

Abstract
Perturbation of intracellular ion homeostasis is a major cellular stress signal for activation of NLRP3 inflammasome signaling that results in caspase-1 mediated production of IL-1β and pyroptosis. However, the relative contributions of decreased cytosolic [K+] versus increased cytosolic [Ca2+] remain disputed and incompletely defined. We investigated roles for elevated cytosolic [Ca2+] in NLRP3 activation and downstream inflammasome signaling responses in primary murine dendritic cells and macrophages in response to two canonical NLRP3 agonists (ATP and nigericin) that facilitate primary K+ efflux by mechanistically distinct pathways or the lysosome-destabilizing agonist Leu-Leu-O-methyl ester (LLME). The study provides three major findings relevant to this unresolved area of NLRP3 regulation. First, increased cytosolic [Ca2+] was neither a necessary nor sufficient signal for the NLRP3 inflammasome cascade during activation by endogenous ATP-gated P2X7 receptor channels, the exogenous bacterial ionophore nigericin, or the lysosomotropic agent LLME. Second, agonists for three Ca2+-mobilizing G protein-coupled receptors (formyl peptide receptor/FPR; P2Y2 purinergic receptor/P2Y2R; calcium-sensing receptor/CaSR) expressed in murine dendritic cells were ineffective as activators of rapidly induced NLRP3 signaling when directly compared to the K+ efflux agonists. Third, the intracellular Ca2+ buffer, BAPTA, and the channel blocker, 2-aminoethoxydiphenyl borate (2-APB), widely used reagents for disruption of Ca2+-dependent signaling pathways, strongly suppressed nigericin-induced NLRP3 inflammasome signaling via mechanisms dissociated from their canonical or expected effects on Ca2+ homeostasis. The results indicate that the ability of K+ efflux agonists to activate NLRP3 inflammasome signaling can be dissociated from changes in cytosolic [Ca2+] as a necessary or sufficient signal.
INTRODUCTION
IL-1β is a primary pro-inflammatory cytokine that activates the acute phase response, induces fever, promotes proliferation of neutrophils in the bone marrow and stimulates adherence of leukocytes to the walls of blood vessels1. Production of biologically active IL-1β requires its proteolytic processing (maturation) by caspase-1. Caspase-1 activation per se involves autocatalytic processing regulated by assembly of inflammasome signaling complexes2. One major type of inflammasome consists of the cytosolic pattern recognition receptor NLRP3, the adaptor protein ASC, and procaspase-12,3. Upon activation, NLRP3 monomers oligomerize into a ring-like structure that recruits ASC monomers to induce formation of ASC filaments or specks4,5. In turn, ASC filaments/specks recruit multiple pro-caspase-1 monomers to facilitate the induced proximity required for autocatalytic proteolysis into the 10- and 20 kDa subunits that assemble into the highly active tetramers of caspase-1. Active caspase-1 mediates both the proteolytic maturation of IL-1β and its release via non-canonical secretion2,3. Caspase-1 also induces pyroptosis, a regulated cell death pathway characterized by the permeabilization of the plasma membrane that facilitates collapse of ionic and osmotic homeostasis and eventual cell lysis6.
The assembly of NLRP3 inflammasomes can be activated by various exogenous stimuli including extracellular ATP (via opening of P2X7 nonselective cation channel receptors), small microbial ionophores such as nigericin or gramicidin, and large bacterial pore-forming protein toxins7,8. All of these NLRP3-activating stimuli directly perturb the permeability of the plasma membrane to K+ with consequent reduction in cytosolic [K+]. Multiple studies have identified decreased cytosolic [K+] as a necessary signal for the induction of NLRP3 inflammasome assembly by those stimuli that directly target plasma membrane permeability9. Importantly, Munoz-Planillo et al. reported that the ability of particulate stimuli, such as monosodium urate, silica, and alum, or small molecule lysosomotropic molecules, such as Leu-Leu-O-methyl ester (LLME), to activate NLRP3 inflammasomes secondary to lysosomal destabilization also requires efflux of K+ 10. However, it remains unclear whether decreased cytosolic [K+] is a sufficient ionic signal in addition to being a necessary signal. In this regard, other studies have implicated elevations in cytosolic [Ca2+] in the NLRP3 inflammasome activation response to several stimuli, including nigericin and ATP-gated P2X7 receptor channels. Several reports indicated that caspase-1 activation and IL-1β release is suppressed in macrophages loaded with BAPTA, a chelator of cytosolic Ca2+ 11,12. Murakami et al. found that NLRP3 inflammasome signaling in response to nigericin and ATP was markedly attenuated by inhibitors of phospholipase C (U73122), IP3-gated Ca2+ release channels (xestospongin C), or store-operated Ca2+ entry (2-aminoethoxydiphenyl borate, 2-APB)13. Finally, agonists for the calcium-sensing GPCR (CaSR) and C5a complement GPCR, which stimulate phospholipase C-mediated increases in cytosolic [Ca2+], have been reported to induce NLRP3-inflammasome dependent IL-1β release from murine or human monocyte/macrophages14–16.
These observations suggest that increased cytosolic [Ca2+] may activate NLRP3 inflammasome signaling either independently of, or synergistically with, decreased cytosolic [K+]. However, no previous studies have directly measured changes in both cytosolic [Ca2+] and [K+] in myeloid leukocytes under the experimental conditions routinely used to interrogate key reactions of the NLRP3 inflammasome signaling cascade. Moreover, the pharmacologic tools for investigating the role of Ca2+ signaling in inflammasome activation can affect the homeostasis of other divalent cations, e.g. Zn2+, or the activity of non-selective channels permeable to both divalent and monovalent cations17–18. In this study, we investigated the role of elevated cytosolic [Ca2+] in NLRP3 activation in murine macrophages and dendritic cells with minimal use of pharmacologic inhibitors and by directly assaying changes in cytosolic [Ca2+] in response to two canonical NLRP3 agonists (ATP or nigericin) that facilitate K+ efflux by mechanistically distinct reactions (Fig. 1A)19,20. Other experiments evaluated whether changes in cytosolic [Ca2+] signaling modulate NLRP3 inflammasome activation by lysosomal destabilization. We assessed possible effects of increased cytosolic [Ca2+] on several discrete phases of the NLRP3 inflammasome signaling cascade: formation of ASC oligomers, processing/release of caspase-1, processing/release of IL-1β, and kinetics of caspase-1 mediated pyroptosis. Our results indicate that the ability of K+ efflux agonists to activate NLRP3 signaling can be dissociated from changes in cytosolic [Ca2+]. Moreover, nigericin-stimulated increases in cytosolic [Ca2+] were temporally correlated with the onset and kinetics of pyroptosis and were absent in DCs isolated from Casp1/11−/−or Nlrp3−/− mice.
Figure 1. Nigericin-induced increases in cytosolic [Ca2+] in LPS-primed BMDC occur downstream of the NLRP3 inflammasome/ caspase-1/ pyroptotic signaling cascade.

A, Diagram of changes in cation homeostasis occurring in myeloid cells in response to treatment with nigericin and ATP/P2X7. B, IL-1β release from LPS-primed WT, Nlrp3−/−, and Casp-1/11−/− BMDC in response to 10μM nigericin and 5mM ATP was measured by ELISA. Data represent mean of 2 independent experiments. C, LPS-primed WT, Nlrp3−/−, and Casp-1/11−/− BMDC were stimulated with 10μM nigericin or 5mM ATP for 30 min. BMDC were lysed with 10% nitric acid and lysates were analyzed by atomic absorption spectroscopy to measure cellular [K+]. Data represent mean of three independent experiments. D–F, WT, Nlrp3−/−, or Casp-1/11−/− BMDC were primed with LPS (1μg/ml) for 4h and loaded with 1μg/ml fluo-4 AM for 30 min. Baseline readings were taken for 5 min and 10μM nigericin (NG) or 5mM ATP were added at t=5 min. Cytosolic [Ca2+] was determined by measuring fluo-4 fluorescence. Data represent a mean of two independent experiments. G–I, LPS-primed WT, Nlrp3−/−, and Casp-1/11−/−BMDC were stimulated with 10μM nigericin or 5mM ATP for 30 min. Onset of pyroptosis was determined by measuring permeability of the cell membrane to propidium2+. Baseline readings were taken for 5 min and nigericin or ATP were added at t=5 min. Data represent a mean of two independent experiments.
MATERIALS AND METHODS
Reagents
Key reagents and their sources were as follows: Escherichia coli LPS serotype O1101:B4 (List Biological Laboratories), Pam3CSK4 (Invivogen), murine rTNFα (Peprotech), murine rMCSF (Peprotech), nigericin (Sigma Aldrich), ATP (Sigma Aldrich), ionomycin (LC Laboratories), N-formyl-Met-Leu-Phe (Sigma Aldrich), UTP (Sigma Aldrich), H-Leu-Leu-OMe·HBr (Bachem), Lidocaine (Sigma Aldrich), R568 (Tocris), thapsigargin (LC Laboratories), disuccinimidyl suberate (Sigma Aldrich), BAPTA-AM (Molecular Probes), 2-APB (Tocris), anti-caspase-1 (P20) mouse monoclonal antibody (Casper-1) (Adipogen), Anti-ASC rabbit polyclonal antibody (N-15) and all HRP-conjugated secondary antibodies (Santa Cruz Biotechnology), murine IL-1β ELISA kit (R&D Systems), Fluo-4 AM (Life Technologies), Pluronic F-127 (Molecular Probes), probenecid (Sigma Aldrich), Propidium iodide (Life Technologies), Cytotoxicity Detection Kit-LDH (Roche). Anti-IL-1β mouse monoclonal antibody was provided by the Biological Resources Branch, National Cancer Institute, Frederick Cancer Research and Development Center (Frederick, MD).
Murine Models
WT C57BL/6 mice were purchased from Taconic. Mice lacking both caspase-1 and caspase-11 on a C57BL/6 background (Casp1/11−/−) have been previously described21. Nlrp3−/− mice were provided by Dr. A. Hise (Case Western Reserve University). All experiments and procedures involving mice were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University. Bone marrow-derived dendritic cells (BMDC) and bone marrow-derived macrophages (BMDM) were isolated from 9–12 week old mice. Mice were euthanized by CO2 inhalation. Femurs and tibiae were removed and briefly sterilized in 10% ethanol. PBS was used to wash out the marrow plugs. Bone marrow cells were resuspended in DMEM (Sigma-Aldrich) supplemented with 10% bovine calf serum (HyClone Laboratories), 100U/ml penicillin, 100μg/ml streptomycin (Invitrogen), 2mM L-glutamine (Lonza) and either 4% J558L cell-conditioned medium (which contains the GM-CSF necessary for BMDC differentiation) or 20ng/ml recombinant M-CSF (necessary for BMDM differentiation). Bone marrow cells were plated onto 150mm plastic petri dishes and cultured in the presence of 10% CO2.
For BMDC, the nonadherent cell population was removed on day 3 post isolation, centrifuged at 300xg for 5 min at room temperature, resuspended in fresh GM-CSF supplemented medium, and replated onto a 150mm petri dish. At 5 days post isolation, the loosely adherent BMDC were collected, pelleted by centrifugation (300xg, 5 min), and resuspended to 1x106 cells/ml in fresh growth medium and plated in 6-well (2ml/well) tissue culture plates for western blot assays, 12-well (1ml/well) plates for K+ atomic absorption assays, and 24-well (0.5ml/well) plates for IL-1β ELISA, fluo-4 assays of cytosolic [Ca2+] changes, and propidium2+ influx assays. All assays of BMDC function were performed between days 7 and 10 post-isolation. For BMDM, 33% of the initial volume of growth medium was removed on day 3 post-isolation and replaced with fresh DMEM containing 20ng/ml recombinant M-CSF. At 7 days post-isolation, the adherent BMDM were detached using PBS supplemented with 5mM EGTA and 4mg/ml lidocaine, pelleted by centrifugation (300xg, 5 min), and resuspended to 1x106 cells/ml in fresh growth medium described above. The BMDM were then plated into 6-, 12-, or 24-well plates as described for the BMDC.
Priming and Stimulation of BMDC and BMDM
Prior to experimental treatments, BMDC-containing TC plates were centrifuged at 300xg for 5 min to prevent loss of the loosely adherent cells; this step was not required for the highly adherent BMDM. The growth/ differentiation medium was removed from BMDC or BMDM-containing plates and replaced with DMEM (10% bovine calf serum, penicillin, streptomycin and L-glutamine) supplemented with either 1μg/ml LPS, 2μg/ml Pam3CSK4 or 100ng/ml TNFα as “signal 1”priming stimuli to induce NFκB-dependent upregulation of pro-IL-1β and NLRP3 expression. The cells were treated with these priming stimuli for 4h at 37°C. Plates with primed BMDC were again centrifuged at 300xg for 5 min before further manipulation. For either BMDC or BMDM cultures, the priming medium was aspirated after 4h and replaced with either Ca2+-containing balanced salt solution (BSS) (130mM NaCl, 4mM KCl, 1.5mM CaCl2, 1mM MgCl2, 25mM NaHEPES, 5mM D-glucose, 0.1% BSA, pH 7.4) Ca2+-free BSS (130mM NaCl, 4mM KCl, 300μM EGTA, 1mM MgCl2, 25mM NaHEPES, 5mM D-glucose, 0.1% BSA, pH 7.4) or high K+ BSS (134mM KCl, 1.5mM CaCl2, 1mM MgCl2, 25mM NaHEPES, 5mM D-glucose, 0.1% BSA, pH 7.4). BMDC and BMDM in BSS were pre-incubated for 5 min at 37°C and then stimulated with 10μM nigericin, 5mM ATP, 1 mM Leu-Leu-O-methyl-ester (LLME) or indicated concentrations of other Ca2+ mobilizing agents for 30 min. Where indicated, signal 1 primed- BMDC and BMDM were treated for 30 min with 300 nM thapsigargin in Ca2+ free BSS to deplete endoplasmic reticulum Ca2+ stores and transferred to either fresh Ca2+-free BSS or Ca2+-containing BSS prior to stimulation with nigericin or ATP.
ELISA Analysis of IL-1β Release
Signal 1-primed BMDC or BMDM in 24 well plates (5x105 cells/0.5 ml BSS/well) were stimulated with nigericin, ATP, LLME, or other Ca2+-mobilizing agents at 37°C. After 30 min, extracellular medium was removed from each well and centrifuged at 10,000xg for 15 sec to pellet detached cells. The cell-free supernatants were then assayed for murine IL-1β by sandwich ELISA (R&D Systems) according to the manufacturer protocol.
Western Blot Analysis of Caspase-1 Processing/Release and IL-1β Processing/Release
Signal 1-primed BMDC in 6 well plates (2x106 cells/1 ml BSS/well) were stimulated with nigericin, ATP, or other Ca2+-mobilizing agents at 37°C. After 30 min, the extracellular media was removed from each well and briefly centrifuged to pellet detached cells. The cell-free supernatant was transferred to a new tube, the extracellular proteins were concentrated by trichloroacetic acid precipitation, and the precipitated proteins processed for SDS-PAGE as previously described22. Any detached cells in the pellet from each extracellular medium sample were dissolved in RIPA buffer and added back to the original well containing the adherent BMDC to generate whole cell lysate samples as described previously22. Cell lysates and matching extracellular medium samples were subjected to SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane for western blot analysis. Primary antibodies were used at the following concentrations: 1μg/ml for caspase-1, 5μg/ml for IL-1β, and 0.4μg/ml for ASC. HRP-conjugated secondary antibodies were used at a concentration of 0.13μg/ml. Chemiluminescent images of western blots were developed and saved using a FluorChemE image processor (Cell Biosciences).
Assay of ASC Oligomerization Using DSS-Crosslinked Detergent Insoluble Lysate Fractions
After stimulation, the extracellular medium from each well was removed, centrifuged at 10,000xg for 15 sec to pellet any detached BMDC, and the cell-free supernatant was aspirated. The adherent BMDC in each well were washed with 1ml ice-cold PBS prior to preparation of whole cell detergent lysates by addition of 150μL of lysis buffer (0.5% sodium deoxycholate, 0.1% SDS, 1% Igepal CA-630 in PBS [pH 7.4], plus protease inhibitor mixture), scraping with a rubber policeman to fully detach the cells, and incubation on ice for 5 min. This cell lysate was pooled with the detached cell pellet, and the resulting whole cell lysate was incubated for an additional 15 min on ice. The cell lysates were then separated into detergent-soluble and detergent-insoluble fractions by centrifugation at 15,000xg for 15 min at 4°C. SDS sample buffer was added to detergent-soluble fractions for extraction at 100°C for 5 min. The detergent-insoluble lysate pellet was washed 2X with 200μL of ice-cold PBS and then suspended in 200 μl of PBS containing 2mM disuccinimidyl suberate (DSS) (from a stock 20mM DSS solution in DMSO). The resuspended detergent-insoluble fractions were incubated with DSS for 30 min at room temperature, re-pelleted by centrifugation at 8,000xg for 15 min at room temperature, and the DSS supernatant solution was removed. The DSS-treated pellets were suspended in SDS-PAGE buffer and extracted at 100°C for 5 min. The DSS-treated fractions were resolved by SDS-PAGE, transferred to PVDF, and analyzed by anti-ASC western blotting.
Atomic Absorption Spectroscopy for Measurement of K+ Efflux
Signal 1-primed BMDC in 12 well plates (106 cells/1 ml BSS/ well) were stimulated for 30 min with nigericin, ATP, LLME, or other Ca2+-mobilizing agents at 37°C. After 30 min, the extracellular medium was removed and centrifuged at 10,00xg for 15 sec to pellet detached cells. Adherent cells were briefly washed with K+-free balanced salt solution (135mM sodium gluconate, 1.5mM CaCl2, 1mM MgCl2, 25mM HEPES, pH 7.4) to remove any residual K+ from BSS treatment medium. The wash solution from each well was used to resuspend the detached cell pellet from that well and this suspension was re-centrifuged at 10,000xg for 15 sec to re-pellet the cells. 1ml of 10% nitric acid was used to resuspend the detached cell pellet and this was then added back to the corresponding well containing the adherent BMDC. This 1 ml nitric acid extract was supplemented with an additional 1ml of 10% nitric acid and the BMDC were extracted into the resulting 2 ml nitric acid volume for 2h to ensure adequate extraction of cellular K+ content. The nitric acid lysates were then transferred into 2ml microcentrifuge tubes and the amount of elemental potassium in each lysate was quantified using a 50AA Atomic Absorption Spectrometer (Agilent Technologies).
Propidium2+ Influx Assay of Pyroptotic Plasma Membrane Permeabilization
Signal 1-primed BMDC or BMDM in 24 well plates (5x105 cells/well) were briefly washed with PBS prior to the addition of 0.5 ml BSS (Ca2+-containing or Ca2+-free) supplemented with 2μg/ml propidium iodide to each well. The plate was placed into a Synergy HT plate reader (BioTek) preheated to 37°C. Baseline fluorescence (540 nm excitation → 620 nm emission at 30 sec intervals) was recorded for 5 min. Cells were then stimulated with 10μM nigericin, 5mM ATP, 1 mM LLME, or 3μM ionomycin for 30 min and changes in 540ex → 620em fluorescence were recorded at 30 sec intervals. Assays were terminated by permeabilization of the BMDC with 1% triton X-100 to quantify maximum fluorescence of the propidium2+/DNA complexes. Where indicated, cells were treated with 300 nM thapsigargin (TG) in Ca2+-free BSS for 30 min to deplete ER Ca2+ stores prior to addition of nigericin or ATP. The agonist-induced increases in fluorescence at each time point were normalized as a percentage of the maximum fluorescence measured after complete permeabilization of all BMDC/ BMDM within the well with 1% triton X-100.
Fluo-4 –Based Assay of Cytosolic [Ca2+]
Signal 1-primed BMDC or BMDM in 24 well plates (5x105 cells/well) were briefly washed with PBS, prior to the addition of 0.5 ml BSS supplemented with 1μM fluo-4 AM, Pluronic F-127 (premixed with fluo-4 AM in 1:1 proportion by volume), and 2.5mM probenecid per well. After incubation at 37°C for 45 min, each well was briefly washed with PBS prior to the addition of a fresh 0.5 ml aliquot of BSS (either Ca2+-containing or Ca2+-free) supplemented with 2.5mM probenecid. The plate was placed into the Synergy HT reader preheated to 37°C. Baseline fluorescence (485 nm excitation → 528 nm emission at 30 sec intervals) was recorded for 10 min. Cells were then stimulated with 10μM nigericin, 5mM ATP, 1 mM LLME, or indicated concentrations of Ca2+ mobilizing agents for 30 min and changes in 485ex → 528em fluorescence were recorded at 30 sec intervals. Where indicated, cells were treated with 300nM thapsigargin (TG) in Ca2+-free BSS for 30 min to deplete ER Ca2+-stores prior to addition of nigericin or ATP. Assays were terminated by permeabilization of the cells with 1% triton X-100 in the presence of excess CaCl2 to quantify the maximum Ca2+-dependent fluorescence (Fmax) of the fluo-4 indicator dye. The wells were then supplemented with 15mM EGTA/50mM Tris to chelate Ca2+ and quantify the minimum Ca2+-independent fluorescence of fluo-4 (Fmin). The Fmax and Fmin values were used to calculate the cytosolic [Ca2+] corresponding to changes in 485ex → 528em fluorescence of fluo-4 within intact cells as described by Tsien et al23.
Assay of Cytotoxicity (LDH Release)
Signal 1-primed BMDC in 24 well plates (5x105 cells/0.5 ml BSS/well) were stimulated for 30 min with 10μM nigericin or 1mM LLME at 37°C. Extracellular medium was removed from each well and centrifuged at 10,000xg for 15 sec to pellet detached cells. The cell-free supernatants were then assayed for lactate dehydrogenase (LDH) enzyme activity using the Cytotoxicity Detection Kit-LDH (Roche) according to the manufacturer protocol. The released LDH was normalized to total LDH content measured in 1% triton X-100-permeabilized samples of BMDC.
Data Processing and Analysis
All experiments were repeated two to ten times with separate BMDC or BMDM preparations. Figures illustrating western blot results are from representative experiments. Figures illustrating quantified changes in IL-1β secretion, intracellular K+ content, cytosolic [Ca2+], or pyroptotic propidium2+ influx represent the means (+/− SE) from two to ten independent experiments. Quantified data were statistically evaluated by one-way ANOVA with Bonferroni post-test using Prism 3.0 software.
RESULTS
Nigericin-induced increases in cytosolic [Ca2+] in LPS-primed BMDC occur downstream of the NLRP3 inflammasome/ caspase-1/ pyroptotic signaling cascade
Millimolar extracellular ATP gates the opening of plasma membrane P2X7 receptor nonselective cation channels that directly facilitate both efflux of cytosolic K+ and a massive influx of extracellular Ca2+ to cause a large increase in cytosolic [Ca2+]19. In contrast, nigericin is a lipophilic ionophore that directly functions as a K+/H+ exchanger upon partitioning into the plasma membrane or intracellular organelles (Fig. 1A). Nigericin is highly selective for monovalent cations and does not directly facilitate transmembrane Ca2+ fluxes20. Thus, any nigericin-induced increases in cytosolic [Ca2+] will reflect secondary changes in the activity of Ca2+ channels, non-selective ion channels, or Ca2+ homeostatic transporters due to altered [K+] or [H+] within the cytosol or organellar compartments. Although ATP and nigericin trigger efflux of cytosolic K+ by distinct mechanisms, both agonists induce similar magnitudes of caspase-1-dependent IL-1β release (Fig. 1B) in LPS-primed murine bone marrow-derived dendritic cells (BMDC). The relative magnitudes of ATP-stimulated versus nigericin-stimulated IL-1β release can vary modestly between different primary BMDC cultures with some showing equivalent responses to the two agonists (Figs. 4C and 5G), others exhibiting slightly higher ATP efficacy (Figs. 2B and 5C), and some showing lower ATP efficacy (Fig. 1B). This likely reflects modest differences in the expression levels of the ionotropic P2X7 receptors. We previously reported that accumulation of extracellular IL-1β by nigericin- and ATP-stimulated BMDC is maximal within 30 min24. The responses are completely abrogated in Casp1/11−/− (these cells are also deficient in caspase-11) and Nlrp3−/− BMDC (Fig. 1B) or in wildtype BMDC stimulated in media with elevated extracellular [K+] to prevent efflux of cytosolic [K+]24. Nigericin and ATP also induced robust decreases in cytosolic [K+] in wildtype, Casp1/11−/−, or Nlrp3−/− cells (Fig. 1C). Thus, key steps in the upstream NLRP3 inflammasome signaling cascade that culminate in IL-1β release are maximally activated within this 30 min interval. To characterize how changes in cytosolic [Ca2+] are temporally regulated during this critical phase of ATP- and nigericin-induced NLRP3 inflammasome activation, we used BMDC loaded with fluo-4 fluorescent Ca2+ sensor dye and assayed under experimental conditions (5x105 cells/ 0.5 ml test medium/ well in 24-well plates at 37°C) identical to those used to measure IL-1β release. Stimulation of LPS-primed BMDC with 5mM ATP induced a ~20-fold increase in cytosolic [Ca2+] from the basal level of 50–60 nM; the change in [Ca2+] peaked within 3 min and then modestly decreased over the next 30 min to values 6- to 10-fold above basal (Fig. 1D). In contrast, cytosolic [Ca2+] did not change during the first 10–12 min after nigericin stimulation and then gradually increased over the next 20 min to values only 3- to 4-fold above basal (Fig. 1D). Notably, ATP stimulated rapid and robust increases in cytosolic [Ca2+] in Casp1/11−/− BMDC (Fig. 1E) and Nlrp3−/−BMDC (Fig. 1F), albeit with somewhat different kinetics. In contrast, the delayed and modest increase in cytosolic [Ca2+] triggered by nigericin in wildtype cells was absent in Casp1/11−/− or Nlrp3−/− BMDC (Figs. 1D and 1E). These findings suggest that the nigericin-induced rise in [Ca2+] observed in wildtype BMDC occurs downstream of NLRP3 inflammasome activation as a result of caspase-1-dependent changes in plasma membrane permeability.
Figure 4. NLRP3 inflammasome signaling responses to K+ efflux agonists are dissociated from release of thapsigargin-sensitive intracellular Ca2+ stores.
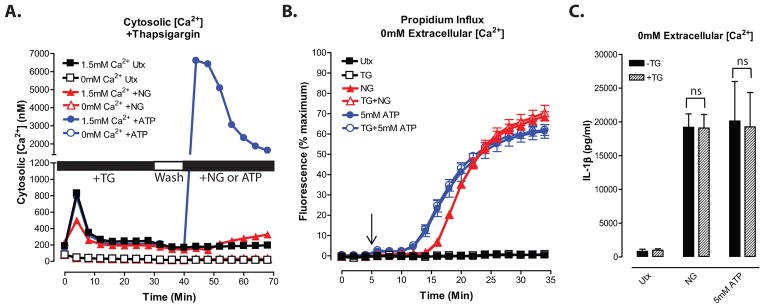
A, LPS-primed BMDC were treated with 300nM thapsigargin (TG) at t=0 in the absence of extracellular Ca2+. BMDC were washed at t=30 min and 10 min of baseline readings were taken prior to addition of 10μM nigericin or 5mM ATP at t=40 min. Cytosolic [Ca2+] was determined as described in figure 1. Data represent a mean of three independent experiments. B and C, LPS-primed BMDC were treated with 300nM thapsigargin in the absence of extracellular Ca2+. BMDC were then stimulated with 10μM nigericin or 5mM ATP in the absence of extracellular Ca2+. B, Onset of pyroptosis was determined by measuring permeability of the cell membrane to propidium2+. Baseline readings were taken for 5 min and nigericin or ATP were added at t=5 min. Data represent a mean of three independent experiments. C, IL-1β release was determined by ELISA. Data represent a mean of six independent experiments.
Figure 5. NLRP3 inflammasome signaling responses to K+ efflux agonists are dissociated from changes in cytosolic [Ca2+] in BMDC primed with TLR2 or TNF receptor agonists.

A and E, BMDC were primed with Pam3CSK4 (2μg/ml) or TNFα (100ng/ml) for 4h. Baseline fluorescence measurements were taken for 5 min prior to addition of 10μM nigericin or 5mM ATP at t=5 min. Cytosolic [Ca2+] was determined as described in figure 1. Data represent a mean of two independent experiments. B–C and F–G, Pam3CSK4-primed or TNFα-primed BMDC were treated with 10μM nigericin or 5mM ATP for 30 min in the presence or absence of 1.5mM extracellular [Ca2+]. B and F, Onset of pyroptosis was determined by measuring permeability of the cell membrane to propidium2+. Baseline readings were taken for 5 min and nigericin or ATP were added at t=5 min. Data represent mean of two independent experiments. C and G, IL-1β release from Pam3CSK4-primed BMDC or TNFα-primed BMDC was determined by ELISA. Data represent a mean of two independent experiments for each panel. D and H, Pam3CSK4-primed or TNFα-primed BMDC were pretreated with 300nM thapsigargin (TG) for 30 min in the absence of extracellular Ca2+. BMDC were washed and stimulated with 10μM nigericin or 5mM ATP for 30 min in the absence of extracellular Ca2+. IL-1β release from Pam3CSK4-primed BMDC or TNFα-primed BMDC was determined by ELISA. Data represent a mean of two independent experiments for each panel.
Figure 2. NLRP3 inflammasome signaling responses to K+ efflux agonists are dissociated from influx of extracellular Ca2+.

A, LPS-primed BMDC were treated with 10μM nigericin (NG) or 5mM ATP for 30 min in the presence/absence of 1.5mM extracellular [Ca2+]. Cytosolic [Ca2+] was determined as described in figure 1. Data represent a mean of three independent experiments. B–E, LPS-primed BMDC were treated for 30 min with 10μM nigericin or 5mM ATP in the presence/absence of 1.5mM extracellular [Ca2+]. B, IL-1β release was determined by ELISA. Data represent a mean of six independent experiments. C, Soluble lysate fraction (Lys) was probed for procaspase-1 and pro-IL-1β, insoluble lysate pellet was crosslinked with DSS and probed for oligomerized ASC, and extracellular medium fraction (ECM) was probed for mature caspase-1 and IL-1β. D, Onset of pyroptosis was determined by measuring permeability of the cell membrane to propidium2+. Baseline readings were taken for 5 min and nigericin or ATP were added at t=5 min. Data represent mean of four independent experiments. E, BMDC were lysed with 10% nitric acid and lysates were analyzed by atomic absorption spectroscopy to measure cellular [K+]. Data represent mean of three independent experiments.
The ability of caspase-1 to alter plasma membrane permeability as part of the pro-inflammatory cell death mechanism of pyroptosis is increasingly recognized as another readout of caspase-1 activity independent of its canonical action as an IL-1β converting enzyme6. Cookson and colleagues have developed assays based on the uptake of small cationic DNA-intercalating dyes, such as ethidium+, to track these caspase-1-dependent changes in membrane permeability during pyroptotic progression in macrophages infected with Salmonella or Anthrax25,26. We reasoned that the nigericin-induced increases in cytosolic [Ca2+] may occur as a secondary consequence of the caspase-1 dependent increase in membrane permeability. We adapted the methods developed by Cookson and colleagues in an assay that measured the onset and progression of this permeability increase under the same experimental conditions used to monitor changes in cytosolic [Ca2+] and IL-1β release. The assay tracks influx of the normally impermeant propidium2+ organic cation (Mr 416 Da as free cation; Mr 668 Da as propidium iodide salt) that undergoes a large increase in 540ex → 620em fluorescence upon intercalation with nuclear DNA.
Nigericin initiated an influx of propidium2+ into wildtype BMDC after a 12–15 min delay following its addition; thereafter, the extent of propidium2+ uptake into the BMDC population rapidly increased with ~80% of the cells accumulating the dye during the 30 min stimulation with nigericin (Fig. 1G). The delay phase and kinetics characterizing nigericin-induced propidium2+ influx were well correlated with the delay phase and kinetics of the nigericin-triggered increase in cytosolic [Ca2+] (Fig. 1D). Although influx of propidium2+ occurred earlier (by ~6–8 min) in response to ATP than to nigericin, the time course of the ATP-stimulated propodium2+ uptake (Fig. 1G) was markedly different from the ATP-induced Ca2+ increase response (Fig. 1D). The propidium2+ influx responses to both nigericin and ATP-gated P2X7 channels were absent in Casp1/11−/− (Fig. 1H) or Nlrp3−/− (Fig. 1I) BMDC and thus reflected induction of the pyroptotic membrane permeability transition. These studies demonstrate that the canonical NLRP3 activator nigericin does induce an increase in cytosolic [Ca2+] in murine BMDC. However, this rise in [Ca2+] occurs downstream of inflammasome activation as a secondary consequence of the pyroptotic membrane permeability transition (Fig. 1A). Enhanced Ca2+ influx may be a general response downstream of diverse caspase-1 inflammasome platforms. Vance and colleagues reported that flagellin activation of NLRC4 inflammasomes in tissue resident mouse macrophages triggered a caspase-1 dependent Ca2+ influx which preceded cytolysis and was required for phospholipase A2-dependent eicosanoid production; this was linked to a Ca2+-dependent ‘eicosanoid storm’ in vivo leading to systemic shock and death of the mice27.
NLRP3 inflammasome signaling responses to K+ efflux agonists or lysosomal destabilization are dissociated from influx of extracellular Ca2+
That the nigericin-induced increases in cytosolic [Ca2+] occur downstream of NLRP3-dependent caspase-1 activation argues against a necessary role for increased [Ca2+] in the regulation of this inflammasome pathway in response to K+ efflux stimuli in the murine BMDC model. However, we directly tested the contribution of extracellular Ca2+ and Ca2+ influx to nigericin- and ATP-induced NLRP3 inflammasome signaling by comparison of BMDC incubated in standard Ca2+-containing (1.5 mM CaCl2) basal salt solution (BSS) versus Ca2+-free BSS (no added CaCl2 plus supplementation with 0.3 mM EGTA). Removal of Ca2+ from the extracellular medium eliminated the increases in cytosolic [Ca2+] triggered by nigericin or ATP (Fig. 2A). Despite the absence of detectable increases in cytosolic [Ca2+], the IL-1β release responses to nigericin or ATP were not inhibited by the removal of extracellular Ca2+ (Fig. 2B and C). Similarly, the processing/release of caspase-1 and the formation of ASC oligomers in response to nigericin or ATP were not attenuated when BMDC were stimulated in Ca2+-free medium (Fig. 2C). Notably, the delay phases characterizing induction of the propidium2+ permeability transition in nigericin- or ATP-stimulated cells were shortened in the absence of extracellular Ca2+ and the rates of propidium2+ influx were accelerated (Fig. 2D). As expected, both nigericin and ATP caused significant decreases in cellular [K+] (Fig. 2E), but this response was similar in either the presence or absence of extracellular Ca2+ and measurable Ca2+ influx. These findings demonstrate that influx of extracellular Ca2+ and the resulting increase in cytosolic [Ca2+] are not necessary signals for NLRP3 inflammasome activation in BMDC stimulated with agents that directly trigger rapid efflux of cytosolic K+. Brough et al. reported the similar observation that Ca2+-free medium did not inhibit, but rather increased, ATP-stimulated IL-1β release from isolated murine peritoneal macrophages11.
We also tested whether changes in cytosolic [Ca2+] are required for NLRP3 inflammasome activation by stimuli that induce lysosomal destabilization. Crystalline particulates or insoluble protein aggregates induce NLRP3 inflammasome assembly via a pathway involving phagocytosis of the particulates, phagosome maturation/fusion with lysosomes, and compromise of lysosome integrity28–29. Because the processes of particulate phagocytosis and phagolysosomal maturation per se may be modulated by perturbation of Ca2+ signaling, we used the soluble lysosomotropic agent, Leu-Leu-O-methyl ester (LLME), to induce rapid and synchronous lysosome disruption in LPS-primed BMDC. LLME enters cells via amino acid transporters, accumulates in lysosomes, and is then converted into membrane-disruptive polyleucine peptides (e.g., Leu4, Leu6) via a reverse condensation reaction catalyzed by the cathepsin C/ dipeptidyl peptidase I30. It has been widely used as an NLRP3 activation stimulus in multiple inflammatory models10. A 30 min incubation with 1 mM LLME induced robust release of IL-1β from wildtype, but not Casp1/11−/− or Nlrp3−/− BMDC (Fig. 3A). Consistent with previous findings in murine macrophages10, LLME stimulated equivalent K+ efflux from wildtype, Casp1/11−/−, and Nlrp3−/− BMDC (Fig. 3B). Although the mechanism by which LLME or particulate lysososomal destabilizing stimuli elicit K+ efflux has not been defined, Zhong et al. reported that alum and silica induce activation of TRMP2 non-selective cation channels in murine macrophages as assayed by Ca2+ influx; there was also partial inhibition of IL-lβ release in Trpm2−/− cells31. Notably, LLME induced Ca2+ influx into BMDC after a 5 min lag period to result in a >10-fold sustained increase in cytosolic [Ca2+] within 20 min (Fig. 3D). This increase was absent when BMDC were stimulated by LLME in Ca2+-free BSS. Despite the absence of a detectable increase in cytosolic [Ca2+] under these latter conditions, the IL-1β release response to LLME was not inhibited, but rather was potentiated, by removal of extracellular Ca2+ (Fig. 3C). LLME also stimulated propidium2+ influx after a 15 min lag period under control (1.5 mM extracellular Ca2+) conditions (Fig. 3E); removal of extracellular Ca2+ modestly shortened the lag phase and increased the rate of propidium2+ influx. Thus, lysosome destabilization, like ATP-gating of P2X7 channels, induces a Ca2+ influx that precedes but is not required for, caspase-1 dependent IL-1β release or the propidium2+ influx response which tracks with caspase-1 activation. Notably, the rates of LLME-stimulated Ca2+ influx were attenuated in Casp1/11−/− and Nlrp3−/− BMDC (Fig. 3F). This suggests that the net Ca2+ influx response induced by LLME in wildtype DC reflects contributions from both an inflammasome-independent cation permeability pathway (that also mediates K+ efflux) and an inflammasome/caspase-1-dependent change in permeability (that also mediates propidium2+ influx).
Figure 3. NLRP3 inflammasome signaling responses to lysosomal destabilization are dissociated from influx of extracellular Ca2+.

A–C, LPS-primed BMDC were stimulated with 1mM LLME for 30 min. A, LPS-primed WT, Nlrp3−/−, and Casp1−/− BMDC were stimulated with LLME and IL-1β release was determined by ELISA. Data represent a mean of two independent experiments. B, LPS-primed WT, Nlrp3−/−, and Casp1−/− BMDC were stimulated with LLME, lysed with 10% nitric acid, and the lysates analyzed by atomic absorption spectroscopy to measure cellular [K+]. Data represent mean of three independent experiments C, LPS-primed WT BMDC were stimulated with LLME in the presence/absence of 1.5mM extracellular [Ca2+] and IL-1β release was determined by ELISA. Data represent a mean of two independent experiments. D, F, Cytosolic [Ca2+] was determined as described in figure 1 in WT (panels D, F) Nlrp3−/−, and Casp1−/− (Panel F) BMDC. Baseline readings were taken for 5 min and LLME was added at t=5 min. Data represent a mean of two independent experiments. E, LLME-induced propidium2+ influx was measured in LPS-primed BMDC in the presence/absence of 1.5mM extracellular [Ca2+]. Baseline readings were taken for 5 min and LLME was added at t=5 min. Data represent mean of two independent experiments..
NLRP3 inflammasome signaling responses to K+ efflux agonists are dissociated from release of thapsigargin-sensitive intracellular Ca2+ stores
The absence of extracellular Ca2+ effectively eliminated the increases in cytosolic [Ca2+] in response to nigericin or ATP but did not attenuate NLRP3 inflammasome activation in BMDC. However, several reports have suggested that mobilization of intracellular Ca2+ stores may be the critical regulatory signal. These studies have proposed that microdomains of the endoplasmic reticulum (ER) membrane system adjacent to mitochondria can release sufficient local Ca2+ to induce mitochondrial dysfunction leading to generation of ROS and release of mitochondrial DNA into the cytosol. The mitochondria-derived ROS and DNA then directly activate NLRP3 conformational changes to drive inflammasome assembly and signaling32. To investigate the role of released ER Ca2+ stores in the activation of NLRP3 inflammasome signaling, we treated BMDC with thapsigargin, an inhibitor of the ER-Ca2+ ATPase, for 30 min prior to stimulation with nigericin or ATP. Submicromolar thapsigargin inhibits all isoforms of the sarco/endoplasmic reticulum Ca-ATPases (SERCA pumps) that actively maintain high intraluminal concentrations (0.3 –1 mM) of free Ca2+ within the ER33. Thapsigargin inhibition of SERCA pump activity facilitates the rapid efflux of this stored Ca2+ into the cytosol via as yet undefined “leak” channels. Ca2+ released into the cytosol is rapidly transported to the extracellular compartment via the combined actions of the plasma membrane Ca2+-ATPase pump (PMCA) and Na+/Ca2+ exchange (NCX) transporters. However, the reduction in intraluminal [Ca2+] in thapsigargin-treated cells also induces oligomerization of STIM-family sensor proteins in the ER membrane and the oligomerized STIM puncta activate conformational changes in Orai-family store-operated Ca2+ influx channels within juxtaposed domains of the plasma membrane34. In the presence of extracellular Ca2+, this STIM-dependent gating of Orai channels facilitates Ca2+ influx to offset the loss of intracellular Ca2+ via PMCA and NCX activity and thereby replenishes and sustains the intracellular Ca2+ pools within the ER. Thus, we routinely treated BMDC with thapsigargin in Ca2+-free BSS (as in Figs. 2–3) to eliminate Ca2+ influx via activated STIM-Orai complexes and any replenishment of the ER Ca2+ stores34. As expected, stimulation of thapsigargin-treated BMDC with nigericin or ATP in the absence of extracellular Ca2+ abolished the increases in cytosolic [Ca2+] induced by these NLRP3 activators (Fig. 4A, open symbols). However, treatment of BMDC with thapsigargin under such Ca2+-free conditions did not delay the onset of pyroptotic membrane permeability transition or inhibit IL-1β release in response to nigercin or ATP (Fig. 4B and C). Positive control experiments using cells treated with thapsigargin in Ca2+-containing BSS verified the efficacy of thapsigargin as a Ca2+-mobilizing agent in this BMDC model system (Fig. 4A, closed symbols). These findings demonstrate that release of ER Ca2+ stores is also not a necessary signal for NLRP3 inflammasome activation in BMDC simulated with K+ efflux agonists. Previously, Menu et al. observed that micromolar concentrations of thapsigargin activate NLRP3 inflammasome signaling via induction of an ER stress response35. In the above experiments we used a lower concentration of thapsigargin (300nM) than in the Menu et al. study and did not observe induction of caspase-1 dependent pyroptosis or significant IL-1β release by BMDC in response to treatment with thapsigargin alone (Fig. 4B and 4C).
NLRP3 inflammasome signaling responses to K+ efflux agonists are dissociated from changes in cytosolic [Ca2+] in BMDC primed with TLR2 or TNF receptor agonists
Although the above findings indicate that increases in cytosolic [Ca2+] are not required for nigericin- or ATP-stimulated NLRP3 activation in BMDC, it is possible that Ca2+ may act as a modulatory signal by increasing the efficiency of productive NLRP3 inflammasome assembly under conditions of submaximal signal 1 priming and/or signal 2 activation. In all experiments described thus far, LPS was used as the priming stimulus to upregulate expression of NLRP3 and pro-IL-1β. Given the ability of TLR4 to drive both MyD88- and TRIF-dependent signaling pathways, LPS is a particularly strong activator of the NFκB-based transcription of these proinflammatory gene products, and we have previously reported that NLRP3 is expressed at high levels in LPS-primed BMDC24. With high levels of expression of inflammasome components, potential modulatory effects of elevated cytosolic [Ca2+] on NLRP3 activation or inflammasome complex assembly may be negligible during stimulation with potent signal 2 agonists such as ATP or nigericin. To test for more subtle modulatory effects of cytosolic [Ca2+] on NLRP3 inflammasome activation, we used less efficacious (relative to LPS) signal 1 priming stimuli, including the synthetic TLR2 agonist Pam3CSK4 and the pro-inflammatory cytokine TNFα. Both nigericin and ATP triggered increases in cytosolic [Ca2+] in BMDC primed with Pam3CSK4 (Fig. 5A) or TNFα (Fig. 5E) that were similar in kinetics to those observed in the LPS-primed cells (Fig. 1D). Control experiments verified that, as expected, nigericin induced similar magnitudes of K+ efflux in Pam3CSK4-and TNFα-primed cells as in unprimed or LPS-primed BMDC. Importantly, the nigericin-induced increases in Ca2+ influx occurred after a 10–15 min delay and correlated with the onset of pyroptotic propidium2+ influx in both the Pam3CSK4-primed (Fig. 5B) and TNFα-primed BMDC (Fig. 5F). Consistent with the reduced efficacy of these signal 1 stimuli, the magnitudes of nigericin- or ATP-stimulated IL-1β production in Pam3CSK4 -primed cells (Fig. 5C: ~12–20 ng/ml/30 min in Ca2+-containing saline), and especially in TNFα-primed cells (Fig. 5G: ~1.5 ng/ml/30 min), were lower than in LPS-primed cells (Figs 1B, 2B, 3C: 15–25 ng/ml/30 min). Similarly, the percentages of BMDC that accumulated proprodium2+ after 30 min of nigericin or ATP stimulation in Ca2+-containing saline were lower with Pam3CSK4 priming (Fig. 5B: 50% with both agonists) and TNFα-priming (Fig. 5F: 45% with nigericin and 15% with ATP) than with LPS priming (Fig. 1G, 2D: 60–80% with both agonists). The absence of extracellular Ca2+ did not inhibit the pyroptotic propidium2+ influx (Figs. 5B and 5F) or IL-1β release (Figs. 5C and 5G) responses to nigericin or ATP in BMDC primed with Pam3CSK4 or TNFα. Rather, as observed in LPS-primed BMDC, removal of extracellular Ca2+ from the Pam3CSK4- or TNFα-primed BMDC during stimulation shortened the delay phases and increased the rates of the propidium2+ uptake responses. In Ca2+-free medium, there was a trend (that did not reach statistical significance) for ATP, but not nigericin, to stimulate more IL-1β release in the TNFα-primed cells (Fig. 5G). Interestingly, thapsigargin treatment (in Ca2+-free saline) produced a modest (20–33%) decreases in the IL-1β release responses to ATP, but not nigericin, in the Pam3CSK4- and TNFα-primed BMDC (Figs. 5D and 5H). Taken together, the experiments with Pam3CSK4- or TNFα-primed BMDC also argue against a necessary role for increased Ca2+ influx and/or mobilization of intracellular Ca2+ stores in the activation of NLRP3 inflammasome signaling by K+ efflux agonists.
NLRP3 inflammasome signaling responses to K+ efflux are dissociated from changes in cytosolic [Ca2+] in murine macrophages
Most studies implicating roles for increased cytosolic [Ca2+] in activation of NLRP3 inflammasome signaling have been performed in murine or human macrophages11–15. Although the key elements of NLRP3 inflammasome composition and function are conserved in macrophage and dendritic cell models, some differences in modulatory signals have been reported. For example, phosphorylation of ASC by Syk markedly potentiates NLRP3 inflammasome activation by nigericin in murine bone marrow derived macrophages (BMDM) and peritoneal macrophages, but not BMDC36. Therefore, we investigated the potential contribution of extracellular Ca2+ and ER Ca2+ stores to NLRP3 inflammasome activation in BMDM. As in BMDC, nigericin and ATP induced increases in cytosolic [Ca2+] with distinctive time courses in BMDM (Fig. 6A). ATP triggered a rapid 20-fold increase from the 50 nM basal [Ca2+] in the macrophages that peaked at 5 min post-stimulation. However, the subsequent decrease was more rapid and robust than that observed in the BMDC (Fig. 1D). As in DC, the ATP-induced Ca2+ influx preceded the induction of propidium2+ influx by ~8 min in the BMDM (Fig 6B). The nigericin-induced increase in macrophage [Ca2+] was defined by a similar 10–12 min delay phase (Fig. 5A) as observed in the DC (Fig. 1D). This was followed by a slow rate of increase that plateaued at 200–300 nM (Fig 6A); the delayed increase in [Ca2+] was also correlated with the onset of pyroptotic propidium2+ influx in the BMDM (Fig. 6B). As in BMDC, removal of extracellular Ca2+ did not suppress either the kinetics or the magnitudes of the propidium2+ influx responses to nigericin and ATP in BMDM (Fig. 6B). Similarly, stimulation of macrophages with nigericin or ATP in Ca2+-free saline did not inhibit, but rather had no effect (nigericin) or modestly enhanced (ATP), the IL-1β release responses (Fig. 6C). The combined removal of extracellular Ca2+ and depletion of ER Ca2+ stores by thapsigargin treatment in BMDM did not significantly change IL-1β release in response to either agonist (Fig. 6D). These findings indicate that increased cytosolic [Ca2+] is a not a necessary signal for NLRP3 inflammasome activation in murine BMDM.
Figure 6. NLRP3 inflammasome signaling responses to K+ efflux are dissociated from changes in cytosolic [Ca2+] in murine macrophages.

A, Murine BMDM were primed with LPS (1μg/ml) for 4h. Baseline readings were taken for 5 min and 10μM nigericin (NG) or 5mM ATP were added at t=5 min. Cytosolic [Ca2+] was determined as described in figure 1. Data represent a mean of two independent experiments. B and C, LPS-primed BMDM were treated for 30 min with 10μM nigericin or 5mM ATP in the presence/absence of 1.5mM extracellular [Ca2+]. B, Onset of pyroptosis was determined by measuring permeability of the cell membrane to propidium2+. Baseline readings were taken for 5 min and nigericin or ATP were added at t=5 min. Data represent mean of four independent experiments. C, IL-1β release was determined by ELISA. Data represent a mean of four independent experiments. D, LPS-primed BMDM were treated with 300nM thapsigargin for 30 min in the absence of extracellular Ca2+, and then stimulated with 10μM nigericin or 5mM ATP for 30 min in the absence of extracellular Ca2+. IL-1β release was determined by ELISA. Data represent a mean of four independent experiments.
Increased cytosolic [Ca2+] induced by Ca2+ ionophore or Ca2+-mobilizing GPCRs is not a sufficient signal for NLRP3 inflammasome activation
Several studies have reported that stimulation of myeloid cells with agonists for certain Gq- or Gi-coupled receptors induces NLRP3 inflammasome activation via phospholipase C (PLC)-mediated generation of inositol trisphosphate (IP3) and release of ER Ca2+ stores 14–16. We compared the efficacy of various Ca2+-mobilizing agonists versus the K+ efflux agonists in the activation of NLRP3 inflammasome signaling by stimulating LPS-primed BMDC under the same test conditions (30 min in 1.5 mM CaCl2-containing saline) routinely used for induction by nigericin or P2X7 channel gating. The Ca2+-mobilizing agonists included: 1) ionomycin, a Ca2+ ionophore that stimulates both influx of extracellular Ca2+ and release of ER Ca2+ stores37; 2) R568, a synthetic agonist of the Gq-coupled Calcium-Sensing Receptor (CaSR) reported to induce robust NLRP3 inflammasome activation in murine and human macrophages via the PLC/ IP3-gated ER Ca2+ mobilization pathway14; 3) ATP or UTP, which at submillimolar concentrations activate the Gq/PLC-coupled P2Y2 nucleotide receptors highly expressed in all myeloid leukocyte subtypes 38 ; 4) fMLP, a synthetic agonist of the Gi/PLC-coupled formyl peptide receptors (FPR) that facilitate chemoattraction of myeloid leukocytes to local accumulations of bacteria-derived formylated proteins39.
As expected, ionomycin mimicked the ability of ATP-gated P2X7 channels to induce a large and immediate increase in cytosolic [Ca2+] that was sustained for up to 30 min (Fig. 7A). Despite this robust elevation in cytosolic [Ca2+], the 30 min treatment with ionomycin did not elicit statistically significant release of IL-1β as indicated by ELISA (Fig. 7B) or western blot for the mature 17 kDa cytokine (Fig. 7G). These observations in BMDC are consistent with Brough et al. 11 and Murakami et al. 13 who reported a similar inability of ionomycin to activate caspase-1 and release of mature IL-1β in murine macrophages. Ionomycin also did not trigger rapid pyroptotic signaling in the BMDC as demonstrated by the lack of propidium2+ influx throughout the ionomycin exposure (Fig. 7E). Ionomycin did induce a modest 25–30% reduction in cellular K+ content (Fig. 7F) that was independent of NLRP3 or caspase-1 expression (Supplemental Fig. 1A). Brough et al. described an ionomycin-induced decrease in cell viability (release of 64% total LDH) in murine macrophages which would necessarily be correlated a decrease in total K+ content11. We observed much less ionomycin-induced cell death (release of 18–20% total LDH within 60 min) in either wildtype or Casp1/11−/− BMDC (Supplemental Fig. 1B), which contrasted with the 70% LDH release in ATP-treated wildtype cells and no LDH release in ATP-treated Casp1/11−/− cells. Thus, the 25–30% decrease in [K+] induced by ionomycin reflects both cell death-dependent and cell death-independent components. The magnitude of this [K+] decrease was significantly smaller than that triggered by nigericin or P2X7 channel gating (Fig. 7F). Notably, this decrease was not sufficient for robust inflammasome assembly as indicated by the barely detectable accumulation of ASC oligomers (Fig. 7G). The findings further suggest that cytosolic [K+] must decrease below a threshold value to entrain the signaling pathways required for conformational activation of NLRP3.
Figure 7. Increased cytosolic [Ca2+] induced by Ca2+ ionophore or Ca2+-mobilizing GPCRs is not a sufficient signal for NLRP3 inflammasome activation.

A, Cytosolic [Ca2+] was determined as described in figure 1. Baseline readings were taken for 5 min and 5mM ATP or 3μM ionomycin were added at t=5 min. Data represent a mean of two independent experiments. B, LPS-primed BMDC were treated with 10μM NG, 5mM ATP or 3μM ionomycin for 30 min. IL-1β release was measured by ELISA. Data represent the mean of ten independent experiments. C, LPS-primed BMDC were treated with 30μM R568, 0.1mM ATP, 1μM N-formyl-Met-Leu-Phe (FMLP), or 0.1mM UTP for 30 min. Cytosolic calcium levels were determined as described in figure 1. Baseline readings were taken for 5 min and GPCR agonists were added at t=5 min. Data are representative of two independent experiments. D, LPS-primed BMDC were treated with calcium mobilizing agents for 30 min. IL-1β release was measured by ELISA. Data represent mean of five independent experiments. E, LPS-primed BMDC were treated with 10μM nigericin, 5mM ATP or 3μM ionomycin. Onset of pyroptosis was determined by measuring permeability of the cell membrane to propidium2+. Baseline readings were taken for 5 min and nigericin, ATP, or ionomycin were added at t=5 min. Data represent a mean of two independent experiments. F, LPS-primed BMDC were treated with 10μM nigericin, 5mM ATP or 3μM ionomycin. Cells were lysed with 10% nitric acid and lysates were analyzed by atomic absorption spectroscopy to measure cellular [K+]. Data represent mean of eight independent experiments. G, LPS-primed BMDC were stimulated with 10μM nigericin, 5mM ATP, 3μM ionomycin or 30μM R568 for 30 min. Soluble lysate fraction (Lys) was probed for procaspase-1 and pro-IL-1β, insoluble lysate pellet was crosslinked with DSS and probed for oligomerized ASC, and extracellular medium fraction (ECM) was probed for mature caspase-1 and IL-1β.
All of the tested GPCR agonists triggered an immediate 4-fold increase in cytosolic [Ca2+] that peaked within 60 seconds and then rapidly decreased over the next few minutes (Fig. 7C). The fMLP-, UTP-, or 100 μM ATP-triggered Ca2+ transients decayed to the basal level within 5–7 min. In contrast, the R568-induced peak in cytosolic [Ca2+] was followed by a sustained 3-fold elevation in cytosolic [Ca2+] over the 30 min test period (Fig. 7C). No statistically significant release of IL-1β was observed in BMDC stimulated for 30 min with fMLP, UTP, 100 μM ATP, or R568 (Fig. 7D). Likewise, R568 did not induce detectable accumulation of mature IL-1β or caspase-1 p20 subunits in the extracellular medium (Fig. 7G) and elicited only weak accumulation of intracellular ASC oligomers (Fig. 7G). These data demonstrate that agonists for Ca2+-mobilizing GPCR do not mimic the ability of K+ efflux agonists to elicit rapid and robust NLRP3 inflammasome activation in the murine BMDC model.
Suppression of nigericin-stimulated NLRP3 inflammasome signaling by BAPTA and 2-APB can be dissociated from perturbation of Ca2+ signaling
Some support implicating Ca2+ signaling in NLRP3 inflammasome activation is based on observations that BAPTA, a strong Ca2+ chelator and buffer of cytosolic Ca2+, and 2-aminoethoxydiphenyl borate [2-APB], an inhibitor of IP3-gated Ca2+ release channels and store-operated calcium entry (SOCE) channels, strongly suppress IL-1β release in response to canonical NLRP3 activators 11–14. Given our finding that removal of extracellular Ca2+ (with or without thapsigargin treatment) eliminates the increases in cytosolic [Ca2+], but not the IL-1β release, stimulated by nigericin or ATP (Figs. 2 and 4), it is possible that the inhibitory actions of BAPTA and 2-APB on NLRP3 signaling may also be dissociated from effects on Ca2+ signaling. We observed that loading LPS-primed BMDC with BAPTA markedly attenuated multiple readouts of nigericin-stimulated NLRP3 inflammasome signaling including total IL-1β release (Fig. 8A), extracellular accumulation of p20 caspase-1 subunit and p17 mature IL-1β (Fig.8D), formation of ASC oligomers (Fig. 8D), and induction of pyroptotic propidium2+ influx (Fig. 8C). It is important to note that all assays were performed in the absence of extracellular Ca2+ which effectively eliminates any nigericin-induced increase in cytosolic [Ca2+] (Fig. 2A). In contrast, the nigericin-stimulated efflux of cytosolic K+ was not inhibited in the BAPTA-loaded cells (Fig. 8B). We verified the efficacy of BAPTA loading and its ability to buffer the delayed increase in cytosolic [Ca2+] induced by nigericin stimulation when BMDC were incubated in 1.5 mM Ca2+-containing basal saline (Supplemental Figure 2). These findings demonstrate that cytosolic BAPTA loading can inhibit IL-1β processing and release independently of its function as a Ca2+ chelator and downstream of the necessary NLRP3-activating K+ efflux signal.
Figure 8. Suppression of nigericin-stimulated NLRP3 inflammasome signaling by BAPTA can be dissociated from perturbation of Ca2+ signaling.

LPS-primed BMDC were loaded with 25μM BAPTA-AM for 45 min per manufacturer’s protocol. Cells were then treated with 10μM nigericin for 30 min in the absence of extracellular Ca2+. A, IL-1β release was measured by ELISA. Data represent a mean of three independent experiments. B, Efflux of cellular K+ was measured by atomic absorption spectroscopy. Data represent a mean of three independent experiments. C. Onset of pyroptosis was determined by measuring permeability of the cell membrane to propidium2+. Baseline readings were taken for 5 min and 10μM nigericin was added at t=5 min. Data represent a mean of four independent experiments. D, LPS-primed BMDC were stimulated with 10μM nigericin for 30 min in the presence/absence of BAPTA loading. Soluble lysate fraction (Lys) was probed for procaspase-1 and pro-IL-1β, insoluble lysate pellet was crosslinked with DSS and probed for oligomerized ASC, and extracellular medium fraction (ECM) was probed for mature caspase-1 and IL-1β.
Previous studies have demonstrated that 2-APB strongly inhibits NLRP3 inflammasome activation in response to nigericin and other stimuli. This suppression has been ascribed to the extensively characterized actions of this reagent as an inhibitor of both IP3-gated Ca2+ release channels in the ER and store-operated Ca2+ influx channels in the plasma membrane13,14. An expectation of this proposed mechanism is that the ability of 2-APB to inhibit nigericin-stimulated IL-1β release and pyroptosis should be highly correlated with suppression of Ca2+ mobilization and influx. We tested the effects of 2-APB on multiple indices of nigericin-activated NLRP3 inflammasome signaling in LPS-primed BMDC incubated in CaCl2-containing medium. Consistent with previous findings13,14, we observed that 2-APB completely suppressed total IL-1β release (Fig. 9A), extracellular accumulation of p20 caspase-1 subunit and p17 mature IL-1β (Fig.9C), formation of ASC oligomers (Fig. 9C), and induction of propidium2+ influx (Fig. 9D) in response to nigericin. In contrast, robust K+ efflux responses were observed in nigericin-treated BMDC in the absence or presence of 2-APB (Fig. 9B). Despite the complete suppression of these multiple readouts of NLRP3 inflammasome activity, 2-APB did not suppress, but rather strongly potentiated, the nigericin-induced increase in cytosolic [Ca2+] (Fig. 9E). Moreover, the initial addition of 2-APB to LPS-primed BMDC elicited an immediate 10–20-fold increase in cytosolic [Ca2+] followed by a decay to a level 5-fold above basal (Fig. 9E) which, even in the absence of a subsequent nigericin stimulus, was steadily maintained for many minutes. These findings demonstrate that 2-APB inhibits NLRP3 inflammasome signaling downstream of K+ efflux signals and independently of its canonical actions as an inhibitor of IP3R/SOCE-mediated increases in [Ca2+]. In contrast to a presumed causal relationship between its inhibitory effects on NLRP3 signaling and the suppression of Ca2+ signaling, 2-APB by itself triggered large increases in cytosolic [Ca2+] and potentiated nigericin-induced Ca2+ influx in LPS-primed murine DC.
Figure 9. Suppression of nigericin-stimulated NLRP3 inflammasome signaling by 2-APB can be dissociated from perturbation of Ca2+ signaling.

A–C, LPS-primed BMDC were incubated with 100μM 2-APB for 20 min prior to treatment with 10μM nigericin for 30 min in the presence of 1.5mM extracellular [Ca2+]. A, IL-1β release was measured by ELISA. Data represent a mean of three independent experiments. B, Efflux of cellular K+ was measured by atomic absorption spectroscopy. Data represent a mean of three independent experiments. C, Soluble lysate fraction (Lys) was probed for procaspase-1 and pro-IL-1β, insoluble lysate pellet was crosslinked with DSS and probed for oligomerized ASC, and extracellular medium fraction (ECM) was probed for mature caspase-1 and IL-1β. D, LPS-primed BMDC were preincubated with 100μM 2-APB for 20 min in the presence of 1.5mM extracellular [Ca2+] and onset of pyroptosis in response to nigericin was assayed by measuring permeability of cells to propidium2+. Baseline readings were taken for 5 min and 10μM nigericin was added at t=5 min. E, Cytosolic [Ca2+] in LPS-primed BMDC was measured using fluo-4 fluorescence in the presence of 1.5mM extracellular [Ca2+]. Baseline readings were taken for 5 min. Cells were treated with 100μM 2-APB at t=5 min (double arrow) and 10μM nigericin (single arrow) at t=15 min. Data are representative of two independent experiments.
DISCUSSION
Perturbation of intracellular ion homeostasis is a major cellular stress signal for activation of NLRP3 inflammasome signaling. However, the relative contributions of decreased cytosolic [K+] versus increased cytosolic [Ca2+] remain disputed and incompletely defined. This study provides three major findings relevant to this unresolved area of NLRP3 regulation. First, increased cytosolic [Ca2+] is neither a necessary nor sufficient signal for the NLRP3 inflammasome cascade induced in murine dendritic cells and macrophages during activation by endogenous ATP-gated P2X7 receptor channels, the bacterial ionophore nigericin, or LLME-induced lysosomal disruption. These stimuli are widely utilized as highly efficacious inducers of NLPR3 inflammasome assembly in murine and human myeloid leukocyte models. Second, agonists for three Ca2+-mobilizing G protein-coupled receptors expressed in murine myeloid leukocytes (FPR, P2Y2R, CaSR) were ineffective as robust activators of NLRP3 signaling when directly compared to the K+ efflux agonists under identical experimental conditions. Third, BAPTA and 2-APB, widely used reagents for disruption of Ca2+-dependent signaling pathways, strongly suppress nigericin-induced NLRP3 inflammasome signaling via mechanisms dissociated from their canonical or expected effects on Ca2+ homeostasis.
We assessed the possible roles for influx of extracellular Ca2+ and/or mobilization of ER Ca2+ stores on multiple steps in the serial NLRP3 signaling pathway including efflux of cytosolic K+, accumulation of stable ASC oligomers, production of active caspase-1, processing and release of mature IL-1β, and induction of pyroptotic changes in plasma membrane permeability. Direct measurements using cells loaded with fluo-4 Ca2+ sensor dye verified the absence of cytosolic [Ca2+] increases during manipulations designed to eliminate Ca2+ influx and mobilization in the ATP- or nigericin-treated cells. Recent reports have indicated that NLRP3 activation triggers the rapid assembly of prion-like ASC aggregates that comprise the critical and essentially irreversible step in coupling conformational changes in the NLRP3 stress sensor to activation of the caspase-1 effector enzyme 4,5. Our findings indicate that ATP and nigericin induce equivalent accumulation of detergent-insoluble ASC aggregates regardless of the presence or absence of increases in cytosolic [Ca2+].
Although our data indicate that Ca2+ is not a critical 2nd messenger for the very proximal steps of the NLRP3 signaling cascade, changes in cytosolic [Ca2+] may modulate reactions downstream of the NLRP3-dependent assembly of the ASC oligomeric platforms for caspase-1 activation, particularly the several non-classical export pathways for release of mature IL-1β and caspase-1 itself. We previously reported that increases in cytosolic [Ca2+] do not affect caspase-1 dependent processing of proIL-1β but can potentiate the release of processed mature IL-1β in some, but not all, cell models40–41. Depending on cell type and activation stimulus, IL-1β can be released in different proportions via three vesicular and one non-vesicular mechanism42. The vesicular mechanisms include: 1) IL-1β trapped within plasma membrane-derived microvesicles that bleb and scission from the cell surface; 2) IL-1β packaged within exosomes contained in multivesicular endosomes that subsequently fuse with the cell surface membrane; and 3) exocytosis of IL-1β that has been internalized within secretory autophagolysosomes. The non-vesicular pathway is via regulated cell lysis as a consequence of caspase-1 driven pyroptosis. In our LPS-primed BMDC and BMDM models, the magnitudes of IL-1β release induced by nigericin or ATP were largely equivalent in the absence or presence of extracellular Ca2+ or in the absence or presence of thapsigargin treatment to deplete ER Ca2+ stores. We also measured propidium2+ influx as an index of the caspase-1 regulated transition in plasma membrane permeability that accompanies pyroptotic cell lysis43. This parameter provides a read-out of caspase-1 activation kinetics independent of IL-1β release by the vesicular mechanisms. Notably, DCs and macrophages stimulated with nigericin or ATP in the absence or presence of extracellular Ca2+ exhibited similar onset and rates of propidium2+ influx. This indicates that the Ca2+-independent pathway predominantly mediates the non-classical export of IL-1β following rapid inflammasome activation by K+ efflux agonists. However, in other inflammasome models with slower progression to pyroptosis, Ca2+- dependent vesicular IL-1β release may be more important. Differential roles for local mobilization of ER Ca2+ stores in downstream IL-1β release, rather than upstream inflammasome activation, may underlie the modest decreases we observed in the IL-1β release responses to ATP, but not nigericin, in the Pam3CSK4- and TNFα-primed BMDC.
Agonists for some, but not all, Ca2+-mobilizing GPCRs expressed in myeloid leukocytes can induce the release of IL-1β via an NLRP3-dependent mechanism14,15. Rossol et al. noted that NLRP3 in human and murine monocytes was a target for CaSR and GPRC6A activated by divalent/ trivalent inorganic cations15. However, those studies indicated that accumulation of extracellular IL-1β was a delayed response with little cytokine release occurring during the initial 3 h and maximal release requiring >8 h of CaSR activation15. Lee et al. described more rapid (within 30–50 min) CaSR-induced activation of NLRP3 signaling in murine BMDM and also used the organic calcimimetic agonist R568 to induce CaSR-dependent NLRP3 inflammasomes14. We compared the relative efficacies of R568 and the K+ efflux agonists to drive rapid NLRP3 inflammasome activation in our LPS-primed BMDC experimental model. Although R568 rapidly induced a sustained increase in cytosolic [Ca2+] over a 30 min test period, this resulted in only very weak accumulation of ASC oligomers that was insufficient to drive significant caspase-1 activation or IL-1β release. Different GPCRs have varying rates of desensitization. For example, P2Y2R desensitize within minutes after activation by the metabolically labile ATP/UTP agonists44. In contrast, the canonical role of the CaSR is to respond to slowly developing increases in serum extracellular [Ca2+] for regulation of parathyroid hormone secretion. The receptor is thus remarkably resistant to desensitization and retains the ability to signal via the Gq /PLC cascade with sustained elevation of cytosolic [Ca2+] for hours after exposure to agonistic stimuli45. Sustained elevation of cytosolic [Ca2+] during prolonged CaSR activation may induce sufficient activity of Ca2+-sensitive K+ channels to decrease cytosolic [K+] to the threshold level required for stable assembly of NLRP3/ASC signaling platforms46. At the single cell level, the assembly of such stable platforms likely comprises an all-or-none response to coincident combinations of critical regulatory signals, such as decreased cytosolic [K+] as well as the ubiquitination or phosphorylation states of NLRP3 and ASC36, 47–48. It would be relevant to test whether sustained stimulation of DC or macrophages with different slowly desensitizing GPCRs gradually increases the number of cells with perinuclear ASC specks and accumulation of active caspase-1.
The Ca2+ chelator BAPTA has been used to implicate increased cytosolic [Ca2+] as a necessary signal for NLRP3 inflammasome activation in response to various stimuli, including nigericin and extracellular ATP11–14. Although our studies confirmed the ability of BAPTA loading to markedly attenuate nigericin-induced NLRP3 signaling in LPS-primed BMDC, this inhibitory action was observed under Ca2+-free stimulation conditions that prevent nigericin-elicited changes in cytosolic [Ca2+]. BAPTA chelates other trace divalent cations, including Zn2+, Fe2+, and Cu2+, that can function as key cofactors for many enzymes, including some implicated in inflammasome regulation. Several reports have implicated roles for intracellular Zn2+ in modulation of NLRP3 inflammasome signaling49,50. One possible specific link is the zinc-dependent metalloprotease, BRCC3, which acts as a K63-specific deubiquitinase and critical regulator of NLRP3. Py et al. found that BRCC3 promotes NLRP3 inflammasome activation by deubiquitinating the LRR domain of NLRP350. In addition to the ubiquitination, other studies have implicated the phosphorylation status of inflammasome components as critical to the efficient assembly of NLRP3/ASC platforms. Martin et al. identified the PP2A phosphatase as a key target for signal 2 stimuli, including ATP and nigericin, which acted to recruit PP2A to complexes of IKKα and ASC47. The recruited PP2A reverses association of IKKα with ASC and thus licenses ASC for interaction with NLRP3. Notably, BAPTA loading attenuated the recruitment of PP2A in response to ATP or nigericin47. Previous studies have identified complex roles for cytosolic Zn2+ in regulation of PP2A activity51,52. Finally, Furuta et al. demonstrated that BAPTA exerts a potent microtubule-depolymerizing activity that is independent of its ability to chelate Ca2+ 17. This is relevant because Misawa et al. have described a microtubule-mediated spatial arrangement of mitochondria in close apposition to the ER that is necessary for optimal NLRP3 inflammasome activation53. The ability of BAPTA to promote microtubule depolymerization might interfere with this organellar juxtaposition and thus attenuate interaction between ASC and NLRP3. Taken together, the combined effects of BAPTA on Zn2+-dependent signaling enzymes and/or microtubule dynamics may underlie its inhibitory actions on NLRP3 signaling independently of its canonical actions on Ca2+ signaling.
Additional support for the involvement of Ca2+ signaling in NLRP3 activation has come from experiments using 2-aminoethoxydiphenyl borate (2-APB) as an inhibitor of both ER Ca2+ release via IP3R channels and influx of extracellular Ca2+ via the Orai-family CRAC channels (Ca2+ release- activated Ca2+ channels) 13,14. We confirmed previously reported observations that pre-treatment of LPS-primed myeloid cells with 2-APB prior to stimulation by nigericin (or ATP) completely suppresses all indices of the activated NLRP3 signaling cascade. Importantly, our experiments also demonstrated that 2-APB inhibits the ability of nigericin to induce the pyroptotic propidium2+ influx as an alternative readout of caspase-1 activation. However, we were unable to correlate these robust inhibitory effects of 2-APB on NLRP3 signaling with the canonical inhibitory actions of 2-APB on elevation of cytosolic [Ca2+] and Ca2+ signaling. Rather, 2-APB per se caused increases in cytosolic [Ca2+] in LPS-primed DCs and also facilitated massive influx of Ca2+ during subsequent stimulation by nigericin (Fig. 9E). Although 2-APB does indeed potently block Ca2+ influx via the Orai1 and Orai2 subtypes of CRAC channels, it has the opposite effect on the Orai3 family member. Several groups have described the ability of 2-APB to allosterically stabilize the open-gated conformation of Orai3 channels and change their permeability properties into nonselective cation channels that facilitate fluxes of Ca2+ and monovalent cations54–59. The ability of 2-APB to stimulate increased cytosolic [Ca2+] in our LPS-primed BMDC model suggests that these cells express significant levels of Orai3 channels and this is supported by our preliminary western blot analyses of BMDC and BMDM. Regardless of how 2-APB-induces Ca2+ influx, our observations indicate that the suppression of NLRP3 inflammasome signaling in 2-APB treated BMDC cannot be ascribed to direct inhibition of Ca2+ signaling. 2-APB does not inhibit the inflammasome signaling cascades initiated by AIM2 14. Given that AIM2, like NLRP3, regulates an ASC-dependent inflammasome, this suggests that 2-APB targets NLRP3 function rather than ASC. Additional studies are required to define the underlying pharmacological mechanism(s) by which 2-APB may inhibit NLRP3 upstream of ASC oligomerization.
In summary, this study provides new insights regarding how perturbation of intracellular ion homeostasis acts as a signal for activation of NLRP3 inflammasome signaling. The data indicate that a rapid decrease in cytosolic [K+] is a highly efficacious signal for initiating the NLRP3 inflammasome cascade regardless of the presence or absence of coincident increases in cytosolic [Ca2+]. Despite this dissociation from Ca2+ signaling, the mechanism by which a decrease in [K+] is coupled to the conformational activation of NLRP3 remains unknown. Moreover, changes in cytosolic [K+] may also act downstream of NLRP3 at the level of ASC given the suppressive effects of elevated [K+] on activation of the AIM2/ASC inflammasome in Francisella-infected macrophages61. Perturbation of multiple mitochondrial functions by disruption of the normal cytosolic [K+]/[Na+] ratio is a relevant area for investigation32,60. However, a recent analysis by Vince and colleagues62 demonstrated normal NLRP3 inflammasome function in ATP- or nigerin-stimulated murine macrophages that lacked expression of different mitochondrial or mitochondria-associated proteins, including cyclophilin D, Bax, Bak, and parkin, previously implicated in the regulation of inflammasome signaling. These findings indicate that linking NLRP3 activation to mitochondrial perturbation by K+ efflux agonists will require consideration of other mitochondrial functions.
Supplementary Material
Footnotes
This work was supported by 13PRE16860052 Predoctoral Fellowship from the American Heart Association (awarded to MAK) and National Institutes of Health Grants R01-GM36387 (GRD), T32-AI089474 (HMR), T32-GM007250 (MAK and HMR), T32-HL105338 (MAK), R25-HL03152L (LGR).
References
- 1.Warren J. Williams Hematology. 8. McGraw Hill Companies; 2010. The Inflammatory Response. Access Medicine. Web. 7 August 2014. [Google Scholar]
- 2.Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 3.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunological Reviews. 2011;243(1):136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 4.Lu A, Magupalli V, Ruan J, Yin Q, Atianand M, Vos M, Schröder G, Fitzgerald K, Wu H, Egelman E. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156(6):1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai X, Chen J, Xu H, Liu S, Jiang Q, Halfmann R, Chen Z. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156(6):1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aachoui Y, Sagulenko V, Miao E, Stacey K. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Current Opinion in Microbiology. 2013;16(3):319–326. doi: 10.1016/j.mib.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mariathasan S, Weiss D, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee W, Weinrauch Y, Monack D, Dixit V. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 8.Muñoz-Planillo R, Franchi L, Miller L, Núñez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. The Journal of Immunology. 2009;183(6):3942–3948. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perregaux D, Gabel C. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. Journal of Biological Chemistry. 1994;269(21):15195–15203. [PubMed] [Google Scholar]
- 10.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith B, Rajendiran T, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brough D, Le Feuvre R, Wheeler R, Solovyova N, Hilfiker S, Rothwell N, Verkhratsky A. Ca2+ stores and Ca2+ entry differentially contribute to the release of IL-1β and IL-1α from murine macrophages. The Journal of Immunology. 2003;170(6):3029–3036. doi: 10.4049/jimmunol.170.6.3029. [DOI] [PubMed] [Google Scholar]
- 12.Rada B, Park J, Sil P, Geiszt M, Leto T. NLRP3 inflammasome activation and interleukin-1β release in macrophages require calcium but are independent of calcium-activated NADPH oxidases. Inflammation Research. 2014:1–10. doi: 10.1007/s00011-014-0756-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer A, Horng T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences. 2012;109(28):11282–11287. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee G, Subramanian N, Kim A, Aksentijevich I, Goldbach-Mansky R, Sacks D, Germain R, Kastner D, Chae J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schöneberg T, Schaefer M, Krügel U, Smajilovic S, Bräuner-Osborne H, Baerwald C, Wagner U. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nature Communications. 2012;3:1329. doi: 10.1038/ncomms2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.An L, Mehta P, Xu L, Turman S, Reimer T, Naiman B, Connor J, Sanjuan M, Kolbeck R, Fung M. Complement C5a potentiates uric acid crystal-induced IL-1β production. European Journal of Immunology. 2014 doi: 10.1002/eji.201444560. Epub ahead of print September 17, 2014. [DOI] [PubMed] [Google Scholar]
- 17.Furuta A, Tanaka M, Omata W, Nagasawa M, Kojima I, Shibata H. Microtubule disruption with BAPTA and dimethyl BAPTA by a calcium chelation-independent mechanism in 3T3-L1 adipocytes. Endocrine Journal. 2009;56(2):235–243. doi: 10.1507/endocrj.k08e-321. [DOI] [PubMed] [Google Scholar]
- 18.Pena F, Ordaz B. Non-selective cation channel blockers: potential use in nervous system basic research and therapeutics. Mini Reviews in Medicinal Chemistry. 2008;8(8):812–819. doi: 10.2174/138955708784912166. [DOI] [PubMed] [Google Scholar]
- 19.Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. American Journal of Physiology: Cell Physiology. 2004;286(5):C1100–1108. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- 20.Pressman B. Biological applications of ionophores. Annual Review of Biochemistry. 1976;45:501–530. doi: 10.1146/annurev.bi.45.070176.002441. [DOI] [PubMed] [Google Scholar]
- 21.Kayagaki N, Warming S, Lamkanfi M, Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee W, Roose-Girma M, Dixit V. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 22.Qu Y, Ramachandra L, Mohr S, Franchi L, Harding C, Nunez G, Dubyak G. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. The Journal of Immunology. 2009;182(8):5052–5062. doi: 10.4049/jimmunol.0802968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grynkiewicz G, Poenie M, Tsien R. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- 24.Antonopoulos C, El Sanadi C, Kaiser W, Mocarski E, Dubyak G. Proapoptotic Chemotherapeutic Drugs Induce Noncanonical Processing and Release of IL-1β via Caspase-8 in Dendritic Cells. The Journal of Immunology. 2013;191(9):4789–4803. doi: 10.4049/jimmunol.1300645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular Microbiology. 2006;8(11):1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 26.Fink S, Bergsbaken T, Cookson B. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proceedings of the National Academy of Sciences. 2008;105(11):4312–4317. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duewell P, Kono H, Rayner K, Sirois C, Vladimer G, Bauernfeind F, Abela G, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald K, Rock K, Moore K, Wright S, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornung V, Bauernfeind F, Halle A, Samstad E, Kono H, Rock K, Fitzgerald K, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunology. 2008;9(8):847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thiele D, Lipsky P. The action of leucyl-leucine methyl ester on cytotoxic lymphocytes requires uptake by a novel dipeptide-specific facilitated transport system and dipeptidyl peptidase I-mediated conversion to membranolytic products. The Journal of Experimental Medicine. 1990;172(1):183–194. doi: 10.1084/jem.172.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala F, Qiao L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nature Communications. 2013;4:1611. doi: 10.1038/ncomms2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends in Immunology. 2014;35(6):253–261. doi: 10.1016/j.it.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Møller J, Olesen C, Winther A, Nissen P. The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Quarterly Reviews of Biophysics. 2010;43(04):501–566. doi: 10.1017/S003358351000017X. [DOI] [PubMed] [Google Scholar]
- 34.Prakriya M. Chapter One - Store-Operated Orai Channels: Structure and Function. In: Murali P, editor. Current Topics in Membranes. Vol. 71. Academic Press; 2013. pp. 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, Tschopp J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death and Disease. 2012;3:e261. doi: 10.1038/cddis.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, Mitsuyama M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nature Immunology. 2013;14(12):1247–1255. doi: 10.1038/ni.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu C, Hermann T. Characterization of ionomycin as a calcium ionophore. Journal of Biological Chemistry. 1978;253(17):5892–5894. [PubMed] [Google Scholar]
- 38.Clifford E, Martin K, Dalal P, Thomas R, Dubyak G. Stage-specific expression of P2Y receptors, ecto-apyrase, and ecto-5′-nucleotidase in myeloid leukocytes. American Journal of Physiology: Cell Physiology. 1997;273(3):C973–987. doi: 10.1152/ajpcell.1997.273.3.C973. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Ye D. Molecular biology for formyl peptide receptors in human diseases. Journal of Molecular Medicine. 2013;91(7):781–789. doi: 10.1007/s00109-013-1005-5. [DOI] [PubMed] [Google Scholar]
- 40.Gudipaty L, Munetz J, Verhoef P, Dubyak G. Essential role for Ca2+ in regulation of IL-1β secretion by P2X7 nucleotide receptor in monocytes, macrophages, and HEK-293 cells. American Journal of Physiology: Cell Physiology. 2003;285(2):C286–299. doi: 10.1152/ajpcell.00070.2003. [DOI] [PubMed] [Google Scholar]
- 41.Qu Y, Franchi L, Nunez G, Dubyak G. Nonclassical IL-1β Secretion Stimulated by P2X7 Receptors Is Dependent on Inflammasome Activation and Correlated with Exosome Release in Murine Macrophages. The Journal of Immunology. 2007;179(3):1913–1925. doi: 10.4049/jimmunol.179.3.1913. [DOI] [PubMed] [Google Scholar]
- 42.Dubyak GR. P2X7 receptor regulation of non-classical secretion from immune effector cells. Cellular Microbiology. 2012;14(11):1697–1706. doi: 10.1111/cmi.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bergsbaken T, Fink S, Cookson B. Pyroptosis: host cell death and inflammation. Nature Reviews Microbiology. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown H, Lazarowski E, Boucher R, Harden T. Evidence that UTP and ATP regulate phospholipase C through a common extracellular 5′-nucleotide receptor in human airway epithelial cells. Molecular Pharmacology. 1991;40(5):648–655. [PubMed] [Google Scholar]
- 45.Conigrave A, Ward D. Calcium-sensing receptor (CaSR): Pharmacological properties and signaling pathways. Best Practice & Research Clinical Endocrinology & Metabolism. 2013;27(3):315–331. doi: 10.1016/j.beem.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Cox D. Ca2+-regulated ion channels. BMB Reports. 2011;44(10):635–646. doi: 10.5483/BMBRep.2011.44.10.635. [DOI] [PubMed] [Google Scholar]
- 47.Martin B, Wang C, Willette-Brown J, Herjan T, Gulen M, Zhou H, Bulek K, Franchi L, Sato T, Alnemri E, Narla G, Zhong X, Thomas J, Klinman D, Fitzgerald K, Karin M, Nuñez G, Dubyak G, Hu Y, Li X. IKKα negatively regulates ASC-dependent inflammasome activation. Nature Communications. 2014;5:4997. doi: 10.1038/ncomms5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri E. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. Journal of Biological Chemistry. 2012;287(43):36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Summersgill H, England H, Lopez-Castejon G, Lawrence CB, Luheshi NM, Pahle J, Mendes P, Brough D. Zinc depletion regulates the processing and secretion of IL-1β. Cell Death and Disease. 2014;5:e1040. doi: 10.1038/cddis.2013.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Py B, Kim M, Helin Vakifahmetoglu-Norberg, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Molecular Cell. 2013;49(2):331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 51.Sun X, Wei Y, Xiong Y, Wang X, Xie A, Wang X, Yang Y, Wang Q, Lu Y, Liu R, Wang J. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A) Journal of Biological Chemistry. 2012;287(14):11174–11182. doi: 10.1074/jbc.M111.309070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiong Y, Jing X, Zhou X, Wang X, Yang Y, Sun X, Qiu M, Cao F, Lu Y, Liu R, Wang J. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiology of Aging. 2013;34(3):745–756. doi: 10.1016/j.neurobiolaging.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature Immunology. 2013;14(5):454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 54.Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. Journal of Physiology. 2008;586:3061–3073. doi: 10.1113/jphysiol.2008.151365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang S, Kozak J, Jiang W, Yeromin A, Chen J, Yu Y, Penna A, Shen W, Chi V, Cahalan M. Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. Journal of Biological Chemistry. 2008;283:17662–17671. doi: 10.1074/jbc.M801536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeHaven W, Smyth J, Boyles R, Bird G, Putney J. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. Journal of Biological Chemistry. 2008;283:19265–19273. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schindl R, Bergsmann J, Frischauf I, Derler I, Fahrner M, Muik M, Fritsch R, Groschner K, Romanin C. 2-aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. Journal of Biological Chemistry. 2008;283:20261–20267. doi: 10.1074/jbc.M803101200. [DOI] [PubMed] [Google Scholar]
- 58.Bergsmann J, Derler I, Muik M, Frischauf I, Fahrner M, Pollheimer P, Schwarzinger C, Gruber H, Groschner K, Romanin C. Molecular determinants within N terminus of Orai3 protein that control channel activation and gating. Journal of Biological Chemistry. 2011;286:31565–31575. doi: 10.1074/jbc.M111.227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amcheslavsky A, Safrina O, Cahalan M. State-dependent block of Orai3 TM1 and TM3 cysteine mutants: insights into 2-APB activation. The Journal of General Physiology. 2014;143(5):621–631. doi: 10.1085/jgp.201411171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawlor K, Vince J. Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochimica and Biophysica Acta. 2014;1840:1433–1440. doi: 10.1016/j.bbagen.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 61.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nature Immunology. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Allam R, Lawlor K, Yu E, Mildenhall A, Moujalled D, Lewis R, Ke F, Mason K, White M, Stacey K, Strasser A, O’Reilly L, Alexander W, Kile B, Vaux D, Vince J. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Reports. 2014;15:982–990. doi: 10.15252/embr.201438463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.