
Table I. Average PSMs of native and decellularized lung by fraction: Peptide spectral matches averaged from two biological- and three technical-replicates analyzed by untargeted LC- MS/MS. PSMs were normalized for protein load and tissue weight and compared at the protein level.
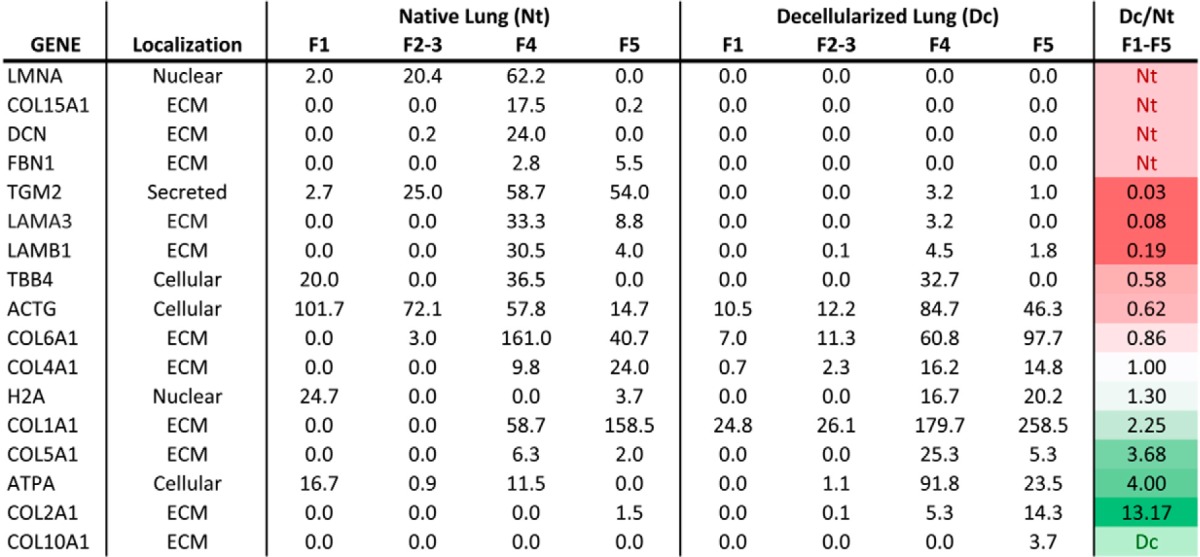
Relative quantification results also indicated over a twofold increase in fibrillar collagens (types I, II, and V) even after normalizations accounting for protein weights and sample loads. Lamin A/C appears to be efficiently depleted during the decellularization process, despite known challenges of completely removing nuclear matrix. In contrast, h H2A, appears enriched postdecellularization, despite reports of efficient DNA depletion (5, 44, 45). Similarly, cellular proteins such as cytoskeletal actins, tubulins, and mitochondrial ATP synthase subunit alpha-1 appear to be inefficiently removed by the decellularization process according to the peptide spectral match comparisons in Table I. These and the above examples are universally inconsistent with previous reports that indicate near complete depletion of intracellular components, while retaining key components of the ECM (4, 44, 46, 47). Due to these significant qualitative and quantitative inconsistencies utilizing untargeted LC-MS/MS, we deemed this method insufficient to accurately measure differences in protein levels between the two lung types.
Development of Quantitative Method
To overcome these limitations, we developed two QconCATs containing a total of 74 peptides selected from 55 ECM, ECM-related, and cellular proteins of interest and one peptide each representing a standard yeast protein (supplemental Table S2). We first developed and optimized SRM assays for each peptide contained within the QconCATs. Collision energy, declustering potential, and retention time were sequentially optimized using the Skyline software to achieve the highest signal intensity for each transition. All optimization was completed in a background of trypsin digested E. coli lysate at a 1/400 ratio (wt./wt.) to mimic the matrix effects of a complex protein sample.
The newly developed scheduled SRM method for each QconCAT (supplemental Fig. S1) was used to test digestion efficiency, stability, LOQ, LOD, and linear dynamic range of the assay. Digestion of all QconCAT peptides was determined to be complete within 1 h of incubation with trypsin, with intensity of the peptide signals remaining stable at 3, 6, and 18 h time points. The LOD was determined to be between 200 and 500 amol, depending on the peptide; an LOQ of 1 fmol was observed for most peptides. Linearity was maintained over at least four orders of magnitude, with all but two peptides maintaining linearity up to a 150:1 ratio (12C/13C). Of the initial 74 QconCAT peptides; 61 were used for quantification of peptides from the lung samples. Three peptides were excluded from the analysis due to variability during LOQ/linear dynamic range experiments, eight were below the LOQ in the biological samples analyzed. The remaining two peptides from Saccharomyces cerevisiae ADH were used as internal controls for quantification. These control peptides allow for normalization of any variability introduced during the sample preparation procedure or LC-MS/MS analysis and yielded a high level of reproducibility as shown in Fig. 3A. Each QconCAT was digested separately for 14 h at seven concentrations between 50 and 500 fmols. The integrated peak area versus our standard ADH peptide resulted in a linear response (R value > 0.99, and an S.E. of < 2%).
Fig. 3.

Quality control for QconCAT peptides. (A) The left and right panel show the concentration curve of the two standard ADH peptides included in QconCAT 1 and 2, respectively. Total QconCAT concentration was determined and then compared with our in-house ADH peptide standard. (B) Dynamic range depicted with a representative peptide from eQ2 shown in the graphs below. The peptide ratios are shown above each graph, with the calculated ratio of the representative peptide shown in italics next to the peak. Over 80% of endogenous peptides quantified with the QconCAT method fell within this range.
One challenge of using SIL peptides to quantify proteins in complex biological samples is the wide range of target protein concentrations encountered. We used a similar approach mentioned above to experimentally characterize the dynamic range of quantification for our QconCAT generated peptides. Ten stepwise ratios, over four orders of magnitude of heavy (13C6) and light (12C6) QconCAT ratios were individually digested in a background of trypsin digested E. coli lysate. SIL labeled QconCATs were added at a constant 60 fmols, while light QconCATs were incrementally varied from 0.01- to 150-fold of the SIL peptides (0.6 to 9000 fmols). Over 80% of peptides quantified in native and decellularized lung fell within the observed range of 2.4 to 1500 fmols per injection. A representative QconCAT peptide is shown in the bottom panel of Fig. 3.
Quantification by Fraction
The quantitative analysis of 49 proteins with a heat map representing the total concentration of protein per fraction is shown in Fig. 4. As expected, the cytoskeletal and nuclear components are abundant in the detergent fractions (F1–3) of the native lung. In contrast, and in line with expectations, CHAPS extraction of the acellular lung yielded very little protein (Fractions 1–3). The overall lack of protein from decellularized lung in Fractions 1–3 reflects their previous removal during the decellularization regimen used to generate the scaffold. Most of the components that make up the ECM scaffold—basement membrane proteins, collagens, glycoproteins, and proteoglycans—are predominantly extracted into Fractions 4 and 5. Focusing on Fraction 4, we note that the basement membrane and collagen proteins detected in the native lung are more abundant when compared the decellularized sample (Fig. 4). This indicates that the decellularization process not only removes cellular components but also solubilizes some of these ECM proteins. This is also true of the glycoproteins and additional proteoglycans evaluated in this study, though the discrepancy is somewhat less striking. Similarity between the iECM fraction (Fraction 5) of native and decellularized lung indicate that the structural proteins (largely fibrillar collagens) are relatively undisturbed by the decellularization process. Together, these data indicate that quantitative information is incomplete for these proteins without analyzing both the soluble and insoluble ECM (Fractions 4 and 5).
Fig. 4.

Protein abundance shifts to the iECM following decellularization. Heat map representing the concentration per fraction of 49 absolutely quantified proteins. Triplicate runs were averaged, and individual protein femtomolar abundances were normalized to z-score for each lung type (native normalized separately from decellularized). Red boxes correlate with higher concentration of protein in a given fraction. * Indicates proteins were detected within 5% of the LOQ T̄ Reporter peptide shared by one or more protein isoforms: ACTA1 includes Act-A1/A2/B/C1/G1/G2, ACTB represents ACTB/G1.
Protein levels appear less consistent between the biological replicates in the decellularized lungs compared with the native lungs (Fig. 4). This variability is likely introduced during the decellularization process, which involves several buffer changes in the bioreactor over the course of several hours. Additionally, normalized z scores depicted in the heat maps can represent minor fluctuations in protein levels if the total range is small. Thus, while it is not surprising that the biological variability is greater in the decellularized lung when compared with the native, these variations may represent a narrow range of the protein levels across fractions.
To determine the total abundance of QconCAT-targeted proteins in each fraction of a native lung, we calculated the total molar quantity, and plotted representative proteins by gene ontology classification (Fig. 5). Cellular proteins were predominantly solubilized in the first detergent and high salt extraction step (Fraction 1), with over 97% of the protein in this fraction for the proteins we monitored. Interestingly, 70–80% of proteins that facilitate cell-ECM adhesion and cross-talk elute in the sECM fraction (Fraction 4), with the remainder solubilized during the initial detergent extractions (Fractions 1–3). This may reflect subpopulations of these proteins based on cellular and matrix location and interaction partners. Proteins of the basement membrane elute in the cellular fraction to a minor extent, while the majority of protein is split between the sECM and iECM fractions. Finally, fibrillar proteins that serve as the structural base of tissue scaffolding remain largely insoluble even in strong chaotrope; nearly 80% of these proteins were observed in the iECM fraction.
Fig. 5.
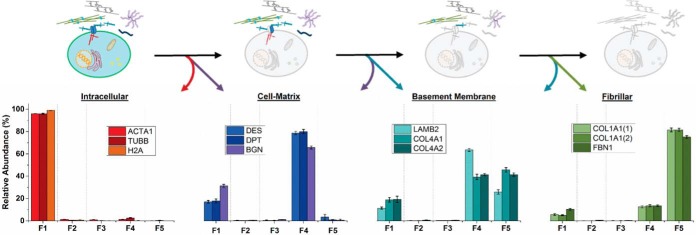
Proteins elute by functional classification. Fractional analysis of protein elution represented as a percentage of the total concentration of that protein in a native lung. Each quantified protein in this graph is represented by a single QconCAT peptide. Error bars are calculated as a standard error of the mean from two biological replicates and three technical triplicates.
To determine the efficiency of our decellularization procedure, we evaluated the composition of lung tissue by protein class for native and decellularized lung (Fig. 6), with the caveat that we are monitoring a subset of ECM and an even smaller set of intracellular proteins. In line with expectations, collagens represent nearly 85% of the total protein weight of the decellularized lung. However, ∼7% of quantified protein weight is accounted for by the few cellular proteins monitored here. Less than 20% of the original ECM proteoglycans and glycoproteins are retained in the decellularized matrix. To further evaluate the efficiency of our decellularization procedure, at the protein level, we calculated the molar quantity per gram of lung for each protein (supplemental Table S3). As expected, decellularized lungs are largely composed of structural collagens. The decellularization process was effective at retaining over 75% of the fibrillar collagens and 30% of the basement membrane components (laminins), and these findings are consistent with a previous report by Petersen et al. (44) Although cytoskeletal and nuclear proteins made up a significant percentage of the decellularized lung, we observed that less than 2% of the total original cellular protein remains. Many of the ECM glycoproteins and proteoglycans are extracted during the decellularization process, with less than 20% identified in the final scaffold.
Fig. 6.

Defining proteins of native and decellularized lung tissue by weight. Chart representing total weight of quantified proteins (supplemental Table 2) sorted by protein class. The third column (diagonal lines) in each protein class represents an estimate of the percentage of each protein class retained after the bioreactor decellularization procedure. T̄ Reporter peptide shared by one or more protein isoforms: ActA1 includes Act-A1/A2/B/C1/G1/G2, Tubb includes Tubb-4a/4b/5, and H2A includes H2A subtypes 1A-K,2,3,4.
DISCUSSION
This work stems from the first successful in vivo lung implantation using an engineered organ based on a scaffold derived from a decellularized organ (4). In this study, successful gas exchange was observed but only for several hours, limited by defects in coagulation and basement membrane barrier function. These studies highlighted the need for higher resolution molecular readouts to guide organ engineering. Understanding the integrity and composition of basement membranes and other aspects of the matrix are critical for successful generation of organs for transplantation.
Accurate characterization of native and decellularized organs has important relevance for tissue engineering efforts because it provides a molecular-level readout for functional variability observed in engineered organ testing. Commonly used methods for protein characterization depend on surrogate analysis of select proteins or class of proteins. For example, collagen levels are estimated using a colorimetric hydroxyproline assay (48). The ability of these methods to generate an accurate representation of the tissue proteome is inadequate. Despite rapid technological and computational advances in proteomics, comprehensive quantification of tissues remains a challenging problem. We set out to overcome two issues hampering our ability to accurately characterize the ECM from tissue engineered lungs: (1) solubilizing the full complement of ECM proteins and (2) accurate quantification to allow for ECM proteome comparison of heterogeneous tissues.
The ability to accurately characterize proteins from tissue is highly dependent on consistent and efficient extraction and solubilization steps (49). The extracellular matrix proteins that make up the structurally rigid scaffolds used for tissue engineering have varying degrees of resistance to common detergent and chaotrope extractions. We used CNBr in strong acid solution to further digest protein in the insoluble pellet (iECM) that remains after detergent (CHAPS) and chaotrope (urea) protein extraction. Historically, chemical digestion with CNBr was used to isolate collagen peptides (50), generate large peptides for middle-down proteomics (51), and more recently to facilitate the identification of membrane proteins (52). The use of CNBr is advantageous for extracting the iECM fraction due to its specificity for cleavage at the C-terminal of methionine within proteins. Additionally, tightly packed proteins can sterically hinder enzymatic digestion facilitated by proteases, which is circumvented with chemical digestion. The robustness of the QconCAT method relies on targeted peptides to be in a known population with minimal heterogeneity; as a result, we avoided methionine-containing peptides in our QconCAT design when possible. This allows for the use of CNBr digestion to generate peptide fragments that can be solubilized and further digested with proteases so as to provide peptides compatible with stable isotope quantitative methods. Other methods for isolation and digestion of collagen and ECM proteins, such as pepsin or collagenase treatment, can be used for traditional bottom-up proteomics but are not amendable to quantitative methods due to their lack of specificity.
The comprehensive comparison of protein abundances between tissue samples is critical to understand the cellular processes that regulate or disregulate the homeostatic equilibrium of an organ. Relative quantitative mass spectrometry by global shotgun MS has emerged over the past decade as a powerful analytical platform to address some aspects of this aim. However, quantitative accuracy and precision of shotgun MS results remains strongly dependent on sample complexity. This is evident in the results shown in Table I where apparent protein levels remaining in the decellularized lung contradict published results attained through orthogonal assays. Sample complexity is known to negatively affect critical parameters for proteome analysis—the dynamic range, LOD, and analysis reproducibility, factors that have been reviewed in depth (53). Even with the combination of constantly evolving instrumentation to increase proteome coverage and the increased ability to multiplex (e.g. iTRAQ/TMT reagents), accurate intrasample quantitative comparisons remains challenging due to a range of factors that affect peptide detection. The QconCAT approach presented here circumvents these issues by providing a semiquantitative measure of protein levels between two heterogeneous tissues, a native and decellularized lung. Protein level concentration within tissues has proven to be an important determinant in producing structurally and functionally competent tissues (54). A quantitative understanding of protein levels throughout the process of organ engineering in combination with detailed understanding of protein composition have the potential to accelerate development of methods to reproducibly generate competent engineered organs.
The use of stable isotope-labeled analytes has long been accepted as the gold standard for quantification in the field of mass spectrometry (55, 56). Stable isotope dilution mass spectrometry allows for absolute quantification since the reporter peptides behave identically (or very similarly depending on the isotope used) to the endogenous peptides being quantified through chromatographic separation, MS ionization, and tandem MS fragmentation (57). In addition, recovery during sample processing can be monitored as the isotope-labeled QconCATs can be added prior to enzymatic digestion. However, this advantage comes at the cost of needing to perform extensive validation experiments. A key component of the validation is ensuring accurate quantification of the QconCAT peptides themselves. To accomplish this, we optimized digestion conditions and utilized standard yeast ADH digests to quantify ADH peptides from each QconCAT to serve as internal controls for quantification. These measures resulted in high analytical precision and allowed for the reliable detection of relatively small changes in protein abundance.
A disadvantage of the QconCAT approach is that generation of peptides with post-translational modifications is not currently possible. Synthetic peptides on the other hand can be synthesized to have virtually any observed modification. While a vast array of synthetic peptides can be purchased commercially, very few derived from ECM proteins are currently available and synthesis of relatively pure peptides in the microgram range quickly becomes cost prohibitive, whereas we are able to generate milligram quantities of highly pure labeled QconCATs for ∼1% of the cost of synthesized peptides using a bacterial expression system. Ultimately, analytical accuracy of quantitative approaches that employ SIL peptides, such as the one presented here, rely on several factors including digestion efficiency and peptide recovery. The local environment of tryptic cleavage sites is different between the QconCAT peptides and the equivalent peptides in the endogenous proteins. Thus, it is important to determine and use digestion conditions that have been optimized for digestion yield.
Quantities of synthetic peptides added to digests can be optimized to yield maximum dynamic range of quantification. However, initial quantification relies on purity and amino acid analysis, when enough material is available, and differential loss during sample processing can confound results (58–60). This is not the case for the QconCATs as the stoichiometry between peptides is known, and with the inclusion of quality control yeast peptides and the corresponding yeast digest standard we obtain intrarun quantification of the labeled peptides. Together, these factors contribute to the utility of the QconCAT approach to provide accurate quantification in a cost-effective manner when compared with traditional methods.
The two QconCATs designed and produced here contain a total of 74 tryptic peptides that facilitate the quantification of 56 proteins. To increase the utility of these constructs, we attempted to select peptides with homology between human sequences and the two most common rodent models used in biomedical research, mouse and rat. One of the main caveats of this method is the assumption that quantification of a peptide directly correlates with the concentration of the parent protein. Some ECM proteins are known to have many splice-variants and are susceptible to proteolytic processing and turnover (61).
Future efforts will be aimed at generating additional QconCATs to increase coverage of the ECM proteome and additional proteins that have the potential to impact recellularization outcomes such as growth factors. Based on our previous work, it is likely that growth factors will be removed during the decellularization process. This supposition is supported by our global proteomics data in which growth factors were not confidently identified in the decellularized lung. However, future application of the methodology will involve quantitative variation with alterations of the decellularization and recellularization procedures, the latter of which will likely include added factors that influence cell signaling, developmental programs, and direct cell attachment. Additional labeled peptides to increase coverage of ECM proteins will not only add a level of statistical confidence to the method but will also lead to isoform specific information, giving a more complete molecular readout of tissue scaffolding.
One of the major challenges of developing engineered organs for transplantation is creating scaffolding that retain specific cues from the local microenvironment that facilitate regeneration of highly specialized structures such as capillaries and alveoli. Thus, the discovery that the iECM fraction not only contains protein but also is composed of a large subset of basement membrane and fibrillar proteins underscores the importance of this fraction.
Depletion of basement membrane proteins such as collagen IV and laminins is relevant to recellularization, as these proteins possess abundant binding sites for both endothelial and epithelial cells. Collagen IV promotes cell adhesion to a greater degree than either fibronectins or laminins (62, 63). In addition to adhesion, these proteins differentially regulate migration and proliferation (64). Collagen IV is the predominant collagen of basement membranes in most tissues and contains multiple cell-binding domains (65, 66). Mesenchymal, epithelial, and endothelial cells all bind to collagen IV. Furthermore, the alpha chains of collagen IV are regionally distributed, both in the lung (67) and the kidney (68), which supports the concept of local ECM niches that may influence cellular phenotype. More than 35% of collagen IV extraction occurs in the iECM fraction, (Fraction 5), highlighting the importance of this fraction in molecular characterization of tissue. Our current protocol can be used to monitor these basement membrane proteins and correlate these levels to recellularization efficiency and transplant outcomes.
Laminins, which are also present in the sECM and iECM fractions, play a prominent role in promoting cell-type specific phenotypes. Laminin-322 (LN-5 old nomenclature) is predominantly found in the basement membrane of alveolar walls and airways in developing and adult lung and absence of the gamma 2 chain results in less organized tracheal epithelial junctions (69). Absence of the laminin alpha-5 gene in knockout mice results in fewer type II cells and a near-absence of type I cells (70). Laminin-chain-specific information of this type has been challenging to obtain, providing evidence that our method outperforms immunohistochemistry methods in several respects. The presence or absence of basement membrane proteins yields key information about the availability of binding sites that may impact the efficiency of recellularization and may ultimately correlate with the ability of the scaffold to promote organ-specific cell phenotypes that lead to successful organ generation. The level of detail provided by our method generates a critical and heretofore unavailable link between in vivo studies and recellularization of acellular scaffolds.
Proteins of the ECM play an important role in defining the mechanical properties of tissues. Glycoproteins and proteoglycans are important to the viscoelastic behavior of the tissue and stabilize the collagen and elastin networks, thereby contributing to the maintenance of overall tissue architecture. Removal of proteoglycans tends to result in “softer” tissue as evidenced by decreased stress per unit strain, though this is somewhat less important than the contribution of fibrillar collagen networks throughout the tissue (71). Fibrillar collagens are largely responsible for the ultimate tensile strength of organs; the apparent preservation of these proteins suggests that the tensile mechanics should be relatively similar between native and decellularized lungs. Indeed, this aligns with our previous work comparing native and CHAPs-decellularized lungs (72). Despite the importance and abundance of the ECM components discussed, a significant percentage of ECM protein is being discarded by methods that only utilize detergent or chaotrope solubilization during sample preparation. Therefore, the methods described here represent an advancement in the characterization of decellularized lung scaffolds, which should be applicable to other decellularized organs, and tissue proteomes in general.
Beyond quantitative accuracy, the presented methodology for fractionation can provide insight into specific ECM microenvironments. For example, it is likely that fibrotic tissue has a larger percentage of several ECM proteins in the insoluble fraction when compared with neighboring nonfibrotic tissue. Likewise, different chemical decellularization approaches will remove variable amounts of soluble and possibly insoluble components. If assay throughput is a priority, the procedure can be modified to pool the sECM and iECM fractions for chemical digestion to create one ECM fraction for analysis. However, many important matricellular ECM proteins from native tissues are predominantly solubilized in chatrope buffer (sECM, Fig. 4), and these proteins can be two or more orders of magnitude lower in concentration than fibrillar proteins found in the iECM fraction. Thus, combining these fractions to increase throughput may result in lower level ECM proteins being below the LOQ.
One advantage of using native tissue over synthetic scaffolds for engineered organ generation is that ECM proteins remaining after decellularization can leave a “footprint” of the previously resident cells (73). This microenvironmental niche may facilitate organ specific features during cellular reseeding. Proteoglycans and glycoproteins of the ECM are known to facilitate many of the complex cell–cell and cell–matrix interactions critical for specialized structures such as capillaries and alveoli. Our results indicate that only 5–25% of these protein classes remain after decellularization using our previously published methods (31). At this time, the importance of retaining these ECM components versus having them generated by resident cells during recellularization is not known. In addition, our results are consistent with previous reports regarding the levels of remaining cellular proteins (less than 2%). The specific profile and amount of cellular protein required to elicit an immune response is poorly defined. The protein composition of the decellularized microenvironment that facilitates optimal recellularization to regenerate a functional organ is not known. The methods presented here can be used to characterize current tissue engineering scaffolds and allow for refinement of decellularization procedures based on protein profiles, both with respect to extent of antigen removal and structural protein conservation.
In summary, by combining a stepwise extraction and digestion procedure with a targeted LC-SRM approach, we were able to establish a rapid method for analysis of proteins from native tissue samples and matched decellularized organs. Our targeted method proved to be more reproducible and sensitive than a more traditional discovery-based proteomic approach. In addition, the chaotrope insoluble fraction was assessed with the use of an extraction step based on chemical digestion. This fraction led to the discovery of an ECM-rich fraction that would have been discarded using standard strong denaturants aimed at solubilizing extracellular matrix proteins. Utilization of the QconCAT method for generating SIL peptides allowed for a level of quantification that would have been difficult to achieve using labeled or label-free relative quantification methods. Refinement and optimization of this approach should provide a valuable assay to the research community by defining organ scaffolds and tissue proteomes at a molecular level never before achieved. Future work will involve using these methods to correlate protein profiles with functional testing and outcomes of transplantation experiments to provide the next generation of engineered organs.
Supplementary Material
Acknowledgments
We would like to thank Dr. Ron Hay and Ellis Jaffray of the University of Dundee for graciously providing the auxotroph used in this study.
Footnotes
Author contributions: R.C.H., E.A.C., M.D., L.E.N., and K.C.H. designed the research; R.C.H. and E.A.C. performed the research; R.C.H. and M.D. contributed new reagents or analytic tools; R.C.H., E.A.C., L.E.N., and K.C.H. analyzed data; R.C.H., E.A.C., L.E.N., and K.C.H. wrote the paper.
* This work was supported by the NIH/NCI IMAT program grant R21CA132741 and NCRR grant S10RR024599 to K.C.H., NIH 1U01HL111016 and R01HL098220, to L.E.N. Additional supported from the NIH/NCATS Colorado CTSA Grant Number UL1 TR001082, and P30 CA046934 to the Proteomic Mass Spectrometry Facility. Contents are the authors' sole responsibility and do not necessarily represent official NIH views. L.E.N. has a financial interest in Humacyte, Inc, a regenerative medicine company. Humacyte did not fund these studies, and Humacyte did not affect the design, interpretation, or reporting of any of the experiments herein.
 This article contains supplemental Tables S1–S3 and Fig. S1.
This article contains supplemental Tables S1–S3 and Fig. S1.
1 The abbreviations used are:
- ECM
- extracellular matrix
- CHAPS
- 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate
- QconCAT
- quantification concatamer
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- SRM
- selected reaction monitoring
- SIL
- stable isotope labeled
- NTA
- nitrilotriacetic acid
- LOD
- limit of detection
- LOQ
- limit of quantification
- CAN
- acetonitrile
- TPCK
- l-1-tosylamido-2-phenylethyl chloromethyl ketone or tosylphenylalanyl chloromethyl ketone
- TCEP
- (tris(2-carboxyethyl)phosphine)
- TEMED
- tetramethylethylenediamine.
REFERENCES
- 1. Orens J. B., Garrity E. R., Jr. (2009) General overview of lung transplantation and review of organ allocation. Proc. Am. Thorac. Soc. 6, 13–19 [DOI] [PubMed] [Google Scholar]
- 2. Christie J. D., Edwards L. B., Kucheryavaya A. Y., Aurora P., Dobbels F., Kirk R., Rahmel A. O., Stehlik J., Hertz M. I. (2010) The Registry of the International Society for Heart and Lung Transplantation: Twenty-seventh official adult lung and heart-lung transplant report—2010. J. Heart Lung Transplant. 29, 1104–1118 [DOI] [PubMed] [Google Scholar]
- 3. Mahida R. Y., Wiscombe S., Fisher A. J. (2012) Current status of lung transplantation. Chronic Respiratory Disease 9, 131–145 [DOI] [PubMed] [Google Scholar]
- 4. Petersen T. H., Calle E. A., Zhao L., Lee E. J., Gui L., Raredon M. B., Gavrilov K., Yi T., Zhuang Z. W., Breuer C., Herzog E., Niklason L. E. (2010) Tissue-engineered lungs for in vivo implantation. Science 329, 538–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ott H. C., Clippinger B., Conrad C., Schuetz C., Pomerantseva I., Ikonomou L., Kotton D., Vacanti J. P. (2010) Regeneration and orthotopic transplantation of a bioartificial lung. Nat. Med. 16, 927–933 [DOI] [PubMed] [Google Scholar]
- 6. Higuchi S., Lin Q., Wang J., Lim T. K., Joshi S. B., Anand G. S., Chung M. C., Sheetz M. P., Fujita H. (2013) Heart extracellular matrix supports cardiomyocyte differentiation of mouse embryonic stem cells. J. Biosci. Bioeng. 115, 320–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daly A., Wallis J., Borg Z., Bonvillain R., Deng B., Ballif B., Jaworski D. M., Allen G. B., Weiss D. J. (2012) Initial binding and recellularization of decellularized mouse lung scaffolds with bone marrow-derived mesenchymal stromal cells. Tissue Eng. Part A 18, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Welham N. V., Chang Z., Smith L. M., Frey B. L. (2013) Proteomic analysis of a decellularized human vocal fold mucosa scaffold using 2D electrophoresis and high-resolution mass spectrometry. Biomaterials 34, 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kliment C. R., Englert J. M., Crum L. P., Oury T. D. (2011) A novel method for accurate collagen and biochemical assessment of pulmonary tissue utilizing one animal. Int. J. Clin. Exp. Pathol. 4, 349–355 [PMC free article] [PubMed] [Google Scholar]
- 10. Bonvillain R. W., Danchuk S., Sullivan D. E., Betancourt A. M., Semon J. A., Eagle M. E., Mayeux J. P., Gregory A. N., Wang G., Townley I. K., Borg Z. D., Weiss D. J., Bunnell B. A. (2012) A nonhuman primate model of lung regeneration: Detergent-mediated decellularization and initial in vitro recellularization with mesenchymal stem cells. Tissue Eng. Part A 18, 2437–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Önnerfjord P., Khabut A., Reinholt F. P., Svensson O., Heinegård D. (2012) Quantitative proteomic analysis of eight cartilaginous tissues reveals characteristic differences as well as similarities between subgroups. J. Biol. Chem. 287, 18913–18924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Brien J. H., Vanderlinden L. A., Schedin P. J., Hansen K. C. (2012) Rat mammary extracellular matrix composition and response to ibuprofen treatment during postpartum involution by differential GeLC–MS/MS analysis. J. Proteome Res. 11, 4894–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boersema P. J., Raijmakers R., Lemeer S., Mohammed S., Heck A. J. R. (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 [DOI] [PubMed] [Google Scholar]
- 14. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 15. Dayon L., Hainard A., Licker V., Turck N., Kuhn K., Hochstrasser D. F., Burkhard P. R., Sanchez J.-C. (2008) Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem. 80, 2921–2931 [DOI] [PubMed] [Google Scholar]
- 16. Callister S. J., Barry R. C., Adkins J. N., Johnson E. T., Qian W. J., Webb-Robertson B.-J. M., Smith R. D., Lipton M. S. (2006) Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res. 5, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Webb-Robertson B.-J., Matzke M. M., Jacobs J. M., Pounds J. G., Waters K. M. (2011) A statistical selection strategy for normalization procedures in LC-MS proteomics experiments through dataset-dependent ranking of normalization scaling factors. Proteomics 11, 4736–4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beynon R. J., Doherty M. K., Pratt J. M., Gaskell S. J. (2005) Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2, 587–589 [DOI] [PubMed] [Google Scholar]
- 19. Miller C., George S., Niklason L. (2010) Developing a tissue-engineered model of the human bronchiole. J. Tissue Eng. Regen. Med. 4, 619–627 [DOI] [PubMed] [Google Scholar]
- 20. Nakayama K. H., Lee C. C., Batchelder C. A., Tarantal A. F. (2013) Tissue specificity of decellularized rhesus monkey kidney and lung scaffolds. PLoS ONE 8, e64134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hansen K. C., Kiemele L., Maller O., O'Brien J., Shankar A., Fornetti J., Schedin P. (2009) An in-solution ultrasonication-assisted digestion method for improved extracellular matrix proteome coverage. Mol. Cell. Proteomics 8, 1648–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vandermarliere E., Mueller M., Martens L. (2013) Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrometry Rev. 32, 453–465 [DOI] [PubMed] [Google Scholar]
- 23. Ericsson C., Nistér M. (2011) Protein extraction from solid tissue. Methods Mol. Biol. 675, 307–312 [DOI] [PubMed] [Google Scholar]
- 24. Harvey A., Yen T.-Y., Aizman I., Tate C., Case C. (2013) Proteomic analysis of the extracellular matrix produced by mesenchymal stromal cells: Implications for cell therapy mechanism. PLoS ONE 8, e79283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilson R., Norris E. L., Brachvogel B., Angelucci C., Zivkovic S., Gordon L., Bernardo B. C., Stermann J., Sekiguchi K., Gorman J. J., Bateman J. F. (2012) Changes in the chondrocyte and extracellular matrix proteome during post-natal mouse cartilage development. Mol. Cell. Proteomics 11, M111.014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lai K. K. Y., Shang S., Lohia N., Booth G. C., Masse D. J., Fausto N., Campbell J. S., Beretta L. (2011) Extracellular matrix dynamics in hepatocarcinogenesis: A comparative proteomics study of PDGFC transgenic and Pten null mouse models. PLoS Genetics 7, DOI: 10.1371/journal.pgen.1002147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geiger T., Velic A., Macek B., Lundberg E., Kampf C., Nagaraj N., Uhlen M., Cox J., Mann M. (2013) Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteomics 12, 1709–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naba A., Clauser K. R., Hoersch S., Liu H., Carr S. A., Hynes R. O. (2011) The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics 11, M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song M.-N., Moon P.-G., Lee J.-E., Na M., Kang W., Chae Y. S., Park J.-Y., Park H., Baek M.-C. (2012) Proteomic analysis of breast cancer tissues to identify biomarker candidates by gel-assisted digestion and label-free quantification methods using LC-MS/MS. Arch. Pharm. Res. 35, 1839–1847 [DOI] [PubMed] [Google Scholar]
- 30. Wallis J. M., Borg Z. D., Daly A. B., Deng B., Ballif B. A., Allen G. B., Jaworski D. M., Weiss D. J. (2012) Comparative assessment of detergent-based protocols for mouse lung de-cellularization and re-cellularization. Tissue Eng. Part C Methods 18, 420–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calle E. A., Petersen T. H., Niklason L. E. (2011) Procedure for Lung Engineering. J. Visual. Experiment. 49, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deutsch E. W., Lam H., Aebersold R. (2008) PeptideAtlas: A resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 9, 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Craig R., Cortens J. P., Beavis R. C. (2004) Open source system for analyzing, validating, and storing protein identification data. J. Proteome Res. 3, 1234–1242 [DOI] [PubMed] [Google Scholar]
- 34. Brownridge P., Holman S. W., Gaskell S. J., Grant C. M., Harman V. M., Hubbard S. J., Lanthaler K., Lawless C., O'Cualain R., Sims P., Watkins R., Beynon R. J. (2011) Global absolute quantification of a proteome: Challenges in the deployment of a QconCAT strategy. Proteomics 11, 2957–2970 [DOI] [PubMed] [Google Scholar]
- 35. Matic I., Jaffray E. G., Oxenham S. K., Groves M. J., Barratt C. L. R., Tauro S., Stanley-Wall N. R., Hay R. T. (2011) Absolute SILAC-compatible expression strain allows Sumo-2 copy number determination in clinical samples. J. Proteome Res. 10, 4869–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pratt J. M., Simpson D. M., Doherty M. K., Rivers J., Gaskell S. J., Beynon R. J. (2006) Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protocols 1, 1029–1043 [DOI] [PubMed] [Google Scholar]
- 37. Lu X., Zhu H. (2005) Tube-gel digestion: A novel proteomic approach for high throughput analysis of membrane proteins. Mol. Cell. Proteomics 4, 1948–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Havlis J., Thomas H., Sebela M., Shevchenko A. (2003) Fast-response proteomics by accelerated in-gel digestion of proteins. Anal. Chem. 75, 1300–1306 [DOI] [PubMed] [Google Scholar]
- 39. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P.-A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H.-J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Green J. M. (1996) Peer reviewed: A practical guide to analytical method validation. Anal. Chem. 68, 305A–309A [Google Scholar]
- 42. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 43. Eyre D. R., Paz M. A., Gallop P. M. (1984) Cross-linking in collagen and elastin. Annu. Rev. Biochem. 53, 717–748 [DOI] [PubMed] [Google Scholar]
- 44. Petersen T. H., Calle E. A., Colehour M. B., Niklason L. E. (2012) Matrix composition and mechanics of decellularized lung scaffolds. Cells Tissues Organs 195, 222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nichols J. E., Niles J., Riddle M., Vargas G., Schilagard T., Ma L., Edward K., La Francesca S., Sakamoto J., Vega S., Ogadegbe M., Mlcak R., Deyo D., Woodson L., McQuitty C., Lick S., Beckles D., Melo E., Cortiella J. (2013) Production and assessment of decellularized pig and human lung scaffolds. Tissue Eng. Part A 19, 2045–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Booth A. J., Hadley R., Cornett A. M., Dreffs A. A., Matthes S. A., Tsui J. L., Weiss K., Horowitz J. C., Fiore V. F., Barker T. H., Moore B. B., Martinez F. J., Niklason L. E., White E. S. (2012) Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 186, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Price A. P., England K. A., Matson A. M., Blazar B. R., Panoskaltsis-Mortari A. (2010) Development of a decellularized lung bioreactor system for bioengineering the lung: The matrix reloaded. Tissue Eng. Part A 16, 2581–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodríguez-Rodríguez P., Arribas S. M., de Pablo A. L., González M. C., Abderrahim F., Condezo-Hoyos L. (2013) A simple dot-blot–Sirius red-based assay for collagen quantification. Anal Bioanal Chem 405, 6863–6871 [DOI] [PubMed] [Google Scholar]
- 49. Piehowski P. D., Petyuk V., Orton D. J., Xie F., Moore R. J., Ramirez-Restrepo M., Engel A., Lieberman A. P., Albin R. L., Camp D. G., Smith R. D., Myers A. J. (2013) (2013) Sources of technical variability in quantitative LC-MS proteomics: Human brain tissue sample analysis. J. Proteome Res. 12, 2128–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Epstein E. H., Jr., Munderloh N. H. (1975) Isolation and characterization of CNBr peptides of human (alpha 1 (III)) 3 collagen and tissue distribution of (alpha 1 (I)) 2 alpha 2 and (alpha 1 (III)) 3 collagens. J. Biol. Chem. 250, 9304–9312 [PubMed] [Google Scholar]
- 51. Kelleher N. L., Lin H. Y., Valaskovic G. A., Aaserud D. J., Fridriksson E. K., McLafferty F. W. (1999) Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry. J. Am. Chem. Soc. 121, 806–812 [Google Scholar]
- 52. Lee J.-E., Kwon J., Baek M.-C. (2011) A combination method of chemical with enzyme reactions for identification of membrane proteins. Biochim. Biophys. Acta 1814, 397–404 [DOI] [PubMed] [Google Scholar]
- 53. Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 54. Zander N. E., Orlicki J. A., Rawlett A. M., Beebe T. P., Jr. (2012) Quantification of protein incorporated into electrospun polycaprolactone tissue engineering scaffolds. ACS Appl. Mater. Interfaces 4, 2074–2081 [DOI] [PubMed] [Google Scholar]
- 55. Holman S. W., Sims P. F., Eyers C. E. (2012) The use of selected reaction monitoring in quantitative proteomics. Bioanalysis 4, 1763–1786 [DOI] [PubMed] [Google Scholar]
- 56. Kirkpatrick D. S., Gerber S. A., Gygi S. P. (2005) The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods 35, 265–273 [DOI] [PubMed] [Google Scholar]
- 57. Ong S.-E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 58. Mirzaei H., McBee J. K., Watts J., Aebersold R. (2008) Comparative evaluation of current peptide production platforms used in absolute quantification in proteomics. Mol. Cell. Proteomics 7, 813–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barnidge D. R., Dratz E. A., Martin T., Bonilla L. E., Moran L. B., Lindall A. (2003) Absolute quantification of the G protein-coupled receptor rhodopsin by LC/MS/MS using proteolysis product peptides and synthetic peptide standards. Anal. Chem. 75, 445–451 [DOI] [PubMed] [Google Scholar]
- 60. Chen J., Turko I. V. (2014) Trends in QconCATs for targeted proteomics. Trends Anal. Chem. 57, 1–5 [Google Scholar]
- 61. Schwarzbauer J. E., DeSimone D. W. (2011) Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 3, DOI: 10.1101/cshperspect.a005041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Olivero D. K., Furcht L. T. (1993) Type IV collagen, laminin, and fibronectin promote the adhesion and migration of rabbit lens epithelial cells in vitro. Invest. Ophthalmol. Vis. Sci. 34, 2825–2834 [PubMed] [Google Scholar]
- 63. Clyman R. I., McDonald K. A., Kramer R. H. (1990) Integrin receptors on aortic smooth muscle cells mediate adhesion to fibronectin, laminin, and collagen. Circ. Res. 67, 175–186 [DOI] [PubMed] [Google Scholar]
- 64. Cameron J. D., Skubitz A. P., Furcht L. T. (1991) Type IV collagen and corneal epithelial adhesion and migration. Effects of type IV collagen fragments and synthetic peptides on rabbit corneal epithelial cell adhesion and migration in vitro. Invest. Ophthalmol. Vis. Sci. 32, 2766–2773 [PubMed] [Google Scholar]
- 65. Han J., Ohno N., Pasco S., Monboisse J.-C., Borel J. P., Kefalides N. A. (1997) A cell binding domain from the α3 chain of type IV collagen inhibits proliferation of melanoma cells. J. Biol. Chem. 272, 20395–20401 [DOI] [PubMed] [Google Scholar]
- 66. Vandenberg P., Kern A., Ries A., Luckenbill-Edds L., Mann K., Kühn K. (1991) Characterization of a type IV collagen major cell binding site with affinity to the alpha 1 beta 1 and the alpha 2 beta 1 integrins. J. Cell Biol. 113, 1475–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hinenoya N., Naito I., Momota R., Sado Y., Kumagishi K., Ninomiya Y., Ohtsuka A. (2008) Type IV collagen alpha chains of the basement membrane in the rat bronchioalveolar transitional segment. Arch. Histol. Cytol. 71, 185–194 [DOI] [PubMed] [Google Scholar]
- 68. Butkowski R. J., Wieslander J., Kleppel M., Michael A. F., Fish A. J. (1989) Basement membrane collagen in the kidney: regional localization of novel chains related to collagen IV. Kidney Int. 35, 1195–1202 [DOI] [PubMed] [Google Scholar]
- 69. Nguyen N. M., Pulkkinen L., Schlueter J. A., Meneguzzi G., Uitto J., Senior R. M. (2006) Lung development in laminin gamma2 deficiency: Abnormal tracheal hemidesmosomes with normal branching morphogenesis and epithelial differentiation. Respir. Res. 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nguyen N. M., Senior R. M. (2006) Laminin isoforms and lung development: all isoforms are not equal. Dev. Biol. 294, 271–279 [DOI] [PubMed] [Google Scholar]
- 71. Cavalcante F. S., Ito S., Brewer K., Sakai H., Alencar A. M., Almeida M. P., Andrade J. S., Jr., Majumdar A., Ingenito E. P., Suki B. (2005) Mechanical interactions between collagen and proteoglycans: Implications for the stability of lung tissue. J. Appl. Physiol. 98, 672–679 [DOI] [PubMed] [Google Scholar]
- 72. Petersen T. H., Calle E. A., Colehour M. B., Niklason L. E. (2012) Matrix composition and mechanics of decellularized lung scaffolds. Cells Tissues Organs 195, 222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Song J. J., Ott H. C. (2011) Organ engineering based on decellularized matrix scaffolds. Trends Mol. Med. 17, 424–432 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.