

Abstract
Protein aggregation is common to dozens of diseases including prionoses, diabetes, Parkinson’s and Alzheimer’s. Over the past 15 years, there has been a paradigm shift in understanding the structural basis for these proteinopathies. Precedent for this shift has come from investigation of soluble Aβ oligomers (AβOs), toxins now widely regarded as instigating neuron damage leading to Alzheimer’s dementia. Toxic AβOs accumulate in AD brain and constitute long-lived alternatives to the disease-defining Aβ fibrils deposited in amyloid plaques. Key experiments using fibril-free AβO solutions demonstrated that while Aβ is essential for memory loss, the fibrillar Aβ in amyloid deposits is not the agent. The AD-like cellular pathologies induced by AβOs suggest their impact provides a unifying mechanism for AD pathogenesis, explaining why early stage disease is specific for memory and accounting for major facets of AD neuropathology. Alternative ideas for triggering mechanisms are being actively investigated. Some research favors insertion of AβOs into membrane, while other evidence supports ligand-like accumulation at particular synapses. Over a dozen candidate toxin receptors have been proposed. AβO binding triggers a redistribution of critical synaptic proteins and induces hyperactivity in metabotropic and ionotropic glutamate receptors. This leads to Ca2+ overload and instigates major facets of AD neuropathology, including tau hyperphosphorylation, insulin resistance, oxidative stress, and synapse loss. Because different species of AβOs have been identified, a remaining question is which oligomer is the major pathogenic culprit. The possibility has been raised that more than one species plays a role. Despite some key unknowns, the clinical relevance of AβOs has been established, and new studies are beginning to point to co-morbidities such as diabetes and hypercholesterolemia as etiological factors. Because pathogenic AβOs appear early in the disease, they offer appealing targets for therapeutics and diagnostics. Promising therapeutic strategies include use of CNS insulin signaling enhancers to protect against the presence of toxins and elimination of the toxins through use of highly specific AβO antibodies. An AD-dependent accumulation of AβOs in CSF suggests their potential use as biomarkers and new AβO probes are opening the door to brain imaging. Overall, current evidence indicates that Aβ oligomers provide a substantive molecular basis for the cause, treatment and diagnosis of Alzheimer’s disease.
Keywords: ADDLs, Antibodies, Diagnostics, Etiology, Immunotherapy, Signaling, Structure, Synapses, Therapeutics
Introduction
Clinical trials for Alzheimer’s disease (AD) that target the buildup of amyloid beta (Aβ) have been a lingering disappointment. Lack of success for over a decade has fueled misgivings about the therapeutic value of Aβ-related targets, and even raised questions about whether Aβ pathology is the key to nerve cell damage and dementia onset. This may, however, be a case of throwing out the proverbial baby with the bathwater. More than 30 years’ worth of evidence has established the critical involvement of Aβ in instigating AD, including the more recent discovery that a different type of mutation in APP, one that reduces Aβ levels, protects carriers from getting the disease [73]. Clinical failures can be attributed to bad timing, poor drugs, and perhaps most importantly, impacting the wrong targets [157]. Widespread studies since 1998 strongly suggest the correct targets comprise toxic amyloid β oligomers (AβOs).
Development of the oligomer hypothesis
Although soluble oligomeric Aβ species were detected in AD brain tissue more than 20 years ago [42], their presence was regarded only as evidence of ongoing fibrillogenesis and not relevant to nerve cell damage and the onset of dementia. However, a different conclusion emerged when experimental methods were found that could give toxic solutions of Aβ that were fibril-free [131]. Soluble AβOs, formed without fibrils using very low doses of Aβ, or through the chaperone-like action of clusterin, were discovered to be potent CNS neurotoxins [95]. AβOs rapidly inhibited LTP in brain slices, and, with increased exposure, they accelerated nerve cell-specific death, through a signaling-dependent mechanism requiring the protein tyrosine kinase Fyn. Based on these findings, it was proposed that early memory loss in AD and the progressively catastrophic dementia were caused by neural signaling dysfunction triggered by soluble AβOs [82, 95].
Over the last decade, the Aβ oligomer hypothesis has generated widespread interest, and numerous reviews have covered major milestones substantiating its applicability to AD [39, 60, 61, 86, 122, 139]. The AβO hypothesis has been described as a small conceptual revolution [60] and is widely regarded as accounting for the onset of neuron damage leading to AD (Fig. 1a). AβOs have been shown to build up in AD brain in a manner distinct from amyloid plaques [76] and to accumulate in AD animal models [14, 17]. AβOs from AD brain and synthetic AβOs appear structurally equivalent [47, 76], they disrupt synaptic plasticity as well as learning and memory when injected into animal models [17, 175], their emergence coincides with onset of memory dysfunction in Tg models [105], and antibodies against AβOs rescue memory performance in those models [183, 194]. Mechanistically, AβOs act like pathogenic gain-of-function ligands, targeting certain cells and synapses on those cells [91]. Although structural questions regarding the nature of toxic AβO species still are under rigorous investigation [5] and not all oligomers are active [16, 93], it has been established that brain-derived and synthetic AβOs share similar toxicology [25, 84]. Moreover, as shown in Fig. 1a, b, the cellular damage that can be instigated by AβOs extends to virtually all major aspects of AD neuropathology. Although not proven, and not uniformly accepted, there is a high probability that AβOs play a critical role in instigating AD.
Fig. 1.

Aβ oligomers (AβOs) instigate neuron damage in Alzheimer’s disease. a Oligomeric Aβ, rather than insoluble amyloid species, instigates neuron damage in AD (adapted from the “2004/2005 Progress Report on Alzheimer’s disease” Health and Human Services). b AD-associated changes attributed to AβOs
Our primary purpose is to focus on recent works and review issues being addressed in the current AβO literature. Given that over 1,200 articles have appeared in the last 3 years alone, it is unfortunate but inevitable that important works have not been included, and we regret the missed opportunities. Due to publication constraints, there is a significant amount of information that can be found in the supplementary material. Our review is organized according to the following themes—AβO etiology; the toxic mechanism; AβO structure and analytics; AβO-directed therapeutics; and AβO-directed diagnostics.
Etiology and mechanism
Why do AβOs show up?
Remarkably little is known about the etiology of AβO accumulation in sporadic AD, which accounts for roughly 95 % of AD cases. A clue to onset may be the peculiar distribution of AβOs seen in very early stages of pathology (Fig. 2). In a field of hundreds of neurons, only about a dozen show the presence of AβOs which, as seen in the inset, distribute peri-somatically. If we could understood what accounts for this cell-specific and sporadic distribution, we might have a better handle on the etiology of AβO.
Fig. 2.

Perisomatic AβOs consistent with synapse binding are present early in human neuropathology. Left Low magnification of human cortical brain section stained with an anti-oligomer antibody. Scattered individual neurons are surrounded by AβOs in early AD, before the appearance of amyloid plaques. The perineuronal distribution of these AβOs (right) is consistent with a binding site within the dendritic arbor. Scale bar 10 µm. Adapted from Lacor et al. [91]
It sometimes is said that AD manifests as multiple diseases. The etiology of AβO buildup may thus involve disparate factors, and in the long run, successful treatment might depend on knowing which etiological triggers are involved. Current investigations concern factors such as pathophysiological co-morbidities, toxic environments, and loss of natural defense mechanisms with aging. Environmental and behavioral factors, including diet choices, will be of particular interest because they can be corrected.
While a broader discussion of etiological factors in AβO buildup can be found in the supplementary material, one rapidly developing area of investigation concerns the defense provided by neuronal insulin signaling, and the relationship between AβOs, diabetes, and resistance to insulin signaling in the AD brain. A detailed review of this relationship has recently become available [22]. One side of the story centers on defense against AβOs: CNS insulin signaling serves to prevent AβO buildup [7] and to block AβO neurotoxic binding [23]. The other side of the story is the vulnerability of the mechanism itself to AβO toxicity: AβOs impair insulin signal transduction on CNS neurons by blocking trafficking of insulin receptors to dendritic membranes [23] and inhibiting the critical effector IRS-1 [111]. By rendering neurons insulin-resistant, AβOs provide a mechanism to explain why AD appears to be a Type 3 diabetes [26, 27]. Consistent with results from cell biology, animals given ICV injections of AβOs show impaired brain insulin signaling and metabolism along with memory loss [57, 135]. This animal model appears to recapitulate insulin neuropathology in the AD brain [8]. Overall, a vicious cycle emerges. As AβOs increase due to impaired CNS insulin signaling, insulin signaling grows even weaker, due to the impact of the toxic AβOs (Fig. 3). Furthermore, when insulin receptors are down, GSK3β activity is up, and this may be germane to pTau elevation [4]. Decreased CNS insulin signaling which appears to occur with age could tip the scales toward AβOs in the struggle for synaptic survival. The section later on Therapeutics discusses the targeting of CNS insulin signaling for AD treatment.
Fig. 3.

Dysfunctional insulin signaling induced by AβOs provides one link to AD etiology. Diabetes causes a reduction in brain insulin and brain insulin signaling as well as an increase in glucose and lipids. This leads to an increase in Aβ production and a reduction in AβO clearance, causing a buildup of oligomers in the brain. As AβO levels rise, they bind synapses and cause neuronal damage, resulting in a decrease in insulin receptors and further reducing insulin signaling in brain cells. This vicious cycle results in cognitive failure and AD
Are AβOs extracellular, intracellular, or both?
A persistent debate is whether AβOs accumulate and instigate neuronal damage extracellularly or intracellularly. The answer is especially relevant given that toxicity assays use exogenously added AβOs. Persuasive evidence has been obtained that supports each view. Subcellular distribution of brain AβOs at very early stages of pathology is consistent with extracellular association of AβOs with surface membranes (Fig. 2). Direct evidence for extracellular accumulation came from older measurements of AβOs in human CSF, which shows increased abundance in AD patients [45] (Fig. 4). A suggested prion-like spread of AβOs, while still being investigated [155], is consistent with an extracellular presence, and the buildup of AβOs in the medium of APP-transfected cells indicates a capacity for natural AβO secretion [17, 175]. The efficacy of antibodies in reducing pathology [132] and rescuing behavior, even in some cases as readily as 24 h [29] further substantiates with the existence of extracellular AβOs. Although antibodies can be internalized, this occurs after binding to cell surface epitopes. On the other hand, immunohistochemistry of many TG models show robust intracellular accumulation of AβOs (see, e.g., Fig. 4), and high-resolution EM studies of human samples show AβOs intracellularly at synaptic terminals [48]. Although AβOs could assemble from extracellular monomers, they also appear to be released from intracellular pools [176]. AβOs additionally appear to accumulate in AD astrocytes, although the form they take is uncertain [96], and it is evident that AβOs are taken up by microglia. Older evidence thus indicates that AβOs can occur in both intracellular and extracellular pools. The possibility that these pools undergo a dynamic exchange would reconcile both views [132].
Fig. 4.

AβOs can accumulate in intracellular and extracellular pools. Intracellular AβOs are detectable in animal models overproducing APP and Aβ; however, the presence of extracellular AβOs on dendrites and in CSF suggests they are also important in AD. Left A representative micrograph of confocal fluorescence labeling of amyloid-β peptide (Aβ)-oligomer-specific antibody NU1 immunoreaction in young, pre-plaque Tg mice shows intracellular localization of AβOs. Adapted from Ferretti et al. [40]. Right A scatter plot from the ultrasensitive scanometric detection of AβOs in cerebrospinal fluid. Adapted from Georganopoulou et al. [45]. The response for the negative human control subject (brain extract) was similar to that observed for the chip control. The data points are averages of several separate experiments normalized for each assay based on the highest response in a series of runs. The mean values for ADDL concentrations (solid lines) are estimated for each group based on a calibration curve
Recent evidence for intracellular AβOs
Evidence for intracellular synthesis and uptake of AβOs continues to come from studies of human neuropathology and transgenic animal models. In human tissue, intracellular AβOs have been identified in cholinergic neurons, suggesting a role in cholinergic deficiency [128]. In Tg2576 mice, intraneuronal AβOs are associated with loss of MAP2 in dendrites and postsynaptic terminals [162], consistent with a previous report of synaptic pTau. The data suggest low MW AβOs precede high MW oligomer and plaque formation. Multiple studies suggest that aggregation and clearance of Aβ are influenced by ApoE isoforms [59, 154]. Co-culture and in vivo experiments demonstrate that ApoE, secreted by astrocytes, is essential for receptormediated uptake of Aβ by neurons. Results showed that a non-toxic ApoE/Aβ interaction antagonist, Aβ12–28P, blocked uptake and blocked synapse protein loss. The peptide was also protective in vivo [90].
Recent evidence for extracellular AβOs
In cell culture models, SH-SY5Y,+/− transfected with Swedish double mutant APP, showed intracellular accumulation of AβO in many membrane-bound organelles as well as cytosolic structures; further data indicate Aβ does not localize to particular organelles and is largely secreted from cells [196]. Overall results are consistent with the pool of extracellular AβOs originating from secreted AβOs as well as a pool that assembles from secreted monomer. Especially convincing new evidence for extracellular pools of AβOs comes from microdialysis experiments. Brain interstitial fluid from living mice showed an age and transgene-dependent presence of AβOs [164]. Acute inhibition of gamma-secretase caused AβO abundance to decrease, a key control that AβOs were derived from expected metabolic pathways. It has been noted in some experimental models (e.g., APP-transfected cell lines) that extracellular pools of AβOs are insensitive to secretase inhibitors [53]. The acute efficacy of therapeutic antibodies further supports the neurological significance of extracellular AβOs [29]. Antibodies against oligomers recently were shown to reduce AβOs and plaques and improve memory function [163]. Data were consistent with earlier conclusions that extracellular and intracellular AβO pools are dynamically related, potentially involving sortilin, and they also indicated that intracellular accumulation of AβOs is upstream of tau pathology. scFv antibodies against Aβ have been found to readily reach the brain following intranasal administration [13], and AβO antibodies delivered through intranasal administration were found to rescue performance on the Morris water maze paradigm [183].
Extracellular prion-like spread?
Data that AβOs can accumulate intracellularly and later be secreted are consistent with a suggested prion-like propagation of AβOs (for history of this idea, see [155]). Oligomers of other non-prion proteins (e.g., tauOs and synucleinOs) also have been reported to show prion-like behavior [55, 88]. Cell-to-cell transfer of AβOs has been seen using a donor–acceptor 3-D co-culture model. Transfer appears to be an early event that occurs before and is independent of cell degeneration [30]. Similar spreading is seen in astrocyte–neuron co-cultures where astrocytes are induced by endogenous Aβ25–35 to make and secrete Aβ42 [20]. Intraneuronal injection of AβOs into primary hippocampal neurons revealed neuron-to-neuron transfer that required direct cellular connections. Intracellular accumulation resulted in tubulin beading and endosomal leakage [127]. In vivo injections into the CA1 region of the hippocampus resulted in time-dependent effects on the expression levels of multiple genes distant from the injection site [34]. With respect to the prion-like potential of cell-to-cell transfer, it has been found that an unusual oligomeric species, large fatty acid-derived AβOs that are 12–18mers and form “off pathway”, can recruit Aβ42 at expense of fibril formation and self-replicate [89]. Another unusual interaction occurs with synuclein, which can interact with Aβ and provide a seed for aggregation in vitro [134]. Oligomers were found to migrate from affected to unaffected neurons, where it was suggested they act as a template to promote formation of larger oligomers in a self-propagating manner [133]. Of particular interest, pyroglutamylated Aβ, which appears to be a pathophysiologically important proteoform, makes especially toxic small oligomers when mixed with Aβ42, more toxic than made by Aβ42 alone. The mixed AβOs show tau-dependent toxicity, and they act as a template for restructuring of Aβ42 into distinct small MW AβOs that propagate in prion-like manner [129].
Targeting synapses
If extracellular pools of AβOs are neurologically active, a key mechanistic question is whether they act with specificity—are particular cells targeted, and if so, how? Early studies showed that AβOs bind only to particular neurons, at most half those present in hippocampal cultures [16, 97]. Less binding occurs in cortical cultures and little to none in cerebellar cultures [97]. This pattern is in harmony with the region-dependent AβO toxicity seen in brain slice preparations [80]. Cell-specific binding correlates with toxic responses, e.g., stimulation of tau hyperphosphorylation [25].
Significantly, exogenous AβOs accumulate at synapses and in particular, at synaptic spines [47, 91], although presynaptic sites may also be targeted [62]. The ligand-like targeting of synapses is evident for both synthetic and brain-derived AβOs (Fig. 5). While additional targets may exist, this highly selective distribution is in harmony with rapid AβO-induced disruption of LTP and LTD [175, 177]. To account for the specificity of AβO targeting, it has been proposed that attachment is mediated by particular cell surface proteins that act as toxin receptors [95]. These receptors would be expressed only on certain cells and would act to convert binding into cell-damaging responses. This section reviews recent evidence regarding the toxin receptor hypothesis and the molecules that have been proposed as candidate receptors. How transduction of binding into toxicity takes place is considered in the next section.
Fig. 5.

Synthetic and brain-derived AβOs are ligands that target synapses. AβOs extracted from AD brain or prepared in vitro show punctate binding to neuronal cell surface proteins. Cultured hippocampal neurons were incubated with synthetic AβOs or soluble extracts of human brain. Binding was visualized by immunofluorescence microscopy by using a polyclonal anti-Aβ oligomer antibody, M93. Synthetic AβOs (Left), soluble extracts of non-AD control brains (Center), and soluble AD-brain extracts (Right) are shown. Small puncta, bound largely along neurites, are evident for AD extracts and synthetic AβOs but not for control extracts. Bar 10 µm. Adapted from Gong et al. [47]
Membrane proteins
The hunt for toxin receptors has been greatly invigorated by identification of cellular prion protein as a receptor candidate, first identified in a screen of a cDNA expression library [101]. Initial evidence strongly indicated binding to PrP was a key step in the mechanism of AβO toxicity [101] (for review, see [100]). Investigations of how externally oriented PrP might bring about intracellular damage indicate coupling to the protein tyrosine kinase Fyn (Fig. 6). This mechanism is consistent with early studies showing Fyn is essential for AβO-induced toxicity [15, 95]. Further details regarding AβO interactions with PrP, which remains somewhat controversial, can be found in the supplementary material.
Fig. 6.
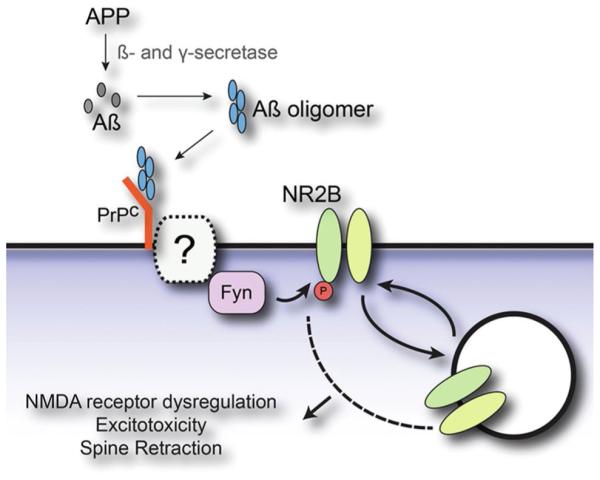
Model for Aβ oligomer-induced synaptic dysfunction involving PrPc and Fyn. Extracellular Aβ monomer, produced through cleavage of the amyloid precursor protein (APP) by both β- and γ-secretase, assembles into toxic Aβ oligomers. These AβOs bind to cellular prion protein (PrPC) and activate Fyn tyrosine kinase, possibly through a yet to be identified transmembrane protein. AβO activation of Fyn signaling drives the tyrosine phosphorylation of NMDA receptors, which in turn produces altered surface expression, dysregulation of receptor function, excitotoxicity and dendritic spine retraction. Adapted from Um and Strittmatter [170]
A number of alternatives to PrP as AβO toxin receptors have emerged, giving what appears to be an oversupply of candidates (Fig. 7). Several prominent candidates are discussed further in the supplementary material. Some of the apparent oversupply may be due to involvement of candidates in responses to alternative forms of AβOs (next section; and see review by De Strooper [5]). It also may be that AβOs are promiscuous ligands, capable of interacting with different proteins expressed by different cells. Additionally, some candidates may have come from studies of AβO–membrane protein interactions that are significant but not as toxin receptors. Multiples types of AβO–membrane protein interactions can be envisioned. (1) AβOs could uniquely attach to one or a small set of high-affinity binding proteins that act as specific toxin receptors. (2) AβOs could attach to a variety of low-affinity toxin receptors and associate subsequently with co-receptors to create a multicomponent high-affinity complex. (3) AβOs, attached to a toxin receptor or embedded within a complex, disrupt additional membrane proteins, whose downstream actions are key to neuronal damage.
Fig. 7.

A surfeit of toxin receptor candidates. Provided is a current list of candidate Aβ/Aβ oligomer receptors that have been proposed over the last 20 years. No single candidate has been shown to be necessary and sufficient to account for all aspects of AβO binding and toxicity
Membrane lipids
A prominent alternative to the receptor hypothesis is that AβOs insert directly into the lipid bilayer and disrupt membranes by acting as a pore [3, 76]. Much of the early support for this hypothesis came from experiments using artificial lipid bilayers. Structural consequences recently have been imaged by AFM, showing damage to POPC/ POPS lipid bilayers caused by Aβ40 in different aggregation states [12]. A study examining the aggregation and lipid interaction properties of Aβ peptide fragments aggregated in the absence or presence of total brain lipid extract bilayers showed that some regions interact with and disrupt bilayers (e.g., Aβ40) but others do not aggregate or interact with bilayers (e.g., Aβ28 and Aβ12–24) [186]. Some reports suggest that oligomers have more membrane affinity than monomers. This is evident in assays of non-neuronal as well as neuronal cells, suggesting that association in this assay is not receptor-dependent. AβO binding did not produce evidence of rapid cell damage [152]. Membrane insertion can be blocked by a pentapeptide from the glycine zipper region of the C-terminal of Aβ. This peptide abolishes synaptotoxicity [137]. Formation of an annular octameric channel of Aβ22–35, which induces a zinc-sensitive Ca2+ influx, is enhanced by cholesterol [28], consistent with a possible lipid raft association. Support for raft domains as primary mediators of Aβ toxicity in AD comes from a study showing that rafts recruit AβOs and that depletion of GM1 blocks interaction of AβOs and their toxicity [37]. On the other hand, data suggest that a moderate increase in membrane cholesterol content may be protective against AβO toxicity [38]. The extensive literature concerning Aβ-lipid membrane interactions and molecular level membrane modeling has recently been reviewed [32], including the possible involvement of metals in the mechanism [179].
One of the difficulties encountered by the bilayer hypothesis for AβO attachment is its inability to account for specificity. Two neurons side-by-side can show completely different ability to accumulate AβOs, one showing robust synaptic accumulation and the other showing virtually none [91]. Toxicity, measured by tau hyperphosphorylation, correlates with binding [91]. A second difficulty is an inability to account for binding saturability [91, 174]. It is possible, however, that rather than lipid bilayers, lipid rafts specific to particular synapses might play a role. It also has been hypothesized that AβOs act through both lipids and proteins, forming pores at the membrane surface and binding as well to specific receptors to induce intracellular responses [69].
Transduction mechanism
How the initial attachment of AβOs to neuronal membranes is transduced to instigate a toxic cascade is uncertain. A simple explanation would be that AβOs are intrinsically toxic. It has long been suggested, e.g., that Aβ and its assemblies could instigate neuronal damage by chemically generating ROS [11], and such localized ROS generation is supported by recent studies as well (e.g., [126]). AβOs also appear capable of creating neurotoxic pores within membranes [3], with current observations continuing to support this concept [28, 99, 140]. However, observations that neurons show highly selective vulnerability to AβO binding and toxicity [91] seem more consistent with indirect mechanisms. Such mechanisms, building on the toxin receptor hypothesis, attribute AβO toxicity to agonist-like actions in which AβOs perturb vital signaling pathways. Although some evidence indicates intracellular AβOs could be responsible for elevated Ca2+, a critical link has been established between elevated ROS and AβO-stimulated Ca2+ through pathways involving metabotropic as well as ionotropic glutamate receptors (see supplementary material).
Protein redistribution: toxic highjacking
A significant mechanistic puzzle is how glutamate receptors are actually stimulated by AβOs. Hypothetically, AβOs might act as simple receptor agonists. The phenomenon, however, is clearly more complex, beginning with binding. Experiments have been done that track movements of individual, quantum dot-labeled AβO molecules on surfaces of live neurons [143]. At first, surface-attached AβOs move freely, but after a few minutes they become immobile, often at synapses (Fig. 8). Significantly, mGluR5 receptors also become immobilized. The AβOs and mGluR5s become co-clustered, and these clusters show a time-dependent increase in size. Hypothetically, this “receptor highjacking” could be a critical part of the transduction mechanism that leads to mGluR5 hyperactivity and excessive Ca2+ mobilization. This is strongly supported by experiments showing that crosslinking and clustering of mGluR5s by antibodies mimics the effects of AβOs. Immobilization of AβOs has been confirmed in recent single molecule tracking experiments [72, 125]. Interestingly, association of AβOs with the surface was blocked by the presence of ATP-independent chaperones.
Fig. 8.
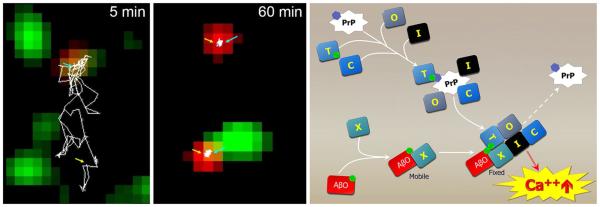
Single molecule trafficking shows AβOs stop diffusion of mGluR5 and “highjack” membrane proteins that can lead to elevated Ca2+. Left panels Dual-color single-particle tracking was used to monitor mGluR5 (red) and biotin-AβO (green) diffusion at syn- apses over time. Following the tracings of mGluR5, mGluR5 diffuses together with an AβO (5 min) outside synapses before both become stabilized at a synaptic site (60 min). Adapted from Renner et al. [143]. Right Clustering of membrane proteins, possibly involving PrPc, leads to AβO binding recruitment and membrane receptor reorganization that instigates toxic signaling. AβO binding to an unidentified receptor, X, and the recruitment of effector protein co-receptors leads to hyperactive Ca2+ signaling and downstream toxicity
An interesting question is how PrP fits as a piece of the puzzle. PrPs and mGluR5s appear to be partners in at least some aspects of AβO toxicity [170]. It is known that PrP abundance increases at the surface in response to AβOs [6]. Whether the anchoring of mGluR5s leads to a magnet-like redistribution of PrPs is possible but unknown. The ability of PrP to organize membrane domains [6, 151] may play a role, with lateral interactions involving a number of proteins that come together to create a potential toxicity domain (Fig. 8). This scaffolding effect can be construed as analogous to formation of focal contacts, but with pathogenic consequences when AβOs become attached. Not only Ca2+ but cAMP levels [24, 156] could be disrupted. It is intriguing that once neurons have reached a fully differentiated state, PrP can enzymatically be removed with little or no impact on AβO accumulation in hot spots and synapses [180].
The current bottom line is that only little is known about how attachment of AβOs to neurons and other brain cells is transduced into pathogenic signaling. On the other hand, a wealth of information exists regarding a more straightforward question—what happens downstream from the initial transduction event? A prominent example is the upstream role of AβOs in instigating tau pathology. As recently demonstrated, tau hyperphosphorylation and mis-sorting induced by AβOs is a significant factor in generating diseased neurons (Fig. 9). This pathway and others are considered in detail in the Supplementary Material.
Fig. 9.

AβOs cause pathological tau redistribution. AβOs (ADDLs) induce missorting of Tau and neurofilaments into the somatodendritic compartment of primary hippocampal neurons that suggests a mechanism for AD nerve cell death. Left, top There is no colocalization of Tau and MAP2 in vehicle-treated control cells. Tau is predominantly localized to the axonal compartment (antibody K9JA—green), while MAP2 is localized to the somatodendritic compartment (antibody AP20—red). Left, bottom In ADDL-treated cells (5 μM for 3 h), Tau is redistributed into soma and dendrites (green), where it colocalizes with MAP2 (red). Arrows indicate colocalization of MAP2 and Tau. Adapted from Zempel et al. [191]. Right Data suggest a mechanism for AD neuronal cell death that involves AβO-induced breakdown of Tau sorting and neuronal polarity. Aβ induces missorting of Tau, which in turn leads to a loss of microtubules, impaired trafficking (e.g., of mitochondria), and loss of spines, ultimately leading to neuronal death. Adapted from Zempel and Mandelkow [190]
Structure
Which forms of AβOs are responsible for instigating AD nerve cell damage is a question of continuing interest [5]. Aβ assembly pathways lead to different outcomes (Fig. 10), and this also is true for assembly pathways that lead to AβOs. As found a decade ago in vitro [16] and in vivo [105], not all AβOs are toxic or neurologically active. Overall, the goal is to obtain structure for native, undenatured disease-relevant AβO species obtained under pathophysiological conditions.
Fig. 10.

Diverse structural outcomes from different aggregation protocols. AFM images revealing a spectrum of Aβ structures are shown. a Fibrils: Aβ1–42 aged in ddH2O for one week contains periodic and smooth fibrils throughout the field of diverse sizes and lengths. b Rings: Aβ1–42 aged in F12 at 37 °C for 24 h and not centrifuged contains many ring-like structures and a few short linear protofibrils. c AβOs/protofibrils: Aβ1–42 aged in PBS for 48 h at RT contains many linear protofibrillar structures and small, spherical structures. Protofibrillar structures appear to contain bead-by-bead assemblies of multiple globular structures. The results suggest that temperature and chemical environment are critical during Aβ aggregation. Scale bar 400 nm. From Chromy et al. [16]
Recent studies have substantiated the influence of structure on oligomer cytotoxicity [161], which is consistent with the specificity of action discussed above. Aβ is a member of the class of proteins known as intrinsically disordered proteins (IDPs)—proteins that lack a stable tertiary structure in physiological conditions [60], and Aβ conformation is highly dependent on its environment [85]. Equilibrium is constantly being reestablished, making it extremely difficult to discern the sizes and abundance of each oligomer present [60, 106]. Methodology itself, including both preparative and analytical methods, influences the outcome. It is difficult to investigate AβOs under pathophysiologically relevant conditions, which themselves are hard to define. Previous investigations often used conditions that alter structure (such as incubations in SDS) or employed insensitive analytical methods requiring high levels of AβOs, at concentrations that influence structure. Supplementary material accompanying this article gives an overview of recent approaches that may be more promising (preparations; immunochemical detection; biochemical analytics; mass spectroscopy; NMR; imaging; X-ray analysis; and molecular dynamic modeling). Comprehensive reviews of AβO methodology are available and described in the supplementary material.
Several articles provide up-to-date overviews of oligomeric structures [5, 60, 77]. Although a bit simplistic, species have been classified by size (low-n oligomers, 2–4mers; high-n oligomers ~12–48mers; and megamers, indefinitely large in size) and by shape (globular, prefibrillar, and annular). It remains uncertain if there is one species of oligomer that uniquely provides a molecular basis for the cause, treatment and diagnosis of AD or whether multiple species are involved. An interesting recent study suggests the latter [41]. In this study, synthetic AβOs were separated into low molecular weight (LMW) and high molecular weight (HMW) species. Injected into brain, LMW oligomers induced a rapid, persistent impairment of memory associated with decreased hippocampal synaptophysin and GluN2B. HMW oligomers induced reversible memory impairment associated with induced oxidative stress but not synapse pathology. Memantine protected against the effects induced by the HMW oligomers, but not against the LMW oligomer. Although not in agreement with earlier findings from cell biology [92], which concluded that HMW AβOs caused synaptic damage, the findings move the field closer to the goal of reconciling differing observations concerning structure and function. Various aspects of AβO structure recently under investigation are considered below as well as in the supplementary material.
Submolecular domains
A number of studies have investigated domains within the sequence of Aβ for their contributions to structure. Smaller AβOs (1–2 nm in size) are highly immunogenic, more so than larger oligomers, because of the exposed nature of their N-terminal [21]. Epitope mapping shows the N-terminus to be immunodominant for MAb production. The N-terminal tail (residues 1–9) of Aβ 40 oligomers, investigated by rapid fluorescence technology and 2-dimensional solid state NMR, also shows great influence over the affinity of oligomers for membranes; the turn region (residues 22–29) additionally influences membrane association. Notably, 10 of 11 FAD Aβ mutants map to these two regions [153]. A toxic turn at positions 22, 23 has been related to the ER stress marker GRP78 [160] and potentially is associated with ROS generation [65]. The nearby Gly25-Ser26 dipeptide, studied using synthetically modified Aβ peptide, appears critical for Aβ42 oligomerization [149, 150]. Comparison of toxic and non-toxic AβOs indicates toxicity increases with the extent of solvent exposure of two hydrophobic domains (aa 16–22 and 30–42), reflecting the importance of conformation in toxicity [93]. Computational models indicate the hydrophobic surfaces of pore-forming AβOs promote insertion into membrane [70], while modeling and experimental studies indicate a C-Terminal turn at Val36-Gly37 (present in Aβ42, not Aβ40) promotes β-pleated sheet formation, rather than the coil seen in Aβ40 [150]. The importance of the beta-pleated sheet is indicated by the ability of the breaker peptide KLVFFK(6) to inhibit AβO formation at substoichiometric doses [193]. β-sheets that are antiparallel are evident in toxic Aβ42Os examined by X-ray diffraction [52], and these, when incubated with an abiotic cyclic d,l-α-peptide, are converted to a non-toxic parallel β-structure [145]. Formation of toxic beta sheet-rich AβOs is enhanced when monomeric Aβ40 inserts into cell surface membranes, which can be prevented by interactions with curcumin, a treatment that prevents the toxic effects of Ca2+ influx [166]
Two major studies have emphasized the importance of out-of-register beta sheets [cylindrins]. These are formed from six antiparallel Aβ protein strands, as seen via X-rayderived atomic analysis [94]. It has been suggested that cylindrins might be part of a toxic amyloid pathway that is distinct and more energetically favored than the in-register amyloid pathway [109].
Exogenous factors influencing oligomerization
The discovery that oligomeric Aβ can be a potent CNS neurotoxin arose from studies of the interaction between Aβ and the chaperone-like action of clusterin/apoJ [95]. Interactions with other proteins are now under study. Interestingly, PrP acts similarly to apoJ. At a substoichiometric 1/20 ratio, PrP traps Aβ in an oligomeric form and inhibits fibril formation. The AβOs are in an antiparallel beta sheet conformation, unlike fibrils, and bind an AβO-specific antibody. The Aβ binding site involves aa 95–113 of PrP [187]. It also has been suggested that transglutaminase (Tg) could play a role in the initiation and development of Aβ aggregates [181]. Some protein interactions appear protective. α-Synuclein, e.g., decreases Aβ42 oligomers and also is neuroprotective by stimulating the PI3K/Act cell survival pathway [144].
A significant interaction takes place between Aβ and Aβ fibrils themselves [18]. A kinetic study using selective radiolabeling and cell viability assays has found that AβOs form following critical accumulation of amyloid fibrils. The fibrils catalyze a secondary nucleation reaction which gives rise to the toxic AβOs. Formation of toxic AβOs from monomers through a fibril-catalyzed secondary nucleation reaction thus couples growth of insoluble amyloid fibrils to the generation of diffusible oligomers. This mechanism is consistent with the halos of AβOs surrounding dense core plaques. The possibility exists that AβO deposits in the brain at loci that lack associated Thioflavin S (thioS) reactivity nonetheless contain trace amounts of fibrillar amyloid [87, 174]. An implication of this fibril-catalyzed mechanism is that perturbation of the secondary nucleation pathway could be an effective strategy to control the proliferation of neurotoxic Aβ42 oligomers. These findings are intriguing in light of the correlation between AβOs and amyloid plaques in cognitively impaired but not in cognitively normal individuals [36].
Divergence of assembly pathways
An emerging concept is that self-association of Aβ proceeds along divergent paths, one toward oligomers and the other toward fibrils [173, 189]. Biophysical analysis shows there are structural similarities and differences between AβOs and fibrils and that, while common β-strand conformations form early, oligomers and fibrils differ in schemes of intermolecular organization [165]. The kinetics of fibril formation suggests three phases: early oligomerization, an intermediate phase, and growth, with conformational change a key rate-limiting step [44]. The conformation of protofibrillar intermediates may be kinetically stable [51]. Existence of pathway differences between oligomer and fibril formation has been suggested from experiments using small organofluorine compounds to disrupt Aβ self-assembly [167]. Also, in addition to secondary nucleation, AβO formation can occur through a nucleated polymerization, consistent with alternative assembly pathways; the form with a toxic turn at aa22–23 appears early in AD, prior to memory loss [160]. Generation of two types of AβOs was further indicated by results with oligomer-specific monoclonal antibodies, which reduced plaque load, tau phosphorylation, and behavior deficits [142]. Dual assembly pathways giving extended versus compact oligomers of Aβ40 were found to be influenced by metal ions, which stabilized the compact form [159]. Different conformations for assemblies of Aβ15–23 peptides were found to depend on the method of analysis [71], while diversity of Aβ dimers results in dual pathways that lead to annular oligomers or fibrils [168].
Divergence of assembly pathways begins virtually as soon as Aβ is put in solution, and the initial products are separable after only several minutes [173]. Fractionation in detergent-free buffer shows that AβOs greater than 50 kDa maintain their oligomeric state with continued incubation, while smaller AβOs show a propensity to form large aggregates and fibrils. Significantly, examined by SDS-PAGE, the early stage large and small AβOs are indistinguishable, underscoring the importance of analytics carried out under conditions that preserve native, pathophysiologically relevant structure. It is interesting that subpopulations are evident even within the larger AβOs. scFv antibodies target a small subset of AβOs that prominently attach to synapses. Polymorphic oligomerization may be a general phenomenon, as alpha-synuclein under conditions that promote amyloid fibril formation generates two well-defined oligomeric species [110]. In analogy with prion species, these different α-synuclein oligomers have been referred to as strains [54].
Therapeutics
A recent review has laid out the case for exploiting AβOs as therapeutic targets [61]. The failure of clinical trials that have targeted Aβ in general do not negate the value of approaches that target AβOs in particular. Whether AβOs are present and have a physiological role in normal brain is not known, but it appears worthwhile to prevent damage caused by excessive AβO levels. In animal models, when AβO levels are brought down, normal behavior goes up [104]. While targeting AβOs seems a rational strategy, there may be no single silver bullet, and what may be required, given the difficulties in contending with AβO toxicity [148], are combinatorial approaches. Different treatments, moreover, may be needed to contend with different etiologies. AβOs instigated by diabetes may require different treatments than those instigated by mechanical injury or hypercholesterolemia. Recent discovery efforts considered below concern vaccines that target AβOs, on targeting the relationship between AβOs, toxin receptor antagonists, inflammation and insulin signaling, and on intriguing findings from behavior modification. Overall, there is an extensive effort underway to find the means to prevent AβO-induced brain damage. The supplementary material summarizes recent attempts to identify natural products and small organic molecules that block accumulation or counter the effects of toxic AβOs.
Immunotherapy—AβO antibodies
Two of the most prominent Alzheimer’s therapeutic antibodies tested to date have been disappointing, but they are not selective for AβOs. Solanezumab, derived from the murine antibody 266, has high preferential affinity for monomer [158]. Removing the precursor to Aβ-derived toxins would require more antibody than targeting the toxins themselves, but modest benefits have nonetheless appeared in the latest analysis [98, 108]. Bapineuzumab, which does bind AβOs and has low affinity for monomers, also attaches to amyloid fibrils [78]. Off-target specificities may account for lack of efficacy of each, and also the micro-hemorrhages caused by bapineuzumab, which can bind vascular amyloid.
Given the promise yet disappointment of Aβ vaccines designed to eliminate amyloid plaques, there have been efforts to obtain antibodies selective for AβOs [75, 96, 102]. The value of AβOs as targets for immunotherapy and the status of clinical candidates have been recently reviewed [49, 124]. Although a caveat has suggested that antibodies bound to AβOs might activate microglia and potentiate AβO neurotoxicity [120], such toxicity is not in harmony with numerous reports of neuroprotection [49]. With respect to safety, an IgG4 antibody against Aβ counters the impact of AβOs in animal models more safely than IgG1 antibodies, and it shows no vasogenic edema in phase I clinical trials [1] AβOs exist in forms that assemble into fibrils and those that do not, and these forms can be distinguished by antibodies [75]. In animal models, antibodies that target non-fibrillar AβOs improve behavior and also reduce the amyloid load [142], consistent with a dynamic equilibrium between AβO and amyloid plaques. These antibodies reduce tau pathology, also seen earlier with the broader-based A11 oligomer antibody [132]. Another AβO antibody (72D9) sequestered both extracellular and intraneuronal AβOs [115]. Sequestering AβOs in older Tg mice attenuates synapse loss near plaques and abolishes loss further away [31]. This location-dependent efficacy is consistent with the proposed capacity of plaques to act as a reservoir that releases AβOs [86]. In a new approach for delivery, AβO antibodies have been administered intranasally, which was found to rescue Morris water maze performance [183]. The therapeutic potential of oligomer-specific monoclonal antibodies for AD therapeutics has led to development of humanized forms. An antibody described as targeting protofibrillar AβOs, promoted by the Arctic mutation [74], reduced AβOs in the brains and CSF of the mouse model with no impact on Aβ monomers [169]. The humanized version (BAN2401) has obtained favorable safety profile in Phase 1 clinical trials. The recently described humanized mAb ACU-193 has high affinity for AβOs with very low affinity for monomers and fibrils [49, 61]. Although effective in model systems, ACU-193 has not been tested in humans.
Single chain antibodies have been identified that target AβOs with great specificity [178]. One human scFv, specific for a small subset of synthetic AβOs [173], greatly reduces AβO binding to synapses despite targeting a negligible fraction of the total AβO pool. Another AβO-specific scFv (h3D6), given in a single intraperitoneal injection to an AD mouse, was found to reduce behavioral deficits [46]. These studies also showed that h3D6 reduced AβO levels in cortex and olfactory bulb but not hippocampus, although it is possible that hippocampal AβOs were neutralized but not eliminated, and reduced nerve cell death in cerebellum. Methods to prepare human Fab phage display libraries and screen for AβO-specific antibodies have been described [188]. Recent developments regarding active vaccines and autoantibodies are presented in the supplementary material.
Toxin receptor antagonists
As discussed above, the identity of AβO toxin receptors is uncertain, but even without definitive receptor identification, it is possible to screen for antagonists using unbiased binding assays. A cell-based assay led to a small organic molecule blocker of AβO binding with potential therapeutic value [66, 67]. The antagonist appears to target Sigma2/ PGRMC1 receptors and it blocks AβO binding and synaptotoxicity. Another unbiased high-throughput screening approach, which uses detergent-extracted synaptic membrane proteins reconstituted in soluble nanoscale membranes, has also shown that small organic molecules can block the protein–protein interactions between AβOs and their receptors (Fig. 11). Interestingly, the AβO antagonist had previously been found to rescue performance in an AD mouse model, although its relation to AβOs had not been determined [103].
Fig. 11.

Capture of receptors for Aβ oligomers in nanodiscs and a high-throughput assay to screen for unknown therapeutic targets. Top Schematic of Nanodisc formation using synaptic plasma membranes. Each Nanodisc consists of a discoidal lipid bilayer stabilized by artificial membrane scaffold proteins (MSP) with His tags. A small fraction of the population contains AβO-binding proteins. His tags on Nanodiscs and biotin on AβOs provide a means for conducting binding assays. Bottom Aurin tricarboxylic acid (ATA) potently reduces synaptic AβO accumulation in culture. ATA was assayed at 1 µM for a preventative effect on AβO accumulation at synapses in cultured rat hippocampal neurons. Images shown are of typical neurons after treatment with AβOs (left panel) or AβOs following ATA pre-treatment (right panel). AβOs are shown in green, neurons identified by β3 tubulin fluorescence are white, and DAPI is blue to indicate nuclei. Selected neurites are enlarged below each image to illustrate the distribution of bound AβO. Scale bar 10 µm. Adapted from Wilcox et al. [180]
Insulin signaling and TNF-α
The relationship between AβOs and impaired brain insulin signaling, although complex (Fig. 3) has helped stimulate efforts to enhance brain insulin signaling for therapeutics. Interest in this possibility already was kindled by earlier work, and clinical trials using intranasal insulin have shown indications of positive benefits [19]. Because AβOs themselves can impair insulin receptor function [23], indirect approaches also are being explored. The diabetes drug rosiglitazone, which lowers AβO binding in culture [23], has been found to rescue synapses and plasticity in vivo via a PPAR gamma pathway [184]. Use of d-chiro-inositol, which also enhances insulin signaling and is now available as an over-the-counter product, also prevents AβO binding [138]. Exendin, an analog of GLP-1 that can substitute for direct insulin signaling, has been shown to have major beneficial effects in multiple preclinical models [8, 111]. Liraglutide, another analog of GLP-1 that reduces AβO impact, is now in clinical trials [117].
An interesting development is the apparent involvement of TNFα as an effector of AβO toxicity, making this inflammatory agent a potentially valuable therapeutic target [111]. AD patients show elevated TNFα in cortex, hippocampus and CSF. Sequestering TNFα was earlier reported to be beneficial to AD patients [50], and, although this approach has not been pursued in the clinic, evidence from AD mouse models suggests potential efficacy [68, 116]. An AD model was found to manifest high levels of TNFα at an early age, when AβOs were present but plaques had not yet formed [43]. Chronic treatment with small molecule inhibitors of TNFα improved working memory performance and brought about reduced brain levels of TNFα mRNA and protein. In culture, exposure of microglia to AβOs leads to increases in TNFα, along with other inflammatory factors [79]. Donepezil surprisingly suppresses these cellular responses, and in AβO-injected mice, it inhibits gliosis and rescues memory function. Curcumin, an ingredient in turmeric that is neuroprotective against AβOs in various models [185], can act to block TNFα action and production [2]. Another natural product found in ginger (6-shogaol) reduces glial cell activation and memory impairment in AβO-injected mice [119]. Mechanistically, TNFα has been identified as critical to AβO-induced insulin resistance, which manifests in IRS-1 inhibition [111] as well as impaired insulin receptor trafficking and surface down-regulation [23, 195]. Two downstream mediators of TNFα have been identified—dsRNAdependent protein kinase (PKR) and eukaryotic translation initiation factor 2alpha (eIF2α-P), which is phosphorylated by PKR [111]. Both effectors of TNFα are elevated in AD brains and in AD animal models, including monkeys given AβOs by icv injections. AβO coupling to eIF2α-P and cognitive impairment is lost in PKR(−/−) and TNFR1(−/−) mice, establishing that proinflammatory signaling mediates AβO-induced IRS-1 inhibition and PKR-dependent synapse and memory loss.
Diet, exercise and enriched environments
Slowing the onset and progression of AβO pathogenesis by behavioral modifications (e.g., changes in diet, exercise or environmental richness) presents an intriguing supplement to pharmaceutical approaches. Several articles above indicate diets rich in compounds such as DHA, curcumin, and ginger would be of value, while diets high in fats or cholesterol would be harmful, fostering AβO accumulation [172]. Effects of diet on cognitive aging have been studied in carnivores, which develop cognitive decline and progressive accumulation of oxidative damage with age. When maintained on an antioxidant diet that includes fruits and vegetables, aged animals show cognitive improvement associated with improved mitochondrial function. Whether such diets benefit non-carnivores such as rabbits, which develop AβOs in response to diabetes [7], is not known.
Exercise increases BDNF levels [64], which are decreased by AβOs [136]. The overall effects of exercise are complex, especially the relationship between exercise and diet. High-fat diet worsens cognitive function in amyloid precursor protein (APP) transgenic mice, but exercise lowers Aβ deposition and memory deficit [112]. Exercise in an enriched environment rescues diet-induced Aβ deposition and memory deficit; however, consumption of a high-fat diet after 10 weeks of exercise reversed the benefits, leading to increased levels of AβOs and impaired memory performance. In aging humans, there is a correlation between reduced onset of AD and elevated blood oxygen, and this may likely be a consequence of the effects of exercise [10].
Intriguingly, enriched environments rescue cognitive function in animal models, a consequence separate from the effects of exercise [107]. The physiology involves enhanced long-term potentiation through activation of beta2-adrenergic receptors and cAMP/PKA signaling. This signaling prevented inhibition of LTP by human brain-derived AβOs.
A diet with a beta-adrenergic agonist in lieu of environmental enrichment also was protective. The conclusion that enriched environments mitigate AβO synaptotoxicity by activating beta-adrenoreceptor signaling substantiates the value of cognitive novelty during aging, perhaps in tandem with oral beta-adrenergic agonists. At an additional level, it has been found that enriched environments increase synthesis of the Aβ-degrading enzyme neprilysin and decreases the abundance of brain AβOs, accompanied by an upward shift of the cortical excitation/inhibition balance [114].
Combination therapy
AD is a complex and multi-faceted disease with various etiological origins that give rise to various co-morbidities. It has been suggested that therapeutic success may therefore depend on combination therapies, such as enhanced insulin signaling in tandem with AβO-specific vaccines [23]. Combination therapies might even be tailored to etiology—AD onset coupled to diabetes might be treated differently than onset coupled to high cholesterol. Use of multiple antibodies for immunotherapy also is a possibility. A new antibody target that has recently emerged is Dickkopf-1 (Dkk1), a secreted Wnt antagonist [141]. Acute treatment of brain slices with AβO induces Dkk1, expression of which is elevated in brains of AD patients and in AD transgenic mouse models. In brain slices, Dkk1-neutralizing antibodies suppress Aβ-induced synapse loss. Roles of TNFα and tau in AβO-instigated pathogenesis suggests further that antibodies targeting these effectors, along with those targeting AβOs, might significantly enhance outcomes [9, 182].
Diagnostics
There is no single rigorous assay for AD. Current recommendations for diagnosing AD include MMSE evaluations, CSF assays for tau and Aβ, MRI for brain volume, and PET scans for Aβ plaques and/or glucose metabolism in the brain. When used in various combinations, the tests are relatively accurate for diagnosing AD and determining disease severity. Given the role of AβOs in pathogenesis, it would be of great value to also detect and measure them. AβOs accumulate early, perhaps the first indicator of AD pathology, so AβO assays would be useful for catching disease onset [91, 121]. And, because AβOs are instigators of critical brain cell damage, their measurement could track the efficacy of disease-modifying treatments [174] (for reviews see [81, 84]). With new evidence that prefibrillar pathological forms of tau can been detected in MCI and early AD [123], it may be that combined assays of AβOs and tauOs will provide definitive tests of disease onset and progression.
CSF assay
New assays have been developed to measure AβOs in CSF, extending early efforts that introduced CSF-AβOs as AD biomarkers [45, 56]. Results, however, have varied widely. CSF-AβOs in AD have been reported to be elevated, decreased, unchanged, or not measureable. Nonetheless, the positive findings suggest cautious optimism (see details in supplementary material). Of particular interest are indications that AβO assays may detect individuals with MCI who have high risk of developing AD. An oligomer antibody-based ELISA found that AβO levels were elevated in AD that later converted to AD; although the assay showed very tight correlation with synthetic AβOs, a large overlap between patient groups limits its current diagnostic potential [63]. Although ELISAs are regarded to be sensitive, the requisite limits of detection for CSF assays are likely to be more demanding. In assays looking specifically at Aβ trimers and 12-mers (Aβ*56), data indicated that these species (both of which increased with age) were elevated in cognitively intact older adults who were at risk for AD [58]. Prior to overt symptoms, one or both of the AβOs, but not fibrillar Aβ, correlated with tau. This correlation diminished when AD advances to symptomatic stage. It also has been reported that annular AβOs appear in a small test of presymptomatic FAD carriers [146].These findings suggest that ultimately it could be possible to prevent progression to AD by knowing which AβO species to target in asymptomatic subjects.
Interestingly, the possible use of plasma AβO levels is suggested from results of a two-target assay measuring AβOs and soluble TNF-R [192]. The large trial assayed plasma from 120 controls, 32 amnestic mild cognitive impairment (aMCI) patients, and 90 mild AD patients. Increases in AβOs and soluble TNF-R levels accurately differentiated mild AD patients from control subjects, and to some extent from aMCI patients, suggesting intriguing potential as diagnostic biomarkers.
Imaging
Precedent for use of brain imaging to detect molecular pathology in AD patients has been established by the revolutionary use of PiB to quantify amyloid plaque load [83]. While brain damage during onset and progression of AD can be assessed by MRI for brain volume and FDG-PET for neuronal health, PiB analysis of amyloid plaques has been especially encouraging because it establishes precedent for uncovering important molecular pathology long before clinical signs of AD [33, 35]. However, it is AβOs rather than amyloid plaques that now are considered the agents of AD onset.
Approaches to target AβOs and other very early biomarkers such as tau-Os are beginning to be described. Pronucleon imaging, using engineered peptides that give readout when associated with beta-rich fibers, has the ability to identify plaques throughout the brain of transgenic mice in vivo and plaque-like structures in Tg mouse brain sections, but it also is regarded as having the potential to detect AβOs [130]. A novel PET probe has been introduced that combines tracer with specific monoclonal antibodies against Aβ protofibrils. The probe produces a signal in AβPP Tg mice that were devoid of Aβ plaques. It locates diffusely within the parenchyma and in older animals can localize around senile plaques (e.g., Fig. 2; [113]). This distribution of AβOs as a halo around dense cores is well known (see, e.g., Fig. 12a). Another antibody-derived PET probe, using the 6E10 N-terminal directed Aβ antibody, was delivered intravenously and found capable of distinguishing AD mice from controls [118]. Use of probes based on antibodies has been extended to molecular MRI, which could provide better spatial resolution and safety than PET [147]. An AβO-targeted MRI probe has been developed using an AβO antibody coupled to a strong T2 contrast agent [174]. These MRI–antibody probes, when delivered intranasally to bypass the blood brain barrier, can reach their hippocampal targets within 4 h and distinguish AD mice from controls (Fig. 12b). Diagnostic results are consistent with the therapeutic benefits found with intranasal delivery of the same antibody [183].
Fig. 12.

Diagnostic assays for Aβ oligomer levels provide AβO-relevant MRI signals in brain. a Sagittal brain sections, 50 µm thick, from 8-month-old 5xFAD and wt mice were probed with 568-NU4 and counterstained with Thioflavin S. Image shows a 5xFAD cortical region stained with both NU4 (red) and thioflavin S (green). NU4 labeling is more abundant than the ThioS staining. Findings demonstrate that NU4 labeling is often associated with, yet distinct from, amyloid plaques. Scale 25 µm. b In vivo imaging of NU4MNS distribution in live mice 4 h after intranasal inoculation shows labeling by the probe in the hippocampal region of the Tg mice, but not the wt controls. Scale bar 5 mm. c Higher magnification of the hippocampal region of the Tg and wt mice shows probe distribution 4 h after inoculation, the changes in distribution 96 h later, and the distribution of the target after re-administering the probe on Day 5. Data suggest that non oligomer-associated probe is clearing the brain. Scale bar 1 mm. Adapted from Viola et al. [174]
Conclusion
Over the past 3 years, well over 1,000 papers have investigated the hypothesis that nerve cell damage leading to Alzheimer’s dementia is instigated by toxic AβOs, a structural archetype that now appears relevant to dozens of diseases of fibrillogenic proteins. New results have substantiated earlier findings that persons with AD, as well as AD animal models, manifest the presence of toxic AβOs. Experiments with multiple in vitro and in vivo models have elucidated the broad pathogenic impact of AβOs, and they have provided significant advances into how cells are targeted and the mechanisms that lead to cell damage. The puzzle of disease-relevant AβO structure is being approached through application of powerful new technologies and the potential of targeting AβOs for early diagnostics and monitoring disease control is beginning to be broached. First insights are being garnered into the etiology of AβO buildup, which ultimately may inform therapeutic strategies that couple the targeting of AβOs to treatments tailored to etiological triggers. Multiple strategies are being explored that target AβOs for therapeutics, including development of AβO-specific antibodies, toxin receptor antagonists, oligomerization blockers and disruptors, and enhanced neurotrophic signaling by behavioral modification as well as pharmacology.
These efforts reflect a widely held belief that AβOs provide a unifying molecular basis for the cause, diagnosis and treatment of AD. Nonetheless, there is no consensus of agreement concerning the AβO hypothesis. Proof demands that disease-modifying AD therapeutics be achieved by targeting of toxic AβOs. This is a significant challenge, as has been discussed. While the new A4 and DIAN trials are promising in their design to treat patients before symptoms develop, what still is missing are trials that specifically target AβOs, the toxins that many investigators think are the true culprits.
Supplementary Material
Acknowledgments
We would like to thank Erika Cline, Henry Weiss, and Kyle Wilcox for their editorial contributions. We would also like to thank our supporters: the National Institutes of Health (AG022547, AG029460 and AG045637), Baxter Healthcare, Inc., and the Northwestern University Clinical and Translational Sciences Institute.
Abbreviations
- Aβ
Amyloid beta
- AβO
Amyloid beta oligomer
- AD
Alzheimer’s disease
- AFM
Atomic force microscopy
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- FAD
Familial Alzheimer’s disease
- HMW
High molecular weight
- IDP
Intrinsically disordered proteins
- LMW
Low molecular weight
- LTP
Long-term potentiation
- MAb
Monoclonal antibody
- MCI
Mild cognitive impairment
- MRI
Magnetic resonance imaging
- MW
Molecular weight
- PET
Positron emission tomography
- PrP
Prion protein
- scFv
Single chain variable fragment
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-015-1386-3) contains supplementary material, which is available to authorized users.
References
- 1.Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J, Chen M, Gogineni A, Weimer RM, Mortensen DL, Friesenhahn M, Ho C, Paul R, Pfeifer A, Muhs A, Watts RJ. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci Off J Soc Neurosci. 2012;32(28):9677–9689. doi: 10.1523/JNEUROSCI.4742-11.2012. doi:10.1523/jneurosci.4742-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Gupta SC, Sung B. Curcumin: an orally bioavailable blocker of TNF and other pro-inflammatory biomarkers. Br J Pharmacol. 2013;169(8):1672–1692. doi: 10.1111/bph.12131. doi:10.1111/ bph.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arispe N. Architecture of the Alzheimer’s A beta P ion channel pore. J Membr Biol. 2004;197(1):33–48. doi: 10.1007/s00232-003-0638-7. doi:10.1007/ s00232-003-0638-7. [DOI] [PubMed] [Google Scholar]
- 4.Bartl J, Meyer A, Brendler S, Riederer P, Grunblatt E. Different effects of soluble and aggregated amyloid beta42 on gene/protein expression and enzyme activity involved in insulin and APP pathways. J Neural Trans Vienna Austria 1996. 2013;120(1):113–120. doi: 10.1007/s00702-012-0852-5. doi:10.1007/s00702-012-0852-5. [DOI] [PubMed] [Google Scholar]
- 5.Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–357. doi: 10.1038/nn.3028. doi:10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 6.Beraldo FH, Arantes CP, Santos TG, Machado CF, Roffe M, Hajj GN, Lee KS, Magalhaes AC, Caetano FA, Mancini GL, Lopes MH, Americo TA, Magdesian MH, Ferguson SS, Linden R, Prado MA, Martins VR. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J Off Publ Fed Am Soc Exp Biol. 2011;25(1):265–279. doi: 10.1096/fj.10-161653. doi:10.1096/ fj.10-161653. [DOI] [PubMed] [Google Scholar]
- 7.Bitel CL, Kasinathan C, Kaswala RH, Klein WL, Frederikse PH. Amyloid-beta and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J Alzheimer’s Dis. 2012;32(2):291–305. doi: 10.3233/JAD-2012-120571. doi:10.3233/jad-2012-120571. [DOI] [PubMed] [Google Scholar]
- 8.Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s diseaseassociated Abeta oligomers. J Clin Investig. 2012;122(4):1339–1353. doi: 10.1172/JCI57256. doi:10.1172/jci57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boutajangout A, Wisniewski T. Tau-based therapeutic approaches for Alzheimer’s disease—a mini-review. Gerontology. 2014;60(5):381–385. doi: 10.1159/000358875. doi:10.1159/000358875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bullain SS, Corrada MM, Shah BA, Mozaffar FH, Panzenboeck M, Kawas CH. Poor physical performance and dementia in the oldest old: the 90 + study. JAMA Neurol. 2013;70(1):107–113. doi: 10.1001/jamaneurol.2013.583. doi:10.1001/jamaneurol.2013.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23(5):655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 12.Canale C, Seghezza S, Vilasi S, Carrotta R, Bulone D, Diaspro A, San Biagio PL, Dante S. Different effects of Alzheimer’s peptide Abeta(1–40) oligomers and fibrils on supported lipid membranes. Biophys Chem. 2013;182:23–29. doi: 10.1016/j.bpc.2013.07.010. doi:10.1016/j.bpc.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Cattepoel S, Hanenberg M, Kulic L, Nitsch RM. Chronic intranasal treatment with an anti-Abeta(30-42) scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2011;6(4):e18296. doi: 10.1371/journal.pone.0018296. doi:10.1371/journal.pone.0018296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang L, Bakhos L, Wang Z, Venton DL, Klein WL. Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer’s disease drug candidate screening. J Mol Neurosci MN. 2003;20(3):305–313. doi: 10.1385/JMN:20:3:305. doi:10.1385/JMN:20:3:305. [DOI] [PubMed] [Google Scholar]
- 15.Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci Off J Society Neurosci. 2004;24(19):4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. doi:10.1523/jneurosci.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry. 2003;42(44):12749–12760. doi: 10.1021/bi030029q. doi:10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 17.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8(1):79–84. doi: 10.1038/nn1372. doi:10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 18.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA. 2013;110(24):9758–9763. doi: 10.1073/pnas.1218402110. doi:10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69(1):29–38. doi: 10.1001/archneurol.2011.233. doi:10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dal Pra I, Chiarini A, Gui L, Chakravarthy B, Pacchiana R, Gardenal E, Whitfield JF, Armato U. Do astrocytes collaborate with neurons in spreading the “infectious” Abeta and tau drivers of Alzheimer’s disease? Neurosci Rev J Bring Neurobiol Neurol Psychiatr. 2014 doi: 10.1177/1073858414529828. doi:10.1177/1073858414529828. [DOI] [PubMed] [Google Scholar]
- 21.Dalgediene I, Lasickiene R, Budvytyte R, Valincius G, Morkuniene R, Borutaite V, Zvirbliene A. Immunogenic properties of amyloid beta oligomers. J Biomed Sci. 2013;20:10. doi: 10.1186/1423-0127-20-10. doi:10.1186/1423-0127-20-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Felice FG, Lourenco MV, Ferreira ST. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimer’s Dement J Alzheimer’s Assoc. 2014;10(1 Suppl):S26–S32. doi: 10.1016/j.jalz.2013.12.004. doi:10.1016/j.jalz.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 23.De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci USA. 2009;106(6):1971–1976. doi: 10.1073/pnas.0809158106. doi:10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Felice FG, Wasilewska-Sampaio AP, Barbosa AC, Gomes FC, Klein WL, Ferreira ST. Cyclic AMP enhancers and Abeta oligomerization blockers as potential therapeutic agents in Alzheimer’s disease. Curr Alzheimer Res. 2007;4(3):263–271. doi: 10.2174/156720507781077287. [DOI] [PubMed] [Google Scholar]
- 25.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Alzheimer’s diseasetype neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29(9):1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. doi:10.1016/j. neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de la Monte SM. Type 3 diabetes is sporadic Alzheimers disease: mini-review. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol. 2014 doi: 10.1016/j.euroneuro.2014.06.008. doi:10.1016/j.euroneuro.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2(6):1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Scala C, Troadec JD, Lelievre C, Garmy N, Fantini J, Chahinian H. Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer beta-amyloid peptide. J Neurochem. 2014;128(1):186–195. doi: 10.1111/jnc.12390. doi:10.1111/jnc.12390. [DOI] [PubMed] [Google Scholar]
- 29.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5(5):452–457. doi: 10.1038/nn842. doi:10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 30.Domert J, Rao SB, Agholme L, Brorsson AC, Marcusson J, Hallbeck M, Nath S. Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol Dis. 2014;65:82–92. doi: 10.1016/j.nbd.2013.12.019. doi:10.1016/j. nbd.2013.12.019. [DOI] [PubMed] [Google Scholar]
- 31.Dorostkar MM, Burgold S, Filser S, Barghorn S, Schmidt B, Anumala UR, Hillen H, Klein C, Herms J. Immunotherapy alleviates amyloid-associated synaptic pathology in an Alzheimer’s disease mouse model. Brain J Neurol. 2014 doi: 10.1093/brain/awu280. doi:10.1093/ brain/awu280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drolle E, Hane F, Lee B, Leonenko Z. Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease. Drug Metab Rev. 2014;46(2):207–223. doi: 10.3109/03602532.2014.882354. doi:10.3109/03602532.2014.882354. [DOI] [PubMed] [Google Scholar]
- 33.Drzezga A, Lautenschlager N, Siebner H, Riemenschneider M, Willoch F, Minoshima S, Schwaiger M, Kurz A. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur J Nucl Med Mol Imaging. 2003;30(8):1104–1113. doi: 10.1007/s00259-003-1194-1. doi:10.1007/ s00259-003-1194-1. [DOI] [PubMed] [Google Scholar]
- 34.Duran-Gonzalez J, Michi ED, Elorza B, Perez-Cordova MG, Pacheco-Otalora LF, Touhami A, Paulson P, Perry G, Murray IV, Colom LV. Amyloid beta peptides modify the expression of antioxidant repair enzymes and a potassium channel in the septohippocampal system. Neurobiol Aging. 2013;34(8):2071–2076. doi: 10.1016/j.neurobiolaging.2013.02.005. doi:10.1016/j.neurobiolaging.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68(7):501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. doi:10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- 36.Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Brody DL. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73(1):104–119. doi: 10.1002/ana.23748. doi:10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evangelisti E, Wright D, Zampagni M, Cascella R, Fiorillo C, Bagnoli S, Relini A, Nichino D, Scartabelli T, Nacmias B, Sorbi S, Cecchi C. Lipid rafts mediate amyloid-induced calcium dyshomeostasis and oxidative stress in Alzheimer’s disease. Curr Alzheimer Res. 2013;10(2):143–153. doi: 10.2174/1567205011310020004. [DOI] [PubMed] [Google Scholar]
- 38.Evangelisti E, Zampagni M, Cascella R, Becatti M, Fiorillo C, Caselli A, Bagnoli S, Nacmias B, Cecchi C. Plasma membrane injury depends on bilayer lipid composition in Alzheimer’s disease. J Alzheimer Dis. 2014;41(1):289–300. doi: 10.3233/JAD-131406. doi:10.3233/jad-131406. [DOI] [PubMed] [Google Scholar]
- 39.Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011;96(4):529–543. doi: 10.1016/j.nlm.2011.08.003. doi:10.1016/j. nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC. Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol Aging. 2012;33(7):1329–1342. doi: 10.1016/j.neurobiolaging.2011.01.007. doi:10.1016/j.neurobiolaging.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, De Felice FG, Ferreira ST. Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by lowmolecular-weight oligomers. J Neurosci Off J Soc Neurosci. 2013;33(23):9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013. doi:10.1523/jneurosci.0482-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frackowiak J, Zoltowska A, Wisniewski HM. Non-fibrillar beta-amyloid protein is associated with smooth muscle cells of vessel walls in Alzheimer disease. J Neuropathol Exp Neurol. 1994;53(6):637–645. doi: 10.1097/00005072-199411000-00011. [DOI] [PubMed] [Google Scholar]
- 43.Gabbita SP, Srivastava MK, Eslami P, Johnson MF, Kobritz NK, Tweedie D, Greig NH, Zemlan FP, Sharma SP, Harris-White ME. Early intervention with a small molecule inhibitor for tumor necrosis factor-alpha prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2012;9:99. doi: 10.1186/1742-2094-9-99. doi:10.1186/1742-2094-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garai K, Frieden C. Quantitative analysis of the time course of Abeta oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Abeta. Proc Natl Acad Sci USA. 2013;110(9):3321–3326. doi: 10.1073/pnas.1222478110. doi:10.1073/pnas.1222478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, Mirkin CA. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc Natl Acad Sci USA. 2005;102(7):2273–2276. doi: 10.1073/pnas.0409336102. doi:10.1073/pnas.0409336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gimenez-Llort L, Rivera-Hernandez G, Marin-Argany M, Sanchez-Quesada JL, Villegas S. Early intervention in the 3xTg-AD mice with an amyloid beta-antibody fragment ameliorates first hallmarks of Alzheimer disease. mAbs. 2013;5(5):665–677. doi: 10.4161/mabs.25424. doi:10.4161/mabs.25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100(18):10417–10422. doi: 10.1073/pnas.1834302100. doi:10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010;119(5):523–541. doi: 10.1007/s00401-010-0679-9. doi:10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goure WF, Krafft GA, Jerecic J, Hefti F. Targeting the proper amyloid-beta neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res Ther. 2014;6(4):42. doi: 10.1186/alzrt272. doi:10.1186/alzrt272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griffin WS. Perispinal etanercept: potential as an Alzheimer therapeutic. J Neuroinflammation. 2008;5:3. doi: 10.1186/1742-2094-5-3. doi:10.1186/1742-2094-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gruning CS, Klinker S, Wolff M, Schneider M, Toksoz K, Klein AN, Nagel-Steger L, Willbold D, Hoyer W. The off-rate of monomers dissociating from amyloid-beta protofibrils. J Biol Chem. 2013;288(52):37104–37111. doi: 10.1074/jbc.M113.513432. doi:10.1074/jbc.M113.513432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gu L, Liu C, Guo Z. Structural insights into Abeta42 oligomers using site-directed spin labeling. J Biol Chem. 2013;288(26):18673–18683. doi: 10.1074/jbc.M113.457739. doi:10.1074/jbc.M113.457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guillot-Sestier MV, Sunyach C, Ferreira ST, Marzolo MP, Bauer C, Thevenet A, Checler F. Alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J Biol Chem. 2012;287(7):5021–5032. doi: 10.1074/jbc.M111.323626. doi:10.1074/jbc.M111.323626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154(1):103–117. doi: 10.1016/j.cell.2013.05.057. doi:10.1016/j. cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286(17):15317–15331. doi: 10.1074/jbc.M110.209296. doi:10.1074/jbc. M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haes AJ, Chang L, Klein WL, Van Duyne RP. Detection of a biomarker for Alzheimer’s disease from synthetic and clinical samples using a nanoscale optical biosensor. J Am Chem Soc. 2005;127(7):2264–2271. doi: 10.1021/ja044087q. doi:10.1021/ja044087q. [DOI] [PubMed] [Google Scholar]
- 57.Han X, Ma Y, Liu X, Wang L, Qi S, Zhang Q, Du Y. Changes in insulin-signaling transduction pathway underlie learning/memory deficits in an Alzheimer’s disease rat model. J Neural Trans Vienna Austria 1996. 2012;119(11):1407–1416. doi: 10.1007/s00702-012-0803-1. doi:10.1007/s00702-012-0803-1. [DOI] [PubMed] [Google Scholar]
- 58.Handoko M, Grant M, Kuskowski M, Zahs KR, Wallin A, Blennow K, Ashe KH. Correlation of specific amyloid-beta oligomers with tau in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol. 2013;70(5):594–599. doi: 10.1001/jamaneurol.2013.48. doi:10.1001/ jamaneurol.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hauser PS, Ryan RO. Impact of apolipoprotein E on Alzheimer’s disease. Curr Alzheimer Res. 2013;10(8):809–817. doi: 10.2174/15672050113109990156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayden EY, Teplow DB. Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Res Ther. 2013;5(6):60. doi: 10.1186/alzrt226. doi:10.1186/alzrt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hefti F, Goure WF, Jerecic J, Iverson KS, Walicke PA, Krafft GA. The case for soluble Abeta oligomers as a drug target in Alzheimer’s disease. Trend Pharmacol Sci. 2013;34(5):261–266. doi: 10.1016/j.tips.2013.03.002. doi:10.1016/j.tips.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 62.Heras-Sandoval D, Ferrera P, Arias C. Amyloid-beta protein modulates insulin signaling in presynaptic terminals. Neurochem Res. 2012;37(9):1879–1885. doi: 10.1007/s11064-012-0800-7. doi:10.1007/s11064-012-0800-7. [DOI] [PubMed] [Google Scholar]
- 63.Holtta M, Hansson O, Andreasson U, Hertze J, Minthon L, Nagga K, Andreasen N, Zetterberg H, Blennow K. Evaluating amyloid-beta oligomers in cerebrospinal fluid as a biomarker for Alzheimer’s disease. PLoS One. 2013;8(6):e66381. doi: 10.1371/journal.pone.0066381. doi:10.1371/journal.pone.0066381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Intlekofer KA, Cotman CW. Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol Dis. 2013;57:47–55. doi: 10.1016/j.nbd.2012.06.011. doi:10.1016/j.nbd.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 65.Izuo N, Kume T, Sato M, Murakami K, Irie K, Izumi Y, Akaike A. Toxicity in rat primary neurons through the cellular oxidative stress induced by the turn formation at positions 22 and 23 of Abeta42. ACS Chem Neurosci. 2012;3(9):674–681. doi: 10.1021/cn300033k. doi:10.1021/cn300033k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, Walko T 3rd, Vaswani A, Wardius M, Syed Z, Ravenscroft J, Mozzoni K, Silky C, Rehak C, Yurko R, Finn P, Look G, Rishton G, Safferstein H, Miller M, Johanson C, Stopa E, Windisch M, Hutter-Paier B, Shamloo M, Arancio O, LeVine H, 3rd, Catalano SM. Alzheimer’s therapeutics targeting amyloid Beta 1-42 oligomers I: abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One. 2014;9(11):e111898. doi: 10.1371/journal.pone.0111898. doi:10.1371/journal.pone.0111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, Yurko R, Look G, Rishton G, Safferstein H, Cruchaga C, Goate A, Cahill MA, Arancio O, Mach RH, Craven R, Head E, LeVine H, 3rd, Spires-Jones TL, Catalano SM. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers ii: sigma-2/pgrmc1 receptors mediate abeta 42 oligomer binding and synaptotoxicity. PLoS One. 2014;9(11):e111899. doi: 10.1371/journal.pone.0111899. doi:10.1371/journal. pone.0111899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janelsins MC, Mastrangelo MA, Park KM, Sudol KL, Narrow WC, Oddo S, LaFerla FM, Callahan LM, Federoff HJ, Bowers WJ. Chronic neuron-specific tumor necrosis factoralpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol. 2008;173(6):1768–1782. doi: 10.2353/ajpath.2008.080528. doi:10.2353/ajpath.2008.080528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jang H, Connelly L, Arce FT, Ramachandran S, Kagan BL, Lal R, Nussinov R. Mechanisms for the insertion of toxic, fibril-like beta-amyloid oligomers into the membrane. J Chem Theory Comput. 2013;9(1):822–833. doi: 10.1021/ct300916f. doi:10.1021/ct300916f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jang H, Connelly L, Arce FT, Ramachandran S, Lal R, Kagan BL, Nussinov R. Alzheimer’s disease: which type of amyloid-preventing drug agents to employ? Phys Chem Chem Phys. 2013;15(23):8868–8877. doi: 10.1039/c3cp00017f. doi:10.1039/c3cp00017f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeong JS, Ansaloni A, Mezzenga R, Lashuel HA, Dietler G. Novel mechanistic insight into the molecular basis of amyloid polymorphism and secondary nucleation during amyloid formation. J Mol Biol. 2013;425(10):1765–1781. doi: 10.1016/j.jmb.2013.02.005. doi:10.1016/j. jmb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 72.Johnson RD, Steel DG, Gafni A. Structural evolution and membrane interactions of Alzheimer’s amyloid-beta peptide oligomers: new knowledge from single-molecule fluorescence studies. Protein Sci Publ Protein Soc. 2014;23(7):869–883. doi: 10.1002/pro.2479. doi:10.1002/pro.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jonsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–99. doi: 10.1038/nature11283. doi:10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 74.Kalimo H, Lalowski M, Bogdanovic N, Philipson O, Bird TD, Nochlin D, Schellenberg GD, Brundin R, Olofsson T, Soliymani R, Baumann M, Wirths O, Bayer TA, Nilsson LN, Basun H, Lannfelt L, Ingelsson M. The Arctic AbetaPP mutation leads to Alzheimer’s disease pathology with highly variable topographic deposition of differentially truncated Abeta. Acta Neuropathol Commun. 2013;1(1):60. doi: 10.1186/2051-5960-1-60. doi:10.1186/2051-5960-1-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. doi:10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science NY. 2003;300(5618):486–489. doi: 10.1126/science.1079469. doi:10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 77.Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimer Dis. 2013;33(Suppl 1):S67–S78. doi: 10.3233/JAD-2012-129001. doi:10.3233/jad-2012-129001. [DOI] [PubMed] [Google Scholar]
- 78.Kerchner GA, Boxer AL. Bapineuzumab. Expert Opin Biol Ther. 2010;10(7):1121–1130. doi: 10.1517/14712598.2010.493872. doi:10.1517/14712598.2010.493872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim HG, Moon M, Choi JG, Park G, Kim AJ, Hur J, Lee KT, Oh MS. Donepezil inhibits the amyloid-beta oligomerinduced microglial activation in vitro and in vivo. Neurotoxicology. 2014;40:23–32. doi: 10.1016/j.neuro.2013.10.004. doi:10.1016/j.neuro.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 80.Kim HJ, Chae SC, Lee DK, Chromy B, Lee SC, Park YC, Klein WL, Krafft GA, Hong ST. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J Off Publ Fed Am Soc Exp Biol. 2003;17(1):118–120. doi: 10.1096/fj.01-0987fje. doi:10.1096/fj.01-0987fje. [DOI] [PubMed] [Google Scholar]
- 81.Klein WL. Synaptotoxic amyloid-beta oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer’s disease? J Alzheimer Dis. 2013;33(Suppl 1):S49–S65. doi: 10.3233/JAD-2012-129039. doi:10.3233/jad-2012-129039. [DOI] [PubMed] [Google Scholar]
- 82.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24(4):219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 83.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–319. doi: 10.1002/ana.20009. doi:10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 84.Klyubin I, Cullen WK, Hu NW, Rowan MJ. Alzheimer’s disease Abeta assemblies mediating rapid disruption of synaptic plasticity and memory. Mol Brain. 2012;5:25. doi: 10.1186/1756-6606-5-25. doi:10.1186/1756-6606-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol. 2007;17(1):48–57. doi: 10.1016/j.sbi.2007.01.007. doi:10.1016/j.sbi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 86.Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer’s disease: synapses gone cold. Mol Neurodegener. 2011;6(1):63. doi: 10.1186/1750-1326-6-63. doi:10.1186/1750-1326-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106(10):4012–4017. doi: 10.1073/pnas.0811698106. doi:10.1073/ pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kovacs GG, Breydo L, Green R, Kis V, Puska G, Lorincz P, Perju-Dumbrava L, Giera R, Pirker W, Lutz M, Lachmann I, Budka H, Uversky VN, Molnar K, Laszlo L. Intracellular processing of disease-associated alpha-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol Dis. 2014;69:76–92. doi: 10.1016/j.nbd.2014.05.020. doi:10.1016/j.nbd.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 89.Kumar A, Paslay LC, Lyons D, Morgan SE, Correia JJ, Rangachari V. Specific soluble oligomers of amyloid-beta peptide undergo replication and form non-fibrillar aggregates in interfacial environments. J Biol Chem. 2012;287(25):21253–21264. doi: 10.1074/jbc.M112.355156. doi:10.1074/jbc.M112.355156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, Guridi M, Asuni AA, Sullivan PM, Holtzman DM, Sadowski MJ. Blocking the interaction between apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am J Pathol. 2013;182(5):1750–1768. doi: 10.1016/j.ajpath.2013.01.034. doi:10.1016/j.ajpath.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’srelated amyloid beta oligomers. J Neurosci Off J Soc Neurosci. 2004;24(45):10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. doi:10.1523/jneurosci.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomerinduced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci Off J Soc Neurosci. 2007;27(4):796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. doi:10.1523/jneurosci.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ladiwala AR, Litt J, Kane RS, Aucoin DS, Smith SO, Ranjan S, Davis J, Van Nostrand WE, Tessier PM. Conformational differences between two amyloid beta oligomers of similar size and dissimilar toxicity. J Biol Chem. 2012;287(29):24765–24773. doi: 10.1074/jbc.M111.329763. doi:10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, Eisenberg D. Atomic view of a toxic amyloid small oligomer. Sci NY. 2012;335(6073):1228–1231. doi: 10.1126/science.1213151. doi:10.1126/ science.1213151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100(1):23–35. doi: 10.1111/j.1471-4159.2006.04157.x. doi:10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- 97.Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79(3):595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- 98.Lannfelt L, Moller C, Basun H, Osswald G, Sehlin D, Satlin A, Logovinsky V, Gellerfors P. Perspectives on future Alzheimer therapies: amyloid-beta protofibrils—a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther. 2014;6(2):16. doi: 10.1186/alzrt246. doi:10.1186/alzrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lasagna-Reeves CA, Glabe CG, Kayed R. Amyloid-beta annular protofibrils evade fibrillar fate in Alzheimer disease brain. J Biol Chem. 2011;286(25):22122–22130. doi: 10.1074/jbc.M111.236257. doi:10.1074/jbc. M111.236257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lauren J. Cellular prion protein as a therapeutic target in Alzheimer’s disease. J Alzheimer Dis. 2014;38(2):227–244. doi: 10.3233/JAD-130950. doi:10.3233/jad-130950. [DOI] [PubMed] [Google Scholar]
- 101.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457(7233):1128–1132. doi: 10.1038/nature07761. doi:10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281(7):4292–4299. doi: 10.1074/jbc.M511018200. doi:10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 103.Lee M, Guo JP, Schwab C, McGeer EG, McGeer PL. Selective inhibition of the membrane attack complex of complement by low molecular weight components of the aurin tricarboxylic acid synthetic complex. Neurobiol Aging. 2012;33(10):2237–2246. doi: 10.1016/j.neurobiolaging.2011.12.005. doi:10.1016/j.neurobiolaging.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 104.Lee SH, Kim Y, Kim HY, Kim YH, Kim MS, Kong JY, Lee MH, Kim DJ, Ahn YG. Aminostyrylbenzofuran directly reduces oligomeric amyloid-beta and reverses cognitive deficits in Alzheimer transgenic mice. PLoS One. 2014;9(4):e95733. doi: 10.1371/journal.pone.0095733. doi:10.1371/journal.pone.0095733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. doi:10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 106.LeVine H., 3rd Alzheimer’s beta-peptide oligomer formation at physiologic concentrations. Anal Biochem. 2004;335(1):81–90. doi: 10.1016/j.ab.2004.08.014. doi:10.1016/j.ab.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 107.Li S, Jin M, Zhang D, Yang T, Koeglsperger T, Fu H, Selkoe DJ. Environmental novelty activates beta2-adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron. 2013;77(5):929–941. doi: 10.1016/j.neuron.2012.12.040. doi:10.1016/j. neuron.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu-Seifert H, Siemers E, Sundell K, Price K, Han B, Selzler K, Aisen P, Cummings J, Raskin J, Mohs R. Cognitive and functional decline and their relationship in patients with mild Alzheimer’s dementia. J Alzheimer Dis. 2014 doi: 10.3233/JAD-140792. doi:10.3233/ jad-140792. [DOI] [PubMed] [Google Scholar]
- 109.Liu C, Zhao M, Jiang L, Cheng PN, Park J, Sawaya MR, Pensalfini A, Gou D, Berk AJ, Glabe CG, Nowick J, Eisenberg D. Out-of-register beta-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci USA. 2012;109(51):20913–20918. doi: 10.1073/pnas.1218792109. doi:10.1073/pnas.1218792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lorenzen N, Nielsen SB, Buell AK, Kaspersen JD, Arosio P, Vad BS, Paslawski W, Christiansen G, Valnickova-Hansen Z, Andreasen M, Enghild JJ, Pedersen JS, Dobson CM, Knowles TP, Otzen DE. The role of stable alpha-synuclein oligomers in the molecular events underlying amyloid formation. J Am Chem Soc. 2014;136(10):3859–3868. doi: 10.1021/ja411577t. doi:10.1021/ja411577t. [DOI] [PubMed] [Google Scholar]
- 111.Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espirito-Santo S, Campello-Costa P, Houzel JC, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, Ferreira ST, De Felice FG. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18(6):831–843. doi: 10.1016/j.cmet.2013.11.002. doi:10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 112.Maesako M, Uemura K, Iwata A, Kubota M, Watanabe K, Uemura M, Noda Y, Asada-Utsugi M, Kihara T, Takahashi R, Shimohama S, Kinoshita A. Continuation of exercise is necessary to inhibit high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. PLoS One. 2013;8(9):e72796. doi: 10.1371/journal.pone.0072796. doi:10.1371/journal. pone.0072796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Magnusson K, Sehlin D, Syvanen S, Svedberg MM, Philipson O, Soderberg L, Tegerstedt K, Holmquist M, Gellerfors P, Tolmachev V, Antoni G, Lannfelt L, Hall H, Nilsson LN. Specific uptake of an amyloid-beta protofibril-binding antibody-tracer in AbetaPP transgenic mouse brain. J Alzheimer Dis. 2013;37(1):29–40. doi: 10.3233/JAD-130029. doi:10.3233/jad-130029. [DOI] [PubMed] [Google Scholar]
- 114.Mainardi M, Di Garbo A, Caleo M, Berardi N, Sale A, Maffei L. Environmental enrichment strengthens corticocortical interactions and reduces amyloid-beta oligomers in aged mice. Front Aging Neurosci. 2014;6:1. doi: 10.3389/fnagi.2014.00001. doi:10.3389/fnagi.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Matsubara E, Takamura A, Okamoto Y, Oono H, Nakata T, Wakasaya Y, Kawarabayashi T, Shoji M. Disease modifying therapies for Alzheimer’s disease targeting Abeta oligomers: implications for therapeutic mechanisms. Biomed Resh Int. 2013;2013:984041. doi: 10.1155/2013/984041. doi:10.1155/2013/984041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, Das P, Golde TE, LaFerla FM, Oddo S, Blesch A, Tansey MG. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloidassociated neuropathology. Neurobiol Dis. 2009;34(1):163–177. doi: 10.1016/j.nbd.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76:57–67. doi: 10.1016/j.neuropharm.2013.08.005. Pt A. doi:10.1016/j.neuropharm.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 118.McLean D, Cooke MJ, Wang Y, Green D, Fraser PE, George-Hyslop PS, Shoichet MS. Anti-amyloid-beta-mediated positron emission tomography imaging in Alzheimer’s disease mouse brains. PLoS One. 2012;7(12):e51958. doi: 10.1371/journal.pone.0051958. doi:10.1371/journal. pone.0051958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moon M, Kim HG, Choi JG, Oh H, Lee PK, Ha SK, Kim SY, Park Y, Huh Y, Oh MS. 6-Shogaol, an active constituent of ginger, attenuates neuroinflammation and cognitive deficits in animal models of dementia. Biochem Biophys Res Commun. 2014;449(1):8–13. doi: 10.1016/j.bbrc.2014.04.121. doi:10.1016/j.bbrc.2014.04.121. [DOI] [PubMed] [Google Scholar]
- 120.Morkuniene R, Zvirbliene A, Dalgediene I, Cizas P, Jankeviciute S, Baliutyte G, Jokubka R, Jankunec M, Valincius G, Borutaite V. Antibodies bound to Abeta oligomers potentiate the neurotoxicity of Abeta by activating microglia. J Neurochem. 2013;126(5):604–615. doi: 10.1111/jnc.12332. doi:10.1111/jnc.12332. [DOI] [PubMed] [Google Scholar]
- 121.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20(11):4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mucke L, Selkoe DJ. Neurotoxicity of amyloid betaprotein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2(7):a006338. doi: 10.1101/cshperspect.a006338. doi:10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mufson EJ, Ward S, Binder L. Prefibrillar tau oligomers in mild cognitive impairment and Alzheimer’s disease. Neurodegener Dis. 2014;13(2–3):151–153. doi: 10.1159/000353687. doi:10.1159/000353687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Murakami K. Conformation-specific antibodies to target amyloid beta oligomers and their application to immunotherapy for Alzheimer’s disease. Biosci Biotechnol Biochem. 2014;78(8):1293–1305. doi: 10.1080/09168451.2014.940275. doi:10.1080/09168451.2014.940275. [DOI] [PubMed] [Google Scholar]
- 125.Narayan P, Ganzinger KA, McColl J, Weimann L, Meehan S, Qamar S, Carver JA, Wilson MR, St George-Hyslop P, Dobson CM, Klenerman D. Single molecule characterization of the interactions between amyloid-beta peptides and the membranes of hippocampal cells. J Am Chem Soc. 2013;135(4):1491–1498. doi: 10.1021/ja3103567. doi:10.1021/ja3103567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Narayan P, Holmstrom KM, Kim DH, Whitcomb DJ, Wilson MR, St George-Hyslop P, Wood NW, Dobson CM, Cho K, Abramov AY, Klenerman D. Rare individual amyloidbeta oligomers act on astrocytes to initiate neuronal damage. Biochemistry. 2014;53(15):2442–2453. doi: 10.1021/bi401606f. doi:10.1021/bi401606f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nath S, Agholme L, Kurudenkandy FR, Granseth B, Marcusson J, Hallbeck M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J Neurosci Off J Soc Neurosci. 2012;32(26):8767–8777. doi: 10.1523/JNEUROSCI.0615-12.2012. doi:10.1523/ jneurosci.0615-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nigh AB, Vahedi S, Davis EG, Weintraub S, Bigio EH, Klein WL, Geula C. Intracellular Amyloid-beta in cholinergic neurons. Brain J Neurol. 2014 In Press. [Google Scholar]
- 129.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Ronicke R, Reymann K, Hutter-Paier B, Alexandru A, Jagla W, Graubner S, Glabe CG, Demuth HU, Bloom GS. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloidbeta. Nature. 2012;485(7400):651–655. doi: 10.1038/nature11060. doi:10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nyborg AC, Moll JR, Wegrzyn RD, Havas D, Hutter-Paier B, Feuerstein GG, Rudolph AS. In vivo and ex vivo imaging of amyloid-beta cascade aggregates with a Pronucleon peptide. J Alzheimer Dis. 2013;34(4):957–967. doi: 10.3233/JAD-122107. doi:10.3233/jad-122107. [DOI] [PubMed] [Google Scholar]
- 131.Oda T, Pasinetti GM, Osterburg HH, Anderson C, Johnson SA, Finch CE. Purification and characterization of brain clusterin. Biochem Biophys Res Commun. 1994;204(3):1131–1136. doi: 10.1006/bbrc.1994.2580. doi:10.1006/bbrc.1994.2580. [DOI] [PubMed] [Google Scholar]
- 132.Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006;281(3):1599–1604. doi: 10.1074/jbc.M507892200. doi:10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- 133.Olanow CW, Brundin P. Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord Off J Mov Disorder Soc. 2013;28(1):31–40. doi: 10.1002/mds.25373. doi:10.1002/ mds.25373. [DOI] [PubMed] [Google Scholar]
- 134.Ono K, Takahashi R, Ikeda T, Yamada M. Cross-seeding effects of amyloid beta-protein and alpha-synuclein. J Neurochem. 2012;122(5):883–890. doi: 10.1111/j.1471-4159.2012.07847.x. doi:10.1111/j.1471-4159.2012.07847.x. [DOI] [PubMed] [Google Scholar]
- 135.Pearson-Leary J, McNay EC. Intrahippocampal administration of amyloid-beta(1-42) oligomers acutely impairs spatial working memory, insulin signaling, and hippocampal metabolism. J Alzheimer Dis. 2012;30(2):413–422. doi: 10.3233/JAD-2012-112192. doi:10.3233/ jad-2012-112192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Peng S, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM, Mount HT, Mufson EJ, Salehi A, Fahnestock M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J Neurosci Off J Socr Neurosci. 2009;29(29):9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009. doi:10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Peters C, Fernandez-Perez EJ, Burgos CF, Espinoza MP, Castillo C, Urrutia JC, Streltsov VA, Opazo C, Aguayo LG. Inhibition of amyloid beta-induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide. Neurobiol Aging. 2013;34(12):2805–2814. doi: 10.1016/j.neurobiolaging.2013.06.001. doi:10.1016/j. neurobiolaging.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 138.Pitt J, Roth W, Lacor P, Smith AB 3rd, Blankenship M, Velasco P, De Felice F, Breslin P, Klein WL. Alzheimer’s-associated Abeta oligomers show altered structure, immunoreactivity and synaptotoxicity with low doses of oleocanthal. Toxicol Appl Pharmacol. 2009;240(2):189–197. doi: 10.1016/j.taap.2009.07.018. doi:10.1016/j.taap.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pozueta J, Lefort R, Shelanski ML. Synaptic changes in Alzheimer’s disease and its models. Neuroscience. 2013;251:51–65. doi: 10.1016/j.neuroscience.2012.05.050. doi:10.1016/j.neuroscience.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 140.Prangkio P, Yusko EC, Sept D, Yang J, Mayer M. Multivariate analyses of amyloid-beta oligomer populations indicate a connection between pore formation and cytotoxicity. PLoS One. 2012;7(10):e47261. doi: 10.1371/journal.pone.0047261. doi:10.1371/journal.pone.0047261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Purro SA, Dickins EM, Salinas PC. The secreted Wnt antagonist Dickkopf-1 is required for amyloid beta-mediated synaptic loss. J Neurosci Off J Soc Neurosci. 2012;32(10):3492–3498. doi: 10.1523/JNEUROSCI.4562-11.2012. doi:10.1523/jneurosci.4562-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rasool S, Martinez-Coria H, Wu JW, LaFerla F, Glabe CG. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Abeta deposition and tau pathology in 3xTg-AD mice. J Neurochem. 2013;126(4):473–482. doi: 10.1111/jnc.12305. doi:10.1111/jnc.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66(5):739–754. doi: 10.1016/j.neuron.2010.04.029. doi:10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Resende R, Marques SC, Ferreiro E, Simoes I, Oliveira CR, Pereira CM. Effect of alpha-synuclein on amyloid betainduced toxicity: relevance to Lewy body variant of Alzheimer disease. Neurochem Res. 2013;38(4):797–806. doi: 10.1007/s11064-013-0982-7. doi:10.1007/ s11064-013-0982-7. [DOI] [PubMed] [Google Scholar]
- 145.Richman M, Wilk S, Chemerovski M, Warmlander SK, Wahlstrom A, Graslund A, Rahimipour S. In vitro and mechanistic studies of an antiamyloidogenic self-assembled cyclic D, L-alpha-peptide architecture. J Am Chem Soc. 2013;135(9):3474–3484. doi: 10.1021/ja310064v. doi:10.1021/ja310064v. [DOI] [PubMed] [Google Scholar]
- 146.Ringman JM, Tomic JL, Coppola G, Elashoff D, Gylys KH, Glabe CG. Conformation-dependent oligomers in cerebrospinal fluid of presymptomatic familial Alzheimer’s disease mutation carriers. Dement Geriatr Cognit Disord Extra. 2012;2(1):652–657. doi: 10.1159/000345771. doi:10.1159/000345771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Robinson R. MRI probe for amyloid-beta oligomers offers potential advantages for detecting Alzheimer’s disease. Neurol Today. 2012;12(24):20–22. doi:10.1097/01.NT.0000425719.88852.88. [Google Scholar]
- 148.Rosenblum WI. Why Alzheimer trials fail: removing soluble oligomeric beta amyloid is essential, inconsistent, and difficult. Neurobiol Aging. 2014;35(5):969–974. doi: 10.1016/j.neurobiolaging.2013.10.085. doi:10.1016/j. neurobiolaging.2013.10.085. [DOI] [PubMed] [Google Scholar]
- 149.Roychaudhuri R, Lomakin A, Bernstein S, Zheng X, Condron MM, Benedek GB, Bowers M, Teplow DB. Gly25-Ser26 amyloid beta-protein structural isomorphs produce distinct Abeta42 conformational dynamics and assembly characteristics. J Mol Biol. 2014;426(13):2422–2441. doi: 10.1016/j.jmb.2014.04.004. doi:10.1016/j.jmb.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roychaudhuri R, Yang M, Deshpande A, Cole GM, Frautschy S, Lomakin A, Benedek GB, Teplow DB. C-terminal turn stability determines assembly differences between Abeta40 and Abeta42. J Mol Biol. 2013;425(2):292–308. doi: 10.1016/j.jmb.2012.11.006. doi:10.1016/j. jmb.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005;169(2):341–354. doi: 10.1083/jcb.200409127. doi:10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sarkar B, Das AK, Maiti S. Thermodynamically stable amyloid-beta monomers have much lower membrane affinity than the small oligomers. Front Physiol. 2013;4:84. doi: 10.3389/fphys.2013.00084. doi:10.3389/ fphys.2013.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sarkar B, Mithu VS, Chandra B, Mandal A, Chandrakesan M, Bhowmik D, Madhu PK, Maiti S. Significant structural differences between transient amyloid-beta oligomers and less-toxic fibrils in regions known to harbor familial Alzheimer’s mutations. Angew Chem Int Ed Engl. 2014;53(27):6888–6892. doi: 10.1002/anie.201402636. doi:10.1002/anie.201402636. [DOI] [PubMed] [Google Scholar]
- 154.Schmidt V, Carlo AS, Willnow TE. Apolipoprotein E receptor pathways in Alzheimer disease. Wiley interdisciplinary reviews Systems biology and medicine. 2014;6(3):255–270. doi: 10.1002/wsbm.1262. doi:10.1002/wsbm.1262. [DOI] [PubMed] [Google Scholar]
- 155.Schnabel J. Amyloid: little proteins, big clues. Nature. 2011;475(7355):S12–S14. doi: 10.1038/475S12a. doi:10.1038/475S12a. [DOI] [PubMed] [Google Scholar]
- 156.Sebollela A, Freitas-Correa L, Oliveira FF, Mendes CT, Wasilewska-Sampaio AP, Camacho-Pereira J, Galina A, Brentani H, Passetti F, De Felice FG, Dias-Neto E, Ferreira ST. Expression profile of rat hippocampal neurons treated with the neuroprotective compound 2,4-dinitrophenol: up-regulation of cAMP signaling genes. Neurotox Res. 2010;18(2):112–123. doi: 10.1007/s12640-009-9133-y. doi:10.1007/s12640-009-9133-y. [DOI] [PubMed] [Google Scholar]
- 157.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17(9):1060–1065. doi: 10.1038/nm.2460. doi:10.1038/ nm.2460. [DOI] [PubMed] [Google Scholar]
- 158.Siemers ER, Friedrich S, Dean RA, Gonzales CR, Farlow MR, Paul SM, Demattos RB. Safety and changes in plasma and cerebrospinal fluid amyloid beta after a single administration of an amyloid beta monoclonal antibody in subjects with Alzheimer disease. Clin Neuropharmacol. 2010;33(2):67–73. doi: 10.1097/WNF.0b013e3181cb577a. doi:10.1097/WNF.0b013e3181cb577a. [DOI] [PubMed] [Google Scholar]
- 159.Sitkiewicz E, Kloniecki M, Poznanski J, Bal W, Dadlez M. Factors influencing compact-extended structure equilibrium in oligomers of abeta1-40 peptide–an ion mobility mass spectrometry study. J Mol Biol. 2014;426(15):2871–2885. doi: 10.1016/j.jmb.2014.05.015. doi:10.1016/j.jmb.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 160.Soejima N, Ohyagi Y, Nakamura N, Himeno E, Iinuma KM, Sakae N, Yamasaki R, Tabira T, Murakami K, Irie K, Kinoshita N, LaFerla FM, Kiyohara Y, Iwaki T, Kira J. Intracellular accumulation of toxic turn amyloid-beta is associated with endoplasmic reticulum stress in Alzheimer’s disease. Curr Alzheimer Res. 2013;10(1):11–20. [PubMed] [Google Scholar]
- 161.Sorgjerd KM, Zako T, Sakono M, Stirling PC, Leroux MR, Saito T, Nilsson P, Sekimoto M, Saido TC, Maeda M. Human prefoldin inhibits amyloid-beta (Abeta) fibrillation and contributes to formation of nontoxic Abeta aggregates. Bio-chemistry. 2013;52(20):3532–3542. doi: 10.1021/bi301705c. doi:10.1021/bi301705c. [DOI] [PubMed] [Google Scholar]
- 162.Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Accumulation of intraneuronal beta-amyloid 42 peptides is associated with early changes in microtubule-associated protein 2 in neurites and synapses. PLoS ONE. 2013;8(1):e51965. doi: 10.1371/journal.pone.0051965. doi:10.1371/journal.pone.0051965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Takamura A, Sato Y, Watabe D, Okamoto Y, Nakata T, Kawarabayashi T, Oddo S, Laferla FM, Shoji M, Matsubara E. Sortilin is required for toxic action of Abeta oligomers (AbetaOs): extracellular AbetaOs trigger apoptosis, and intraneuronal AbetaOs impair degradation pathways. Life Sci. 2012;91(23–24):1177–1186. doi: 10.1016/j.lfs.2012.04.038. doi:10.1016/j.lfs.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 164.Takeda S, Hashimoto T, Roe AD, Hori Y, Spires-Jones TL, Hyman BT. Brain interstitial oligomeric amyloid beta increases with age and is resistant to clearance from brain in a mouse model of Alzheimer’s disease. FASEB J Off Publ Fed Am Soc Exp Biol. 2013;27(8):3239–3248. doi: 10.1096/fj.13-229666. doi:10.1096/fj.13-229666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tay WM, Huang D, Rosenberry TL, Paravastu AK. The Alzheimer’s amyloid-beta(1-42) peptide forms off-pathway oligomers and fibrils that are distinguished structurally by intermolecular organization. J Mol Biol. 2013;425(14):2494–2508. doi: 10.1016/j.jmb.2013.04.003. doi:10.1016/j.jmb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thapa A, Vernon BC, De la Pena K, Soliz G, Moreno HA, Lopez GP, Chi EY. Membrane-mediated neuroprotection by curcumin from amyloid-beta-peptide-induced toxicity. Langmuir : the ACS journal of surfaces and colloids. 2013;29(37):11713–11723. doi: 10.1021/la4020459. doi:10.1021/la4020459. [DOI] [PubMed] [Google Scholar]
- 167.Torok B, Sood A, Bag S, Kulkarni A, Borkin D, Lawler E, Dasgupta S, Landge S, Abid M, Zhou W, Foster M, LeVine H, 3rd, Torok M. Structure-activity relationships of organofluorine inhibitors of beta-amyloid self-assembly. Chem Med Chem. 2012;7(5):910–919. doi: 10.1002/cmdc.201100569. doi:10.1002/cmdc.201100569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tsigelny IF, Sharikov Y, Kouznetsova VL, Greenberg JP, Wrasidlo W, Gonzalez T, Desplats P, Michael SE, Trejo-Morales M, Overk CR, Masliah E. Structural diversity of Alzheimer’s disease amyloid-beta dimers and their role in oligomerization and fibril formation. J Alzheimer Dis. 2014;39(3):583–600. doi: 10.3233/JAD-131589. doi:10.3233/jad-131589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tucker S, Moller C, Tegerstedt K, Lord A, Laudon H, Sjodahl J, Soderberg L, Spens E, Sahlin C, Waara ER, Satlin A, Gellerfors P, Osswald G, Lannfelt L. The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-arcswe mice. J Alzheimer Dis. 2014 doi: 10.3233/JAD-140741. doi:10.3233/jad-140741. [DOI] [PubMed] [Google Scholar]
- 170.Um JW, Strittmatter SM. Amyloid-beta induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion. 2013;7(1):37–41. doi: 10.4161/pri.22212. doi:10.4161/pri.22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron. 2013;79(5):887–902. doi: 10.1016/j.neuron.2013.06.036. doi:10.1016/j.neuron.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Umeda T, Tomiyama T, Kitajima E, Idomoto T, Nomura S, Lambert MP, Klein WL, Mori H. Hypercholesterolemia accelerates intraneuronal accumulation of Abeta oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci. 2012;91(23–24):1169–1176. doi: 10.1016/j.lfs.2011.12.022. doi:10.1016/j. lfs.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 173.Velasco PT, Heffern MC, Sebollela A, Popova IA, Lacor PN, Lee KB, Sun X, Tiano BN, Viola KL, Eckermann AL, Meade TJ, Klein WL. Synapse-binding subpopulations of Abeta oligomers sensitive to peptide assembly blockers and scFv antibodies. ACS Chem Neurosci. 2012;3(11):972–981. doi: 10.1021/cn300122k. doi:10.1021/ cn300122k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Viola KL, Sbarboro J, Sureka R, De M, Bicca MA, Wang J, Vasavada S, Satpathy S, Wu S, Joshi H, Velasco PT, MacRenaris K, Waters EA, Lu C, Phan J, Lacor P, Prasad P, Dravid VP, Klein WL. Towards non-invasive diagnostic imaging of early-stage Alzheimer’s disease. Nat Nanotechnol. 2014 doi: 10.1038/nnano.2014.254. doi:10.1038/ nnano.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. doi:10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 176.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39(35):10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 177.Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924(2):133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- 178.Wang XP, Zhang JH, Wang YJ, Feng Y, Zhang X, Sun XX, Li JL, Du XT, Lambert MP, Yang SG, Zhao M, Klein WL, Liu RT. Conformation-dependent single-chain variable fragment antibodies specifically recognize beta-amyloid oligomers. FEBS Lett. 2009;583(3):579–584. doi: 10.1016/j.febslet.2008.12.064. doi:10.1016/j.febslet.2008.12.064. [DOI] [PubMed] [Google Scholar]
- 179.Watt AD, Villemagne VL, Barnham KJ. Metals, membranes, and amyloid-beta oligomers: key pieces in the Alzheimer’s disease puzzle? J Alzheimer Dis. 2013;33(Suppl 1):S283–S293. doi: 10.3233/JAD-2012-129017. doi:10.3233/jad-2012-129017. [DOI] [PubMed] [Google Scholar]
- 180.Wilcox KC, Marunde MR, Das A, Velasco PT, Kuhns B, Marty MT, Jiang H, Luan C-H, Sligar SG, Klein WL. Nanoscale synaptic membrane mimetic allows unbiased high throughput screen that targets binding sites for Alzheimer’s-associated Aβ oligomers. PloS One. 2015 doi: 10.1371/journal.pone.0125263. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wilhelmus MM, de Jager M, Bakker EN, Drukarch B. Tissue transglutaminase in Alzheimer’s disease: involvement in pathogenesis and its potential as a therapeutic target. J Alzheimer Dis. 2014;42:S289–S303. doi: 10.3233/JAD-132492. doi:10.3233/jad-132492. [DOI] [PubMed] [Google Scholar]
- 182.Wisniewski T, Goni F. Immunotherapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):499–507. doi: 10.1016/j.bcp.2013.12.020. doi:10.1016/j. bcp.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Xiao C, Davis FJ, Chauhan BC, Viola KL, Lacor PN, Velasco PT, Klein WL, Chauhan NB. Brain transit and ameliorative effects of intranasally delivered anti-amyloid-beta oligomer antibody in 5XFAD mice. J Alzheimer Dis. 2013;35(4):777–788. doi: 10.3233/JAD-122419. doi:10.3233/jad-122419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xu S, Liu G, Bao X, Wu J, Li S, Zheng B, Anwyl R, Wang Q. Rosiglitazone prevents amyloid-beta oligomer-induced impairment of synapse formation and plasticity via increasing dendrite and spine mitochondrial number. J Alzheimer Dis. 2014;39(2):239–251. doi: 10.3233/JAD-130680. doi:10.3233/jad-130680. [DOI] [PubMed] [Google Scholar]
- 185.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280(7):5892–5901. doi: 10.1074/jbc.M404751200. doi:10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 186.Yates EA, Owens SL, Lynch MF, Cucco EM, Umbaugh CS, Legleiter J. Specific domains of Abeta facilitate aggregation on and association with lipid bilayers. J Mol Biol. 2013;425(11):1915–1933. doi: 10.1016/j.jmb.2013.03.022. doi:10.1016/j.jmb.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 187.Younan ND, Sarell CJ, Davies P, Brown DR, Viles JH. The cellular prion protein traps Alzheimer’s Abeta in an oligomeric form and disassembles amyloid fibers. FASEB J Off Publ Fed Am Soc Exp Biol. 2013;27(5):1847–1858. doi: 10.1096/fj.12-222588. doi:10.1096/fj.12-222588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yuan Z, Du M, Chen Y, Dou F. Construction of human Fab library and screening of a single-domain antibody of amyloid-beta 42 oligomers. Neural Regener Res. 2013;8(33):3107–3115. doi: 10.3969/j.issn.1673-5374.2013.33.004. doi:10.3969/j.issn.1673-5374.2013.33.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zahs KR, Ashe KH. beta-Amyloid oligomers in aging and Alzheimer’s disease. Front Aging Neurosci. 2013;5:28. doi: 10.3389/fnagi.2013.00028. doi:10.3389/fnagi.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trend Neurosci. 2014 doi: 10.1016/j.tins.2014.08.004. doi:10.1016/j.tins.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 191.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30(36):11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. doi:10.1523/jneurosci.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang J, Peng M, Jia J. Plasma Amyloid-beta Oligomers and Soluble Tumor Necrosis Factor Receptors as Potential Bio-markers of AD. Current Alzheimer research. 2014 doi: 10.2174/1567205011666140317103222. [DOI] [PubMed] [Google Scholar]
- 193.Zhang L, Yagnik G, Peng Y, Wang J, Xu HH, Hao Y, Liu YN, Zhou F. Kinetic studies of inhibition of the amyloid beta (1-42) aggregation using a ferrocene-tagged beta-sheet breaker peptide. Anal Biochem. 2013;434(2):292–299. doi: 10.1016/j.ab.2012.11.025. doi:10.1016/j. ab.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhao M, Wang SW, Wang YJ, Zhang R, Li YN, Su YJ, Zhou WW, Yu XL, Liu RT. Pan-amyloid oligomer specific scFv antibody attenuates memory deficits and brain amyloid burden in mice with Alzheimer’s disease. Curr Alzheimer Res. 2014;11(1):69–78. doi: 10.2174/15672050113106660176. [DOI] [PubMed] [Google Scholar]
- 195.Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J Off Publ Fed Am Soc Exp Biol. 2008;22(1):246–260. doi: 10.1096/fj.06-7703com. doi:10.1096/ fj.06-7703com. [DOI] [PubMed] [Google Scholar]
- 196.Zheng L, Calvo-Garrido J, Hallbeck M, Hultenby K, Marcusson J, Cedazo-Minguez A, Terman A. Intracellular localization of amyloid-beta peptide in SH-SY5Y neuroblastoma cells. J Alzheimer’s Dis. 2013;37(4):713–733. doi: 10.3233/JAD-122455. doi:10.3233/jad-122455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.