

Abstract
Background
Idiopathic Pulmonary arterial Hypertension (IPAH) is a cardiopulmonary disease characterized by cellular proliferation and vascular remodeling. A more recently recognized characteristic of the disease is dysregulation of glucose metabolism. The primary link between altered glucose metabolism and cell proliferation in IPAH has not been elucidated. We aimed to determine the relationship between glucose metabolism and smooth muscle cell proliferation in IPAH.
Methods and Results
Human IPAH and control patient lung tissues and pulmonary artery smooth muscle cells (PASMCs) were used to analyze a specific pathway of glucose metabolism, the hexosamine biosynthetic pathway (HBP). We measured the levels of O-linked N-acetylglucosamine modification (O-GlcNAc), O-GlcNAc transferase (OGT), and O-GlcNAc hydrolase (OGA) in control and IPAH cells and tissues. Our data suggests that the activation of the HBP directly increased OGT levels and activity triggering changes in glycosylation and PASMC proliferation. Partial knockdown of OGT in IPAH PASMCs resulted in reduced global O-GlcNAc levels and abrogated PASMC proliferation. The increased proliferation observed in IPAH PASMCs was directly impacted by proteolytic activation of the cell cycle regulator, host cell factor-1 (HCF-1).
Conclusions
Our data demonstrate that HBP flux is increased in IPAH and drives OGT-facilitated PASMC proliferation through specific proteolysis and direct activation of HCF-1. These findings establish a novel regulatory role for OGT in IPAH, shed a new light on our understanding of the disease pathobiology, and provide opportunities to design novel therapeutic strategies for IPAH.
Keywords: Idiopathic pulmonary arterial hypertension, O-GlcNAc, OGT, hexosamine biosynthetic pathway, vascular cell proliferation, HCF-1
INTRODUCTION
Idiopathic pulmonary arterial hypertension (IPAH) is a rapidly progressive disease with poor prognosis that affects the heart and lungs1–4. Presently, IPAH is considered a vasculopathy that results from the structural and morphological changes of the vasculature within the lung. Altered metabolic functions, including dysregulated glucose uptake/metabolism have been described in IPAH5–7 and have the potential to impact several key processes implicated in the pathogenesis of the disease, including cellular proliferation. Understanding the molecular mechanisms and pathways that link dysregulation in glucose metabolism to cell proliferation can lead to a better understanding of the disease and the development of novel IPAH therapies.
The hexosamine biosynthetic pathway (HBP) serves as a sensor for metabolic flux and is a precursor for glycosylation pathways8–10. Once activated, the HBP generates the sugar nucleotide UDP-N-acetyl-glucosamine (UDP-GlcNAc), which is a substrate for hyaluronan (HA) and CMP-sialic acid synthesis, and for the O-linked β-N-acetylglucosamine (O-GlcNAc) modification of proteins11. O-GlcNAc modification is a cellular process similar, and often reciprocal, to protein phosphorylation with occupation of the same serine/threonine residues when the phosphate is removed12. Typically, O-GlcNAc modification has an inverse functional relationship with phosphorylation and is important in the regulation of the target protein’s function13.
In the presence of UDP-GlcNAc, transfer of the ‘GlcNAc’ moiety to serine and threonine residues within proteins is governed by the O-GlcNAc transferase (OGT)14, 15. Conversely, the removal of the residue is governed by the cytosolic or nuclear β-N-acetylglucosaminidase (O-GlcNAc hydrolase, OGA)16, 17. Recently, OGT was recognized for its dual role in the hyper O-GlcNAc modification and subsequent proteolytic activation of the cell cycle master regulator, host cell factor-1 (HCF-1), indicating a direct role for OGT in cell proliferation18, 19. Indeed, OGT synergizes with the HBP to regulate both nuclear and cytoplasmic protein functions through the O-GlcNAc modification.
We previously demonstrated increased levels of HA in plasma and tissues in IPAH patients20, suggesting augmented glucose flux through the HBP to increase UDP-GlcNAc, a substrate for HA synthesis. However, the functional role of the HBP and the underlying mechanisms of IPAH pulmonary artery smooth muscle cell (PASMC) proliferation have not been determined. We hypothesized that IPAH is characterized by increased HBP flux and OGT upregulation, which promotes and perpetuates PASMC proliferation. We report here that HBP flux is indeed enhanced in IPAH and OGT levels and activity are increased. Together, these changes promote protein glycosylation leading to PASMC proliferation. The increased proliferation observed in IPAH PASMCs is directly impacted by the OGT activation of host cell factor-1 (HCF-1), a phenomenon recently described in proliferating cancer cells18. These new insights into the mechanism of PASMC proliferation can lead to the identification of novel therapeutic targets in IPAH.
MATERIALS AND METHODS
Lung Tissue, PASMC Isolation, and Culture Conditions
All explanted lungs were collected either at the Cleveland Clinic through an Institutional Review Board approved protocol or they were provided by Baylor, Stanford, Vanderbilt, University of Alabama, and Allegany College of Maryland under the Pulmonary Hypertension Breakthrough Initiative (PHBI). Funding for the PHBI was provided by the Cardiovascular Medical Research and Education Fund (CMREF). Human lung tissues used in this study were from 8 donor lung explants not suitable for lung transplantation and 8 idiopathic PAH patients (Table 1). Human PASMCs and pulmonary arterial endothelial cells (PAECs) were isolated from elastic pulmonary arteries dissected from both control and IPAH lungs obtained at explantation (Table 1) using a previously described method21. Briefly, human pulmonary arteries were minced and digested overnight in Hanks’ balanced salt solution containing collagenase and DNase and Hepes buffer (Sigma, St. Louis, MO). Upon the removal of the PAECs, smooth muscle cells were released from the artery tissue, filtered with a 100-μm-pore nylon cell strainer (BD Falcon, Bedford, MA), cultured in DMEM/F-12 medium supplemented with 10% FBS (Bio-Whittaker, Walkersville, MD) and antibiotics, and incubated at 37°C, 5% CO2 with 90% humidity follow by media changes at 24 hours and every 4 days until confluence. The PASMCs were confirmed routinely through positivity staining for α-smooth muscle cell actin (Sigma/Aldrich, St. Louis, MO).
Table 1.
Demographic information for lung explants and isolated cells.
| IPAH Lung Tissue (n) | 8 |
| Age, years | 38.3 ± 16.0 |
| Female | 4 (50.0%) |
| PAH category | |
| Idiopathic PAH | 7 (87.5%) |
| Heritable PAH | 1 (14.3%) |
|
| |
| IPAH Vascular Cells* (n) | 4 |
| Age, years | 36.3 ± 12.3 |
| Female | 4 (100%) |
| PAH category | |
| Idiopathic PAH | 2 (50.0%) |
| Heritable PAH | 2 (50.0%) |
|
| |
| Control Lung Tissue (n) | 8 |
| Age, years | 43.3 ± 20.0 |
| Female | 2 (25.0%) |
|
| |
| IPAH Vascular Cells* (n) | 4 |
| Age, years | 49.5 ± 6.45 |
| Female | 3 (75.0%) |
Denotes PASMCs and PAECs
Immunohistochemistry, Immunofluorescence, and hematoxylin and Eosin (H&E) Stains
Sample Preparation and Western blotting
Frozen human lung tissue20 and PASMCs22 were prepared as previously described with PUGNAC (50 μM; Sigma, St. Louis, MO) + Thiamet G (25 nM, Sigma) added to block removal of the O-GlcNAc modification and subjected to Western blot analysis. Nitrocellulose membranes were probed with antisera for the following: 1) O-GlcNAc (1:1,000; clone CTD 110.6, generous gift from the lab of Dr. Gerald Hart) and HCF-1 (1:1000; Abcam, Cambridge, MA, USA) probed blots were developed using enhanced chemiluminescence (ECL, Amersham, Pittsburgh, PA, USA), and 2) blots probed for GFAT1 (1:2,000; Abcam, Cambridge, MA, USA), OGT (1:5,000, Hart Lab), OGA (1:5,000 Hart Lab), β-Actin (1:10,000; Santa Cruz, CA, USA), GLUT1 (1:2,000; Millipore, Billerica, MA) and GLUT4 (1:2,000; Millipore) were blocked, washed, and imaged using an Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE, USA).
Fluorophore Assisted Carbohydrate Electrophoresis (FACE) UDP-Sugar Analysis
PASMCs were washed, collected, and centrifuged in cold PBS. Cells were fixed with 75% cold ethanol, sonicated and subjected to FACE analysis (see Supplemental Materials and Methods).
siRNA Transfection and OGT Inhibitor Experiments
PASMCs were subjected to siRNA knockdown with a scrambled, an antisense siRNA oligo-nucleotide against OGT, or an OGT specific inhibitor (see Supplemental Materials and Methods).
Cell Proliferation and Flow Cytometry Analysis
PASMCs were pulsed for 2 hours with 10 μM 5-bromo-2′-deoxyuridine (BrdU) (Sigma, St. Louis, MO, USA) and subjected to Flow Cyotmetric analysis (see Supplemental Materials and Methods). The gating strategy and the relevant controls for flow cytometric analysis are shown in Supplemental Figure 1.
Study Population for Erythrocyte Collection
Patients were recruited from the Pulmonary Vascular Program at Cleveland Clinic. Blood was drawn from patients and deposited into our biorepository. Demographic and clinical characteristics of the PAH individuals are listed in Table 2. Pulmonary hypertension was confirmed by right heart catheterization. PAH categories were identified based on the updated clinical classification of pulmonary hypertension23. Healthy controls were defined as patients with no history of pulmonary/cardiac disease or symptoms. All participants signed a consent form that was approved by the Cleveland Clinic Institutional Review Board (IRB) prior to participation in the study.
Table 2.
Baseline clinical features of PAH patients (RBC analysis)
| PAH (n) | 86 |
| Age, years | 45.9 ± 12.3 |
| Female | 70 (81%) |
|
| |
| PAH category | |
| Idiopathic PAH | 63 (73.0%) |
| Heritable PAH | 7 (8.0%) |
| CTD-PAH | 6 (7.0%) |
| CHD-PAH | 10 (11.6%) |
|
| |
| NYHA class (n=80) | |
| I–II | 45 (56%) |
| III–IV | 35 (44%) |
|
| |
| 6MWD, meters (n=85) | 410.0 ± 128.0 |
| BNP, pg/mL (n=81) | 38 (13.5–107.5) |
| RAP, mmHg (n=85) | 9.0 ± 5.8 |
| Mean PAP, mmHg | 52.2 ± 14.2 |
| CI, L/min/m2 (n=80) | 2.5 ± 0.9 |
| PVR, Wood units (n=83) | 10.2 ± 6.4 |
Data presented as mean +/− SD (except for BNP where the data is presented as Median (25th and 75th percentile) or number (%) as appropriate. Abbreviations: RAP, right atrial pressure; CI, cardiac index; PVR, pulmonary vascular resistance; 6MWD, six-minute walk distance; BNP, B-type natriuretic peptide; CHD = Congenital heart disease; CTD = connective tissue disease; mPAP = mean Pulmonary artery pressure; PAH = pulmonary arterial hypertension
Erythrocyte Experiments and Western Blotting Procedures
An initial set of human PAH (n=9) and control (n=9) RBCs were lysed and hemoglobin depleted using a procedure described previously24 to compare the levels of O-GlcNAc (CTD 110.6, Hart Lab) and OGT (Hart Lab) by Western blot analysis. OGT and the O-GlcNAc modification were normalized to β-Actin and quantitated using ImageJ25 software.
OGT association with Clinical Outcomes Using PAH RBCs
PAH RBC hemoglobin depleted lysates (n=86) were subjected to Western blot analysis to determine the levels of OGT for each patient. After quantitation, OGT levels were correlated to clinical parameters of disease severity, including the functional class, walk distance, brain natriuretic peptide (BNP) levels, echocardiographic parameters (Right Ventricular dilation and dysfunction, pericardial effusion), and hemodynamic measurements. Data were obtained from the IRB-approved Cleveland Clinic Pulmonary Hypertension registry. Clinical events were ascertained since the time of venous blood sampling by review of medical records and query of the Social Security Death index. During the median follow up of 24.5 months (IQR 9 to 58.3) there were 38 hospitalizations, 3 lung transplantations and 1 death. However, hospitalizations were the earliest event for the death and transplant events, so the total number of events for the analysis was 38.
Statistical Analysis
Summary statistics reported for continuous variable are ‘mean ± standard error of mean (SEM)’ or ‘median (25th, 75th percentiles)’, with the latter used for skewed distributions. Two-group comparisons with respect to continuous variables were compared with the Wilcoxon rank sum test. A p-value less than or equal to 0.05 was considered as significant, except when comparisons were performed as multiple comparisons among more than two groups, in which case a Bonferroni correction was used to adjust for the number of primary two-group comparisons. Given the anticipated skewed distribution of OGT levels, its associations with continuous clinical parameters were assessed using the Spearman correlation coefficient, and with categorical parameters using the Wilcoxon test. We also investigated the association between OGT levels and clinical worsening (defined as disease-related hospitalizations, lung transplantation or all-cause mortality) inferentially using Cox proportional hazard models with respect to OGT in its continuous form, and descriptively using Kaplan-Meier curves. All statistical analysis was done using JMP version 10.0.0 for Microsoft Windows or R version 3.0.1 (www.R-project.org).
RESULTS
Glycosylation changes in the pulmonary vasculature of IPAH patients
To determine if altered glycosylation is observed in IPAH, we stained tissues from control and IPAH explanted lungs with wheat-germ agglutinin (WGA), a lectin that specifically recognizes N-acetylglucosamine (GlcNAc) and sialic acid. IPAH tissues had an increased intensity of WGA lectin staining suggesting changes in glycosylation (Figure 1A–B and Supplemental Figure 2). The increased glycosylation was much more evident in the occluded pulmonary vessels and plexogenic lesions in IPAH than in control tissues (Figure 1 A–D). These data indicate that glycosylation changes are prominent in IPAH.
Figure 1.
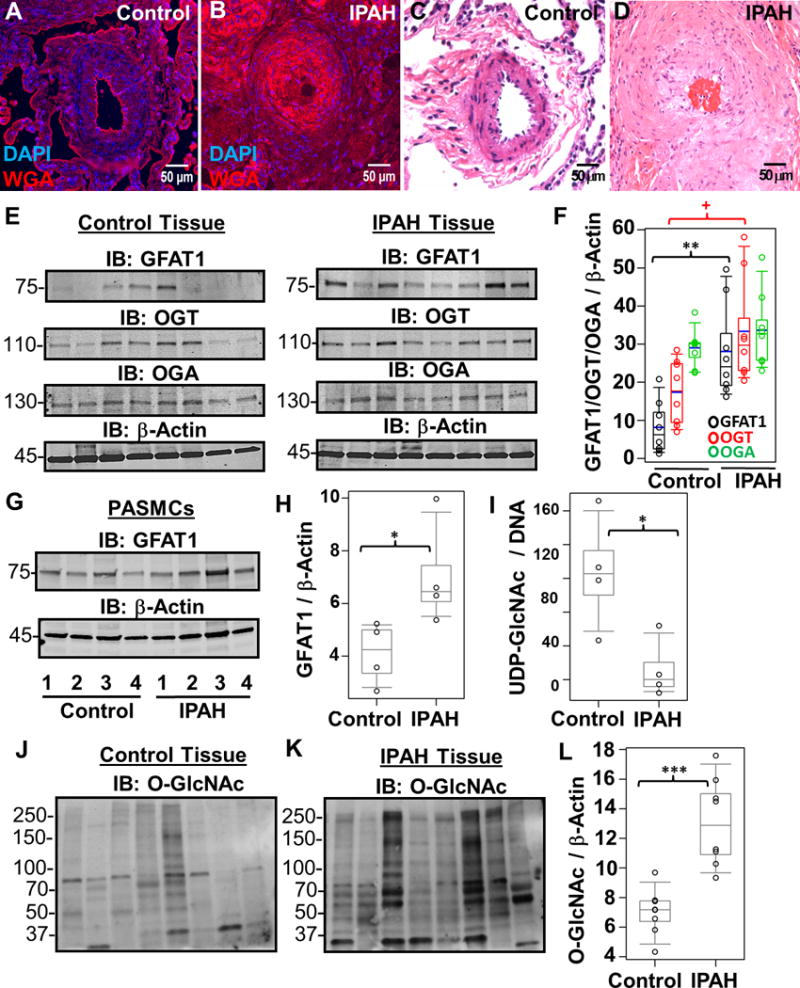
Increased hexosamine biosynthesis results in global glycosylation changes in IPAH. Representative WGA lectin (A and B) and H&E (C and D) stains of a control (A and C) and IPAH (B and D) occluded vessel within a plexogenic lesion from serial sections of the same lung tissue. (E) Characteristic Immunoblots of GFAT1, OGT, and OGA from multiple control and IPAH human tissues isolated from explant lungs (n=8). Lysates were normalized to total protein and β-Actin was used as a loading control. (F) Densitometry quantitation of each protein compared to β-Actin. (G) GFAT1 was also probed in multiple control and IPAH human PASMCs isolated from pulmonary arteries (n= 4) and quantitated (H). (I) UDP-GlcNAc pools were isolated (see Methods) and analyzed from multiple control and IPAH PASMCs (n=3). UDP-GlcNAc levels were normalized to total DNA concentration. In addition, total O-GlcNAc levels were analyzed in control (J) and IPAH (K) lung tissue. The global protein O-GlcNac levels were normalized to β-Actin (E) and quantified (L). As a reactivity control, the O-GlcNAc antibody was pre-treated with 250 mM GlcNAc before primary incubation (Supplemental Figure 3). The p-values were calculated based on a Wilcoxon test (see Materials and Methods) determined from the independent experiments (+, p=0.027; *, p<0.05; **, p<0.01; and ***, p<0.001).
HBP flux in IPAH
Since IPAH has increased HA levels and altered glycosylation, we examined the activation state of the HBP. As shown in Figure 1E–F, glutamine:fructose-6-phosphate aminotransferase-1 (GFAT1), the HBP rate-limiting enzyme, was increased in IPAH lung tissue (control, 8.2±7.0; IPAH, 28.1±12.4, p=0.002). A similar finding was determined in IPAH pulmonary arterial smooth muscle cells (PASMCs) isolated from the lung explants (Figure 1G–H, GFAT1: control, 4.1±1.2; IPAH, 7.1±2.0, p=0.02). Importantly, UDP-GlcNAc pools were reduced in IPAH PASMCs compared to controls (Figure 1I; control, 103.7±34.0; IPAH, 46.1±17.8, p=0.04). Collectively, these data suggest that the HBP is augmented in IPAH.
Global increase in OGT-facilitated O-GlcNAc modification of proteins in human IPAH lungs and PASMCs
To determine the outcome of HBP flux on the dynamic post translational modification by O-GlcNAc, we assessed the changes in global protein O-GlcNAc levels, as well as the levels of OGT and OGA in IPAH. The global O-GlcNAc modification on proteins was increased in IPAH human lung tissue compared to controls (Figure 1J–L; control, 7.05±1.6; IPAH, 13.0±2.9, p=0.001; and Supplemental Figure 3). Consistent with the O-GlcNAc changes, OGT levels were also increased in IPAH lung tissue compared to controls (Figure 1E–F; control, 17.4±8.5; IPAH, 33.4±13.9, p=0.027) without alterations in OGA levels (Figure 1E–F; control, 29.1±5.0; IPAH, 33.7±9.9, p=0.25), suggesting the O-GlcNAc elevation is a function of increased OGT.
Immunohistochemical staining for the O-GlcNAc modification of proteins within the lung tissue validated the increased O-GlcNAc levels observed in IPAH patients (Figure 2A–H). Higher magnification of the lung tissue revealed increased O-GlcNAc staining within the pulmonary vasculature (including plexogenic lesions) of IPAH patients compared to controls (Figure 2A–H). These findings are consistent with our WGA lectin stains (Figure 1A–B) and Immunoblot data (Figure 1J–L), suggesting increased glycosylation events within IPAH lung tissue.
Figure 2.
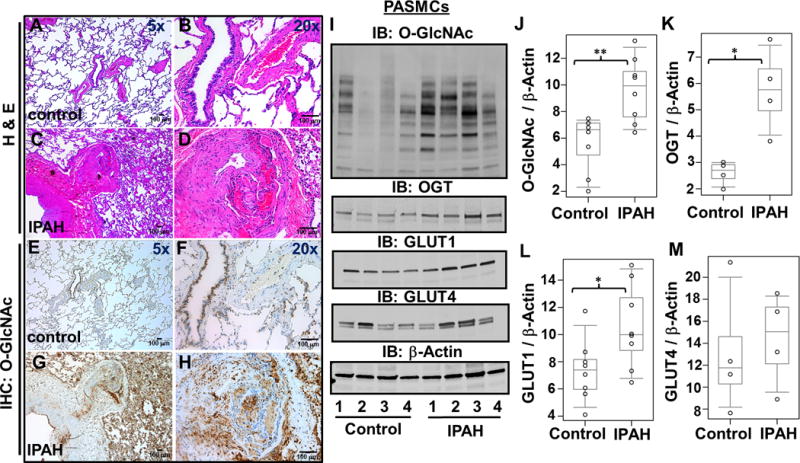
The O-GlcNAc modification of proteins is increased in the IPAH lung parenchyma and pulmonary vasculature. Representative H&E (A–D) as well as Immunohistochemical (E–H) staining for O-GlcNAc within paraffin embedded control (A–B, E–F) and IPAH (C–D, G–H) lung tissue. (I) Global O-GlcNAc, OGT, GLUT1, and GLUT4 were examined in multiple control and IPAH PASMCs (n=4). (J–M) Protein amounts were quantitated as described in Figure 1. IPAH patients 2 and 3 have genetic BMPR mutations. *, p<0.05 and **, p<0.01
Since the O-GlcNAc modification was more prevalent in the thickened tunica media of the pulmonary arteries in IPAH, we investigated the nature of this region using primary IPAH PASMCs isolated from explanted lungs. Western blot analysis determined that the levels of O-GlcNAc modified proteins were also elevated in IPAH PASMCs (Figure 2I and 2J, control, 5.7±2.1; IPAH, 9.6±2.4, p=0.01) and demonstrated consistently higher levels of OGT (Figure 2I and 2K; control, 2.6 ± 0.5; IPAH, 5.8±1.6, p=0.021) similar to the lung tissue observations (Figure 1E–F and 1J–L) and IPAH PAEC isolates (Supplemental Figure 4). In addition, GLUT1 levels were elevated in the IPAH PASMCs (Figure 2I and 2L; control, 7.4±2.3; IPAH, 10.6±3.1, p=0.03) analogous to reports indicating increased glucose uptake5, 26, 27. However, the insulin sensitive transporter GLUT4 was not significantly different (Figure 2I and 2M; control, 13.1±2.9; IPAH, 14.4±2.1, p=0.56). This finding is consistent with previously published reports that described GLUT1 as the predominant transporter for glucose uptake in primary PASMCs28. Collectively, these findings suggest that the excessive HBP flux consequentially results in increased OGT transfer of O-GlcNAc within the IPAH pulmonary vasculature.
OGT directs cell proliferation in primary PASMCs
Next, we determined whether OGT can mediate PASMC proliferation in IPAH. We used siRNA facilitated knock-down (KD) of OGT in IPAH PASMCs and compared to control levels. Gene silencing caused a reduction in OGT [Figure 3A; 7.14±0.33 (IPAH untreated), 2.50±0.56 (control); p=0.05 and 7.14±0.33 (IPAH untreated), 2.64±0.66 (IPAH 60nM OGT siRNA); p=0.05] and O-GlcNAc levels (Figure 3A) in IPAH PASMCs. The consequences of OGT KD on cell density 48 hours post-siRNA transfection are shown in Figure 3B–C [(Control, 5.33×105 ± 2.10×104; IPAH (Untreated), 9.86×105 ± 3.32×104; IPAH (Scramble), 1.02×106 ± 6.97×104; IPAH (60 nM OGT siRNA), 3.18×105 ± 4.41×104; p= 0.05, respectively]. In addition, cell proliferation was assessed by cellular BrdU incorporation, which confirmed a significant decline [Figure 3D–E; 32.2±3.12 (IPAH untreated), 14.7±0.59 (IPAH 60 nM OGT siRNA); p=0.05]. Strikingly, IPAH PASMC proliferation was decreased to control rates after OGT KD (Figure 3D–E; 32.2±3.12 (IPAH untreated), 14.1±1.75 (control); p=0.05). A reduction in PAH PASMC proliferation to control levels was also determined using an OGT inhibitor (Alloxan monohydrate), consistent with the siRNA KD to OGT (Supplemental Figure 5). These data indicate that the reduction in OGT levels results in decreased PASMC proliferation in IPAH.
Figure 3.
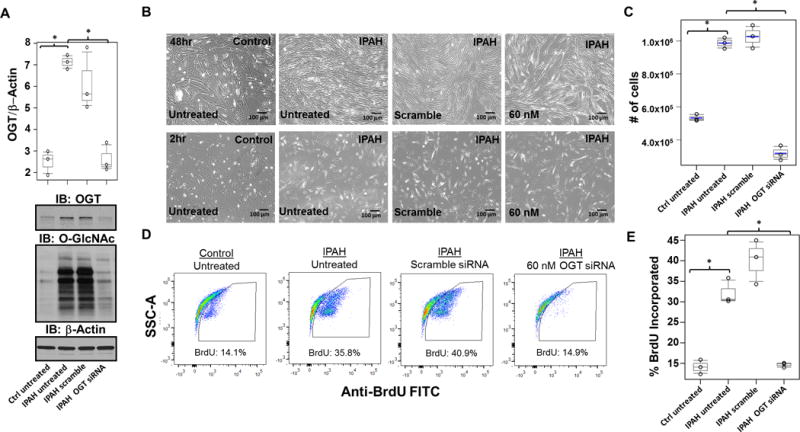
OGT governs proliferation in IPAH PASMCs. (A) Representative Immunoblots of OGT and O-GlcNAc from control and IPAH human PASMCs are shown. OGT/□-Actin was quantitated based on the densitometry determined using ImageJ software. (B) Twenty four hours after transfection, PASMCs were trypsinized and reseeded at 150,000 cells/mL, followed by snapshot assessment of cell density at the times indicated post transfection. A graph of the cell density from triplicate experiments determined at 48 hours post transfection is shown (C). (D–E) Flow cytometric analysis was used to determine the percentage of cellular BrdU incorporation in control and IPAH PASMCs treated for 2hr following different gene silencing strategies. Flow cytometric analysis of IPAH and control PASMCs was quantitated and percentage of cellular BrdU incorporation was compared to untreated IPAH (E). siRNA transfections were performed in triplicate experiments and calculated as described in statistical analysis section (see Materials and Methods). BrdU incorporation assays are shown as a representative pseudo-color side scatter (SSC) plot shown for each condition (D). *, p<0.05.
Increased OGT activates HCF-1 in IPAH PASMCs
To determine whether increased OGT expression corresponds to enhanced activity, HCF-1 cleavage, and subsequent activation in IPAH, we reduced OGT levels by siRNA KD or its activity by administration of an OGT inhibitor (TT40) in IPAH PASMCs. Reduced OGT expression resulted in a marked decrease in HCF-1 cleavage products comparable to control (Figure 4A and Supplemental Figure 6A). Similar results were observed upon reducing OGT activity with TT40 in IPAH PASMCs (Figure 4B and Supplemental Figure 6B), which is analogous to the reduction in HCF-1 cleavage observed in cancer cells upon OGT inhibition (Supplemental Figure 7). Collectively, these data provide a mechanism whereby OGT promotes PASMC proliferation in IPAH.
Figure 4.
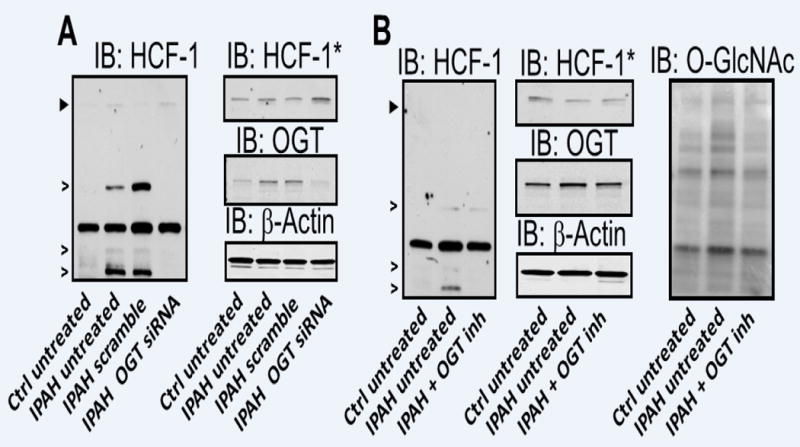
Increased OGT levels impact HCF-1 cleavage/activation in IPAH. (A) siRNA mediated KD of OGT and assessment of HCF-1 in IPAH and control. (B) IPAH PASMCs were cultured for 24 hours with and without OGT inhibitor and compared to untreated control PASMCs. OGT and O-GlcNAc levels were used to determine the level of siRNA KD (A) or OGT inhibition (B). siRNA KD of OGT and western blots were performed in triplicate. Asterisks denote a longer exposure of the Immunoblot for HCF-1 shown in panel A and B (see Supplemental Figure 6). Arrowheads represent the precursor HCF-1 and open arrows indicate HCF-1 specific cleavage products generated by OGT. A non-specific, but antibody reactive band is not marked in figure.
OGT associates with clinical worsening in PAH
Increased OGT levels have been shown to associate with poor outcomes and disease aggressiveness in prostate cancer patients29, 30, suggesting that increased OGT activity contributes to the disease progression. In addition, a previous report screened multiple patient red blood cells (RBCs) for changes in global O-GlcNAc, OGT, and OGA as potential indicators of disease24. Based on these studies, we analyzed global O-GlcNAc, OGT, and OGA levels from PAH and control RBCs by Western blot. Our initial assessment demonstrated an increase in the overall levels of OGT and O-GlcNAc in PAH RBCs compared to control with no significant change in OGA levels (Figure 5A–E), which is consistent with Figure 1E–F and 1J–L. Since OGT levels were higher in PAH RBCs, we measured OGT levels by Western blot analysis in 86 PAH RBC patient samples and associated with clinical outcomes in these patients. Data are expressed as OGT/beta actin ratios. Table 2 shows the baseline clinical characteristics of this cohort. Inverse correlations between OGT expression and right heart stroke volume (rs=−0.31, p<0.01,) and the 6MWD (rs=−0.21, p<0.05) were observed. OGT was higher in patient NYHA functional class III–IV symptoms compared to NYHA class I–II (median 0.48, IQR 0.35–0.58vs0.36, IQR 0.23–0.48, respectively, p<0.05). Finally, OGT levels were associated with a higher probability to clinical worsening (Figure 5F), defined as hospitalization for PAH, lung transplantation, or death (3.71, 95% CI 1.05 – 13.2; for an OGT/Beta-actin ratio greater than or equal to 0.396; p = 0.043 for OGT in its continuous form). These data suggest that OGT may be a useful marker of disease severity in PAH.
Figure 5.
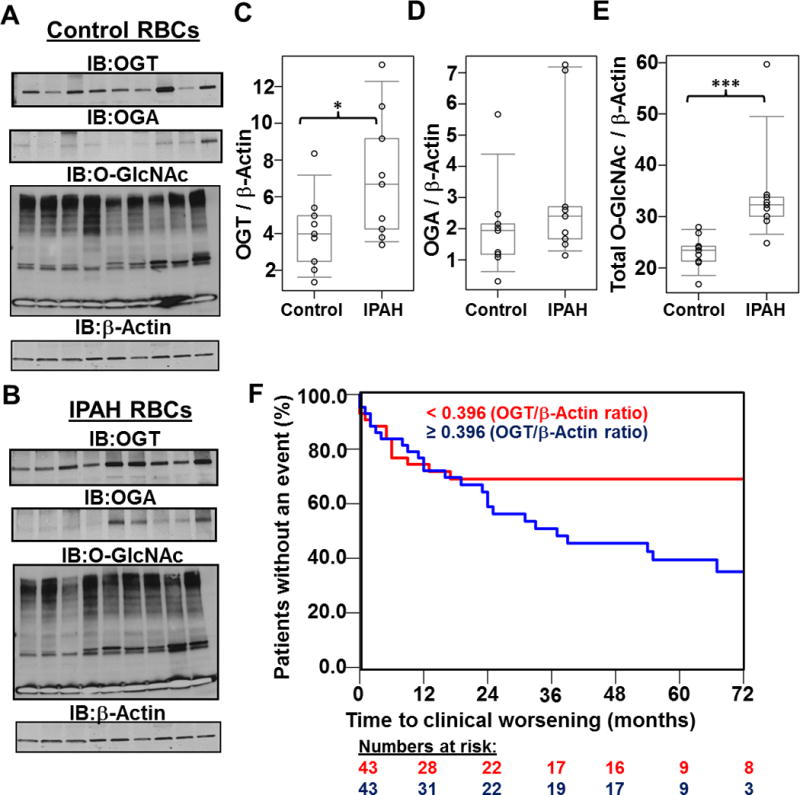
Analysis of multiple PAH patient RBCs identifies OGT as a potential marker for clinical worsening in PAH. Levels of OGT, OGA, and O-GlcNAc were determined in control (A) and PAH (B) samples (n=9) and subsequently quantitated (C–E). OGT: control, 4.1 ± 2.1; PAH, 7.0 ± 3.4, p = 0.04. OGA: control, 2.0 ± 1.5; IPAH, 2.8 ± 1.9, p = 0.25. O-GlcNAc: control, 23.1 ± 3.3; PAH, 34.3 ± 10.6, p = 0.001. The p-values were determined using a Wilcoxon test (see Material and Methods; *, p<0.05 and **, p<0.01). (F) Higher OGT levels determined from PAH RBCs (n=86) associate with poor clinical outcomes. Kaplan-Meier curves show time to clinical worsening according to the median OGT/β-actin cut off of 0.396, which was associated with a hazard radio of 3.71, 95% CI 1.05 – 13.2 (based on OGT in its continuous form, p=0.043).
DISCUSSION
IPAH is a rapidly progressive cardiopulmonary disease characterized by vasoconstriction and aberrant vascular cell proliferation. Current therapies mainly target vasoconstriction. The future of IPAH therapy, however, depends on our ability to identify and target the molecular underpinnings of cell proliferation in this devastating disease. In this report, we demonstrate that PASMC proliferation, a major component in the pathobiology of IPAH, is directed by increased HBP flux coupled with enhanced OGT activity, causing proteolytic activation of HCF-1. To our knowledge, this is the first description of altered HBP flux in IPAH resulting in global O-GlcNAc modification of proteins and direct activation of HCF-1 by OGT.
IPAH shares several disease signatures with cancer, including cell proliferation and dysregulated glucose metabolism and utilization31. These changes in energy production and metabolic dysregulation are hallmarks of hyperproliferative cells. These cells disproportionately utilize metabolites (such as glucose and glutamine) to survive in energy-starved environments. In 1924, Otto Warburg found that highly proliferative cancer cells metabolize glucose via the less energy efficient glycolysis pathway despite the presence of oxygen32. For almost a century, scientists have speculated on the reason for this phenomenon. The current consensus is that glycolysis provides not only energy, but also the building blocks required for cell proliferation33.
Once the proliferating cells have switched to aerobic glycolysis, they produce less ATP than cells utilizing glucose oxidation and, in turn, more rapidly take up glucose when sufficient levels are available34. The extensive energy requirements of highly proliferative cells often result in excess utilization of metabolites (namely glucose, glutamine, acetyl-CoA, and uridine triphosphate) through multiple pathways, including the HBP. Our studies are in agreement with this notion and suggest that the increased HBP flux observed in IPAH patients (Figure 1–2) is an effect of the metabolic switch to aerobic glycolysis observed in proliferating cells. Studies in cancer cells have demonstrated a similar role of HBP flux, OGT, and hyper O-GlcNAc modification of proteins involved in cancer cell proliferation30, 35, 36. Our data (Figure 3) demonstrates a direct role for OGT in IPAH PASMC proliferation, which corresponds with the cancer cell reports30, 35, 36.
As a nutrient sensor of the cell, the HBP9, 10, 37 integrates the metabolism of carbohydrates, amino acids, fat, and nucleotides for the synthesis of UDP-GlcNAc, the final product of the pathway. Recently, Wellen et al. demonstrated that treatment of glucose-starved cells with GlcNAc (to maintain hexosamine biosynthesis) rescued cell growth, indicating the importance of the HBP in cell proliferation38. The effect of the HBP on cell proliferation is most likely mediated by OGT, which utilizes UDP-GlcNAc for the O-GlcNAc modification of proteins. This finding is consistent with the observed low levels of UDP-GlcNAc determined in the IPAH patients (Figure 1) in our study despite the increased HBP flux and GFAT1 protein expression.
Modulating O-GlcNAc levels has already been shown in cancer cells to contribute to cancer cell survival, proliferation, and metastasis30, 39. OGT, through protein O-GlcNAc modification, can control many cellular events including cell cycle12, transcription40, signal transduction41, nutrient sensing10, and cell stress responses42. Interestingly, OGT can also effect cell proliferation through a completely different and distinct mechanism. Herr and colleagues recently demonstrated that OGT specifically cleaves the precursor form of the cell cycle master regulator, HCF-118, 19. Upon cleavage by OGT, the N-terminal and C-terminal subunits of HCF-1 are functionally activated18 and facilitate G1 phase cell cycle progression43 as well as mitosis and cytokinesis44. In line with these findings, our data suggests that cleavage of HCF-1 is directly facilitated by OGT in IPAH (Figure 4) and represents the first evidence for this phenomenon in a disease other than cancer. Moreover, this result underscores a more direct involvement of OGT in the cell proliferation process. While the HBP and O-GlcNAc have been implicated in pathogenesis of other non-cancerous diseases including heart disease45–47, type II diabetes41, 48–51, and insulin resistance37, 52. In addition, increased OGT levels have been shown to associate with poor outcomes and disease persistence in prostate cancer patients29, 30, suggesting that the increased O-GlcNAc protein modification contributes to the disease progression. However, these findings have not been previously reported in IPAH. We found increased HBP flux, elevated OGT, and global O-GlcNAc modification of proteins in IPAH. In particular, we observed increased OGT and O-GlcNAc levels in RBCs PAH patients (Figure 5), which is similar to the results from the IPAH lung (Figure 1). Also, we showed that increased OGT levels in RBCs associate with poor PAH clinical outcomes (Figure 5). This data suggests that OGT levels from RBCs may be useful to gauge the severity of PAH. More importantly, the altered OGT and O-GlcNAc levels observed in PAH RBCs and lung suggests that the metabolic changes may be a systemic phenomenon. Others have reported glucose intolerance, insulin resistance, and metabolic dysregulation in PAH2, 6, 53–55 consistent with the notion that IPAH patients may have systemic metabolic derangements. In one report, Pugh et al. demonstrated that higher hemoglobin A1c levels correlated with the 6-minute walk test for IPAH patients6. Thus, it is possible that our findings may be related to or a consequence of the well-documented dysregulation of glucose metabolism in this disease. Still, more research is needed to determine the systemic nature of the dysregulated metabolism in IPAH.
Fundamentally, this report demonstrates OGT as: (i) an activator of HCF-1 and (ii) a regulator of cell proliferation in IPAH. These findings in IPAH, along with published findings in cancer, collectively demonstrate a direct role for OGT in linking nutrient sensing of the HBP to cell cycle progression and proliferation, which may have clinical implications for a wide array of diseases beyond IPAH including cardiovascular disease, cancer, and diabetes.
Combining the previous reports on the characterization of HA in IPAH20, 22 along with the observations described here in this report, we put forth a model whereby an activated HBP regulates PASMC proliferation in IPAH as outlined in Figure 6. This model establishes a regulatory role for OGT in IPAH, sheds a new light on our understanding of the disease pathobiology, and provides opportunities to design novel therapeutic strategies for IPAH.
Figure 6.
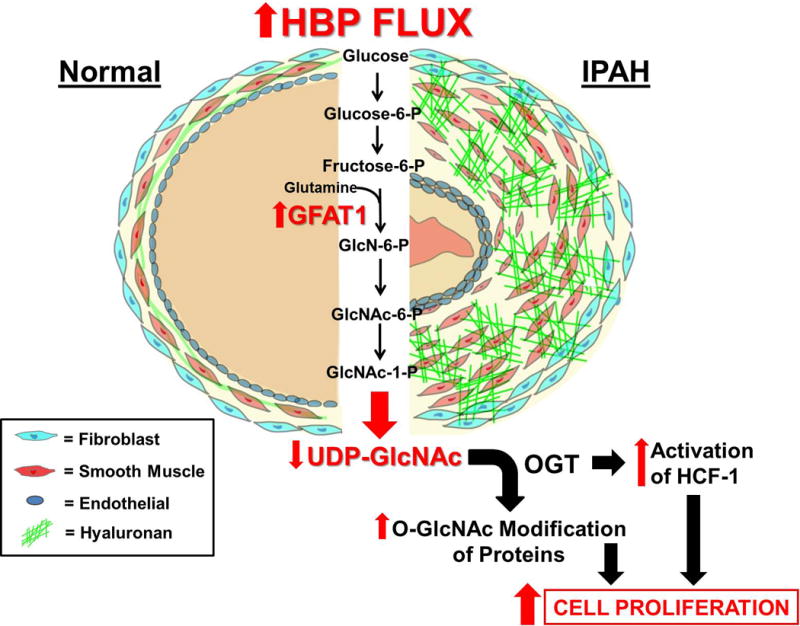
A Model illustrating the functional consequences of excessive HBP Flux in IPAH. In IPAH, increased HBP flux leads to increased UDP-GlcNAc production that is vastly utilized by OGT for the O-GlcNAc modification of proteins and for the synthesis of HA. Both of these changes have been demonstrated in the cell proliferation process. As shown here, the consumption of UDP-GlcNAc by OGT facilitates enhanced O-GlcNAc modification of proteins and HCF-1 activation, resulting in an increase in vascular cell proliferation, including PASMCs. Abbreviations: P = Phosphate, GlcN = Glucosamine
Supplementary Material
Acknowledgments
We wish to thank Natasha Zachara (NHLBI P01 HL107153 CoreC4) for providing the O-GlcNAc (CTD 110.6), OGT, and OGA antibodies, The Cleveland Clinic Programs of Excellence in Glycoscience (CCPEG) Resource Core for their technical help with the FACE UDP-Sugar analysis and the Cleveland Clinic Imaging and Histology Cores for the use of the microscopes and services for histology experiments. Competing interests: There are no competing interests for any of the authors. Tissue samples were provided by Baylor, Stanford, Vanderbilt, University of Alabama, and Allegany College of Maryland under the Pulmonary Hypertension Breakthrough Initiative (PHBI). Funding for the PHBI is provided by the Cardiovascular Medical Research and Education Fund (CMREF).
Funding Sources: This project was supported by the following grants from the National Institutes of Health: the Ruth L Kirschstein F32 postdoctoral fellowship (F32HL120629 to J.B.) and the Programs of Excellence in Glycosciences (1P01HL10714 to R.A.D.) both from NHLBI. In addition, the explanted lungs tissue and pulmonary artery smooth muscle cell isolation was supported by the translational Program Project Grant and Program Project Grant P01HL103453 and P01HL081064, respectively (both to S.A.A.C and R.A.D). Kewal Asosingh is a Scholar of the International Society for Advancement of Cytometry.
Footnotes
Disclosures: None.
References
- 1.Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J. 2012;40:1555–65. doi: 10.1183/09031936.00046612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. 2014;115:148–64. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 3.Tuder RM, Archer SL, Dorfmuller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D4–12. doi: 10.1016/j.jacc.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115:189–202. doi: 10.1161/CIRCRESAHA.115.303404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, Chen CT, Archer SL. Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:670–9. doi: 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pugh ME, Robbins IM, Rice TW, West J, Newman JH, Hemnes AR. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J Heart Lung Transplant. 2011;30:904–11. doi: 10.1016/j.healun.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104:1342–7. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005;2005:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- 9.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. 1991;266:4706–12. [PubMed] [Google Scholar]
- 10.Wells L, Vosseller K, Hart GW. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell Mol Life Sci. 2003;60:222–8. doi: 10.1007/s000180300017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varki A. Essentials of glycobiology. (2) [Google Scholar]
- 12.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–22. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 13.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–58. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–15. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- 15.Lubas WA, Frank DW, Krause M, Hanover JA. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 1997;272:9316–24. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–45. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 17.Wells L, Gao Y, Mahoney JA, Vosseller K, Chen C, Rosen A, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J Biol Chem. 2002;277:1755–61. doi: 10.1074/jbc.m109656200. [DOI] [PubMed] [Google Scholar]
- 18.Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell. 2011;144:376–88. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 19.Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science. 2013;342:1235–9. doi: 10.1126/science.1243990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lauer ME, Aytekin M, Comhair SA, Loftis J, Tian L, Farver CF, Hascall VC, Dweik RA. Modification of hyaluronan by heavy chains of inter-alpha-inhibitor in idiopathic pulmonary arterial hypertension. J Biol Chem. 2014;289:6791–8. doi: 10.1074/jbc.M113.512491. Epub 2014 Jan 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Comhair SA, Xu W, Mavrakis L, Aldred MA, Asosingh K, Erzurum SC. Human primary lung endothelial cells in culture. Am J Respir Cell Mol Biol. 2012;46:723–30. doi: 10.1165/rcmb.2011-0416TE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aytekin M, Comhair SA, de la Motte C, Bandyopadhyay SK, Farver CF, Hascall VC, Erzurum SC, Dweik RA. High levels of hyaluronan in idiopathic pulmonary arterial hypertension. Am J Physiol. 2008;295:L789–99. doi: 10.1152/ajplung.90306.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 24.Park K, Saudek CD, Hart GW. Increased expression of beta-N-acetylglucosaminidase in erythrocytes from individuals with pre-diabetes and diabetes. Diabetes. 2010;59:1845–50. doi: 10.2337/db09-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rehman J, Archer SL. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the Warburg model of pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:171–85. doi: 10.1007/978-1-60761-500-2_11. [DOI] [PubMed] [Google Scholar]
- 27.Lundgrin EL, Park MM, Sharp J, Tang WH, Thomas JD, Asosingh K, Comhair SA, DiFilippo FP, Neumann DR, Davis L, Graham BB, Tuder RM, Dostanic I, Erzurum SC. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc. 2013;10:1–9. doi: 10.1513/AnnalsATS.201206-029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rufino M, Hernandez D, Barrios Y, Salido E. The GLUT-1 XbaI gene polymorphism is associated with vascular calcifications in nondiabetic uremic patients. Nephron Clin Pract. 2008;108:c182–7. doi: 10.1159/000118940. [DOI] [PubMed] [Google Scholar]
- 29.Kamigaito T, Okaneya T, Kawakubo M, Shimojo H, Nishizawa O, Nakayama J. Overexpression of O-GlcNAc by prostate cancer cells is significantly associated with poor prognosis of patients. Prostate Cancer Prostatic Dis. 2014;17:18–22. doi: 10.1038/pcan.2013.56. [DOI] [PubMed] [Google Scholar]
- 30.Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem. 2012;287:11070–81. doi: 10.1074/jbc.M111.302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrell NW, Archer SL, Defelice A, Evans S, Fiszman M, Martin T, Saulnier M, Rabinovitch M, Schermuly R, Stewart D, Truebel H, Walker G, Stenmark KR. Anticipated classes of new medications and molecular targets for pulmonary arterial hypertension. Pulm Circ. 2013;3:226–44. doi: 10.4103/2045-8932.109940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 33.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 34.Diaz-Ruiz R, Rigoulet M, Devin A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta. 2011;1807:568–76. doi: 10.1016/j.bbabio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem. 2013;288:15121–30. doi: 10.1074/jbc.M113.470047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taniguchi N. A sugar-coated switch for cellular growth and arrest. Nat Chem Biol. 2007;3:307–9. doi: 10.1038/nchembio0607-307. [DOI] [PubMed] [Google Scholar]
- 37.Einstein FH, Fishman S, Bauman J, Thompson RF, Huffman DM, Atzmon G, Barzilai N, Muzumdar RH. Enhanced activation of a “nutrient-sensing” pathway with age contributes to insulin resistance. FASEB J. 2008;22:3450–7. doi: 10.1096/fj.08-109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24:2784–99. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaidyanathan K, Durning S, Wells L. Functional O-GlcNAc modifications: Implications in molecular regulation and pathophysiology. Crit Rev Biochem Mol Biol. 2014;49:140–63. doi: 10.3109/10409238.2014.884535. Epub 2014 Feb 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou TY, Dang CV, Hart GW. Glycosylation of the c-Myc transactivation domain. Proc Natl Acad Sci U S A. 1995;92:4417–21. doi: 10.1073/pnas.92.10.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 2002;99:5313–8. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–42. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- 43.Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27:107–19. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 44.Julien E, Herr W. Proteolytic processing is necessary to separate and ensure proper cell growth and cytokinesis functions of HCF-1. EMBO J. 2003;22:2360–9. doi: 10.1093/emboj/cdg242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–6. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jensen RV, Zachara NE, Nielsen PH, Kimose HH, Kristiansen SB, Botker HE. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc Res. 2013;97:369–78. doi: 10.1093/cvr/cvs337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLarty JL, Marsh SA, Chatham JC. Post-translational protein modification by O-linked N-acetyl-glucosamine: its role in mediating the adverse effects of diabetes on the heart. Life Sci. 2013;92:621–7. doi: 10.1016/j.lfs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010;36:423–35. doi: 10.1016/j.diabet.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 49.Dentin R, Hedrick S, Xie J, Yates J, 3rd, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science. 2008;319:1402–5. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- 50.Ball LE, Berkaw MN, Buse MG. Identification of the major site of O-linked beta-N-acetylglucosamine modification in the C terminus of insulin receptor substrate-1. Mol Cell Proteom. 2006;5:313–23. doi: 10.1074/mcp.M500314-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McClain DA, Lubas WA, Cooksey RC, Hazel M, Parker GJ, Love DC, Hanover JA. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci U S A. 2002;99:10695–9. doi: 10.1073/pnas.152346899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta. 2010;1800:80–95. doi: 10.1016/j.bbagen.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, Rabinovitch M, Doyle RL. Insulin resistance in pulmonary arterial hypertension. Eur Respir J. 2009;33:318–24. doi: 10.1183/09031936.00000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brunner NW, Skhiri M, Fortenko O, Hsi A, Haddad F, Khazeni N, Zamanian RT. Impact of insulin resistance on ventricular function in pulmonary arterial hypertension. J Heart Lung Transplant. 2014;33:721–6. doi: 10.1016/j.healun.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 55.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115:1275–84. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.