

Abstract
Local modulation of glucocorticoid action in adipocytes regulates adiposity and systemic insulin sensitivity. However, the specific cofactors that mediate glucocorticoid receptor (GR) action in adipocytes remain unclear. Here we show that the silencing mediator of retinoid and thyroid hormone receptors (SMRT) is recruited to GR in adipocytes and regulates ligand-dependent GR function. Decreased SMRT expression in adipocytes in vivo increases expression of glucocorticoid-responsive genes. Moreover, adipocytes with decreased SMRT expression exhibit altered glucocorticoid regulation of lipolysis. We conclude that SMRT regulates the metabolic functions of GR in adipocytes in vivo. Modulation of GR-SMRT interactions in adipocytes represents a novel approach to control the local degree of glucocorticoid action and thus influence adipocyte metabolic function.
Keywords: Corepressor, nuclear receptor, glucocorticoid, adipocyte
Introduction
Glucocorticoids exhibit diverse metabolic and anti-inflammatory functions. While the anti-inflammatory effects of glucocorticoids can be beneficial clinically, therapeutic doses of glucocorticoids lead to severe metabolic dysfunction (1). In particular, excess glucocorticoid action leads to obesity, insulin resistance, and Type 2 diabetes mellitus. Recent work suggests that increased local production of glucocorticoids in adipocytes leads to metabolic dysfunction. For example, an increase in adipocyte 11β-hydroxysteroid dehydrogenase Type 1 (11βHSD1), which increases the conversion from inactive to active glucocorticoids, results in insulin resistance, obesity, hypertension, and dyslipidemia (2, 3), whereas adipocyte-specific knock-out of 11βHSD1 leads to resistance to both obesity and insulin resistance (4). Thus, identification of the pathways leading to local activation of glucocorticoid action presents a promising target for the treatment of obesity and the metabolic syndrome.
Glucocorticoids act by binding to the glucocorticoid receptor (GR). In the absence of ligand, the GR is predominantly located in the cytoplasm. Recent studies have suggested a model for GR action (5), in which upon glucocorticoid binding, the GR translocates to the nucleus and binds to regions of DNA known as glucocorticoid response elements (GREs). Glucocorticoid binding to GR also induces a conformational change that allows the GR dimer to recruit a class of nuclear proteins termed coactivators. The recruitment of coactivators to the GR, via their effects on histone acetyltransferase activity and other pathways, then leads to increased DNA transcription of target genes.
There exists another class of nuclear proteins, termed corepressors, whose role in GR action is much less clear. Corepressors, such as the silencing mediator of retinoid and thyroid hormone receptors (SMRT) and the nuclear corepressor protein (NCoR), classically repress the transcriptional activity of a variety of nuclear receptors, such as the thyroid hormone receptor (TR) and the retinoic acid receptor (RAR) (6–8). More recent work suggests that SMRT and NCoR also modulates peroxisome proliferator-activated receptor gamma (PPARγ) action in adipocytes, leading to a derepression of the adipogenic program (9–12). However, the role of corepressors in GR action is much less clear, in part because the GR is predominantly located in the cytoplasm in the absence of ligand and thus does not exert basal (ligand-independent) repression.
A few studies have suggested that corepressors modulate GR transcriptional activity (13–18), but these studies have been mainly limited to an evaluation of the effects of SMRT on GR action in immortalized cell lines, and have not been able to determine how SMRT modulates GR action in metabolism. We have developed a SMRT knock-out (KO) mouse model that allows an evaluation of SMRT function on GR action in adipocytes. Although SMRT −/− mice die in utero, SMRT +/− mice survive, and exhibit increased adiposity when challenged with a high-fat diet (HFD) (19). The enhanced adiposity in the mice is due in part to greater caloric intake, however, there was also enhanced adipogenesis, likely due to the absence of SMRT repression of PPARγ. The increased activation of PPARγ also led to a modest increase in the insulin sensitivity of the adipocytes. The strong effects of the knockout on adipogenesis contrasted with the relatively mild effects on insulin sensitivity, and led us to suspect that SMRT modulates the activities of a variety of nuclear receptors, some of which may enhance insulin sensitivity and others that impair this process. Here we show that SMRT regulates GR action in adipocytes, and that altering the ability of SMRT to repress adipocytic GR action represents a novel mechanism to alter local adipocyte GR signaling and modulate adipocyte insulin sensitivity.
Materials and Methods
Mouse Treatment and Care
Mice were housed in a specific pathogen-free barrier facility with a 12-h light/dark cycle, with free access to water and a standard chow diet at the University of Chicago. We used age-matched males for each experiment and littermates when possible. All animal husbandry and animal experiments were approved by the University of Chicago Institutional Animal Care and Use Committee. After weaning at 4 weeks of age, ear clippings were obtained for genotyping by PCR. At 12–16 weeks of age, male mice were sacrificed using isofluorane and cervical dislocation.
RNA Analyses
Total RNA from epididymal fat pads was extracted using e.Z.N.A Total RNA Kit II according to manufacturer’s directions (Omega) before cDNA synthesis was performed using the QScript cDNA Super Mix (Quanta). Experiments were performed as described in the manual for SybrGreen (Quanta) with minor modifications. Each cDNA sample in triplicate was subjected to at least three individual PCR analyses using primers specific to Gilz, Lipin, or 18s RNA. RNA expression was normalized using 18s RNA, and 1.0 is defined as relative gene expression using WT adipose tissue. The primers used were Lipin: 5′-CCCTCGATTTCAACGTACCC and 5′-GCAGCCTGTGGCAATTCA. (20) Gilz: 5′-CAGCAGCCACTCAAACCAGC and 5′-ACCACATCCCCTCCAAGCAG. 18s RNA: 5′-GTAACCCGTTGAACCCCATT and 5′-CCATCCAATCGGTAGTAGCG. (21)
MEF Isolation
Mouse embryonic fibroblasts were isolated from embryonic day 13.5 embryos as previously described (19). MEF culture media included high-glucose DMEM, 8 mg/L biotin, 4 mg/L pantothenate, L-glutamine, 10% FBS, and Pen/Strep antibiotics.
Immunoprecipitation
Protein isolated from epididymal fat pads was incubated overnight with protein A/G magnetic beads (Millipore) and an Anti-SMRT (Millipore #17-10057) or Anti-GR (Santa Cruz #sc-1004) antibody. In the morning the beads were washed with RIPA buffer three times, then denatured in Laemmle buffer at 95°C. The resulting proteins were run on a western blot and probed with Anti-GR (Santa Cruz #sc-1004), Anti-CBP (#sc-369), Anti-Med1 (#sc-5334), Anti-PPARγ (#sc-7273), or Anti-SMRT (Millipore #17-10057) antibodies. Blots were performed at least three times.
Chromatin Immunoprecipitation (ChIP)
Epididymal fat pads were isolated from WT animals, minced, and incubated in DMEM with or without 100 nM Dexamethasone for two hours. Cells were crosslinked with Formaldehyde for 10 minutes and the reaction was quenched with glycine. The fat was then washed with PBS, homogenized with a polytron homogenizer, and sonicated. The resulting chromatin was incubated overnight with protein A/G magnetic beads and mouse IgG, Anti-SMRT, or Anti-GR antibodies. Beads were washed and eluted by shaking in buffer with proteinase K at 60°C for 2 hours. DNA was purified from eluent using DNA spin columns and was analyzed by running PCR with primers specific to the glucocorticoid response element (GRE) of Gilz or Lipin. The primers used were Lipin (22): 5′-TTCCCGGCTCGCATAAAGTA and 5′-AGGGAGTAGGCAGCCAAAGTC. Gilz (23): 5′-GAGCCCTTGAGAAACCAGTG and 5′-AGCTCTGGCAGAAAACGAAG. Representative blots from three ChIP assays are shown.
Luciferase Assay
MEFs were isolated from WT and KO embryos as described previously (19). Cells were grown to 60% confluence in 6 well plates and transfected with plasmids containing a luciferase tagged GRE (a generous gift of Ella Atlas), the glucocorticoid receptor (GR), and a β-galactosidase- expressing vector. The GRE sequence was GGTACATTTTGTTCTAGCCAG, and 2 copies were cloned into the pGL2 luciferase vector (24). The transfection was carried out using Transit-2020 (Mirus). 5 hours post transfection cells were treated with 0, 1, 10, or 100 nM of Dexamethasone (or ethanol) in DMEM + .5% FBS and allowed to incubate overnight. In the morning cells were washed and scraped. Luciferase activity was measured in 200 μL of cell slurry using a Lumat LB 9507 luminomator. Transfection efficiency was controlled by β-galactosidase activity, which was measured using Galacto-Light Plus (Applied Biosystems). The data presented are an average of three experiments, and each experiment was performed in triplicate.
Primary Adipocyte Isolation
Epididymal and subcutaneous WAT pads were minced in Krebs-Ringer bicarbonate HEPES buffer (KRBH) with 3% fatty acid free bovine serum albumin, 5 mM glucose, and 2 mg/ml type I collagenase (Worthington 49J11380). Samples were incubated at 37 °C, with agitation at 80 rpm for 1 hour. Tubes were then centrifuged at 1200 rpm for 1 min to separate the stromal vascular fraction and isolated primary adipocytes were transferred to 15-ml conical tubes and washed three times with KRBH + 500 nM adenosine.
Lipolysis Assay
Glycerol release from primary adipocytes was monitored as a measure of lipolysis. Primary adipocytes isolated from 12–16 week-old animals as described above, were used in lipolysis experiments. Freshly isolated primary adipocytes were incubated in KRBH buffer supplemented with 3% bovine serum albumin, 5 mM glucose, and 1U/mL adenosine deaminase were treated overnight in triplicate in four treatment groups, Basal with 100 nM PIA, 100 nM Dex, 100 ng/mL TNFα, or 100 nM Dex + 100 ng/mL TNFα. The infranatant medium was aspirated for measurement of glycerol, which was measured using Free Glycerol Reagent (Sigma). For each treatment condition, there were 4 biological replicates, and when the glycerol assay was performed it was done with technical triplicates. Statistics were performed on the biological replicates. 1.0 is defined as glycerol measurement in WT mice after overnight culture without added dexamethasone or TNF.
LDH Assay
LDH cytotoxicity assays were performed after overnight culture (conditions as per lipolysis experiments) using the Pierce Kit (Thermo Scientific #88953), per manufacturer’s instructions. Three mice per genotype were used for the experiments, and each LDH assay was performed in triplicate. 1.0 is defined as LDH measurement in WT mice after overnight culture without added dexamethasone or TNF. The experiment was also performed at baseline, but there was minimal LDH detected in all conditions at that time point.
Statistics
Statistical comparisons were made by the 2 tailed 2 sample equal variance student t test. Statistical significance is noted for p<0.05.
Results
To determine if SMRT regulates GR function, we initially isolated mouse embryonic fibroblasts (MEFs) lacking SMRT. Although SMRT homozygous deletion is embryonic lethal, we were able to generate SMRT −/− MEFs and compare them to wild-type (WT) MEFs as previously described (19). MEFs were transfected with GR and GRE-luciferase plasmids. As shown in Figure 1, dexamethasone (DEX) increases luciferase expression in WT MEFs. However, the ability of DEX to increase luciferase activity is dramatically upregulated in SMRT −/− MEFs, suggesting that SMRT represses GR action in primary cells.
Figure 1. SMRT Deficiency Increases GR-Mediated Transactivation.
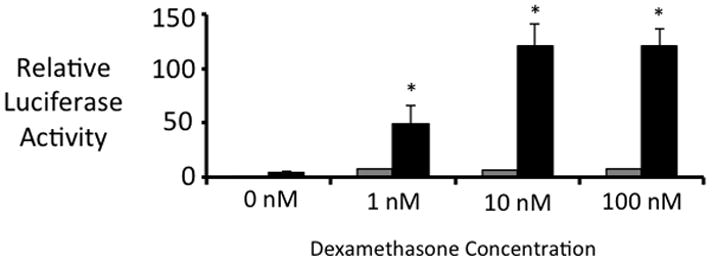
GRE-Luciferase and pSG5-GR plasmids were transfected into wild-type (black bars) or SMRT −/− (gray bars) mouse embryonic fibroblasts, along with a CMV-b-galactosidase plasmid to assess transfection efficiency. 5 hrs after transfection, the indicated amount of dexamethasone was added (0 nM – 100 nM). 24 hrs after transfection, luciferase activity was assessed. Transfection efficiency was controlled for by measuring beta-galactosidase activity. Dexamethasone statistically increased luciferase expression in WT MEFs. Dexamethasone had a significantly larger effect in SMRT −/− MEFs. The statistical evaluation noted in the figure compares luciferase expression in WT versus that in SMRT −/− cells for each dose of dexamethasone. * p<0.05 (compared to WT cells), using 2 tailed 2 sample equal variance student t test. Three experiments were performed in triplicate.
To define the mechanism by which SMRT regulates GR action, we first aimed to determine if SMRT is recruited to the GR complex in primary wild-type adipocytes by using co-immunoprecipitation experiments. We initially incubated cell extracts with an anti-SMRT antibody and pulled-down proteins that interacted with SMRT in cells, and then performed a Western blot with anti-GR or anti-PPARγ antibodies. As expected, we were able to blot for PPARγ, consistent with our prior results suggesting that SMRT is recruited to the PPARγ complex (Figure 2A). We were also able to pull down the GR with the SMRT antibody, showing that SMRT and GR also form a complex in adipocytes. We next performed the opposite experiments, in which we pulled down a complex associated with the GR. As shown in Figure 2B, we were able to identify SMRT as a member of this complex. These experiments indicate that SMRT is recruited to the GR complex in primary adipocytes.
Figure 2. SMRT is recruited to GR in adipocytes.
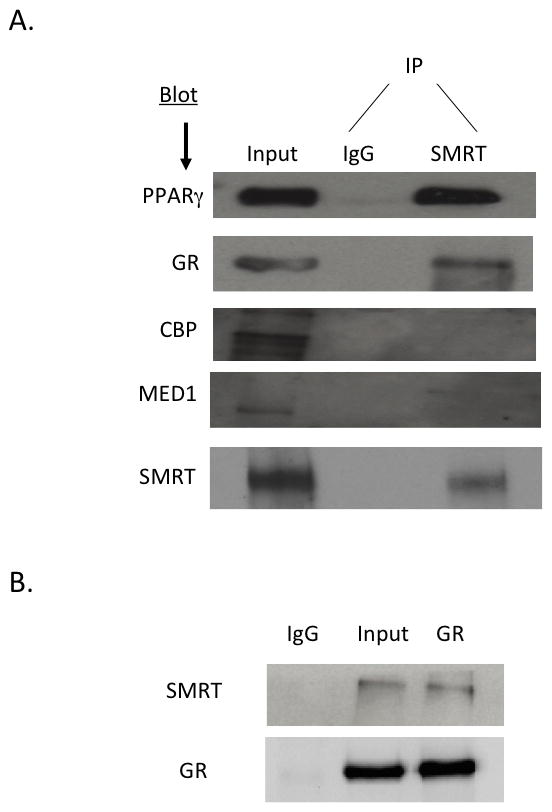
(A) Protein extracts from wild-type primary epididymal adipose tissue were incubated with an anti-SMRT antibody overnight and then assessed by Western blot using anti-GR, anti-PPARγ, anti-CBP, and anti-MED antibodies. (B) Co-immunoprecipitation experiment performed as in (A) but initially an anti-GR antibody was used to precipitate adipose tissue extracts, and an anti-SMRT antibody was used in Western blot.
To act as a corepressor of the GR, SMRT must be recruited by the GR to glucocorticoid response elements (GREs). Thus, in the next series of experiments, we took advantage of well-characterized GREs in the Lipin and Gilz gene promoters. Primary adipocytes were isolated from the epididymal depot of WT mice, and incubated with and without 100 nM DEX. As shown in Figure 3, chromatin immunoprecipitation (ChIP) experiments indicate that SMRT is recruited to the GREs of both Lipin (Fig 3A) and Gilz (Fig 3B). Interestingly, this recruitment occurs in the presence of ligand, differing from pre-existing models of SMRT repression of nuclear receptor activity.
Figure 3. SMRT is recruited to GREs in the presence of dexamethasone.
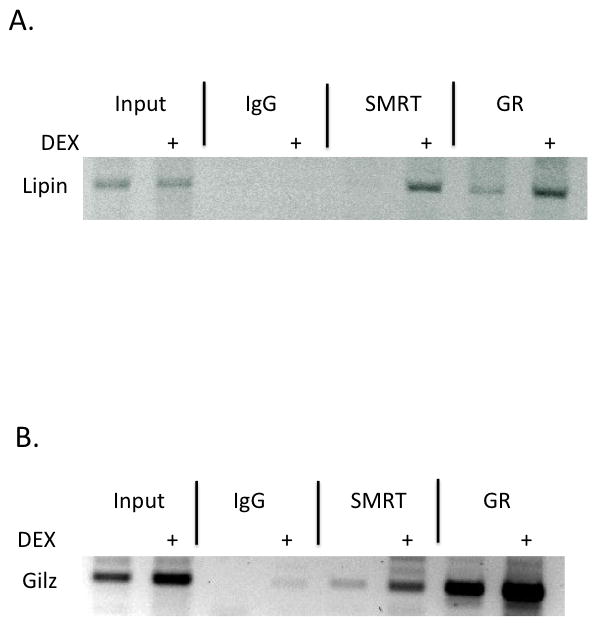
Chromatin immunoprecipitation (ChIP) experiments were performed by incubating anti-SMRT or anti-GR antibodies with extracts derived from epididymal adipose tissue treated with (+) or without 100 nM dexamethasone for 2 hours. After extensive washing, DNA was analyzed using PCR primers to glucocorticoid response elements (GREs) in the Lipin (A) or Gilz (B) genes. Three experiments were performed, and a representative experiment is presented.
To determine if SMRT regulates GR action in primary adipocytes, we isolated primary adipocytes from the epididymal depots of WT and SMRT +/− mice and assayed the amount basal gene expression of Lipin1 and GILZ, two representative glucocorticoid-responsive genes, and the ones that were evaluated in our ChIP assays. Of note, these experiments were performed from epididymal adipose tissue immediately removed WT and SMRT +/− mice, and thus represent gene expression in the setting of endogenous levels of glucocorticoids. As shown in Figure 4, the expression levels of both genes were significantly up-regulated in SMRT +/− adipocytes compared to WT adipocytes. These data suggest that SMRT influences GR action in adipocytes, as SMRT is recruited to GREs and represses the expression of down-stream genes.
Figure 4. Glucocorticoid-responsive genes are upregulated in the setting of decreased SMRT.
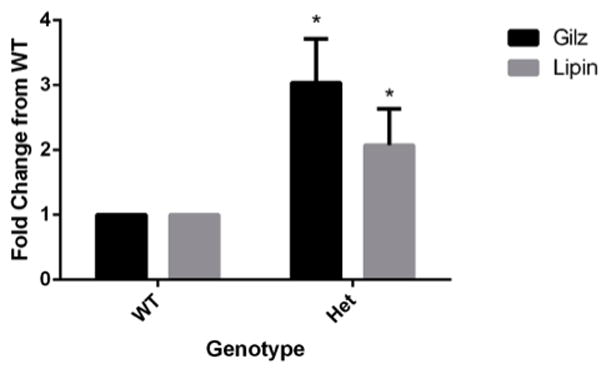
Total RNA from epididymal fat pads was extracted and cDNA synthesis was performed. Experiments were performed as described in the manual for SybrGreen (Quanta) with minor modifications. Each cDNA sample in triplicate was subjected to three individual PCR analyses using primers specific to Gilz, Lipin, or 18s RNA. Gene expression corrected using 18s data, and 1.0 was defined as the relative gene expression in WT adipose tissue. Data is expressed as fold change from WT. * p<0.05, using 2 tailed 2 sample equal variance student t test of relative gene expression when compared with WT. Data was collected from at least three experiments, and each reaction was performed in triplicate.
To further characterize the functional effects of SMRT deficiency on GR action in the adipocyte, we studied the ability of adipocytes to undergo lipolysis. It has previously been shown that glucocorticoids impair the ability of cytokines such as TNFα to stimulate lipolysis (25). As shown in Figure 5A, DEX decreased TNF-stimulated lipolysis in WT cells, as measured by glycerol release, though the effect did not reach statistical significance. In contrast, the DEX effect was much stronger in SMRT +/− adipocytes, suggesting that glucocorticoids exhibit enhanced physiologic activity in the setting of diminished SMRT expression. We performed LDH assays of both WT and SMRT +/− adipocytes, using the various treatment conditions, and found that cell death was the same in each treatment group (Fig. 5B), and did not differ between treatment groups prior to incubation (data not shown). Thus, differences in glycerol release were not due to alterations in cell viability. In our hands, treatment with DEX alone did not alter glycerol release (data not shown).
Figure 5. SMRT deficiency enhances glucocorticoid suppression of TNF-stimulated lipolysis.
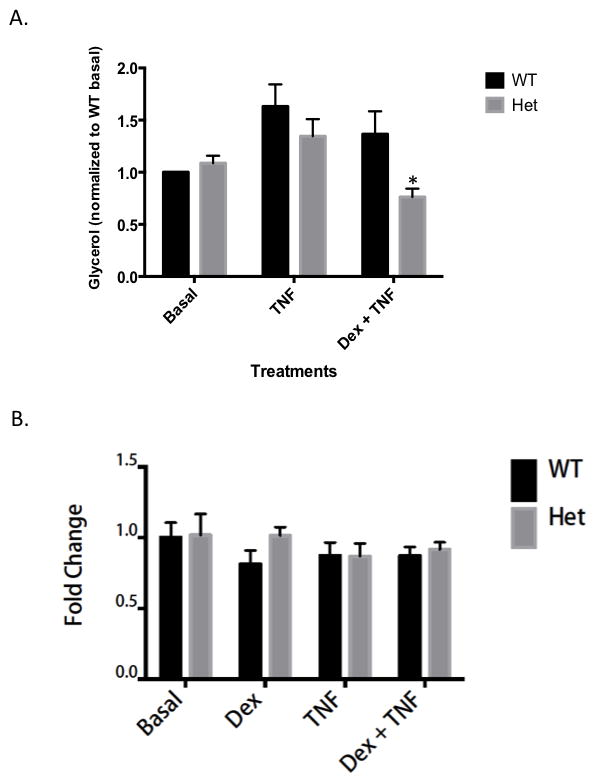
A. Glycerol release from primary adipocytes was monitored as a measure of lipolysis. Primary adipocytes isolated from 12–16 week-old animals. Freshly isolated primary adipocytes were treated overnight in triplicate in four treatment groups, Basal with 100 nM PIA, 100 nM Dex, 100 ng/mL TNFα, or 100 nM Dex + 100 ng/mL TNFα. * p<0.05, using 2 tailed 2 sample equal variance student t test, of SMRT +/− adipocytes compared with WT adipocytes. Data was collected from four mice per genotype, and experiments were performed in triplicate. 1.0 is defined as glycerol release by WT cells in the absence of dexamethasone and/or TNF. B. Primary adipocytes were obtained and treated in conditions as noted above. LDH was measured from the culture media as per manufacturers’ protocol (Thermo Scientific). Adipocytes from three mice per genotype were used, and experiments were performed in triplicate. 1.0 is defined as LDH value from WT cells in the absence of dexamethasone and/or TNF after overnight incubation.
Discussion
Although it has long been known that excess systemic glucocorticoid exposure leads to obesity and insulin resistance, it has only recently become clear that an increase in local adipocyte glucocorticoid action has dramatic systemic consequences. This has been most clearly shown with modulation of 11β-HSD1 activity. Increased adipocyte 11β-HSD1 leads to enhanced local production of active corticosterone, resulting in the systemic metabolic abnormalities usually associated with the metabolic syndrome (2, 3). In contrast, deletion of 11β-HSD1 in adipocytes increases systemic insulin sensitivity (4). Therefore, there has been great excitement in the possibility of developing selective glucocorticoid agonists or other ways that could differentially modulate glucocorticoid activity. This led us to hypothesize that corepressors might alter glucocorticoid action in adipocytes. Though glucocorticoids work via binding and activation of the GR and recruitment of coactivators, the role of corepressors in this process are less well established. While some recent studies have supported a role for corepressors in GR action (13–18), these studies have been limited by the use of immortalized cell lines and have not been designed to study the metabolic role of GR. In this study, we took advantage of our SMRT +/− knock-out mouse. We and others have shown that SMRT is recruited to the nuclear receptor PPARγ (9, 11, 12), and some of the beneficial effects of SMRT deficiency are likely mediated by this interaction. However, data by our group and others suggest that SMRT influences adipocyte actions by PPARγ-independent actions as well (26). We therefore undertook an evaluation of the effect of SMRT on GR function, since GR is known to play such an important role in differentiation and in the mature adipocyte.
Our studies suggest that SMRT and GR form a complex in adipocytes, and that SMRT represses GR transcriptional activity. Interestingly, our ChIP data also suggests that SMRT is recruited to GREs in the presence of ligand. This is very different from nuclear receptors such as TR, RAR, and PPAR, which preferentially recruit SMRT in the absence of ligand. Furthermore, this situation is different from ER, which recruits corepressors particularly in the presence of antagonists (27). Therefore, glucocorticoid binding to the GR causes the GR to translocate to the nucleus, bind DNA, and recruit both coactivators and corepressors. The binding of corepressors to liganded GR is expected to function as a brake on GR action. Although perhaps counterintuitive, recent in vivo evidence suggests that corepressor and coactivator binding to NRs is an active process, and should not be construed as an all-or-none phenomenon. It has been shown that NCoR deficiency results in an increase not only in ligand-independent TR action, but also ligand-dependent stimulation as well (28). Our studies suggest that like the TR, the GR is exposed to both coactivators and corepressors when bound to ligand.
This raises the question of how SMRT regulates GR function. The genes we evaluated (Lipin and GILZ) are positively regulated by glucocorticoids. Our data suggest that SMRT represses gene transcription in the presence of ligand, at least for these genes. We therefore suggest that the transcriptional activity of ligand-activated GR depends on the balance of forces of coactivators and corepressors, and thus may be different in each individual and/or cell type. This may explain why certain patients are more sensitive to the detrimental metabolic effects of glucocorticoids than others. We would hypothesize that such individuals might exhibit decreased levels of corepressors in their adipocytes. In addition, recent work has also suggested that certain genes that are repressed by glucocorticoids may harbor so-called negative GREs (29). It may be that these GREs are structured in such a way to lead to the more efficient recruitment of SMRT, though a further examination of such a phenomenon is beyond the scope of the current study.
In summary, our data suggest that GR recruits SMRT in adipocytes to regulate metabolic function. Liganded GR enters the nucleus, binds to DNA, and recruits both coactivators and corepressors. The balance of coactivators and/or corepressors in the particular cell, or the relative levels induced to bind the GR in the setting of a particular GRE, will dictate the degree of transcriptional activation induced by local levels of glucocorticoids. Modulation of SMRT levels or SMRT activity represents a potential approach to influence local GR function in adipocytes.
Highlights.
SMRT is a corepressor for nuclear receptors
SMRT represses ligand-dependent glucocorticoid receptor (GR) activation
SMRT regulates glucocorticoid effects on adipocyte function
These experiments highlight a novel role for SMRT in metabolism
Design of selective GR ligands must take into account the role of SMRT in GR action
Acknowledgments
The authors would like to acknowledge the NIH (NIDDK R01DK078125) and American Diabetes Association (7-13-BS-033) for grant support. We would like to thank Ella Atlas (Health Canada) for the GRE-Luciferase plasmid. We would also like to thank Alen Blagajcevic for technical support.
Abbreviations
- SMRT
Silencing Mediator of Retinoid and Thyroid Hormone Receptors
- NCoR
Nuclear Corepressor Protein
- GR
Glucocorticoid Receptor
- PPAR
Peroxisome Proliferator-Activated Receptor
- GRE
Glucocorticoid Response Element
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedman TC, Mastorakos G, Newman TD, Mullen NM, Horton EG, Costello R, Papadopoulos NM, Chrousos GP. Carbohydrate and lipid metabolism in endogenous hypercortisolism: shared features with metabolic syndrome X and NIDDM. Endocrine journal. 1996;43:645–655. doi: 10.1507/endocrj.43.645. [DOI] [PubMed] [Google Scholar]
- 2.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 3.Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90. doi: 10.1172/JCI17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kershaw EE, Morton NM, Dhillon H, Ramage L, Seckl JR, Flier JS. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes. 2005;54:1023–1031. doi: 10.2337/diabetes.54.4.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. Glucocorticoid receptor physiology. Reviews in endocrine & metabolic disorders. 2007;8:321–330. doi: 10.1007/s11154-007-9059-8. [DOI] [PubMed] [Google Scholar]
- 6.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 7.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 8.Sande S, Privalsky ML. Identification of TRACs (T3 receptor-associating cofactors), a family of cofactors that associate with, and modulate the activity of, nuclear hormone receptors. Mol Endocrinol. 1996;10:813–825. doi: 10.1210/mend.10.7.8813722. [DOI] [PubMed] [Google Scholar]
- 9.Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA. Corepressors selectively control the transcriptional activity of PPARgamma in adipocytes. Genes Dev. 2005;19:453–461. doi: 10.1101/gad.1263305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, Sears DD, Talukdar S, Oh D, Chen A, Bandyopadhyay G, Scadeng M, Ofrecio JM, Nalbandian S, Olefsky JM. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell. 2011;147:815–826. doi: 10.1016/j.cell.2011.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nofsinger RR, Li P, Hong SH, Jonker JW, Barish GD, Ying H, Cheng SY, Leblanc M, Xu W, Pei L, Kang YJ, Nelson M, Downes M, Yu RT, Olefsky JM, Lee CH, Evans RM. SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis. Proc Natl Acad Sci U S A. 2008;105:20021–20026. doi: 10.1073/pnas.0811012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem. 2005;280:13600–13605. doi: 10.1074/jbc.M409468200. [DOI] [PubMed] [Google Scholar]
- 13.Blackford JA, Jr, Guo C, Zhu R, Dougherty EJ, Chow CC, Simons SS., Jr Identification of location and kinetically defined mechanism of cofactors and reporter genes in the cascade of steroid-regulated transactivation. J Biol Chem. 2012;287:40982–40995. doi: 10.1074/jbc.M112.414805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Q, Blackford JA, Jr, Song LN, Huang Y, Cho S, Simons SS., Jr Equilibrium interactions of corepressors and coactivators with agonist and antagonist complexes of glucocorticoid receptors. Mol Endocrinol. 2004;18:1376–1395. doi: 10.1210/me.2003-0421. [DOI] [PubMed] [Google Scholar]
- 15.van der Laan S, Lachize SB, Vreugdenhil E, de Kloet ER, Meijer OC. Nuclear receptor coregulators differentially modulate induction and glucocorticoid receptor-mediated repression of the corticotropin-releasing hormone gene. Endocrinology. 2008;149:725–732. doi: 10.1210/en.2007-1234. [DOI] [PubMed] [Google Scholar]
- 16.Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS, Jr, Louw A, Hapgood JP. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. 2009;299:219–231. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Hong W, Baniahmad A, Li J, Chang C, Gao W, Liu Y. Bag-1M inhibits the transactivation of the glucocorticoid receptor via recruitment of corepressors. FEBS letters. 2009;583:2451–2456. doi: 10.1016/j.febslet.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Bush KA, Krukowski K, Eddy JL, Janusek LW, Mathews HL. Glucocorticoid receptor mediated suppression of natural killer cell activity: identification of associated deacetylase and corepressor molecules. Cellular immunology. 2012;275:80–89. doi: 10.1016/j.cellimm.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sutanto MM, Ferguson KK, Sakuma H, Ye H, Brady MJ, Cohen RN. The silencing mediator of retinoid and thyroid hormone receptors (SMRT) regulates adipose tissue accumulation and adipocyte insulin sensitivity in vivo. J Biol Chem. 2010;285:18485–18495. doi: 10.1074/jbc.M110.107680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem. 2007;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Lu Y, Yu D, Wang Y, Chen F, Yang H, Zheng SJ. Enhanced resistance of restraint-stressed mice to sepsis. J Immunol. 2008;181:3441–3448. doi: 10.4049/jimmunol.181.5.3441. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P, O’Loughlin L, Brindley DN, Reue K. Regulation of lipin-1 gene expression by glucocorticoids during adipogenesis. J Lipid Res. 2008;49:1519–1528. doi: 10.1194/jlr.M800061-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neel BA, Brady MJ, Sargis RM. The endocrine disrupting chemical tolylfluanid alters adipocyte metabolism via glucocorticoid receptor activation. Mol Endocrinol. 2013;27:394–406. doi: 10.1210/me.2012-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atlas E, Pope L, Wade MG, Kawata A, Boudreau A, Boucher JG. Bisphenol A increases aP2 expression in 3T3L1 by enhancing the transcriptional activity of nuclear receptors at the promoter. Adipocyte. 2014;3:170–179. doi: 10.4161/adip.28436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee MJ, Fried SK. Glucocorticoids antagonize tumor necrosis factor-alpha-stimulated lipolysis and resistance to the antilipolytic effect of insulin in human adipocytes. Am J Physiol Endocrinol Metab. 2012;303:E1126–1133. doi: 10.1152/ajpendo.00228.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghav SK, Waszak SM, Krier I, Gubelmann C, Isakova A, Mikkelsen TS, Deplancke B. Integrative genomics identifies the corepressor SMRT as a gatekeeper of adipogenesis through the transcription factors C/EBPbeta and KAISO. Mol Cell. 2012;46:335–350. doi: 10.1016/j.molcel.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 27.Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 28.Astapova I, Lee LJ, Morales C, Tauber S, Bilban M, Hollenberg AN. The nuclear corepressor, NCoR, regulates thyroid hormone action in vivo. Proc Natl Acad Sci U S A. 2008;105:19544–19549. doi: 10.1073/pnas.0804604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]