

Abstract
Diets high in cruciferous vegetables are associated with lower risk of incidence of prostate cancer, including aggressive forms of this disease. Human intervention studies with cruciferous vegetable-rich diets also demonstrate modulation of gene expression in important pathways in prostate cells. Sulforaphane is a constituent of these foods postulated to harbor the anti-neoplastic activity based on multiple tumor models. Our own work demonstrates that sulforaphane inhibits AR signaling in prostate cancer cells. Here, we report results from the first clinical trial of sulforaphane-rich extracts in men with prostate cancer. We treated 20 patients who had recurrent prostate cancer with 200μmoles/day of sulforaphane-rich extracts for a maximum period of 20 weeks and determined the proportion of patients with ≥50% PSA declines, the primary endpoint. Only one subject experienced a ≥50% PSA decline. Thus, the primary endpoint was not achieved. Seven patients experienced smaller PSA declines (<50%). There was also a significant lengthening of the on-treatment PSA doubling time (PSADT) compared with the pre-treatment PSADT [6.1 months pre-treatment vs. 9.6 months on-treatment (p=0.044)]. Finally, treatment with sulforaphane-rich extracts was safe with no Grade 3 adverse events.
Treatment with 200μmoles/day of sulforaphane-rich extracts did not lead to ≥50% PSA declines in the majority of patients. However, because of the safety of treatment and the effects on PSADT modulation, further studies, including those with higher doses, may be warranted to clarify the role of sulforaphane as a prevention agent or treatment agent.
Keywords: Sulforaphane, prostate cancer, biochemical recurrence
INTRODUCTION
Prostate cancer is the most common cancer in men in the United States and the second-leading cause of cancer-related mortality.[1] This underscores the urgent need to identify new approaches to prevent and treat this disease. Epidemiological data suggest that men who consume diets high in cruciferous vegetables have a lower risk of developing prostate cancer, including locally-advanced forms of this disease.[2–4] Additionally, prospective studies with diets rich in the cruciferous vegetable broccoli demonstrate modulation of expression of genes in important pathways in prostate tissue in subjects without cancer.[5] Sulforaphane is an isothiocyanate compound found in these foods, and sulforaphane treatment has anti-tumor activity in pre-clinical cancer models, including prostate cancer.[6–10]
We previously determined that sulforaphane suppresses expression of the androgen receptor (AR) protein, the central signaling pathway in prostate cancer; this effect was due to inhibition of the cytoplasmic protein deacetylase HDAC6.[9] Additionally, others have demonstrated that sulforaphane also inhibits nuclear HDAC proteins whose primary substrates are histones.[11,10] Moreover, studies with sulforaphane-containing broccoli sprouts in normal human volunteers demonstrated inhibition of HDAC function in peripheral blood mononuclear cells (MCs) in several subjects.[10] However, the anti-tumor activity, pharmacokinetics, and pharmacodynamics of sulforaphane in men with prostate cancer were unknown.
Sulforaphane is excreted as a glutathione conjugate, a product of glutathione S transferase M1 (GSTM1) enzyme-mediated metabolism among other enzymes.[12] Nearly half of individuals do not express GSTM1 due to homozygous deletion of the gene.[13] There is conflicting data from epidemiological studies on the role of GSTM1 genotype on the modulation of prostate cancer risk with consumption of diets high in cruciferous vegetables.[14,15] Further, prior studies in normal human volunteers demonstrate that individuals with GSTM1 null genotypes have higher area under the curve (AUC) than GSTM1 intact individuals when fed sulforaphane-containing foods.[16] Another study demonstrated differences in modulation of gene expression in prostatic tissue based upon GSTM1 genotype.[5] However, the effect of GSTM1 genotype on metabolism of the sulforaphane extracts studied herein remained unknown.
In this single arm trial, we investigated the anti-tumor efficacy, safety, pharmacokinetics, and pharmacodynamics of daily treatment with 200μmoles of sulforaphane-rich broccoli sprout extracts in 20 men with biochemical (PSA) recurrence. The selected dose was chosen because pilot data demonstrated that treatment with this dose of these extracts led to low micromolar intra-prostatic concentrations of sulforaphane (personal communication P Talalay and J Fahey) similar to those that lead to anti-tumor activity in prostate cancer cells in pre-clinical studies.[9] Second, prior studies of shorter duration with similar doses of these extracts also showed tolerability and safety.[17,18] Finally, it was not feasible to treat with higher doses of these sulforaphane-rich extracts. This is because the necessary pre-clinical animal toxicological studies required for dose escalation clinical trials had not yet been completed.
MATERIALS AND METHODS
Patients
All subjects had pathologically confirmed prostate adenocarcinoma that had been treated with a prostatectomy or radiation. All patients had biochemical (PSA-only) recurrence after local therapy with study inclusion determined using a protocol-specific Prostate Cancer Working Group 2 (PCWG2) criteria for increasing PSA.[19] The absence of metastases prior to study entry was confirmed in all subjects by bone scans and either CT scans or MRI scans. All patients had three rising PSA values, with the most recent PSA at a minimum of 1ng/ml for post-surgical patients and a minimum of 2ng/ml for post-radiotherapy patients. Prior androgen deprivation therapy was allowed as long as the patient did not progress while on therapy, and all patients had a non-castrate testosterone level at the time of enrollment.[19] Baseline characteristics for all subjects are shown in Table 1. The study was approved by Oregon Health & Science University’s Institutional Review Board, and all patients provided written informed consent. The study was registered on clinicaltrials.gov (NCT01228084) prior to enrollment of the first subject.
Table 1.
Patient Demographics
| # of Participants | |
|---|---|
| Race/ethnicity | |
| Non-Hispanic, white | 19 |
| Non-Hispanic, Asian | 1 |
| Gleason sum at diagnosis | |
| 6 | 4 |
| 7 | 9 |
| 8 | 3 |
| 9 | 3 |
| 10 | 1 |
| Primary therapy | |
| Prostatectomy | 15 |
| Radiation | 1 |
| Brachytherapy | 4 |
| Salvage therapy | |
| Prostatectomy | 1 |
| Radiation | 13 |
| Cryotherapy | 1 |
| Pre-treatment PSA (ng/mL) | |
| Mean | 4.94 |
| Median | 2.60 |
| GSTM1 genotype | |
| Null | 8 |
| Intact | 12 |
Study Design
We conducted a single arm trial of sulforaphane-rich broccoli sprout extracts in 20 patients with biochemical recurrence. Patients were instructed to take 200μmoles (four capsules) of sulforaphane extracts daily by mouth prior to their morning meal for up to 20 weeks. Dosing compliance was monitored through comparison of patient reported dosing (via diary) to pill counts at each visit. Patients were seen in clinic every four weeks for a physical exam and toxicity assessment. Treatment on study could be held and delayed for up to 14 days. Study drug was to be held for ≥Grade 3 toxicity if possibly related to sulforaphane. If the adverse event resolved to ≤Grade 1 within 14 days, patients were to resume study treatment with a 50% dose reduction. Only one dose reduction was permitted. Other systemic therapies for prostate cancer were not allowed on study.
Serum PSA was measured every four weeks during the 20 week treatment period. PSA progression was not a criterion for study discontinuation. All subjects were scheduled for a post-study visit within 28 days of completing therapy.
Antitumor Activity
The primary endpoint was the proportion of patients who experienced a ≥50% decline in PSA levels within 20 weeks of sulforaphane treatment. Secondary endpoints included percent change in PSA from baseline, the effect of polymorphisms in the GSTM1 enzyme, and pharmacodynamic measurements of HDAC inhibition in MCs. Finally, we also analyzed pre- vs. on-treatment PSA trajectory [log2(PSA) rate of change] to determine PSA doubling time (PSADT).
Safety
Side effects and laboratory values that were deemed clinically significant were graded based on the NIH CTEP Common Terminology Criteria for Adverse Events Version 4.0 [CTCAE v4.0 (http://ctep.cancer.gov)]. We measured complete blood counts (CBC) with platelet and automated differential, complete metabolic panels (CMP), TSH levels, and testosterone at screening and weeks 1, 5, 9, 13, 17, and 21, and 14–30 days after the final dose. Testosterone levels were confirmed to be non-castrate (>150ng/dL) in all subjects prior to enrollment.
Blood Collection
Blood samples were drawn from all patients’ IV catheters at baseline and at the following times post-dose: 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 10 hours. Peripheral blood MC fractions were isolated at the same timepoints. Plasma samples for pharmacokinetic analysis were centrifuged in lithium heparin-coated BD Vacutainer tubes for 10 minutes at 1200xg at 25°C. Plasma was aliquoted and frozen on dry ice within 30 minutes of collection and stored at −80°C.
Pharmacodynamic Assays
Blood samples for MC fractions were collected in EDTA-coated BD Vacutainer tubes. Diluted blood was fractionated using Ficoll-Paque PLUS (Stemcell Technologies cat #07967), according to the manufacturer’s instructions and using a single wash step. MC pellets were washed in 1X SigmaFast protease inhibitor cocktail (Sigma cat #S8820), and stored at −80°C. Protein lysates were prepared from these pellets in RIPA buffer with 1X protease inhibitor. Histone acetylation and levels of the control beta-actin were assayed by SDS-PAGE and Western blotting (rabbit anti-acetylated histone H3, Millipore cat #06-599, mouse anti-beta-actin, Sigma cat #A5441).
GSTM1 Genotyping
Whole blood was collected for GSTM1 genotyping. DNA was extracted using the Gentra Puregene Blood Kit (Qiagen cat #158445) according to the manufacturer’s instructions. GSTM1 genotype was determined by PCR using primers and conditions described previously[20] on 25ng of genomic DNA. PCR products for GSTM1 and B-globin loading control were resolved by agarose gel electrophoresis.
Each patient was classified as GSTM1 genotype intact (at least one GSTM1 allele present) if a band was present in the PCR reaction vs. GSTM1 null (homozygous deletion) if a band was absent in the PCR.
Preparation of Sulforaphane-Rich Broccoli Sprout Extracts
Preparation of extracts was essentially as described previously.[21–24] Briefly, broccoli (Brassica oleracea var. italica) sprouts were grown from selected seeds with adequate levels of glucoraphanin (GR; the precursor of sulforaphane) to yield three-day-old fresh green sprouts with levels of at least 6μmol of GR per gram. Seeds were surface-disinfected, and grown in a commercial green-sprouting facility that adheres to U.S. Food and Drug Administration (FDA)-mandated sanitary regulations for sprout production. After three days of growth in which water and light were the only inputs, an aqueous extract was prepared in a steam-jacketed kettle at a food processing facility (Oregon Freeze Dry, Albany, OR). Sprouts were plunged into boiling deionized water and maintained at >95ºC for 30 minutes, and the sprout residues removed by filtration. The aqueous extract containing ~5μmoles of GR per ml was cooled to 37°C, and treated with myrosinase, an enzyme released from a small amount of daikon (Raphanussativus) sprouts, for 4 hours in order to convert the GR to sulforaphane. The levels of total isothiocyanate, sulforaphane, and residual GR were then quantified by cyclocondensation[25] and by direct HPLC, respectively.[24,26] The hydrolyzed aqueous extract was frozen rapidly, and lyophilized in industrial freeze-driers at Oregon Freeze Dry. The bulk powders were reanalyzed, bioassayed to determine induction potency for nicotinamide quinone oxidoreductase (NQO1) activity, tested for microbial contaminants, shipped to Johns Hopkins University (Baltimore, MD) and stored in sealed bags in a locked, dedicated freezer until use.[27] Confirmatory microbiological analyses (e.g., total aerobic plate count, yeast, mold, absence of specific pathogens) were performed by a commercial laboratory (Eurofins - Strasberger and Siegel, Hanover, MD), as were a suite of chemical analyses (in order to fully characterize and ensure against unintentional contaminants), all according to standard methods. Before clinical use, the sulforaphane-rich broccoli sprout extracts were re-analyzed for sulforaphane content. Doses of the powder were aliquoted by a commercial pharmacy (ALFA Pharmacy, Columbia, MD) into opaque, purple gel-caps delivering 218mg of powder (containing 50μmol sulforaphane) per gel-cap, and stored in dry conditions in the presence of desiccant, at (−20°C) until delivery to subjects.
LC-MS/MS Analysis of Sulforaphane
Chemicals and Reagents
The sulforaphane standard was purchased from Toronto Research Chemicals (North York, Ontario, Canada). Blood products were purchased from BioReclamation (www.Bioreclamation.com). Methanol and water (GC-MS grade) were obtained from Burdick and Jackson (Muskegon, MI). Sample vials were obtained from Sun Sri (a subdivision of Fisher Scientific, Rockwood, TN). Formic acid was from J.T. Baker (Phillipsburg, NJ), and 0.45 micron sample spin filters were from Millipore Corp. (Bedford, MA).
Preparation of Sulforaphane and Metabolite Calibrators and Blood Samples
Sulforaphane and metabolite standards were prepared by dissolving compounds in methanol at 1mg/ml and making appropriate dilutions in methanol before adding known concentrations of all compounds in relevant concentrations. The internal standard, sulforaphane-NAC-d3 in methanol was diluted to 1ng/10μl and added to each sample and standard. All plasma and standards were stored at −80°C.
Aliquots (100μl) of plasma were placed into an 1.7ml Eppendorf tube and 10μl of the internal standard in methanol was added. At the same time the naïve plasma samples were spiked with both the internal standard and the pre-diluted standard curve stocks. The standard curve concentrations prepared ranged from 0 to 20ng/ml. The lower limit of detection as judged by a signal to noise of 3:1, was 0.5ng/ml and the lower limit of quantification was 2ng/ml, exhibiting a signal to noise of 10:1 and a relative standard deviation of replicate samples of less than 20%. All samples were acidified with 5μl of 88% formic acid and touch vortexed before 300μl of ice cold methanol was added to all tubes. Tubes were then vortexed on a hands-free pulsing vortexer for 1 minute. The samples were then placed on ice for 15 minutes, then centrifuged at 10,000xg for 15 minutes at 4°C. 200μl of clear supe rnatant was removed and placed into a glass tube and dried at 35°C for 25 minutes under r educed pressure. The residue was dissolved in 100μl of 0.1% formic acid in water, vortexed and then briefly spun to aide in recovery of the sample and transferred to Millipore spin filters. The spin filters were centrifuged at 10,000xg for 5 minutes at 4°C. The filtered samples were then pi petted into 200μl inserts in autosampler vials and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS).
LC-MS/MS Analysis of Sulforaphane
The methods for LC-MS/MS analysis were adapted from the methods of Al Janobi[28] and Clarke.[29] Plasma samples were acidified with formic acid after the addition of internal standard, sulforaphane-NAC-d3, followed by three volumes of ice cold methanol. Following centrifugation the supernatant was dried, and the residue was dissolved in 100μl of 0.1% formic acid in water and analyzed by LC-MS/MS. Chromatographic separation was achieved using a Shimadzu Prominence high performance liquid chromatography (HPLC) system with autosampler and column oven interfaced to an Applied Biosystems/SCIEX Q-TRAP 5500 hybrid triple quadrupole/linear ion trap mass spectrometer instrument. HPLC separation of sulforaphane was done using a Hypersil Gold column (2.1×100mm, 3μm) (ThermoFisher) with a mobile phase of 0.1% formic acid (A) and methanol with 0.1% formic acid (B) at a flow rate of 0.3ml/min and kept at 30°C. Solvent B was increased from 5% to 43% over 10 minutes, then increased to 90% over 1 minute, held at 90% for 1 minute, then decreased to 5% over 1 minute, and then held at 5% for 5 minutes to re-equilibrate. Analytes were detected using multiple reaction monitoring (MRM). The MRM transitions monitored were: m/z 178/114 for sulforaphane; m/z 341/178 for sulforaphane-N-acetylcysteine (sulforaphane-NAC); m/z 484.9/179 for sulforaphane glutathione (sulforaphane-GSH); m/z 299/136 for sulforaphane cysteine (sulforaphane-CYS); and m/z 344/178 for sulforaphane-NAC-d3. Data were acquired and analyzed using Analyst 1.5.1 software. The lower limit of detection as judged by a signal to noise of 3:1 was 0.5ng/ml, and the lower limit of quantification was 2ng/ml, exhibiting a signal to noise of 10:1 and a relative standard deviation of replicate samples of less than 20%.
Pharmacokinetic Analysis
For determination of pharmacokinetic parameters, plasma sulforaphane concentrations were analyzed by noncompartmental methods using WinNonLin (v6.1; Pharsight, Mountain View, CA). Maximum plasma concentrations (Cmax) and time to maximum concentration (Tmax) were observed values. AUC was calculated from time zero to ten hours (AUC0-t) using the linear trapezoidal rule and then extrapolated to infinity (AUC0-∞) which provides a more accurate calculation of drug clearance.[30] AUC0-∞ is measured as a sum of AUC0-t and ‘excess area’ which is computed as a ratio of last observed concentration (Clast) to terminal elimination rate constant (λz). Drug half-life (t1/2), oral clearance (CL), and volume of distribution (VD) were estimated using standard pharmacokinetic calculations .
Statistical Analysis
A sample size of 20 patients was chosen to provide >90% power to detect 20% (desired) vs. 1% PSA response (≥50% PSA decline) rate using a binomial test with one-sided 5% significance level. This endpoint was chosen based upon previous placebo-controlled trials in this disease state that did not show ≥50% PSA declines with placebo treatment.[31,32] Given the toxicities and lack of known survival benefit with hormonal therapy, a PSA response rate of 20% was considered promising.
An intent-to-treat population included all patients who signed the study consent form and were enrolled in the study. A response evaluable population includes patients with at least one PSA value available on-study to measure response. The response evaluable population was used for the analysis of primary and secondary endpoints. A safety analysis population included patients who took at least one dose of the study drug.
We summarized demographic and clinical characteristics of patients using descriptive statistics (proportion, mean, standard deviation, median, range). A binomial distribution was used to estimate the proportion of PSA response with their exact confidence intervals. Waterfall plots were used to demonstrate the maximal decline in PSA on-study compared to baseline for each subject.[19] A mixed effects model was used to examine PSADT prior to and while on study. Specifically, a linear regression model was fitted to each patient’s log2(PSA) values, allowing random intercept and slopes (pre-study slope and on-study slope). The predicted slopes were obtained for the group, as well as for each individual patient. PSADT was estimated by the reciprocal of the predicted slopes for each individual patient. Predicted pre- and on-study slopes were then correlated with GSTM1 genotype and histone acetylation response using Wilcoxon rank sum test and with PK parameters using Spearman correlation coefficient.
Fisher’s exact test was used to assess an association between PSA decline while on study and histone acetylation response. Wilcoxon rank sum test was used to determine differences in PK/PD parameters between GSTM1 null vs. intact genotypes.
Toxicity data were tabulated and summarized for each major organ category and grade according to CTCAE v4.0.
RESULTS
Patient Characteristics and Disposition
Twenty patients were enrolled on this study, which met its accrual goal. Table 1 contains the demographic information. Sixteen patients completed the full 20 weeks of treatment. Two patients discontinued treatment at the discretion of the treating investigator due to rising PSA levels and two patients discontinued study treatment due to gastrointestinal (GI) side effects or inability to comply with required study visits.
Efficacy
PSA was measured every four weeks while on study. Figure 1 shows the maximal PSA decline during study treatment using a waterfall plot. Only one subject (5%) achieved a ≥50% PSA decline while on study (95% CI: 0–15%). Thus, the study did not meet the primary efficacy criteria. Seven subjects achieved lesser PSA declines ranging from 3% to 20%. Overall, a total of eight subjects (40%) experienced any degree of PSA decline while on study (95% CI: 19%, 64%). End of study PSA values were lower vs. baseline in three subjects while 17 subjects had higher end of study PSA values vs. baseline.
Fig 1.

Waterfall plot depicting maximal PSA decline on study compared to baseline
Pre- and on-study log2(PSA) trajectories (rates of change) were estimated using a mixed effects model. When evaluating the study population as a whole, the PSA rate of change was significantly modulated with the predicted doubling time increased from 6.1 months pre-treatment to 9.6 months on-treatment (p=0.044). This translates to an average of 57% increase in PSADT while on study.
Safety
Adverse events were all Grade 1 with exception of Grade 2 constipation that was experienced by one subject. The most common adverse events occurring in >15% of subjects were GI disorders. Four subjects experienced bloating, diarrhea, and dyspepsia, and seven subjects experienced flatulence. There were three reports of general GI pain. Only one patient discontinued study treatment due to side effects from treatment – for Grade 1 GI pain. No clinically significant changes in testosterone, CBC, CMP, or thyroid function lab values were seen on study (data not shown).
Effects of GSTM1 Genotype on PSA Modulation
We measured GSTM1 genotype in all patients prior to treatment. Twelve of the subjects were GSTM1 intact. Eight subjects had a GSTM1 null genotype. This matches prior reports, which suggest a similar frequency of these two genotypes in the population.[13] Neither GSTM1 genotype was associated with a greater likelihood of PSA decline on study (data not shown). Additionally, there were no significant differences in pre- and on-study PSA trajectories based on GSTM1 genotype (data not shown).
Pharmacokinetics
We measured pharmacokinetic parameters of sulforaphane and its metabolites (Table 2). Most sulforaphane pharmacokinetic parameters were similar between the two GSTM1 genotypes with the exception of sulforaphane half-life that was significantly longer in the GSTM1 null/null group (median 2.59 hours vs. 2.09 hours, p<0.05). No differences were seen in pharmacokinetic parameters of the sulforaphane metabolites: sulforaphane-CYS, sulforaphane-NAC, or sulforaphane-GSH according to GSTM1 genotype (Table 2).
Table 2. Pharmacokinetic parameters of sulforaphane and metabolites.
Data is presented as mean ± S.D. or median (25%, 75%).
| All | Null/Null | +/+ or +/Null | |
|---|---|---|---|
| SFN | |||
| AUC0-inf (h*ng/mL) | 121.6 ± 36.1 | 115.1 (96.6, 139.8) | 114.4 (97.8, 128.5) |
| Cmax (ng/mL) | 36.7 ± 9.8 | 35.8 ± 9.5 | 37.3 ± 10.3 |
| Tmax (h) | 1.47 ± 0.78 | 1.02 (0.90, 2.00) | 1.02 (1.00, 2.00) |
| T1/2 (h) | 2.55 ± 0.95 | 2.59 (2.18, 3.9) | 2.09 (1.91, 2.39)# |
| CL/F (L/h) | 315 ± 84 | 308 ± 76 | 319 ± 93 |
| Vz (L) | 1196 ± 676 | 931 (822, 2099) | 1029 (792, 1249) |
| SFN-CYS | |||
| AUC0-inf (h*ng/mL) | 514. 4 ± 146.9 | 494.8 ± 173.7 | 527.4 ± 132.7 |
| Cmax (ng/mL) | 165.2 ± 48.6 | 159.8 ± 62.7 | 168.8 ± 39.3 |
| Tmax (h) | 1.58 ± 0.78 | 1.51 (0.90, 2.02) | 1.51 (1.00, 2.00) |
| T1/2 (h) | 2.16 ± 0.34 | 2.27 ± 0.29 | 2.06 ± 0.37 |
| SFN-NAC | |||
| AUC0-inf (h*ng/mL) | 443.5 ± 136.9 | 448.9 ± 198.3 | 439.8 ± 85.6 |
| Cmax (ng/mL) | 75.1 ± 16.5 | 67.1 (59.8, 86.8) | 73.3 (65.8, 85.0) |
| Tmax (h) | 3.20 ± 1.01 | 3.99 (2.00, 4.00) | 4.00 (2.00, 4.00) |
| T1/2 (h) | 2.59 ± 0.66 | 2.59 (2.26, 3.19) | 2.46 (2.04, 2.71) |
| SFN-GSH | |||
| AUC0-inf (h*ng/mL) | 475.1 ± 224.2 | 555.5 ± 274.3 | 421.5 ± 176.3 |
| Cmax (ng/mL) | 160.9 ± 73.0 | 197.6 ± 78.8 | 136.5 ± 60.3 |
| Tmax (h) | 1.35 ± 0.79 | 1.02 (0.875, 2.00) | 1.00 (1.00, 1.51) |
| T1/2 (h) | 2.07 ± 0.57 | 1.86 (1.719, 2.400) | 1.82 (1.74, 2.31) |
Significantly different from Null/Null; p<0.05
Pharmacodynamics
Baseline and on-treatment histone acetylation levels were measured with day one MC samples. Histone acetylation varied between patients. Time course from six patients showed a similar pattern of increasing acetylation post-dose, peaking at 30 minutes to one hour and again at six to eight hours post-dose. Representative Western blots are shown in Figure 2. Time courses from 14 patients showed no increase in histone acetylation compared to baseline. There was no correlation between increase in MC histone acetylation and PSA declines, although the small sample size and low number of patients with ≥50% PSA declines limited this analysis (data not shown).
Fig 2. Sulforaphane treatment and histone acetylation changes in mononuclear cells.
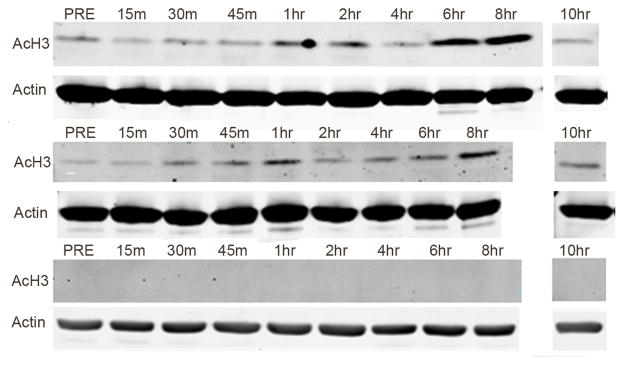
Proteins were extracted from mononuclear cells drawn at the indicated time points before (PRE) or after sulforaphane treatment on Day 1. Western blots were used to measure levels of acetylated histone H3 (AcH3) and Actin. Blots from three patients are shown
DISCUSSION
The results of this study demonstrate that treatment with 200μmoles of sulforaphane-rich broccoli sprout extracts daily does not lead to ≥50% PSA declines in a high proportion of men with recurrent prostate cancer. Indeed, only one patient out of 20 had a ≥50% PSA decline, the primary endpoint of this study. Smaller declines in PSA were seen in seven patients. Despite the lack of efficacy, treatment with 200μmoles of sulforaphane daily was well-tolerated with no Grade 3 or 4 AEs, suggesting that the dose tested is well below the maximally tolerated dose. The primary endpoint for this study was chosen based on prior pre-clinical work, including our own. That work demonstrated that sulforaphane treatment suppressed AR expression, expression of the AR target gene KLK3 that encodes for PSA, and prostate cancer cell survival.[9] Additionally, ≥50% PSA declines were not seen with placebo treatment in prior placebo-controlled studies in this patient population.[32,31] Thus, achieving a ≥50% PSA decline in this single arm trial would provide evidence for sulforaphane activity at this dose level.[32,31]
There are several possible explanations for the low observed rate of ≥50% PSA decline in our study. First, it is possible that the dose of sulforaphane we chose was too low. Pre-clinical studies demonstrate a dose-dependent anti-tumor effect for sulforaphane in cancer cells.[9,7] However, the necessary toxicological studies to perform a dose-escalation trial had not been performed with the sulforaphane formulation we tested. For that reason, we choose to test a dose of 200umoles of sulforaphane – the highest dose that had been used clinically and the dose that the U.S. FDA granted us permission to test. Treatment at this dose was well-tolerated. Indeed, no Grade 3 adverse events were seen in our study, which matches prior studies using a similar dose of this same sulforaphane formulation.17,18 Therefore, it is clear that further dose escalation of sulforaphane is possible. However, before these studies may proceed, the necessary pre-clinical animal toxicology studies with dose escalation must be completed with the sulforaphane extracts used herein or another sulforaphane formulation.
Second, it is possible that our sulforaphane treatment schedule was not optimal. Indeed, our PK studies demonstrate that sulforaphane was rapidly cleared from the bloodstream with a half-life of 2.55 hours. Thus, more frequent dosing of sulforaphane than the daily dosing schedule we chose may lead to more sustained drug levels.
PSA declines that did not meet the primary endpoint were seen in an additional seven subjects of our 20 patient cohort and ranged from 2% to 20% declines in PSA levels. Three of 20 subjects had lower PSA levels at the end of study vs. baseline, while 17 subjects had a higher end of study PSA levels vs. baseline. We also measured PSADT in our study because PSADT is a strong predictor of risk of metastases and death in patients with PSA-only recurrence of prostate cancer.[33] We observed a significant increase in PSADT for all 20 patients on-study compared with their pre-treatment PSADT [6.1 months pre-treatment vs. 9.6 months on-treatment (p=0.044)]. Prior placebo-controlled trials in this patient population have been conducted with celecoxib or rosiglitazone; both placebo and study drug treatment on those trials led to a similar lengthening of PSADT.[32,31] Thus, while these PSADT results are intriguing, the lack of a placebo control arm in our study limits their interpretability.
To understand the role of the sulforaphane clearance enzyme GSTM1 in sulforaphane clearance and treatment effect, we measured GSTM1 genotype in all subjects prior to study treatment. The prior epidemiological literature demonstrated conflicting information on the risk of prostate cancer with high consumption of cruciferous vegetables based on GSTM1 genotype[14,15] Other feeding showed higher sulforaphane blood concentrations amongst individuals with GSTM1 null genotypes, and dietary studies showed differential gene modulation in human prostate tissue based on GSMT1 genotype.[16,5] In our study, fully soluble sulforaphane-rich broccoli sprout extracts rather than whole foods were administered. We also saw a small but statistically significant increase in sulforaphane half-life in GSTM1 null individuals (median 2.59 hours vs. 2.09 hours, p<0.05) (Table 2). Despite the longer sulforaphane half-life, the clearance of sulforaphane was similar between both GSTM1 groups, and levels of the sulforaphane metabolites were also similar in both GSTM1 genotype groups (Table 2). It is possible that the dosing schedule, discussed above, precluded us from detecting the impact of GSTM1 genotype on drug exposure. However, the lack of relationship between GSTM1 genotype and sulforaphane metabolites, especially sulforaphane-GSH, also demonstrates the importance of identifying other pharmacogenetic factors that contribute to sulforaphane metabolism and clearance.
We also examined the association with GSTM1 genotype and ≥50% PSA declines or PSA trajectories, and no associations were seen (data not shown). Less is known about other pharmacogenetic determinants of sulforaphane metabolism in humans. However, these data demonstrate the importance of identifying other patient factors that contribute to sulforaphane metabolism and clearance.
Finally, prior work demonstrates that sulforaphane suppresses the function of HDAC enzymes in cancer cells and in human MCs.[9–11] Therefore, we measured levels of histone acetylation in MCs. Western blots from MCs in six of our subjects showed increased histone acetylation with sulforaphane treatment; we did not see an effect on histone acetylation in the other 14 subjects, and most of these patients had undetectable levels of histone acetylation in their MCs to begin with (Figure 2). Increase in histone acetylation was not associated with either GSTM1 genotype or plasma levels of sulforaphane, although this study was not powered to adequately assess these correlations (data not shown). However, the fact that MC histone acetylation did not correlate with PSA declines suggests that measuring MC histone acetylation has limited utility as a marker of sulforaphane on-target effects, at least with this dose of sulforaphane.
None of the patients in our study had tumors suitable for biopsy, so we were unable to correlate MC histone acetylation changes with those in tumor cells or to identify other markers of sulforaphane treatment effect. There are several ongoing studies (NCT01265953 and NCT00946309) that are using the same sulforaphane-rich extracts studied herein. These trial focus on sulforaphane treatment in patients prior to prostate biopsy or radical prostatectomy and will help to clarify the anti-tumor efficacy, target engagement, and achievable tissue levels of sulforaphane. These studies may also identify other valuable, readily measurable pharmacodynamics markers of sulforaphane treatment effect. If those studies, like ours, fail to show significant anti-tumor activity or target engagement, dose escalation studies of sulforaphane may be warranted before abandoning sulforaphane as a cancer treatment or prevention agent.
In summary, our results demonstrate that treatment with 200μmoles of sulforaphane does not lead to large reductions in PSA levels in the vast majority of patients. While this trial did not meet its primary endpoint, sulforaphane treatment was safe, and we further clarified the pharmacokinetics of treatment with 200μmoles of sulforaphane-rich broccoli sprout extracts. We look forward to ongoing and future sulforaphane studies that build on these results and that further clarify the safety and anti-tumor efficacy of sulforaphane treatment.
Acknowledgments
Financial Support: This clinical study was supported by a Kuni Foundation Clinical Trial Award (No#) (JA); a Kuni Foundation Scholar Award (No#) (JA); a Prostate Cancer Foundation Young Investigator Award (No#) (JA); Oregon Clinical and Translational Research Institute (OCTRI), grant numbers (UL1TR000128,KL2TR000152) from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Biostatical support was in part provided by the Biostatistics Shared Resource of the Knight Cancer Institute (P30 CA069533) (MM). Finally, we also thank Platt Electric, Bruce Burns, and The Burns Family Fund of the Oregon Community Foundation for their philanthropic support of this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors thank Jed Fahey, Sc.D (Johns Hopkins University) and Paul Talalay, MD (Johns Hopkins University) for providing and preparing the extracts, Emily Ho, PhD (Oregon State University) for providing the internal sulforaphane standard, Evan Yu, MD (University of Washington) for referring numerous patients for this trial, and all the patients who volunteered to take part in this study. Infrastructure support for conduct of the trial was provided by a Prostate Cancer Clinical Trials Consortium Award (W81XWH-09-1-0140).
Footnotes
Conflicts of Interest: None to disclose
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64 (1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Cohen JH, Kristal AR, Stanford JL. Fruit and vegetable intakes and prostate cancer risk. J Natl Cancer Inst. 2000;92 (1):61–68. doi: 10.1093/jnci/92.1.61. [DOI] [PubMed] [Google Scholar]
- 3.Kristal AR, Lampe JW. Brassica vegetables and prostate cancer risk: a review of the epidemiological evidence. Nutrition and cancer. 2002;42 (1):1–9. doi: 10.1207/S15327914NC421_1. [DOI] [PubMed] [Google Scholar]
- 4.Kirsh VA, Peters U, Mayne ST, Subar AF, Chatterjee N, Johnson CC, Hayes RB. Prospective study of fruit and vegetable intake and risk of prostate cancer. J Natl Cancer Inst. 2007;99 (15):1200–1209. doi: 10.1093/jnci/djm065. [DOI] [PubMed] [Google Scholar]
- 5.Traka M, Gasper AV, Melchini A, Bacon JR, Needs PW, Frost V, Chantry A, Jones AM, Ortori CA, Barrett DA, Ball RY, Mills RD, Mithen RF. Broccoli consumption interacts with GSTM1 to perturb oncogenic signalling pathways in the prostate. PLoS One. 2008;3 (7):e2568. doi: 10.1371/journal.pone.0002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A. 1992;89 (6):2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27 (4):811–819. doi: 10.1093/carcin/bgi265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh SV, Warin R, Xiao D, Powolny AA, Stan SD, Arlotti JA, Zeng Y, Hahm ER, Marynowski SW, Bommareddy A, Desai D, Amin S, Parise RA, Beumer JH, Chambers WH. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009;69 (5):2117–2125. doi: 10.1158/0008-5472.CAN-08-3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibbs A, Schwartzman J, Deng V, Alumkal J. Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc Natl Acad Sci U S A. 2009;106 (39):16663–16668. doi: 10.1073/pnas.0908908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Experimental biology and medicine. 2007;232 (2):227–234. [PMC free article] [PubMed] [Google Scholar]
- 11.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64 (16):5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Kolm RH, Mannervik B, Talalay P. Reversible conjugation of isothiocyanates with glutathione catalyzed by human glutathione transferases. Biochemical and biophysical research communications. 1995;206(2):748–755. doi: 10.1006/bbrc.1995.1106. S0006291X85711060 [pii] [DOI] [PubMed] [Google Scholar]
- 13.Seidegard J, Vorachek WR, Pero RW, Pearson WR. Hereditary differences in the expression of the human glutathione transferase active on trans-stilbene oxide are due to a gene deletion. Proc Natl Acad Sci U S A. 1988;85 (19):7293–7297. doi: 10.1073/pnas.85.19.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brennan P, Hsu CC, Moullan N, Szeszenia-Dabrowska N, Lissowska J, Zaridze D, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Gemignani F, Chabrier A, Hall J, Hung RJ, Boffetta P, Canzian F. Effect of cruciferous vegetables on lung cancer in patients stratified by genetic status: a mendelian randomisation approach. Lancet. 2005;366 (9496):1558–1560. doi: 10.1016/S0140-6736(05)67628-3. [DOI] [PubMed] [Google Scholar]
- 15.Joseph MA, Moysich KB, Freudenheim JL, Shields PG, Bowman ED, Zhang Y, Marshall JR, Ambrosone CB. Cruciferous vegetables, genetic polymorphisms in glutathione S-transferases M1 and T1, and prostate cancer risk. Nutrition and cancer. 2004;50 (2):206–213. doi: 10.1207/s15327914nc5002_11. [DOI] [PubMed] [Google Scholar]
- 16.Gasper AV, Al-Janobi A, Smith JA, Bacon JR, Fortun P, Atherton C, Taylor MA, Hawkey CJ, Barrett DA, Mithen RF. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. The American journal of clinical nutrition. 2005;82(6):1283–1291. doi: 10.1093/ajcn/82.6.1283. 82/6/1283 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Riedl MA, Saxon A, Diaz-Sanchez D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin Immunol. 2009;130 (3):244–251. doi: 10.1016/j.clim.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, Ye L, Coady JL, Wang JB, Wu Y, Sun Y, Zhang QN, Zhang BC, Zhu YR, Qian GS, Carmella SG, Hecht SS, Benning L, Gange SJ, Groopman JD, Talalay P. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People's Republic of China. Cancer Epidemiol Biomarkers Prev. 2005;14 (11 Pt 1):2605–2613. doi: 10.1158/1055-9965.EPI-05-0368. [DOI] [PubMed] [Google Scholar]
- 19.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, Eisenberger MA, Higano C, Bubley GJ, Dreicer R, Petrylak D, Kantoff P, Basch E, Kelly WK, Figg WD, Small EJ, Beer TM, Wilding G, Martin A, Hussain M Prostate Cancer Clinical Trials Working G. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26 (7):1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timofeeva M, Jager B, Rosenberger A, Sauter W, Wichmann HE, Group KS, Bickeboller H, Risch A. A multiplex real-time PCR method for detection of GSTM1 and GSTT1 copy numbers. Clinical biochemistry. 2009;42 (6):500–509. doi: 10.1016/j.clinbiochem.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 21.Dinkova-Kostova AT, Fahey JW, Benedict AL, Jenkins SN, Ye L, Wehage SL, Talalay P. Dietary glucoraphanin-rich broccoli sprout extracts protect against UV radiation-induced skin carcinogenesis in SKH-1 hairless mice. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2010;9 (4):597–600. doi: 10.1039/b9pp00130a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egner PA, Chen JG, Wang JB, Wu Y, Sun Y, Lu JH, Zhu J, Zhang YH, Chen YS, Friesen MD, Jacobson LP, Munoz A, Ng D, Qian GS, Zhu YR, Chen TY, Botting NP, Zhang Q, Fahey JW, Talalay P, Groopman JD, Kensler TW. Bioavailability of Sulforaphane from two broccoli sprout beverages: results of a short-term, cross-over clinical trial in Qidong, China. Cancer Prev Res (Phila) 2011;4 (3):384–395. doi: 10.1158/1940-6207.CAPR-10-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shapiro TA, Fahey JW, Dinkova-Kostova AT, Holtzclaw WD, Stephenson KK, Wade KL, Ye L, Talalay P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: a clinical phase I study. Nutrition and cancer. 2006;55 (1):53–62. doi: 10.1207/s15327914nc5501_7. [DOI] [PubMed] [Google Scholar]
- 24.Tang L, Zhang Y, Jobson HE, Li J, Stephenson KK, Wade KL, Fahey JW. Potent activation of mitochondria-mediated apoptosis and arrest in S and M phases of cancer cells by a broccoli sprout extract. Molecular cancer therapeutics. 2006;5 (4):935–944. doi: 10.1158/1535-7163.MCT-05-0476. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Wade KL, Prestera T, Talalay P. Quantitative determination of isothiocyanates, dithiocarbamates, carbon disulfide, and related thiocarbonyl compounds by cyclocondensation with 1,2-benzenedithiol. Analytical biochemistry. 1996;239 (2):160–167. doi: 10.1006/abio.1996.0311. [DOI] [PubMed] [Google Scholar]
- 26.Wade KL, Garrard IJ, Fahey JW. Improved hydrophilic interaction chromatography method for the identification and quantification of glucosinolates. Journal of chromatography A. 2007;1154 (1–2):469–472. doi: 10.1016/j.chroma.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fahey JW, Dinkova-Kostova AT, Stephenson KK, Talalay P. The “Prochaska” microtiter plate bioassay for inducers of NQO1. Methods in enzymology. 2004;382:243–258. doi: 10.1016/S0076-6879(04)82014-7. [DOI] [PubMed] [Google Scholar]
- 28.Al Janobi AA, Mithen RF, Gasper AV, Shaw PN, Middleton RJ, Ortori CA, Barrett DA. Quantitative measurement of sulforaphane, iberin and their mercapturic acid pathway metabolites in human plasma and urine using liquid chromatography-tandem electrospray ionisation mass spectrometry. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2006;844 (2):223–234. doi: 10.1016/j.jchromb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Clarke JD, Hsu A, Williams DE, Dashwood RH, Stevens JF, Yamamoto M, Ho E. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharmaceutical research. 2011;28 (12):3171–3179. doi: 10.1007/s11095-011-0500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowland M, Tozer TN. Clinical Pharmacokinetics: concepts and applications. Lea & Febiger; Philadelphia: 1980. [Google Scholar]
- 31.Smith MR, Manola J, Kaufman DS, Oh WK, Bubley GJ, Kantoff PW. Celecoxib versus placebo for men with prostate cancer and a rising serum prostate-specific antigen after radical prostatectomy and/or radiation therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24 (18):2723–2728. doi: 10.1200/JCO.2005.03.7804. [DOI] [PubMed] [Google Scholar]
- 32.Smith MR, Manola J, Kaufman DS, George D, Oh WK, Mueller E, Slovin S, Spiegelman B, Small E, Kantoff PW. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer. 2004;101 (7):1569–1574. doi: 10.1002/cncr.20493. [DOI] [PubMed] [Google Scholar]
- 33.Freedland SJ, Humphreys EB, Mangold LA, Eisenberger M, Dorey FJ, Walsh PC, Partin AW. Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. JAMA : the journal of the American Medical Association. 2005;294 (4):433–439. doi: 10.1001/jama.294.4.433. [DOI] [PubMed] [Google Scholar]