

Abstract
Resistance to androgen deprivation therapies and increased androgen receptor (AR) activity are major drivers of castration resistant prostate cancer (CRPC). Although prior work focused on targeting AR directly, co-activators of AR signaling—which may represent new therapeutic targets—are relatively underexplored. Here we demonstrate that the mixed-lineage leukemia (MLL) complex, a well-known driver of MLL-fusion-positive leukemia, acts as a co-activator of AR signaling. AR directly interacts with the MLL complex via the menin MLL subunit. Menin expression is higher in castration resistant prostate cancer compared to hormone naïve prostate cancer and benign prostate and high menin expression correlates with poor overall survival. Treatment with a small molecule inhibitor of the menin-MLL interaction blocks AR signaling and inhibits the growth of castration resistant tumors in vivo in mice. Taken together, this work identifies the MLL complex as a critical co-activator of AR and a potential therapeutic target in advanced prostate cancer.
Keywords: Androgen deprivation, prostate cancer, MLL complex, menin, androgen receptor
For prostate cancer, androgen deprivation therapies (ADT) are front-line treatments in addition to surgery and radiotherapy for patients with high-risk localized disease, and second-generation anti-androgens such as abiraterone and enzalutamide have recently been shown to benefit patients with advanced disease1–5. However, lack of cure for patients who progress to the hormone-refractory castrate-resistant disease results in a high mortality rate6.
Androgen receptor (AR) and its downstream signaling play a critical role in the development and progression of both localized and castrate-resistant prostate cancer (CRPC)7. Despite androgen ablation therapies, castrate-resistant tumors restore AR signaling through several mechanisms, including AR gene amplification and activating mutations8–10. Substantial efforts are being invested to fully understand the regulation of AR in CRPC, and to discover novel ways to target the AR pathway11.
Mixed-lineage leukemia (MLL), a homolog of Drosophila melanogaster trithorax (trxG), is a component of a large SET-1-like histone methyl transferase (HMT) complex that possesses an inherent histone 3 lysine 4 (H3K4) methyl transferase activity12. The MLL–HMT complex consists of highly conserved core proteins including MLL, ASH2L, RbBP5 and WDR5, which are essential for the enzymatic activity of the complex13–15. Frequent translocation of the MLL gene in acute leukemia results in the formation of chimeric proteins with aberrant transcriptional activity12. However, the chimeric proteins depend on direct interaction with menin for their oncogenic activity16.
The 67 kDa Menin protein which binds to the N-terminus of MLL is essential for MLL target genes expression14,16,17,18. Small molecule inhibitors of menin-MLL interaction can block MLL fusion protein–mediated leukemic transformation19. The lack of a DNA binding motif in menin protein, is overcome by its direct interaction with MLL as described above or with other transcription factors like c-Myb and chromatin associated proteins such as lens epithelium-derived growth factor (LEDGF)20,21. The function of menin and its ability to coordinate oncogenic behavior in other cell types remains an area of active research. For example, in breast cancer the direct binding of menin to activated estrogen receptor (ER) facilitates MLL recruitment thereby modulating ER transcriptional response22. Interestingly, an oncogenic role of menin in ER positive breast cancers was suggested, as patients with high menin expression show poor outcomes22,23. Similarly menin expression is also correlated with poor prognosis in hepatocellular carcinoma24. In addition, a recent study identified menin as a potential therapeutic target in pediatric gliomas harboring H3.3K27M mutations25 and a drug screen identified MI-2, a small molecule inhibitor of the menin-MLL interaction18, which suppressed tumor growth. Taken together these studies suggest an oncogenic role of menin in solid tumors.
Here we describe a functionally important interaction between AR, menin and the MLL complex in advanced prostate cancer. We found that AR associates with the MLL histone methyltransferase complex through a direct interaction with menin. Furthermore, the MLL complex is required for AR-mediated gene expression and can be targeted with small molecule menin-MLL inhibitors, suggesting that therapies in development for MLL fusion-positive leukemia’s may have utility for castrate-resistant prostate cancer.
RESULTS
AR interacts with the MLL complex
Using co-immunoprecipitation (co-IP) assays in the AR-dependent prostate cancer cell line VCaP, we previously reported that AR interacts with proteins of the MLL complex26. To further study the nature of this interaction, we fractionated VCaP cell nuclear extracts by size-exclusion chromatography and measured the presence of AR and MLL complex proteins by immunoblot analysis. AR eluted in a fraction that contains high-molecular weight complexes, akin to the elution pattern of MLL complex components including MLL, MLL4, WDR5, ASH2L, and menin (Fig. 1a). Next we co-immunoprecipitated endogenous ASH2L, menin and AR from VCaP and another AR-dependent prostate cancer cell line, LNCaP, to confirm an association between AR and MLL complex proteins. Subsequent immunoprecipitation with AR, ASH2L and menin antibodies followed by immunoblot analysis for AR and MLL complex proteins demonstrated their association (Fig. 1b,c). To test the robustness of this interaction, we performed co-IP experiments in VCaP cells under stringent condition (350 mM NaCl), and we used a different AR antibody; in both instances, MLL complex proteins co-immunoprecipitated with AR (Supplementary Fig. 1a,b). Confocal immunofluorescence microscopy in VCaP cells also demonstrated that ASH2L and menin co-localize with AR in the nucleus (Fig. 1d). To corroborate this interaction in situ, we stained sections from benign, localized and metastatic human prostate cancer tissue with antibodies against menin, MLL and AR; AR and menin staining was predominantly nuclear in epithelial cells with some staining observed in the stroma (Supplementary Fig. 1c,d). Likewise, most of the MLL staining was primarily limited to epithelial cells; however, some smooth muscle cells also showed nuclear staining (Supplementary Fig. 1e). Collectively, these results show that AR physically associates with the MLL complex in prostate cancer cells and tissues.
Figure 1.
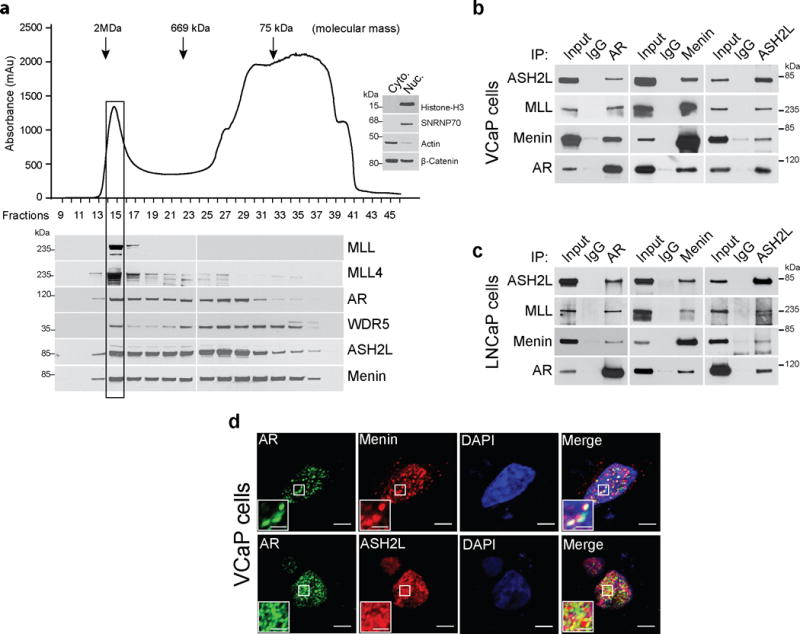
Androgen receptor interacts with MLL complex proteins. (a) VCaP nuclear lysate fractions from Superose-6 gel filtration column were immunoblotted using indicated antibodies. Arrows indicate the approximate mass of the complexes eluted. Distribution of cytoplasmic or nuclear proteins demonstrates the efficiency of lysate fractionation (inset western blot). Shown are representative blots (n=3). (b–c) VCaP (b) and LNCaP (c) nuclear lysates were immunoprecipitated (IP) with AR, ASH2L and menin antibodies followed by immunoblotting (IB) using indicated antisera. Shown are representative blots from three independent experiments. (d) VCaP cells were fixed and immunostained using AR (green) and ASH2L (red) or menin (red) antibodies. Confocal microscopic images were analyzed by ImageJ software. Co-localization is represented by yellow puncta. Bar = 10μm. Inset bar = 2.5μm. Shown are representative images from three independent experiments.
MLL complex is required for AR signaling and cell growth
Next we conducted knockdown experiments to study the role of MLL complex in AR-driven transcription. Compared to VCaP cells treated with two independent siRNAs against the MLL subunit ASH2L, cells treated with control siRNA showed higher induction of AR target gene expression after treatment with synthetic androgen (R1881), as revealed by microarray and gene set enrichment analysis (GSEA) with an AR gene signature (Fig. 2a,b, Supplementary Fig. 2a, Supplementary table S2). Similar effects of ASH2L knockdown on AR signaling (both at transcript and protein levels) were observed in LNCaP cells (Fig. 2c and Supplementary Fig. 2b–e). Next, we assessed the role of menin on AR signaling using qPCR and immunoblotting. Analogous to ASH2L, knockdown of menin resulted in a significant decrease in expression of DHT induced AR target genes (Fig. 2d,e). This was further confirmed by negative enrichment of the AR target gene signature in menin knockdown cells (Supplementary Fig. 2f,g).
Figure 2.
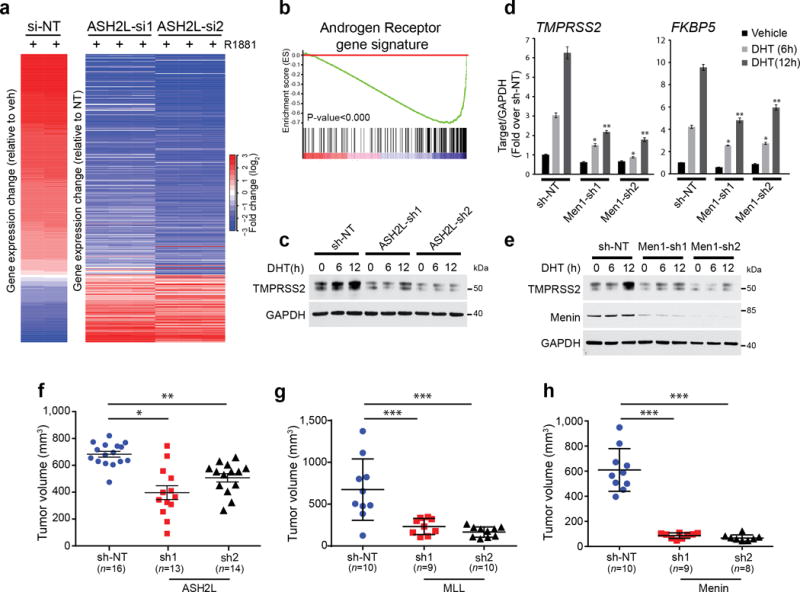
MLL complex proteins are important for AR signaling and cell growth. (a) Microarray profiling of ASH2L knockdown (two independent siRNAs) and control non-target (NT) VCaP cells following 24 hr androgen (R1881) treatment. Heatmap displays the altered expression of the androgen-induced genes upon ASH2L knockdown. (b) Gene set enrichment analysis (GSEA) of microarray data shows enrichment of AR target gene signature. (c) Immunoblots for TMPRSS2 protein expression in VCaP cells stably expressing NT shRNA or one of two independent ASH2L shRNAs after DHT stimulation for indicated time points. Shown are representative blots (n=2). (d) Expression of FKBP5 and TMPRSS2 AR-target genes was measured using qPCR in LNCaP cells stably expressing non-targeting (NT) shRNA or one of two independent menin shRNAs, after DHT stimulation for indicated time points. * P < 0.01, **P < 0.001, compared with vehicle by one-way ANOVA. (n = 3, mean ± s.e.m) (e) Immunoblots for TMPRSS2 protein expression in VCaP cells stably expressing NT shRNA or one of two independent menin shRNAs after DHT stimulation for indicated time points. Shown are representative blots (n=3). (f,g) VCaP cells expressing NT shRNA or one of two independent ASH2L (f), MLL (g) or menin (h) shRNAs respectively were injected subcutaneously into both sides of mouse dorsal flank. Tumor volumes of xenografts were measured at 7 weeks post implantation. Each dot represents individual tumors. *, P<0.01; **, P<0.001, ***P<0.0001, compared with shNT by one-way ANOVA. All graphs are shown with mean ± s.e.m.
Since menin is a crucial component of the MLL-MLL4 complex and not the MLL2-MLL3 complex, we examined the effects of MLL and MLL4 knockdown on AR signaling. Knockdown of either MLL or MLL4 using siRNA significantly attenuated the transcription of known AR target genes both in LNCaP and VCaP cells (Supplementary Fig. 3a,b). Similar results were obtained by GSEA analysis with the AR gene signature in VCaP cells expressing MLL shRNA (Supplementary Fig. 3c). Knockdown of both MLL and MLL4 did not have a synergistic effect when compared to independent knockdowns of either MLL or MLL4, suggesting that both MLL and MLL4 are necessary for MLL complex activity in this context.
Next we investigated the role of MLL in the proliferation of AR-driven prostate cancer cells. Stable ASH2L knockdown using shRNA reduced AR-mediated gene expression as evidenced by microarray (Supplementary Fig. 3d) and decreased proliferation of VCaP cells in vitro (Supplementary Fig. 4a), supporting a potential oncogenic role of MLL. Importantly, ASH2L knockdown VCaP cells generated smaller xenograft tumors in vivo compared to control VCaP cells (Fig. 2f). Similarly knockdown of menin, MLL or MLL4 decreased the proliferation of prostate cancer cells in vitro (Supplementary Fig. 4b–d) as well as growth of VCaP xenografts in vivo (Fig. 2g,h). Taken together our data suggests that MLL complex proteins are required for the AR transcriptional program and tumor growth.
The MLL complex is localized on AR target genes
Given the role of MLL complex proteins in AR transcriptional regulation, we hypothesized that the MLL complex may co-localize with AR on a genome-wide scale. To investigate this, we identified genome-wide ASH2L binding by chromatin immunoprecipitation coupled with high throughput sequencing (ChIP-Seq) in VCaP cells upon synthetic androgen (R1881) or vehicle-treatment and compared the data with published AR ChIP-Seq data27. First, we noted an overlap between ASH2L binding sites and androgen-stimulated AR binding sites (Fig. 3a,b). Next we identified a total of 15,637 distinct genome-wide individual AR peaks (FDR < 0.05) out of which 12,243 peaks increased upon androgen stimulation (Fig. 3c). For ASH2L, we identified a total 30,114 peaks (FDR < 0.05) out of which 2,187 showed increased binding upon androgen stimulation (Supplementary Fig. 5a). Importantly, we noted a significant overlap of 1,410 target regions (64.4% of ASH2L-binding sites) where both ASH2L and AR are concomitantly recruited following androgen stimulation (Fig. 3c). Representative gene promoter with overlapping AR and ASH2L binding patterns is shown in Fig. 3d, and others are shown in Supplementary Fig. 5b–e. To investigate the presence of potential cis regulatory elements among ASH2L genomic binding regions, we performed de novo motif discovery using Multiple EM for Motif Elicitation (MEME)39, on the ASH2L ChIP-Seq data. We identified significant enrichment of two androgen responsive element (ARE) half sites in the ASH2L binding site, further supporting the overlap observed between ASH2L and AR binding in AR dependent cell lines (Fig. 3e). We next examined the expression profile of genes that were within 10kb of androgen-induced ASH2L peaks, and observed a significant decrease in their expression upon ASH2L knockdown (Supplementary Fig. 5f). Similar to ASH2L, we observed an enrichment of MLL and menin on AR target genes by ChIP-PCR (Supplementary Fig. 6a–c). Taken together these data suggest that upon androgen stimulation the MLL complex is co-recruited to direct AR targets and modulates their transcriptional activation.
Figure 3.
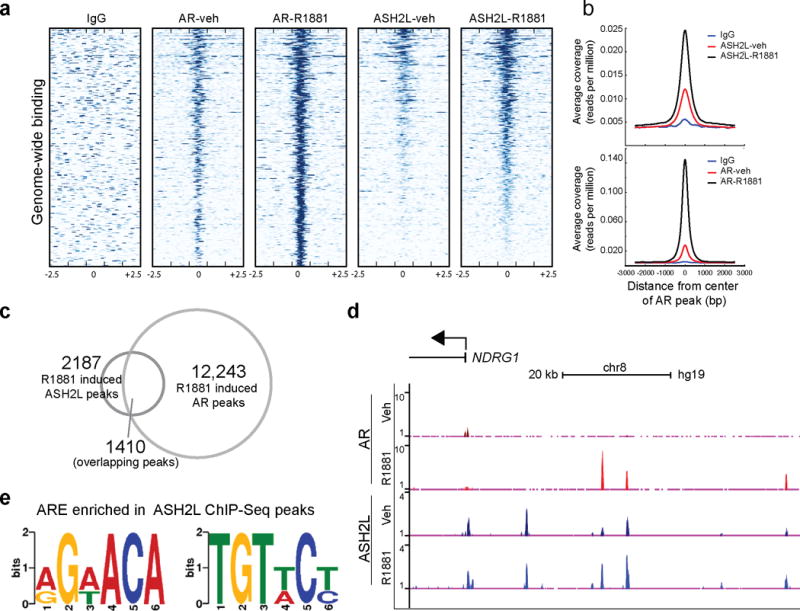
AR and ASH2L are recruited to the same genomic loci upon androgen stimulation. (a) A heat map representation of AR and ASH2L binding to promoter regions, 2.5kb flanking transcriptional start sites (TSS, indicated by 0) in vehicle and R1881 stimulated VCaP cells as assessed by ChIP-Sequencing. Gene promoters are rank-ordered by the level of AR enrichment at the TSS. (b) Average TSS-aligned profiles of AR and ASH2L occupancy for all annotated genes before and after vehicle or R1881 stimulation. (c) The overlap between R1881-induced AR and ASH2L peaks. (d) Representative gene (NDRG1) co-occupied by AR and ASH2L before and after AR stimulation. (e) De novo motif detection with MEME program identified enrichment of two half-androgen response elements (ARE) among ASH2L binding sites. (MEME E value 2.1e-025).
AR directly interacts with menin
Having demonstrated the recruitment of MLL complex proteins to AR bound chromatin regions; we sought to further characterize this interaction. We performed in vitro pull-down experiments and detected direct interaction between AR and menin (Fig. 4a). Further, we observed binding between purified untagged menin and Halo-AR (Fig. 4b). Immunoprecipitation of purified menin also pulled down Halo-AR (Fig. 4c). Next, to fine-map the AR-menin interaction, we generated deletion constructs for Halo-tagged AR (Fig. 4d), and found that menin interacts with the N-terminal domain of AR (Fig. 4e), specifically to amino acids 469–559 of the AR NTD (Fig. 4f). We examined the effect of AR stimulation on distribution of menin and saw no change (Supplementary Fig. 7a,b). Both AR and menin were mostly localized to the nucleus. Taken together, our experiments suggest a direct interaction between AR and menin.
Figure 4.
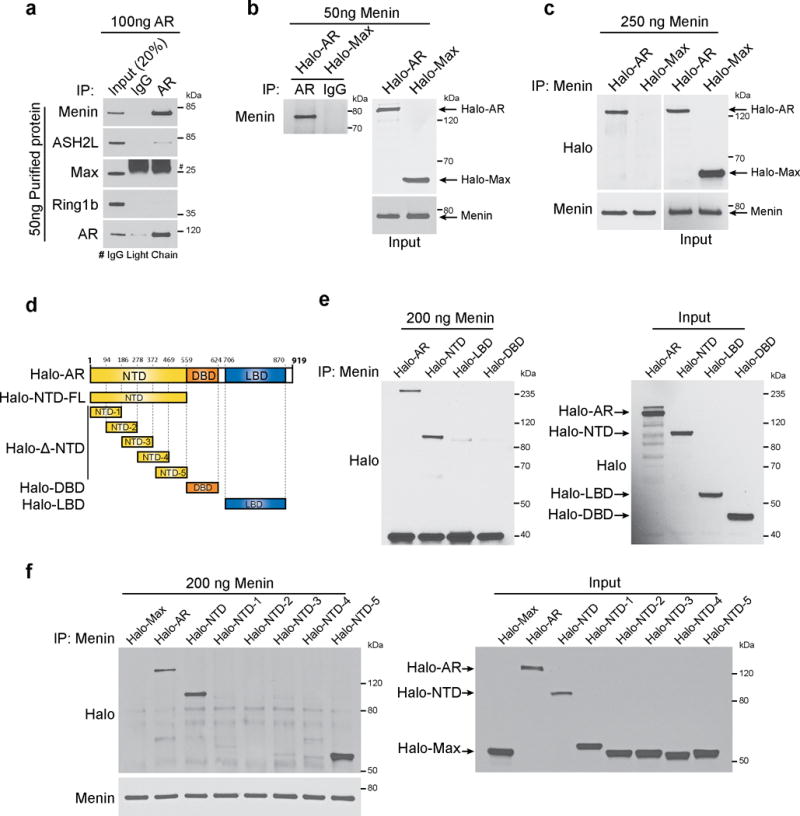
AR directly interacts with menin. (a) Purified untagged full-length (FL) AR was incubated with purified menin, ASH2L, Max or Ring1b proteins. Anti-AR antibody or control IgG immunoprecipitates (IP) were subjected to immunoblot (IB) analysis using indicated antibodies. (b) Halo-tagged FL-AR or FL-Max was incubated with purified menin. AR was immunoprecipitated using anti-AR and immunoprecipitates were subjected to immunoblot analysis with anti-menin. Immunoblots were stripped and re-probed using anti-Halo. (c) Purified menin was incubated with Halo-tagged FL-AR and FL-Max. Menin was immunoprecipitated using anti-menin and binding to AR was analyzed by immunoblotting with anti-Halo. (d) Schematic representation of Halo-tagged AR truncation mutants used in this study. (e,f) Halo-tagged FL and truncation mutants of AR were incubated with purified menin. Menin was immunoprecipitated and binding to either FL or truncation mutants of AR was analyzed by immunoblot analysis using anti-Halo. The amounts of Halo-tagged proteins used in the pull-down experiments were estimated by anti-Halo immunoblot analysis. All panels show representative blots of three independent experiments.
Menin expression is elevated in human prostate cancer
Given the importance of the MLL complex in solid tumors23–25, we examined menin expression in a set of human prostate cancer tissue samples. Using RNA-Seq we observed that menin expression was associated with disease progression with significantly elevated levels in metastatic prostate cancer compared to hormone naïve prostate cancer and benign prostate (Fig. 5a). We validated this observation using prostate cancer samples from The Cancer Genome Atlas (TCGA); that also demonstrated menin up-regulation in prostate cancer compared to benign controls (Fig. 5b). Notably, among other members of the MLL complex, while WDR5 transcript levels was elevated in metastatic prostate cancer MLL, MLL4, ASH2L and RBBP5 were not differential (Supplementary Fig. 8a–e). We next analyzed expression of menin in published microarray datasets using the Oncomine database29. Similar to the RNA-Seq data, menin expression was elevated in localized and metastatic prostate cancer in multiple published studies (Fig. 5c–d). Similarly, Menin protein was also elevated in during prostate cancer progression, with notably higher protein levels in metastatic compared to localized disease (Fig. 5e).
Figure 5.
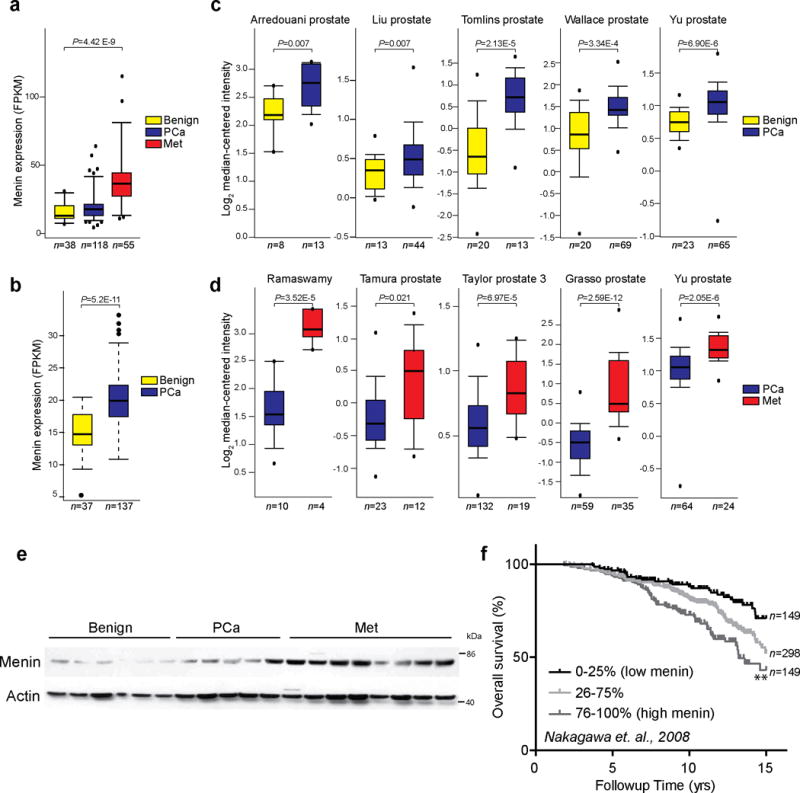
Menin is upregulated in both localized and metastatic castration resistant prostate cancer. (a,b) Menin expression in RNA-Sequencing (RNA-Seq) data from benign (n=38), localized (PCa) (n=118) and metastatic tumor (Met) (n=55) prostate tissues in Michigan Center for Translational Pathology (MCTP) cohort (a) and in benign (n=37) and localized tumor (n=137) prostate tissue samples in The Cancer Genome Atlas (TCGA) (b). The y-axis denotes Fragments Per Kilobase of transcript per Million mapped reads (FPKM). P values are calculated using one-way ANOVA (a) or t-test (b) (c,d) Menin transcript expression in multiple prostate cancer microarray studies from the Oncomine database. Datasets were analyzed for menin expression in benign vs localized prostate cancer (PCa) (c) and PCa vs metastatic castrate resistant prostate cancer (Met) (d). Study first author, statistical significance and number of samples are indicated. P values are calculated using two-sample, one-tailed Welch’s t-test. (e) Expression of menin protein in benign (n=6), PCa (n=5) and CRPC (n=8) tissues was assessed by immunoblotting. Loading control, β-actin (f) Menin mRNA expression correlates with poor overall survival by Kaplan-Meier analyses of prostate cancer outcome in the Nakagawa study. Samples were divided into quartiles based on menin expression. Expression of menin in the middle two quartiles was merged (25–75%). ** P<0.001, compared to low menin expressers based on log-rank (Mantel-Cox) test.
To assess whether menin expression is associated with poor prognosis, we used outcomes data from a large published prostate cancer study30 to carry out Kaplan-Meier analysis. We found menin mRNA overexpression was predictive of poor patient survival (Fig. 5f). Taken together, these data establish that menin is up-regulated both at the transcript and protein level in localized and metastatic prostate cancer and its expression is associated with poor survival.
Inhibition of the menin-MLL complex suppresses AR signaling
Since the MLL-HMT complex mediates AR signaling and menin is a key player in recruiting the complex to AR targets, we hypothesized that inhibiting the interaction between menin and MLL would block AR signaling and tumor growth. We utilized MI-136, a variant of a previously described inhibitor that can specifically inhibit the menin-MLL interaction19 (Supplementary Fig. 9a). AR positive cell lines such as VCaP, LNCaP and 22RV1 were sensitive to MI-136, as assessed by in-vitro cell viability assays. (Supplementary Fig. 9b). Furthermore, treatment with MI-136 blocked DHT-induced cell proliferation in AR-dependent cell lines (LNCaP and VCaP) (Supplementary Fig. 9c). The effect of MI-136 on cell proliferation was similar to MDV-3100, a second-generation FDA-approved anti-androgen for patients with refractory prostate cancer.
Next we monitored the effect of MI-136 on the AR transcriptional program using qRT-PCR on VCaP cells treated with MI-136, MDV-3100, or MI-nc (non-active control). Treatment with MI-136 and MDV-3100 inhibited DHT-induced expression of AR target genes (Supplementary Fig. 9d). Inhibition of PSA protein expression was also observed in both VCaP and LNCaP cells (Supplementary Fig. 9e,f). To examine the effects of menin inhibition on global AR signaling, we performed microarray analysis on DHT stimulated VCaP cells pre-treated with MI-136. GSEA revealed that MI-136 treatment blocked the induction of AR-upregulated genes (Fig. 6a,b). Treatment with MI-136 also inhibited the expression of genes that were bound to ASH2L after AR stimulation (Supplementary Fig. 9g, Supplementary Table 2). We further showed that treatment with MI-136 induced apoptosis of VCaP cells as evidenced by PARP (cPARP) cleavage (Supplementary Fig. 9h).
Figure 6.
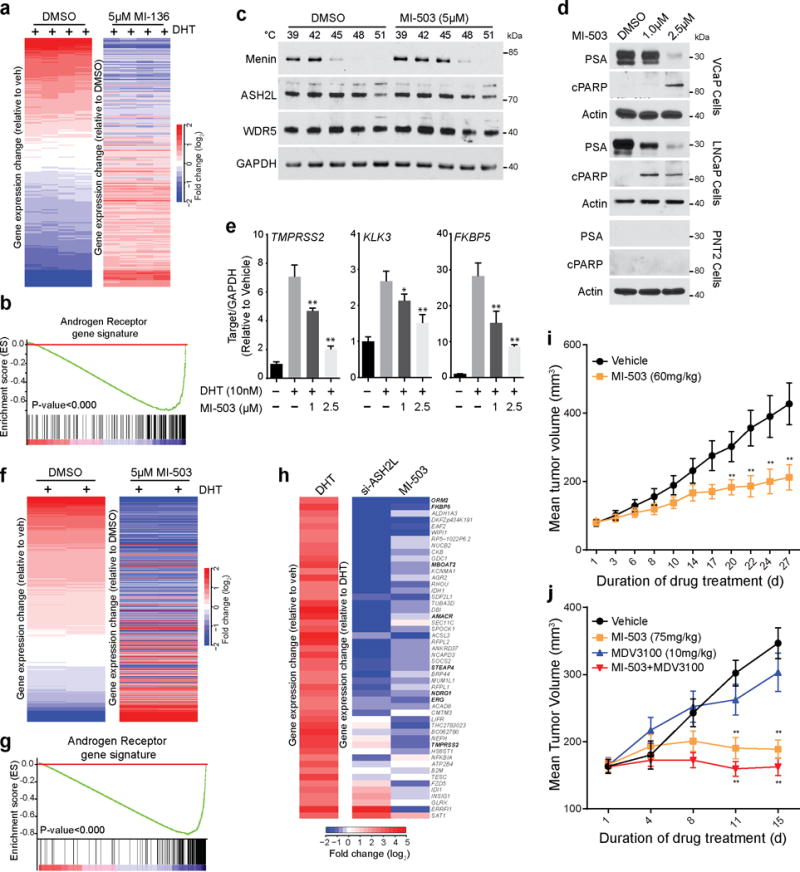
A Menin-MLL small molecule inhibitor impairs prostate cancer growth in mice. (a) Heat map representation of the impact of 5μM MI-136 treatment on DHT induced genes in VCaP cells as assessed by microarray. (b) GSEA was performed using an AR target gene signature (supplementary table S2). (c) VCaP cells were treated with 5μM MI-503 and incubated at indicated temperatures. Cells were lysed and soluble proteins were detected by immunoblotting. Shown are representative blots (n=2). (d) VCaP, LNCaP or PNT2 cells were treated with either DMSO or MI-503 for 48 hours and the effects on protein levels of PSA and cleaved PARP were determined by immunoblotting Shown are representative blots (n=3). (e) The effect on AR target gene expression (TMPRSS2, FKBP5, and KLK3) was quantified by qPCR in VCaP cells pre-treated with either DMSO or MI-503 and subsequently stimulated with 10nM DHT for 6 hours. *, P<0.01; **, P<0.001, compared with untreated by one-way ANOVA. (n = 3, mean ± s.e.m) (f) Heat map representation of the impact of 5μM MI-503 on DHT induced genes in VCaP cells was assessed by microarray. (g) GSEA was performed using an AR target gene signature (supplementary table S2). (h) Heat map representation demonstrates the effects on gene expression after ASH2L KD and MI-503 treatment. (i) Impact of MI-503 on growth of LNCaP-AR xenografts in castrated mice. LNCaP-AR xenografts were implanted in castrated mice. Once the tumors reach 80–100mm3, mice were treated daily with vehicle (n=20) or 60mg/kg MI-503 (n=18) intraperitoneally. (j) Impact of MI-503 on growth of castrate resistant VCaP xenografts. Castrate resistant VCaP tumors were generated as described in online methods. Once tumors reach 80–100mm3, mice were treated with vehicle (n=20), MI-503 (75mg/kg, n=16), MDV-3100 (10mg/kg, n=20) or combination (n=20). MI-503 was given intraperitoneally and MDV-3100 was given by oral route. Tumors are measured using caliper measurements taken bi-weekly. *, P<0.05; **, P<0.005. Compared to vehicle by a Student’s t test.
Next, we looked into the mechanism of action of MI-136. In leukemic cells, treatment with MI-136-like compounds inhibits the menin-MLL interaction19. Similarly, in prostate cancer cells, while MI-136 concentrations as low as 10 μM inhibited menin-MLL interaction (Supplementary Fig. 10a) the menin-AR interaction was retained even at 100 μM. Similar results were also seen in an in-vitro purified protein pull-down experiment (Supplementary Fig. 10b). Next, we looked at the recruitment of the MLL complex to AR target genes in the presence of MI-136. Consistent with the interaction data, we observed that recruitment of ASH2L (but not AR) to TMPRSS2 and PSA promoters was significantly decreased in the presence of MI-136 (Supplementary Fig. 10c). Together these results suggest that MI-136 inhibits AR-mediated transcription by blocking MLL recruitment predominantly at the level of the menin-MLL interaction.
Menin-MLL inhibitor reduces tumor growth in vivo
We next examined the efficacy of MI-136 in inhibiting tumor growth in vivo using VCaP xenografts31. Treatment of VCaP tumor-bearing mice with MI-136 (40mg/kg) led to a modest but significant reduction in tumor volume (Supplementary Fig. 10d) with no effect on mouse body weight (Supplementary Fig. 10e). Next, we investigated the impact of MI-136 treatment in the context of murine castration, which deprives the AR-dependent VCaP cells of circulating murine androgens (Supplementary Fig. 12c),11,32. We castrated mice bearing VCaP xenografts and when the tumors reached their original volume (approx. 100mm), these mice were treated with MI-136 (40mg/kg) and tumor regrowth was monitored. Treatment with MI-136 led to a significant decrease in the growth of castration-resistant VCaP tumors compared to vehicle controls (Supplementary Fig. 10f), confirming an important role for the menin-MLL complex in the biology of hormone-refractory prostate cancers.
But as the effects of MI-136 on VCaP tumor growth were modest, we examined the efficacy of a variant compound (MI-503), which has better solubility and bioavailability profile and is derived from the same scaffold (Supplementary Fig 11a)33. We first evaluated the target binding specificity of MI-503 using the cellular thermal shift assay (CETSA)34. Treatment of VCaP cells with MI-503 increased the levels of menin protein at 45°C (Fig. 6c), while most menin protein precipitated at 45°C in untreated cells, indicating MI-503 stabilizes the menin protein upon binding. Similar thermal stability of menin was also seen in LNCaP cells (Supplementary Fig 11b). In vitro, MI-503 had modestly lower IC50 values than MI-136 (Supplementary Fig. 11c). Like MI-136, MI-503 inhibited AR signaling, as determined by its ability to reduce expression of PSA protein (Fig. 6d) and canonical AR induced genes (Fig. 6e, Supplementary Fig. 11d) and inhibited interaction between menin and MLL (Supplementary Fig. 11e). MI-503 also induced apoptosis in VCaP and LNCaP cells as determined by PARP cleavage (Fig. 6e). We next assessed the effect of MI-503 on global AR mediated gene regulation by performing gene expression microarrays in VCaP cells. MI-503 repressed \DHT mediated gene transcription (Fig. 6f,g), and importantly had a superior effect when compared to MI-136 (Supplementary Fig. 11f). To assess if MI-503 treatment phenocopies ASH2L knockdown, we compared the microarray data obtained from VCaP cells treated with either ASH2L siRNA or MI-503 and observed significant overlap in AR target gene repression (Fig. 6h).
Next, we investigated the effect of MI-503 treatment on in vivo growth of LNCaP-AR xenograft model5. Once the LNCaP-AR xenografts were established in castrated mice, MI-503 (60mg/kg) was injected through intraperitoneal route 7 days a week, and tumor growth was monitored for 27 days (Supplementary Fig. 12a). Treatment with MI-503 significantly impeded tumor growth (Fig 6i, Supplementary Fig. 12b). Additionally, we evaluated the effect of MI-503 on VCaP xenograft growth in murine castration model, similar to MI-136 (Supplementary Fig. 12c). Treatment with MI-503 (75mg/kg) leads to a significant decrease in the growth of castrate resistant VCaP xenografts without any effect on mouse body weight (Fig. 6j, Supplementary Fig. 12d). We also compared the efficacy of MI-503 with MDV-3100 in this model. Although MDV-3100 had a less pronounced effect on tumor growth compared to MI-503, a combination treatment (MDV-3100 and MI-503) demonstrated a slightly stronger reduction in tumor growth (Fig. 6j). Next, we evaluated the post-treatment status of neuroendocrine differentiation markers in both LNCaP and VCaP xenografts. No significant change in the mRNA expression of synaptophysin and chromogranin-A was seen (Supplementary Fig. 13a–d). Taken together, these findings show that inhibition of the AR-menin interaction may enable a novel therapy for hormone-refractory prostate cancers.
DISCUSSION
Mammalian SET-domain containing proteins such as MLL1, MLL2 and SET7/9 mediate nuclear hormone receptor signaling through their ability to promote gene activation35,36. Exploring our prior observation on AR-MLL interaction26, we now reveal a mechanism of AR gene regulation mediated by MLL complex members ASH2L and menin. Specifically, our work addresses the key question of how AR signaling remains critical despite anti-androgen treatment, aiding in progression to CRPC. During this process, we establish the key regulatory role of MLL complex in AR transcriptional program, thereby uncovering a potential therapeutic angle for CRPC treatment.
In this study, we find that prostate cancer patients with menin overexpression show poor overall survival. Although menin has been extensively characterized as a tumor suppressor in multiple endocrine neoplasia type 117, our data, and previous literature on estrogen receptor, strongly argues that menin can facilitate oncogenic gene activation through hormone receptor signaling in a contextual manner.
Given that menin has a known oncogenic role in MLL-associated leukemias where small molecule inhibitors targeting menin-MLL interaction have been pursued16,19,37, we envisioned a similar strategy to disrupt AR-mediated signaling to cause tumor growth impairment. We confirmed the utility of menin-MLL inhibition both in vitro and in vivo using AR-dependent prostate cell lines and castration-resistant xenograft models.
Interestingly, inhibition of menin by shRNA or small molecules also suppressed the growth of the AR negative cell line Du145 (data not shown). This would suggest that menin could utilize other transcription factors in these cells to recruit the MLL complex. Although, there is currently a lack of experimental evidence to support this hypothesis, we speculate that the menin inhibitors would be efficacious on neuroendocrine type prostate cancer. More detailed studies directed towards understanding the role of menin and MLL complex in AR negative prostate cancer is required.
Collectively, our study proposes a model in which binding of menin to AR recruits the MLL complex to target genes modulating AR-dependent gene activation (Supplementary Fig 13e). We therefore propose menin as a key mediator of aggressive prostate cancer, and provide a rationale for the refinement of menin small molecule inhibitors as a novel therapeutic strategy for patients with advanced castration-resistant prostate cancer.
Online Methods
Cell lines, lentiviral and siRNA transfections
VCaP prostate cancer cells were cultured in DMEM (Invitrogen, Carlsbad, CA) with Glutamax (Gibco), LNCaP and 22RV1 cells in RPMI 1640 (Invitrogen), DU145 in DMEM; all the culture media were supplemented with 10% FBS (Invitrogen). LNCaP-AR cells were a gift from Dr. Charles Sawyers, Memorial Sloan Kettering Cancer Center, New York, NY. All the cell lines were cultured in a 5% CO2 cell culture incubator. To ensure the identity, all the cell lines were genotyped at the University of Michigan Sequencing Core using Profiler Plus (Applied Biosystems) and compared with the short tandem repeat (STR) profiles of respective cell lines available in the STR Profile Database (ATCC). All cell lines were tested and found to be free of mycoplasma contamination.
Lentiviral plasmid encoding shRNA targeting ASH2L (RHS4430-98851191, RHS4430-99881709) or control shRNA (GIPZ, RHS4346) were from OpenBiosystems/Dharmacon. shRNA targeting menin (TRCN0000338331, and TRCN0000040141) and control shRNA were from Sigma. shRNA targeting MLL (TL311462) and MLL4 (TL315696) and control shRNA (TR30021) were from Origene. Lentiviral particles were generated by the University of Michigan Vector Core. VCaP and LNCaP cells were infected with lentiviruses and stable cell lines were generated by selection with 5μg/ml puromycin.
Knockdown of ASH2L, MLL and MLL4 in VCaP and LNCaP cells was accomplished by RNA interference using commercially available siRNA duplexes for ASH2L (Dharmacon, Cat# J-019831-05 and J-019831-08) and MLL (Dharmacon; Cat# L-009914-00-0010), MLL4 (Dharmacon; Cat# L-009670-00-0010. Transfections were performed with OptiMEM (Invitrogen) and Oligofectamine (Invitrogen) following manufacturers instruction.
Inhibitors
Discovery, chemical synthesis and characterization of MI-136 and MI-503 compounds are described previously33.
Antibodies and immunoblot analyses
Cell lysates were separated on 4% to 12% SDS polyacrylamide gels (Novex) and transferred onto Nitrocellulose Membrane (Novex) using wet transfer (30V overnight). The membrane was incubated for one hour in blocking buffer [Tris-buffered saline, 0.1% Tween (TBS-T), 5% nonfat dry milk] and incubated overnight at 4°C with indicated antibodies. Blots were developed using enhanced chemiluminescence (ECL Prime), per the manufacturer’s protocol. All the antibodies used in this study are described in Supplementary Table 1a.
Gel Filtration Chromatography
VCaP nuclear extracts were obtained using NE-PER nuclear extraction kit (Thermo Scientific), and dialyzed against FPLC buffer (20 mM Tris-HCl, 0.2 mM EDTA, 5 mM MgCl2, 0.1 M KCl, 10% (v/v) glycerol, 0.5 mM DTT, 1 mM benzamidine, 0.2 m MPMSF, pH7.9). 5mg of nuclear protein was concentrated to 500ul using Microcon centrifugal filter (Millipore) and then applied to a Superose 6 size exclusion column (10/300 GL GE Healthcare) pre-calibrated using the Gel Filtration HMW Calibration Kit (GE Healthcare). 500 μl elute was collected for each fraction at a flow rate of 0.5ml/min, and eluted fractions were subjected to SDS-PAGE and western blotting. To assess the purity of fractionation, equal amounts of cytoplasmic and nuclear fractions were analyzed by SDS-PAGE.
Immunoprecipitation
For endogenous immunoprecipitation experiments, nuclear extracts from the prostate cancer cell lines LNCaP and VCaP were obtained using NE-PER nuclear extraction kit (Thermo Scientific). Nuclei obtained were lysed in the IP buffer (20 mM Tris-HCl, pH 7.5, 1% Triton-X, 150 mM NaCl and complete protease inhibitor cocktail (Roche)). Nuclear lysates (0.5-1.0mg) were then pre-cleaned by incubation with protein G Dynabeads (Life Technologies) for 1 hour at 4°C. 5μg antibody was added to the pre-cleared lysates and incubated on a rocker at 4°C overnight prior to the addition of protein G Dynabeads for 1hr. Beads were washed thrice in IP buffer, resuspended in 40 μL of 2× loading buffer and boiled at 90°C for 10 minutes for separation of the protein and beads. Samples were then analyzed by immunoblotting as described above.
To assess the effect of MI-136 on interaction between MLL complex and AR, VCaP cells were treated with varying concentrations of MI-136 for 24 hrs and nuclear extracts were prepared as described above. Immunoprecipitation was performed using anti-menin antibody exactly as described above followed by immunoblotting using anti-AR and anti-MLL antibody.
Immunofluorescence analysis
VCaP cells were fixed with 3.7% paraformaldehyde, and then permeabilized with 0.1% (w/v) saponin for 15 min. Cells were co-incubated with primary antibodies against AR and menin or ASH2L for 12hr at 4°C, followed by incubating with appropriate Alexa-Fluor-conjugated secondary antibodies for 30 min at 37°C. Cells were washed and mounted onto glass slides using VectaShield mounting medium containing DAPI. Samples were analyzed using a Nikon A1 laser-scanning confocal microscope equipped with a Plan-Apo ×63/1.4 numerical aperture oil lens objective. Acquired images were then analyzed using ImageJ software (version 1.41o). Scale bar: 10μm.
Gene Expression microarray
Total RNA extracted using RNeasy Mini Kit (Qiagen) and treated with RNase free DNAse (Qiagen). Integrity of RNA was assessed using bioanalyzer. Expression profiling was performed using the Agilent Whole Human Genome Oligo Microarray (SantaClara, CA) according to the manufacturer’s protocol. Microarrays were analyzed using Bioconductor and limma38. Briefly, raw Agilent data files where imported and normalized both within (method=”normexp”, offset=200) and between (method=”Aquantile”). All arrays where used for between array normalization, but log2 fold-changes were not quantile normalized as differences in the magnitude of responses are expected for the various data sets. Control probes and probes for which the log2 fold-change could not be estimated where removed. Heatmaps represent log2 fold-changes estimated by limma (all default parameters for “lmFit” and “eBayes”, using design matrices that contrast the treatment (e.g. drug + DHT) with vehicle (e.g. vehicle + DHT). For display purposes of replicate drug treatment reproducibility the mean log2 fold-changes of vehicle are subtracted from the log2 fold-changes of drug treatment. i.e. the presented log2 fold-changes are relative to the vehicle treated log2 fold-changes, which approximates statistical modeling procedure above. For GSEA analysis genes were ranked according to the shrunken limma log2 fold-changes and the GSEA tool was used in “pre-ranked” mode with all default parameters. Multiple hypothesis correction was not necessary as only the AR signature11 was used.
To assess the effect of MI-136 and MI-503 on AR signaling, VCaP cells were treated with DMSO or 5 μM MI-136 or 5 μM MI-503 for 48 hours. Cells were serum starved by replacing the media with DMEM containing 5% charcoal-striped serum and MI-136 or MI-503 for 48 hrs. Cells were then stimulated with 10nM DHT for 12 hrs and RNA was isolated and processed for expression microarrays as described above.
Quantitative RT-PCR Assay
RNA was isolated from cell lysates by the RNeasy Micro Kit (Qiagen), and cDNA was synthesized from 1 μg RNA using SuperScript III (Invitrogen) and Random Primers (Invitrogen), per the manufacturer’s protocol. qRT-PCR was carried out on the ABI7900 HT Fast Real time system (Applied Biosystems) using gene-specific primers designed with Primer3 software and synthesized by IDT Technologies. qRT-PCR data were analyzed using the relative quantification method and plotted as average fold-change compared with the control. GAPDH and actin were used as an internal reference.
To evaluation the effect of ASH2L and MLL knockdown on AR signaling, cells were first hormone starved and treated with indicated siRNAs against ASH2L, menin or MLL. After 48 hours, cells were treated with 10nM DHT and RNA isolation and qPCR was performed essentially as described above using Power SYBR Green Mastermix (Applied Biosystems). The primers used for qPCR are described in Supplementary Table 1b.
Cell Viability Assay
Cells were seeded in 96-well plates at 2000–10,000 cells/well (optimum density for growth) in a total volume of 100μl media containing 10% FBS. Serially diluted compounds in 100μl media were added to the cells 12hr later. Following 5 days of incubation, cell viability was assessed by Cell-Titer GLO (Promega). The values were normalized and IC50 was calculated using GraphPad Prism 6 software.
Chromatin Immunoprecipitation (ChIP)
The ChIP assays were performed using HighCell ChIP kit (Diagenode) according to manufacturer’s protocol. Briefly, VCaP cells were grown in charcoal-stripped serum containing media for 48 hrs followed by stimulation with 10nM DHT or 100nM R1881 for 12 hrs. Next, cells were cross-linked for 10 min with 1% formaldehyde. Cross-linking was terminated by the addition of 1/10 volume 1.25M glycine for 5 min. at room temperature followed by cell lysis and sonication (Bioruptor, Diagenode), resulting in an average chromatin fragment size of 300bp. Chromatin equivalent to 5×106 cells were used for ChIP using different antibodies. ChIP DNA was isolated from samples by incubation with the antibody at 4°C overnight followed by wash and reversal of cross-linking.
ChIP-Seq library construction and sequencing analysis
DNA was purified for library preparation using the IPure Kit (Diagenode). The ChIP-seq sample preparation for sequencing was performed according to the manufacturer’s instructions (Illumina). ChIP-enriched DNA samples (1–10 ng) were converted to blunt-ended fragments using T4 DNA polymerase, E.coli DNA polymerase I large fragment (Klenow polymerase) and T4 polynuleotide kinase (New England BioLabs, NEB). A single A-base was added to fragment ends by Klenow fragment (3′ to 5′ exo minus; NEB) followed by ligation of Illumina adaptors (Quick ligase, NEB). The adaptor-modified DNA fragments were enriched by PCR using the Illumina Barcode primers and Phusion DNA polymerase (NEB). PCR products were size selected using 3% NuSieve agarose gels (Lonza) followed by gel extraction using QIAEX II reagents (QIAGEN). Libraries were quantified with the Bioanalyzer 2100 (Agilent) and sequenced on the Illumina HiSeq 2000 Sequencer (100 nucleotide read length). ChIP-Seq data were mapped to human genome version hg19 using BWA. The MACS program was used to generate coverage map files to visualize the raw signal on the UCSC genome browser. HPeak, a Hidden Markov model (HMM)-based peak-calling software 1 designed for the identification of protein-interactive genomic regions, was employed for ChIP-seq peak determination. For enrichment plots shown in Fig. 3a and b, identified peaks for each sample are centered by peak summit and average coverage per million was counted within 1500bp relative to the peak center. The overlap of AR and ASH2L enriched regions were calculated by BEDtools. The significance of overlap between AR and ASH2L binding was calculated using hypergeometric test based on the derived number of associated genes. The heatmap for AR peak enrichment was generated using python-based script on raw data and visualized using JavaTreeView3. Motif analysis was performed using MEME motif analysis39.
Cell free protein-protein interaction studies
Full-length AR and AR truncation mutants were cloned into pFN21A vectors (Promega) in accordance to the manufacturer’s instructions. After cloning, the fusion proteins were expressed using the TNT® SP6 High-Yield Wheat Germ Reaction cell-free transcription and translation system (Cat.# L5030, Promega) following the manufacturer’s protocol. For each reaction, protein expression was confirmed by immunoblotting. Purified menin protein was kindly provided by Dr. Tomek Cierpicki. Purified AR was purchased from Prolias Technologies (R1089-2), purified ASH2L was purchased from Cayman Chemicals (Cat. No. 10946), Purified Max and Ring1B protein was kindly provided by Dr. Xiaoju Wang.
For in-vitro purified pull-down experiments, 100ng AR was incubated with 20ng purified protein in binding buffer (20mM Tris-Cl (pH 7.5), 150mM NaCl, 0.1% Triton X-100 (vol/vol), 1 mM dithiothreitol and protease inhibitors) and incubated at 4°C for 1 hr. AR was then immunoprecipitated and bound proteins were analyzed by western blotting as described above.
For interaction between menin and AR truncation mutants, 200–250ng purified menin was incubated with Halo-tagged FL-AR and truncation mutants in binding buffer. Menin was immunoprecipitated and bound proteins were analyzed by immunoblotting with anti-Halo antibody.
Cellular Thermal Shift Assay
Cellular thermal shift assay (CETSA) was performed as described before33. Briefly, cells were plated at a density of 10×106 (VCaP) or 5×106 (LNCaP) in a 10 cm Poly-D-Lysine coated plate (Corning 354469) and incubated overnight (37°C, 5% CO2). Cells were treated the following morning with DMSO or 5 uM MI-503 for 3 hours at 37°C, 5% CO2. Following the incubation the cells were washed twice with PBS to remove residual drug and detached from the surface using TrypLE solution (Life Technologies). Cells were resuspended in PBS with HALT protease inhibitors (Promega) and equal amounts of cell suspensions were aliquoted into a 0.2 mL PCR plate. The plate was heated individually at different temperatures (Veriti thermal cycler, Applied Biosystems/Life Technologies) for 4 minutes followed by cooling at room temperature for 3 minutes. Cells were lysed by 2 cycles of freeze-thawing with liquid nitrogen and soluble fractions were centrifuged at 20000 × g for 20 minutes. Soluble proteins were measured by western blotting.
Prostate Tumor Xenograft Model and Drug studies
Four to six week-old male CB17 Severe combined immunodeficiency (SCID) mice were from University of Michigan ULAM-Breeding Colony. Based on power calculation (http://www.biomath.info/power/index.htm), we have determined that more that 6 mice per group is sufficient to detect significant differences in tumor volume with high statistical power. Parental prostate cancer cells VCaP or LNCaP-AR (3 to 4×106 cells) were injected subcutaneously in 50% Matrigel (BD Biosciences, Becton Drive, NJ) into both sides of the dorsal flank of mice. Knockdowns of ASH2L, menin, and MLL genes using targeting shRNAs in VCaP background were studied for their effects on tumor growth. The efficacy of small molecule menin inhibitors (MI-136, MI-503) was similarly studied. Tumors were measured biweekly using digital calipers following the formula (π/6) (L × W2), where L = length of tumor and W = width. University of Michigan University Committee on use and care of animals (UCUCA) approved all in vivo studies. All in vivo experiments were done in a blinded fashion. The person performing the measurements was blinded to the treatment groups.
To study the effect of ASH2L, MLL and menin knockdown, VCaP cells infected with lentivirus encoding control shRNA or shRNA targeting specific genes (see Cell lines, lentiviral and siRNA transfections section above) were injected subcutaneously into both sides of the dorsal flank of mice. Tumors were measured biweekly as described above.
To study at the effect of menin inhibitors (MI-136) on tumor growth, VCaP xenografts were grown as described above. Once the tumors reached a palpable stage (80mm3), the animals were randomized and treated with either vehicle (15% DMSO and 25% PEG + PBS) (n=8) or 40mg/kg MI136 (n=9) intraperitoneally daily for the first 2 weeks, then five days a week thereafter. Before randomization, mice were excluded from the study based on three criteria. 1. If there was no tumor uptake. 2. If the tumors growth was static after 23 days of initial uptake. 3. If tumors size at the time of randomization was more than twice the SD of the mean. Tumor volumes were measured as describe above.
To assess the efficacy of MI-136 on growth of castrate resistant VCaP xenografts (CRPC model), mice were injected with VCaP cells. Once the tumors were approximately 200-300mm3 in size, tumor-bearing mice were physically castrated and tumors were observed for regression, and regrowth to approximately ~150mm3. Mice were then randomized (10 mice/group) and treated IP with vehicle or 40mg/kg MI-136.
To assess the efficacy of MI-503, the VCaP CRPC model was generated as described above. Mice were then randomized (10 mice/group) and treated with vehicle, 75mg/kg MI-503, 10mg/kg MDV3100 or 75mg/kg MI-503 and 10mg/kg MDV3100. MDV3100 was given by oral gavage and MI-503 was administered intraperitoneally.
To assess the effect of MI-503 on growth of LNCaP-AR xenografts, four week-old male SCID mice (CB.17. SCID) were surgically castrated and allowed to recover for 2-3 weeks. LNCaP-AR cells were inoculated subcutaneously and once the tumors reached 80-100mm3, mice were randomized at 10 mice/group and treated IP with vehicle or 60mg/kg MI-503 daily for 27 days.
Supplementary Material
Acknowledgments
We thank Anton Poliakov, Javed Siddiqui and Sudhanshu Shukla for helpful discussions. Bhavna Malik, Shruthi Subramanian, Kari Wilder-Romans, Sahr Yazdani, and Vijaya L. Dommeti for technical assistance. Karen Giles and Christine Betts for critically looking over the manuscript and submission of documents. University of Michigan Viral vector core for generating the lentiviral constructs and Microscopy and Image-analysis Laboratory (MIL) for help with immunofluorescence microscopy. We thank Charles Sawyers (Memorial Sloan-Kettering Cancer Center) for the LNCaP-AR cell line. This work was supported in part by the NIH Prostate Specialized Program of Research Excellence grant P50CA186786, the Early Detection Research Network grant UO1 CA111275. A.M.C. is supported by the Prostate Cancer Foundation, and the Howard Hughes Medical Institute. A.M.C. is an American Cancer Society Research Professor and a Taubman Scholar of the University of Michigan. J.G. is supported by NIH R01 grant (1R01CA160467) and LLS TRP grant (6116-12). T.C. is supported by ACS Research Scholar grant (RSG-11-082-01-DMC). A.N. is supported by R01-GM-094231. R.M is supported by Department of Defense Post-doctoral award (W81XWH-13-1-0284). R.M, I.A.A and M.C are supported by a Prostate Cancer Foundation Young Investigator Award.
Footnotes
ACCESSION CODE
Gene-expression profiling and ChIP-Seq data have been deposited in Gene Expression Omnibus under accession number GSE60842
AUTHOR CONTRIBUTIONS
R.M., A.P.K. and A.M.C. conceived and designed the research. R.M. performed most experiments with the help of A.P.K., I.A.A., J.R.P., X.W., Y.Q., and P.M.K. R.M. and X.W. carried out in vitro interaction studies. X.J. performed microarray and M.C. analyzed the data. M.K.I analyzed ChIP-Seq data. M.K.I. and Y.S.N performed gene expression analysis. J.E.-W., R.S. and F.Y.F. performed mouse xenograft studies. Y-M.W. generated ChIP-Seq libraries and X.C. performed the sequencing. N.P and L.P.K performed IHC. A.I.N., A.K.Y. and D.M. assisted with data analysis. D.B, J.G. and T.C. provided inhibitors. R.M. and A.M.C. wrote the manuscript with help from S.M.D, I.A.A and A.P.K. All authors discussed the results and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The University of Michigan has filed a patent on the menin inhibitors described in this study and J.G. and T.C. are named as co-inventors.
References
- 1.Damber JE, Aus G. Prostate cancer. Lancet. 2008;371:1710–1721. doi: 10.1016/S0140-6736(08)60729-1. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.de Bono JS, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 5.Tran C, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith MR, Cook R, Lee KA, Nelson JB. Disease and host characteristics as predictors of time to first bone metastasis and death in men with progressive castration-resistant nonmetastatic prostate cancer. Cancer. 2011;117:2077–2085. doi: 10.1002/cncr.25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 8.Chen CD, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 9.Sun S, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taplin ME, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 11.Asangani IA, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dou Y, Hess JL. Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int J Hematol. 2008;87:10–18. doi: 10.1007/s12185-007-0009-8. [DOI] [PubMed] [Google Scholar]
- 13.Dou Y, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 14.Hughes CM, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 15.Steward MM, et al. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama A, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 17.Chandrasekharappa SC, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 18.Caslini C, et al. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67:7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grembecka J, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin S, et al. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest. 2010;120:593–606. doi: 10.1172/JCI38030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreijerink KM, et al. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006;66:4929–4935. doi: 10.1158/0008-5472.CAN-05-4461. [DOI] [PubMed] [Google Scholar]
- 23.Imachi H, et al. Menin, a product of the MENI gene, binds to estrogen receptor to enhance its activity in breast cancer cells: possibility of a novel predictive factor for tamoxifen resistance. Breast Cancer Res Treat. 2010;122:395–407. doi: 10.1007/s10549-009-0581-0. [DOI] [PubMed] [Google Scholar]
- 24.Xu B, et al. Menin promotes hepatocellular carcinogenesis and epigenetically up-regulates Yap1 transcription. Proc Natl Acad Sci U S A. 2013;110:17480–17485. doi: 10.1073/pnas.1312022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014 doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grasso CS, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prensner JR, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29:742–749. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhodes DR, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa T, et al. A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS One. 2008;3:e2318. doi: 10.1371/journal.pone.0002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korenchuk S, et al. VCaP, a cell-based model system of human prostate cancer. In Vivo. 2001;15:163–168. [PubMed] [Google Scholar]
- 32.Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009;69:6027–6032. doi: 10.1158/0008-5472.CAN-09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borkin D, et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. doi: 10.1016/j.ccell.2015.02.016. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez Molina D, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 35.Mo R, Rao SM, Zhu YJ. Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J Biol Chem. 2006;281:15714–15720. doi: 10.1074/jbc.M513245200. [DOI] [PubMed] [Google Scholar]
- 36.Shi L, et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci U S A. 2011;108:7541–7546. doi: 10.1073/pnas.1017374108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou H, et al. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein-protein interaction. J Med Chem. 2013;56:1113–1123. doi: 10.1021/jm3015298. [DOI] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCE
- 38.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43 doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.