

Abstract
Shigellae cause significant diarrheal disease and mortality in humans, as there are approximately 163 million episodes of shigellosis and 1.1 million deaths annually. While significant strides have been made in the understanding of the pathogenesis, few studies on the genomic content of the Shigella species have been completed. The goal of this study was to characterize the genomic diversity of Shigella species through sequencing of 55 isolates representing members of each of the four Shigella species: S. flexneri, S. sonnei, S. boydii, and S. dysenteriae. Phylogeny inferred from 336 available Shigella and Escherichia coli genomes defined exclusive clades of Shigella; conserved genomic markers that can identify each clade were then identified. PCR assays were developed for each clade-specific marker, which was combined with an amplicon for the conserved Shigella invasion antigen, IpaH3, into a multiplex PCR assay. This assay demonstrated high specificity, correctly identifying 218 of 221 presumptive Shigella isolates, and sensitivity, by not identifying any of 151 diverse E. coli isolates incorrectly as Shigella. This new phylogenomics-based PCR assay represents a valuable tool for rapid typing of uncharacterized Shigella isolates and provides a framework that can be utilized for the identification of novel genomic markers from genomic data.
INTRODUCTION
Shigellae are intracellular Gram-negative pathogens that cause a wide range of illnesses, from mild abdominal discomfort to death, in humans and nonhuman primates (1). The estimated 165 million cases of shigellosis that occur annually (2) result in the deaths of ∼1.1 million people, most in the developing world. An additional 500,000 cases of shigellosis occur in travelers from developed countries (3). In the recent landmark Global Enteric Multisite Study (GEMS), Shigella species were identified as being among of the pathogens most associated with mortality (4, 5). Additionally, the GEMS suggested that houseflies could contribute to the spread of Shigella, introducing a novel route of transmission of this human pathogen (6). There are four species of Shigella that can cause diarrheal disease in humans, S. boydii, S. dysenteriae, S. flexneri, and S. sonnei, formally termed serotypes A to D (7). These species are currently defined by serotyping based on components of the O-specific side chain of the lipopolysaccharide.
Shigella species are frequently identified in the laboratory by their lack of both motility and lactose fermentation, but those biochemical assays often cannot differentiate Shigella from some enteroinvasive Escherichia coli (EIEC) isolates (8). Additionally, clinical symptoms often cannot differentiate Shigella infection from Escherichia coli infection or distinguish between Shigella species (8). Further confounding accurate identification, some O antigens present in Shigella are identical to those found in E. coli (9). Serotyping is the current gold standard method for Shigella species determination, but cross-reactivity among Shigella isolates, and E. coli isolates, may confound results (10).
The diversity of the isolates within the species, as well as the technical aspects of serotyping, make the identification of a molecular diagnostic an important objective for accurate study of the impact of this important human pathogen. In an attempt to circumvent the challenges associated with serotyping, several genetic methods, including ribotyping (11), restriction fragment length polymorphism (12), multilocus sequence typing (MLST) (13), multilocus variable-number tandem-repeat (VNTR) analysis (14), and multiplex PCR assays, have been explored to identify Shigella isolates. These approaches remain overly labor-intensive or are not sensitive enough and are unable to discriminate between Shigella species isolates or even certain E. coli isolates.
Previous MLST-based phylogenetic studies indicated that the Shigella species have emerged at least seven separate times from E. coli (15). However, MLST methods interrogate a relatively small number of conserved housekeeping genes (16). Furthermore, phylogenies inferred from concatenated MLST sequences have been demonstrated to not always be representative of phylogenies inferred using the entire conserved genomic core (17). Incongruities with gene-based trees from the sequences from a limited number of loci have been attributed to the high recombination rate observed in E. coli (18).
The genomic diversity of the Shigella species has not been studied in detail. This report contributes 55 draft Shigella assemblies to the community for the broadest analysis of Shigella species genomics to date. The goals of this study were to compare a significant number (n = 69) of Shigella genomes in order to (i) determine the phylogeny of a diverse set of Shigella isolates compared to sequenced E. coli and Shigella genomes using whole-genome sequence data, (ii) identify genomic differences between Shigella and E. coli genomes, and (iii) develop a molecular assay that identifies unknown Shigella isolates and classify them in a phylogenetic context. The use of comparative genomics to identify and validate Shigella-specific phylogenetic markers has provided an opportunity to accurately and rapidly identify these important human pathogens. Additionally, this report provides a framework for the use of genomic data in the development of diagnostics for any species.
MATERIALS AND METHODS
Strain selection.
A total of 55 Shigella genomes were selected from our culture collection for sequencing in an effort to capture a broad range of genomic, geographic, and temporal diversity (Table 1); 30 of these genomes were sequenced as part of the NIAID Genome Sequencing Center for Infectious Diseases (GSCID) project (http://gscid.igs.umaryland.edu/wp.php?wp = emerging _diarrheal_pathogens). Additional sequenced isolates included a collection of Shigella isolates (n = 12) from Nyanza Province, western Kenya, collected by the Kenya Medical Research Institute (KEMRI)/Centers for Disease Control and Prevention (CDC) Research and Public Health Collaboration that were associated with lethal clinical outcomes (19), a set of isolates from Chile (n = 2) used in previous studies (20, 21), and a collection of Canadian isolates (n = 11) obtained from the Public Health Agency of Canada through routine surveillance from 2008 to 2011; all Shigella isolates were identified based on serological analyses. A list of 281 additional genomes downloaded from GenBank, including completed genomes as well as draft assemblies, as well as the reads for an additional 96 S. flexneri genomes for comparative analyses are listed in Table S1 in the supplemental material.
TABLE 1.
Strains examined in this study
| Isolate name | Speciesa | Cladeb | MLST typec | Predicted O antigenc | Data setd | No. of contigs | Total no. of bp | GenBank accession no. |
|---|---|---|---|---|---|---|---|---|
| SB_4444-74 | S. boydii | S1 | 145 | O53 | GSCID | 314 | 4,976,495 | AKNB00000000 |
| SB_08_0009 | S. boydii | S1 | 145 | S. boydii type 2 | Canada | 165 | 4,864,228 | AMJZ00000000 |
| SB_08_2671 | S. boydii | S1 | 145 | O53 | Canada | 185 | 4,817,878 | AMKB00000000 |
| SB_S6614 | S. boydii | S1 | 145 | O150 | Kenya | 479 | 4,610,666 | AMJU00000000 |
| SB_S7334 | S. boydii | S1 | 145 | S. boydii type 2 | Kenya | 249 | 4,711,626 | AMJX00000000 |
| 248-1B | S. boydii | S1 | 145 | S. boydii O | Chile | 166 | 4,788,006 | AMKG00000000 |
| SB_08_0280 | S. boydii | S1 | 243 | S. dysenteriae type 9 | Canada | 124 | 4,835,559 | AMKA00000000 |
| SB_08_6341 | S. boydii | S1 | 243 | O164 | Canada | 138 | 4,800,746 | AMKD00000000 |
| SB_09_0344 | S. boydii | S1 | 243 | S. boydii type 2 | Canada | 174 | 4,821,210 | AMKE00000000 |
| SB_08_2675 | S. boydii | S1 | 243 | S. boydii type 2 | Canada | 335 | 4,832,830 | AMKC00000000 |
| SB_3594-74 | S. boydii | S1 | 1,130 | S. dysenteriae O | GSCID | 96 | 4,634,068 | AFGC00000000 |
| SB_965-58 | S. boydii | S3 | 250 | S. boydii type 15 | GSCID | 96 | 5,184,598 | AKNA00000000 |
| SB_5216-82 | S. boydii | S3 | 1,748 | O40 | GSCID | 75 | 4,882,454 | AFGE00000000 |
| SD_1617 | S. dysenteriae type 1 | S4 | 146 | S. dysenteriae O | GSCID | 67 | 4,613,558 | ADUT00000000 |
| SD_225-75 | S. dysenteriae type 2 | S1 | 148 | S. dysenteriae type 3 | GSCID | 111 | 4,813,171 | AKNG00000000 |
| SD_S6554 | S. dysenteriae type 2 | S1 | 243 | O150 | Kenya | 555 | 4,260,325 | AMJS00000000 |
| SD_S6205 | S. dysenteriae type 2 | S3 | 147 | S. dysenteriae type 2 | Kenya | 582 | 5,069,695 | AMJQ00000000 |
| SD_155-74 | S. dysenteriae type 2 | S3 | 288 | S. dysenteriae O | GSCID | 114 | 5,162,699 | AFFZ00000000 |
| SF_CCH060 | S. flexneri | S1 | 145 | O13 | GSCID | 82 | 4,771,928 | AKMW00000000 |
| SF_1485-80 | S. flexneri | S1 | 145 | F6 | GSCID | 82 | 4,680,138 | SRX024343 |
| SF_K-315 | S. flexneri | S1 | 1,512 | O13 | GSCID | 79 | 4,564,844 | AKMY00000000 |
| SF_2457T | S. flexneri | S5 | 245 | O13 | GSCID | 94 | 4,807,953 | ADUV00000000 |
| SF_K-218 | S. flexneri | S5 | 245 | O13 | GSCID | 74 | 4,885,634 | AFGV00000000 |
| SF_K-304 | S. flexneri | S5 | 245 | O13 | GSCID | 104 | 4,698,223 | AFGZ00000000 |
| SF_K-404 | S. flexneri | S5 | 245 | O13 | GSCID | 91 | 4,836,578 | AKMZ00000000 |
| SF_K-671 | S. flexneri | S5 | 245 | O13 | GSCID | 82 | 4,702,647 | AFHA00000000 |
| SF_2747-71 | S. flexneri | S5 | 245 | O13 | GSCID | 50 | 4,656,186 | AFHB00000000 |
| SF_2930-71 | S. flexneri | S5 | 245 | O13 | GSCID | 50 | 4,644,642 | AFHD00000000 |
| SF_4343-70 | S. flexneri | S5 | 245 | O13 | GSCID | 63 | 4,320,710 | AFHC00000000 |
| SF_S5644 | S. flexneri | S5 | 245 | O13 | Kenya | 491 | 4,689,099 | AMWM00000000 |
| SF_S5717 | S. flexneri | S5 | 245 | O13 | Kenya | 222 | 4,805,235 | AMJP00000000 |
| SF_S6585 | S. flexneri | S5 | 245 | O13 | Kenya | 274 | 4,815,370 | AMJT00000000 |
| SF_S6678 | S. flexneri | S5 | 245 | O13 | Kenya | 247 | 4,798,162 | AMJV00000000 |
| SF_S6764 | S. flexneri | S5 | 245 | O13 | Kenya | 402 | 4,673,236 | AMJW00000000 |
| SF_S7737 | S. flexneri | S5 | 245 | O13 | Kenya | 288 | 4,583,415 | AMJY00000000 |
| SF_K-227 | S. flexneri | S5 | 628 | O13 | GSCID | 77 | 4,804,544 | AFGY00000000 |
| SF_K-272 | S. flexneri | S5 | 628 | O13 | GSCID | 71 | 4,510,649 | AFGX00000000 |
| SF_2850-71 | S. flexneri | S5 | 628 | O13 | GSCID | 53 | 4,787,407 | AKMV00000000 |
| SF_J1713/17B | S. flexneri | S5 | 629 | O13 | GSCID | 52 | 4,729,738 | AFOW00000000 |
| SF_S6162 | S. flexneri | S5 | 630 | O13 | Kenya | 869 | 4,669,004 | ANAN00000000 |
| SF_6603-63 | S. flexneri | S5 | 1,022 | O13 | GSCID | 66 | 4,626,909 | SRX023788 |
| SF_VA-6 | S. flexneri | S5 | 1,025 | O13 | GSCID | 70 | 4,679,117 | AFGW00000000 |
| SF_K-1770 | S. flexneri | S5 | 1,025 | O13 | GSCID | 101 | 4,814,276 | AKMX00000000 |
| MT1457 | S. flexneri | S5 | 1,753 | O58 | Chile | 341 | 4,552,076 | AMKF00000000 |
| SS_Moseley | S. sonnei | S2 | 152 | O58 | GSCID | 115 | 5,145,982 | SRX024338 |
| SS_53G | S. sonnei | S2 | 152 | O58 | GSCID | 176 | 5,188,167 | ADUU00000000 |
| SS_3226-85 | S. sonnei | S2 | 152 | O58 | GSCID | 108 | 4,976,082 | AKNC00000000 |
| SS_3233-85 | S. sonnei | S2 | 152 | O58 | GSCID | 72 | 4,997,922 | AKND00000000 |
| SS_4822-66 | S. sonnei | S2 | 152 | O13 | GSCID | 91 | 4,710,354 | AKNE00000000 |
| SS_08_7765 | S. sonnei | S2 | 152 | O58 | Canada | 55 | 4,885,496 | AMKI00000000 |
| SS_08_7761 | S. sonnei | S2 | 152 | O58 | Canada | 95 | 4,893,159 | AMKH00000000 |
| SS_09_1032 | S. sonnei | S2 | 152 | O58 | Canada | 110 | 4,924,296 | AMKJ00000000 |
| SS_09_4962 | S. sonnei | S2 | 152 | O58 | Canada | 86 | 4,883,415 | AMKL00000000 |
| SS_09_2245 | S. sonnei | S2 | 152 | O58 | Canada | 92 | 4,862,336 | AMKK00000000 |
| SS_S6513 | S. sonnei | S2 | 152 | O58 | Kenya | 234 | 4,843,790 | AMJR00000000 |
Data were determined by serology.
Clade names are from the current study.
Data were determined informatically.
GSCID, Genome Sequencing Center for Infectious Diseases.
DNA extraction.
For genomic sequencing, DNA was extracted with standard methods reported previously (17). For the multiplex PCR assay, genomic DNA was prepped with a GenElute kit (SigmaAldrich). As a proof of concept, DNA was also isolated by heating 100 μl of overnight culture in a thermocycler for 10 min at 94°C; cell debris was then briefly pelleted at 4,000 × g for 5 min. These extractions were used for screening the large culture collections.
Genome sequencing and assembly.
Genomic DNA was sequenced at the Genome Resource Center at the Institute for Genome Sciences (http://www.igs.umaryland.edu/resources/grc/). As part of the GSCID project, 454 paired-end reads (3-kb insertion size) were assembled with the Celera assembler (22). Paired-end Illumina reads from a GA-II platform were also assembled with a reference-guided approach (AMOScmp [23]); contigs were further processed with ABACAS (24) and IMAGE (25) to generate more-contiguous assemblies. Raw sequence reads were then mapped to the reference-guided assembly with bwa (26). To identify potential genome sequences not present in the reference genome, raw reads that failed to map to the reference-guided assembly were quality trimmed with sickle and assembled with Velvet (27). Contigs from the two methods (reference guided, de novo) were concatenated, and sequencing errors were corrected with iCORN (28).
Whole-genome alignment and phylogeny.
To identify the conserved genomic core, conserved regions in isolates from E. coli and Shigella species were identified from a Mugsy (29) alignment of a diverse set of reference genomes (n = 40) (30). These conserved genomic regions were then extracted from 336 E. coli and Shigella genomes (see Table S1 in the supplemental material) with BLASTN (31), aligned with MUSCLE (32), and concatenated. A tree was inferred on the basis of reduced alignment with FastTree2 (33), with the following settings: -spr 4 -mlacc 2 -slownni.
A total of 69 Shigella genomes were aligned with Mugsy and processed as reported previously (17); this set included the 55 genomes sequenced in this study as well as 14 reference genomes. From the whole-genome alignment, subtractive methods were used to identify blocks of sequence from the output that are unique to monophyletic Shigella lineages. This was accomplished by identifying blocks of sequence that were conserved only in the targeted Shigella lineage and were absent from all other Shigella genomes.
Multigene phylogenies.
For a comparison to the whole-genome phylogeny, gene-based trees were also inferred from a concatenation of multilocus sequence typing (MLST) markers (34) (see Table S2 in the supplemental material) informatically extracted from 336 E. coli and Shigella genomes; a tree was also inferred with FastTree2 from 7 markers described in a previous study of Shigella evolution (15). Recombination of aligned markers was tested with Phi (35).
Multiplex PCR screening.
PCR primers for the multiplex assay (see Table S3 in the supplemental material) were designed in Primer3 (36) based on Shigella phylogenomic markers identified with Mugsy. All PCRs were performed with GoTaq master mix (Promega). For the multiplex PCR, primers were combined as a single mixture with a final concentration of 0.14 μM for each primer set; the primer set for clade S1 was added for a final concentration of 0.28 μM. The touchdown PCR program consisted of an initial denaturation at 94°C for 5 min, followed by 2 cycles of 94°C for 45 s, 68°C for 45 s, and 72°C for 1 min; this was followed by 2 cycles with an annealing temperature of 64°C and then 28 cycles with an annealing temperature of 60°C, keeping all other parameters constant. To determine the specificity of each Shigella phylogenomic marker, 6 strain collections were screened with the multiplex assay: the collection of isolates sequenced in this study, a collection of isolates (n = 42) from western Kenya (see description above), a collection from the Public Health Agency of Canada (n = 39), a collection from Chile (n = 106), the environmental E. coli collection (n = 72) (ECOR) (37), and the diarrheagenic E. coli (DECA) collection (n = 79) (http://www.shigatox.net/stec/cgi-bin/deca) (38); the isolates from the ECOR collection were characterized by multilocus enzyme electrophoresis, and the DECA isolates have all been sequenced and deposited in public databases.
BSR analysis.
A comparison of genes between the Shigella and E. coli genomes was performed with a BLAST score ratio (BSR) analysis (39, 40). Coding region sequences (CDSs) were predicted independently with Prodigal (41) for 69 E. coli and 69 Shigella genomes (see Table S1 in the supplemental material); the E. coli genomes were randomly subsampled from all available E. coli genomes with a Python script (https://gist.github.com/jasonsahl/115d22bfa35ac932d452). All CDSs were translated with BioPython (42) and then clustered with USEARCH v6 (43) at an identity value (ID) of 0.9 to dereplicate the data set. Each unique cluster was then translated with BioPython, and peptides were aligned against their nucleotide sequences with TBLASTN in order to obtain the maximum alignment bit score. The alignment bit score for each gene was divided by the maximum bit score for all genomes in order to obtain the BSR. For the pan-genome calculation, peptides from all genomes were clustered with USEARCH (43) over a range of IDs (0.1 to 1.0). The number of clusters at each identity threshold was calculated and plotted. This procedure was applied to all Shigella genomes (n = 69) and a subset of randomly selected E. coli genomes (n = 69) (see Table S1).
O antigen typing of each newly sequenced Shigella genome.
The nucleotide sequences for all annotated Shigella O antigens (9) were downloaded. Each genome was assigned a bioinformatically derived O antigen type based on the BLAST hit most similar to previously characterized O antigen sequences.
Nucleotide sequence accession numbers.
Nucleotide sequence data determined in this work have been deposited in GenBank (see Table 1 for accession numbers).
RESULTS
Pan-genome comparisons between Shigella and E. coli.
The predicted size of the Shigella pan-genome, based on an analysis of 69 draft and finished genomes, is ∼10,000 genes (USEARCH ID threshold of ∼0.40% to ∼40% protein identity over 100% of the peptide) (Fig. 1); this number is relatively small, considering that each Shigella genome contains ∼5,050 predicted coding regions as determined on the basis of default settings in Prodigal (41). The pan-genome for E. coli, based on the same threshold, is significantly larger (∼17,000 genes) (44). This difference may be indicative of the large number of environments where E. coli can be isolated, in contrast to Shigella species, which are primarily identified as pathogens of humans.
FIG 1.
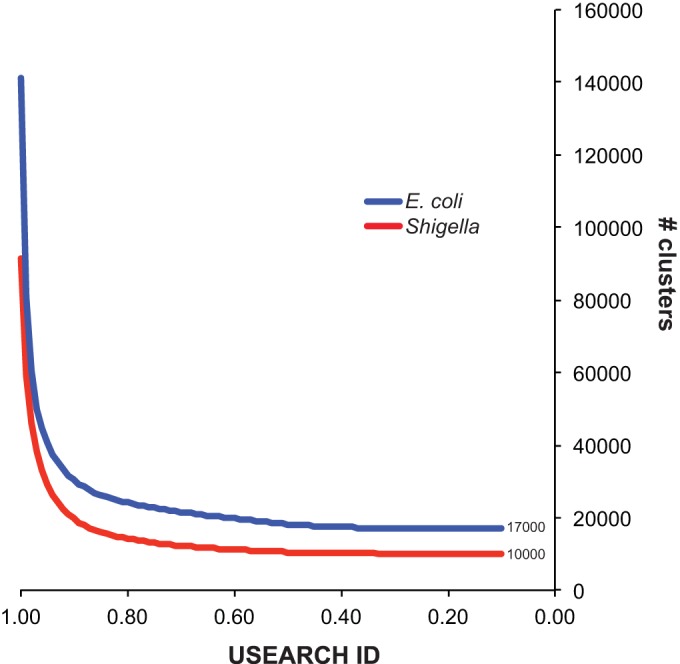
A plot of unique gene clusters at different levels of identity. Coding regions for 69 Shigella genomes or 69 E. coli genomes were predicted with Prodigal (41). Coding regions were translated with BioPython (42) and concatenated. USEARCH (43) was then used to cluster all peptides at different levels of identity. The number of unique clusters at each identity threshold was plotted for both groups. The results demonstrate the smaller pan-genome size for Shigella compared to E. coli genomes.
The numbers and compositions of genes were also compared using BLAST score ratio (BSR) analysis (40, 45). The core genome of E. coli, based on an analysis of 69 genomes, is ∼2,155 genes (BSR ≥ 0.80 in 100% of the genomes). This number is consistent with a previous calculation (44, 46) based on a smaller number of genomes. The core conserved Shigella genome consists of ∼1,880 genes (BSR ≥ 0.80 in 100% of the genomes); a list of accession numbers for the genes that are conserved in the E. coli and Shigella pan-genome is provided in Table S4 in the supplemental material. A comparison of genes between both pan-genomes demonstrates that only 1,447 genes are shared by all E. coli and Shigella isolates. This finding provides the insight that Shigella species do not have the same genomic profile as E. coli.
Genomic comparison of E. coli and Shigella genomes.
To identify genes differentially distributed between the E. coli and Shigella genomes, a large-scale BSR (LS-BSR) analysis was performed on 69 E. coli and 69 Shigella genomes (45). The results demonstrate that several genes, primarily associated with metabolism, are conserved in E. coli isolates and largely absent (n < 2) in Shigella isolates (Table 2); this stands in contrast to a recent study which suggested that no genes could be used to distinguish the two groups (47). In fact, some of the genes identified as being differentially present in E. coli and not in Shigella have been previously identified as being pathoadaptive for Shigella (48), suggesting that the analysis is valid. Genes were also identified that are differentially conserved in Shigella genomes; these include those encoding a siderophore receptor, an invasion plasmid antigen, and several hypothetical proteins (Table 2). These genes appear to be involved in pathogenesis (49), suggesting a niche specialization of Shigella compared to E. coli. The BSR matrix for these comparisons is publically available (https://github.com/jasonsahl/shigella_BSR_matrix).
TABLE 2.
Differences in BSR values in features between the E. coli and Shigella genomes
| Locus tag | Avg BSRa |
Annotation | |
|---|---|---|---|
| E. coli (n = 69) | Shigella (n = 69) | ||
| EcE24377A_0358 | 0.98 | 0.04 | 2-Methylcitrate dehydratase |
| ECO5905_06979 | 0.98 | 0.06 | Cytosine permease |
| EcHS_A0402 | 0.98 | 0.06 | Cytosine deaminase |
| HMPREF9530_02672 | 0.97 | 0.14 | Methylisocitrate lyase |
| ECNG_01839 | 0.86 | 0.08 | Hypothetical protein |
| ECSTEC94C_0398 | 0.9 | 0.11 | Lactose permease |
| ECAA86_00424 | 0.96 | 0.23 | 2-Methylcitrate synthase |
| ECSE_0359 | 0.99 | 0.26 | Propionyl-CoA synthetase |
| UTI89_C0362 | 0.98 | 0.28 | Hypothetical protein |
| EIQ72748 | 0.97 | 0.24 | Protein PrpR |
| EcSMS35_0562 | 0.99 | 0.22 | Ureidoglycolate dehydrogenase |
| SBO_4341 | 0.24 | 0.97 | Ferric siderophore receptor |
| SDY_P140 | 0.11 | 0.83 | Invasion plasmid antigen |
| SbBS512_E0714 | 0.41 | 0.99 | Hypothetical protein |
| SFK671_1049 | 0.29 | 0.89 | Hypothetical protein |
| Sd1012_0960 | 0.26 | 0.93 | Hypothetical protein |
Boldface indicates values that are ≥0.8 in one group and <0.4 in the other, indicating genes that are highly conserved in one group and absent or significantly divergent in the other.
Multigene phylogenies.
Previous conclusions regarding Shigella evolution were based on a concatenation of a small set of conserved genomic loci. To evaluate the topology of trees inferred from concatenated multigene alignments, a phylogeny was inferred from ∼3.5 kb of concatenated MLST sequences using the pubMLST system (34). The resulting MLST-based phylogeny indicates that Shigella has emerged from E. coli on five separate occasions (Fig. 2A). A phylogeny inferred from seven concatenated markers (∼7 kb) used in a previous study of Shigella evolution (15) indicates that the Shigella genotype has emerged from E. coli on a minimum of four separate occasions (Fig. 2B). These findings highlight the variability in phylogenetic placement based on the composition of the input sequence data.
FIG 2.
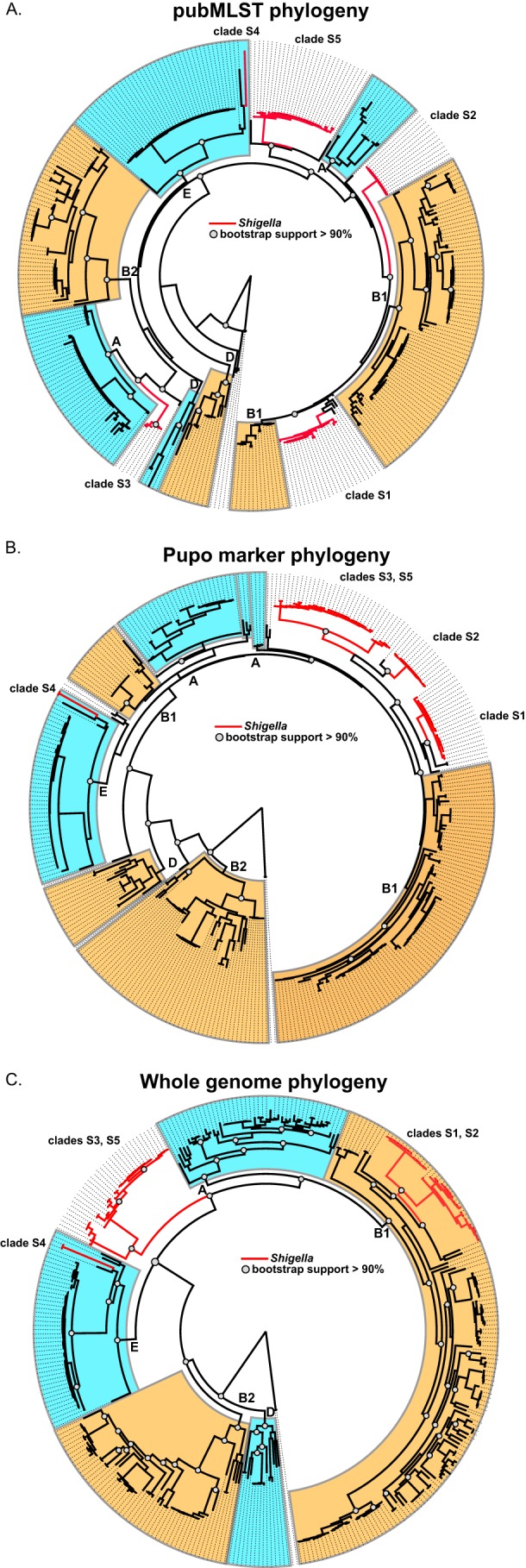
Phylogenies inferred from a diverse set of E. coli and Shigella genomes (n = 336). (A) A phylogeny inferred from a concatenation of sequences from multilocus sequence typing markers (see Table S3 in the supplemental material) from the E. coli pubMLST system (34). Conserved sequences were extracted from BLAST (31) alignments and were aligned with MUSCLE (32). The phylogenetic tree was inferred with FastTree2 (33), with 1,000 bootstrap replicates. (B) A phylogeny was inferred with FastTree2 from a concatenation of sequence markers (see Table S2) used in a previous study of Shigella evolution (15). (C) A phylogenetic tree of E. coli and Shigella isolates using whole-genome sequence data. Conserved genomic fragments were first identified in a core set of 40 E. coli genomes aligned with Mugsy (29). Conserved genomic regions were extracted by BLASTN, aligned with MUSCLE, and concatenated. A tree was then inferred on this alignment with FastTree2, with 1,000 bootstrap replicates.
Whole-genome-alignment phylogeny.
A whole-genome-based phylogeny was inferred for all E. coli and Shigella genomes, including the 55 new Shigella genomes sequenced in this study and 14 Shigella assemblies in GenBank (Fig. 2C). The resulting phylogeny illustrates the phylogenetic placement of Shigella genomes in the context of a diverse set of E. coli genomes. Clades S1 and S2 form a monophyletic clade, as do clades S3 and S5. Clade S4, which includes only S. dysenteriae type 1 isolates, is closely related to O157:H7 enterohemorrhagic E. coli (EHEC) isolates, as has been demonstrated previously (46, 50).
The conserved genomic core, based on a whole-genome alignment of 69 Shigella genomes, consists of ∼2.4 Mb of homologous sequence data. A phylogeny based on this whole-genome alignment demonstrates the presence of five clearly defined monophyletic Shigella clades (S1 to S5) (Fig. 3). The S. flexneri, S. boydii, and S. dysenteriae isolates, as defined by serology studies, did not follow a monophyletic genomic distribution within these five clades. Although Shigella isolates grouped into three clades in the comparative studies with E. coli, the Shigella-only comparisons provide subclade designations that allow improved discrimination of Shigella genomes based on genomic content.
FIG 3.

Whole-genome phylogeny. A whole-genome phylogenetic tree of 69 sequenced Shigella genomes, including 55 sequenced as part of this study, is shown. The tree was inferred with FastTree2 (33) on a Mugsy (29) whole-genome alignment, as has been done previously (17). Labels at branch nodes indicate the clade-naming convention developed in this study. Bootstrap support values from 100 replicates are shown at nodes. This tree demonstrates that Shigella genomes group into 5 monophyletic lineages (S1 to S5) and that there is a mixing of species, based on serology, in clades S1 and S3.
Identification of Shigella clade-specific genomic regions.
When the 69 Shigella genomes were aligned using Mugsy (29), no universally conserved genomic regions could be identified for any S. flexneri, S. boydii, or S. dysenteriae isolates. Therefore, an approach was employed to consider gene conservation in each of the five phylogenomic clades in the Shigella-only whole-genome phylogeny (Fig. 3), regardless of species designations based on previous identification by serotyping. Genomic regions were identified from the Mugsy alignment that were unique to each of the five clades.
The phylogenetic reconstruction clearly demonstrates that clades S1 and S3 contain a mixture of Shigella species, as defined by traditional typing, including serological methods (Fig. 3). To confirm that these anomalous genomes had not been mistyped, the closest O antigen for each genome was determined informatically (9) (Table 1). The results demonstrate that the bioinformatics-based serotyping is congruent with the laboratory-determined serotype. For example, there are four S. flexneri genomes that are included in clade S1 (Fig. 2A). A BLAST search demonstrated that each of these genomes contains the S. flexneri 6 O antigen (9), while all S. flexneri isolates from clade S5 contain the O13 antigen. The phylogeny demonstrates that genomes with the S. flexneri 6 O antigen are not closely related to sequenced S. flexneri genomes with the O13 antigen as determined on the basis of genomic content. This example highlights the difference between the phenotypic markers and the genotypic markers, which we can now integrate into the identification algorithm.
To identify the conservation of phylogenomic markers, a set of 96 S. flexneri genomes from a separate study (see Table S1 in the supplemental material) were queried with conserved S. flexneri regions identified in this study. All 96 genomes contained the S5 marker, a finding which supports the idea of the specificity of this marker for S. flexneri genomes in this genomic context (Fig. S1 in the supplemental material).
PCR assay development.
PCR assays were developed to amplify conserved genomic regions from each clade in the Shigella phylogeny (Fig. 3; see also primer sequences in Table S3 in the supplemental material). A PCR assay of the 55 isolates sequenced in this study demonstrated that a single amplicon was produced for each isolate as visualized by gel electrophoresis; the size of the band corresponded to the conserved genomic fragment designed for each clade (Fig. 3 and 4). To investigate the specificity and sensitivity of the Shigella PCR assay, additional culture collections were examined, and the results demonstrated that the conserved markers were present in temporally and geographically diverse Shigella isolates (Table 3). However, a PCR screen of two E. coli isolate collections demonstrated positive amplification of some target regions in a small number of E. coli isolates (Table 3). Therefore, the assay was redesigned to increase the specificity for Shigella genomes by adding a second, species-specific amplicon.
FIG 4.
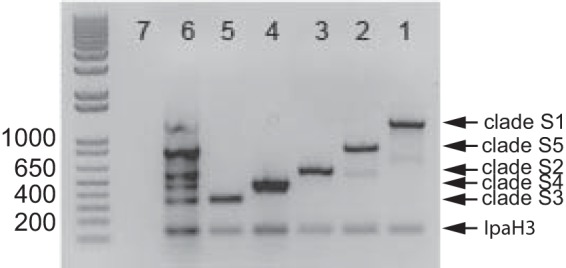
Shigella biomarker development. A gel electrophoresis image of amplicons from the 5 major clades identified in this study (lanes 1 to 5) is shown; two bands, one genus targeted (ipaH3) and one clade targeted (S1 to S5), indicate a positive reaction. Lane 6 shows a coinfection reaction with all 5 clades, plus the universally conserved ipaH3 marker. Numbers on the left represent numbers of base pairs in the DNA ladder.
TABLE 3.
Results of the multiplex PCR assay
| Isolate collection or parametera | No. of isolates |
|||
|---|---|---|---|---|
| Total | 0 bands | 1 band | 2 bands | |
| Kenyan | 42 | 0 | 0 | 42 |
| Canadian | 39 | 0 | 0 | 39 |
| Chilean | 106 | 1 | 2 | 103 |
| MSU/STEC Center | 34 | 0 | 0 | 34 |
| ECOR | 72 | 70 | 2 | 0 |
| DECA | 79 | 75 | 4 | 0 |
| Total no. of isolates screened | 372 | |||
MSU, Michigan State University; STEC, Shiga-toxigenic Escherichia coli; ECOR, environmental E. coli collection; DECA, diarrheagenic E. coli collection.
From the Mugsy alignment, genomic fragments were identified that were conserved in all 69 sequenced Shigella genomes and absent in E. coli. A BLAST search of these putative markers against a curated database of E. coli genomes identified a number of conserved Shigella markers in all 69 Shigella sequences and absent in the curated E. coli collection. One genomic fragment (240 bp) of the invasion antigen IpaH3 was found to be conserved in all Shigella genomes and also enteroinvasive E. coli (EIEC) isolates 53638 (NZ_AAKB00000000) and LT-68 (ADUP00000000). However, these EIEC isolates do not contain any of the Shigella clade-specific markers.
PCR primers designed from the IpaH3 marker were added to a mixture containing primers for all five clades. In this new multiplex assay, two bands, one for the IpaH3 marker and an additional band for the phylogenomic clade-specific marker, are required for identification of the isolate as positive for Shigella (Fig. 4). The assay can also potentially identify coinfections, where isolates from multiple clades are potentially present in a single sample (Fig. 4, lane 6). Six strain collections, totaling 372 isolates, were PCR screened with this multiplex PCR assay. The results demonstrated that, of 221 Shigella isolates collected from diverse geographic locations, 218 produced two distinct bands, indicating identification of both “genus” and phylogenomic clade. Furthermore, of 151 E. coli isolates examined, none produced two amplicons (Table 3). Three putative Shigella isolates failed to produce 2 bands, which gives a false-negative rate of 1.4% and a sensitivity of 98.6%; two of these negative isolates were serotyped as S. boydii and one isolate was typed as S. dysenteriae. Phylogenetic analysis and further genome sequencing are required to confirm the identity of these isolates.
Subclade typing.
In addition to the five major Shigella clades, PCR assays were also designed to identify Shigella genomes that did not group with other representative genomes of the same species (see Table S3 in the supplemental material). An additional PCR screen of 105 Shigella isolates from the Chilean collection identified three isolates that were typed as S. flexneri but belong to clade S1, which contains S. boydii, S. dysenteriae, and S. flerxneri, based on the multiplex assay. A PCR assay using primers designed for the S. flexneri 6 O antigen biosynthetic cluster demonstrated positive amplification for each of the three S. flexneri clade S1 isolates. PCR assays were also designed for S. dysenteriae genomes in clade S1, S. boydii genomes in clade S3, and S. dysenteriae genomes in clade S3. Although these markers are not unique to targeted genomes in each clade, they may be used for differentiation of the species, as defined by traditional serotyping, within a given phylogenomic clade.
DISCUSSION
Shigella species are intracellular human pathogens that can cause serious, potentially lethal intestinal disease, primarily in the developing world, with wide-ranging clinical manifestations, including tenesmus, abdominal pain, and bloody, mucous-like, or watery diarrhea (1) (4). On the basis of sequencing of a small number of genomic loci, Shigella species have been thought to have emerged from Escherichia coli on at least seven separate occasions. However, by analysis of the core, conserved genome, a higher-resolution analysis of evolution can be performed. The results of a whole-genome alignment and phylogenetic method utilizing 336 E. coli and Shigella genomes (Fig. 2C) clearly demonstrate that all Shigella isolates sequenced to date group into 3 monophyletic clades; this demonstrates that Shigella clades S1 and S2 are more similar to each other than they are to those of other E. coli isolates. Figure 2 also demonstrates the close relatedness of Shigella genomes, especially within clades S3 and S5.
A study by Pupo et al. divided Shigella isolates into 3 monophyletic clades, with 5 outliers, based on a phylogenetic analysis of ∼7 kb of concatenated sequence (15). Those authors concluded that the Shigella phenotype has arisen seven times, not counting the divergent S. boydii 13 isolate (15). Recent evidence has demonstrated that S. boydii 13 is not invasive and is therefore likely not similar or related to other Shigella species (51). In the current study, markers used in the study by Pupo et al. were informatically extracted from genome assemblies and used to infer a phylogeny from 336 E. coli and Shigella genomes (Fig. 2B). Four E. coli genomes were identified in this phylogeny that grouped with Shigella clades and that did not group with Shigella clades in the whole-genome phylogeny (Fig. 2B and C). Using the Phi test for recombination (35), the current study demonstrated that at least two of the markers (thrC and trpC) show signs of recombination (P value < 0.001), which may explain this incongruent topology. This highlights the difficulty with using small amounts of genetic material for the inference of genomic relatedness.
In one other study, a phylogeny was inferred from a concatenation of 345 coding regions in 25 genomes that did not show evidence of recombination; the phylogeny revealed that the Shigella genomes fell into 2 defined lineages (52). Additionally, a recent study used k-mer frequency clustering of 36 finished genomes to infer a phylogeny and showed that Shigella genomes grouped into two monophyletic clades (53). Although our results suggest the presence of three clades, all methods suggest a less diverse evolutionary history for Shigella in the broader context of a significant number of E. coli isolates. The MLST phylogeny did a relatively poor job of recapitulating the whole-genome Shigella phylogeny (Fig. 2A), as has been demonstrated previously (17).
Typically, Shigella identification is based on serological or biochemical measures in the field or laboratory (1). Based on the species concept utilized by ecologists, a species of Shigella would be expected to follow a monophyletic history (54). However, the results of our phylogenetic reconstruction based on genomic content demonstrate that Shigella species, based on serotype analysis, are not restricted to a particular phylogenomic clade. A previous study also demonstrated mixing of Shigella species across phylogenetic clades (15). This finding may have been due to a lack of consensus in strains chosen for antiserum grouping (10) and demonstrates the need for a more comprehensive genomic-based assay to understand the phylogenetic history of Shigella.
In addition to the examination of the evolutionary history, whole-genome sequencing and comparative genomic analyses have provided the opportunity to develop a robust PCR-based typing assay. In the present study, a single multiplex PCR assay, designed to produce one genus-targeted and one phylogenomic-clade-targeted amplicon, was developed based on a large-scale comparative genomics analysis. Based on the PCR screening of 218 Shigella isolates, the assay appears to universally and specifically amplify Shigella. This assay will be a valuable tool to examine both new clinical isolates and existing Shigella culture collections. One limitation to this assay is that new and emergent Shigella isolates may lack one or more of these genetic markers; however, this is the same limitation that would exist for serotyping isolates with previously uncharacterized O antigens. Additional genome sequencing will improve the understanding of the conservation and distribution of genetic markers, which will help in the continued design and verification of PCR primers for diverse and emergent isolates.
PCR assays have been used previously to detect Shigella in a variety of media (55). A recent study proposed a multiplex PCR assay to differentiate only S. sonnei and S. flexneri isolates (56) but did not factor in the remaining species. The assay presented in our study improves on this proposed multiplex assay by generating a single amplicon per genomic clade and targeting specific genomic sequences that are conserved in all Shigella species. The IpaH invasion antigen targets used in this study have previously been used to amplify and quantify Shigella isolates (57, 58). The primer set developed in this study was utilized because it amplifies a larger product than the previously published primer pair.
Shigella isolates contain a genome significantly smaller than most related E. coli genomes (50); the loss of genes is characteristic of an intracellular pathogenic lifestyle (59). In Shigella, genes that potentially interfere with pathogenesis are prone to deletion (60). Many deleted genes have been associated with cellular metabolism (15); these observations were verified by a comparative genomics analysis conducted in the current study (Table 2).
Whole-genome sequence data are an invaluable tool for the study of bacterial pathogens. In this study, genome sequence data were used to refine the evolutionary history of Shigella and focus the design of a multiplex PCR assay to characterize isolates. This method represents a new paradigm in which genome sequence data are utilized to better characterize and monitor important human pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Miles Majcher and Helen Tabor for assistance in provision of strains as well as the Canadian provincial laboratories that provided strains: British Columbia Centre for Disease Control Public Health Microbiology & Reference Laboratory, Alberta ProvLab, Cadham Provincial Laboratory (Manitoba), Ontario Public Health laboratories, and the hospital laboratories of New Brunswick.
This project was funded in part by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract number HHSN272200900009C and NIH grant number 1U19AI090873. C.R.M. was a trainee under Institutional Training Grant T32AI007540 from the National Institute of Allergy and Infectious Diseases. Additionally, J.W.S., C.R.M., and D.A.R. are supported by funds from the state of Maryland.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.03527-14.
REFERENCES
- 1.Niyogi SK. 2005. Shigellosis. J Microbiol 43:133–143. [PubMed] [Google Scholar]
- 2.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ 77:651–666. [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. 2009, posting date Initiative for vaccine research (IVR): diarrhoeal diseases. WHO, Geneva, Switzerland. [Google Scholar]
- 4.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 5.Kotloff KL, Blackwelder WC, Nasrin D, Nataro JP, Farag TH, van Eijk A, Adegbola RA, Alonso PL, Breiman RF, Faruque AS, Saha D, Sow SO, Sur D, Zaidi AK, Biswas K, Panchalingam S, Clemens JD, Cohen D, Glass RI, Mintz ED, Sommerfelt H, Levine MM. 2012. The Global Enteric Multicenter Study (GEMS) of diarrheal disease in infants and young children in developing countries: epidemiologic and clinical methods of the case/control study. Clin Infect Dis 55(Suppl 4):S232–S245. doi: 10.1093/cid/cis753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farag TH, Faruque AS, Wu Y, Das SK, Hossain A, Ahmed S, Ahmed D, Nasrin D, Kotloff KL, Panchilangam S, Nataro JP, Cohen D, Blackwelder WC, Levine MM. 2013. Housefly population density correlates with shigellosis among children in Mirzapur, Bangladesh: a time series analysis. PLoS Negl Trop Dis 7:e2280. doi: 10.1371/journal.pntd.0002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ewing WH. 1949. Shigella nomenclature. J Bacteriol 57:633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson JR. 2000. Shigella and Escherichia coli at the crossroads: Machiavellian masqueraders or taxonomic treachery? J Med Microbiol 49:583–585. [DOI] [PubMed] [Google Scholar]
- 9.Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Wang Q, Reeves PR, Wang L. 2008. Structure and genetics of Shigella O antigens. FEMS Microbiol Rev 32:627–653. doi: 10.1111/j.1574-6976.2008.00114.x. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre J, Gosselin F, Ismail J, Lorange M, Lior H, Woodward D. 1995. Evaluation of commercial antisera for Shigella serogrouping. J Clin Microbiol 33:1997–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faruque SM, Haider K, Rahman MM, Abdul Alim AR, Ahmad QS, Albert MJ, Sack RB. 1992. Differentiation of Shigella flexneri strains by rRNA gene restriction patterns. J Clin Microbiol 30:2996–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu PY, Lau YJ, Hu BS, Shyr JM, Shi ZY, Tsai WS, Lin YH, Tseng CY. 1995. Analysis of clonal relationships among isolates of Shigella sonnei by different molecular typing methods. J Clin Microbiol 33:1779–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang J, Nie H, Chen L, Zhang X, Yang F, Xu X, Zhu Y, Yu J, Jin Q. 2007. Revisiting the molecular evolutionary history of Shigella spp. J Mol Evol 64:71–79. doi: 10.1007/s00239-006-0052-8. [DOI] [PubMed] [Google Scholar]
- 14.Gorgé O, Lopez S, Hilaire V, Lisanti O, Ramisse V, Vergnaud G. 2008. Selection and validation of a multilocus variable-number tandem-repeat analysis panel for typing Shigella spp. J Clin Microbiol 46:1026–1036. doi: 10.1128/JCM.02027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pupo GM, Lan R, Reeves PR. 2000. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc Natl Acad Sci U S A 97:10567–10572. doi: 10.1073/pnas.180094797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahl JW, Steinsland H, Redman JC, Angiuoli SV, Nataro JP, Sommerfelt H, Rasko DA. 2011. A comparative genomic analysis of diverse clonal types of enterotoxigenic Escherichia coli reveals pathovar-specific conservation. Infect Immun 79:950–960. doi: 10.1128/IAI.00932-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dykhuizen DE, Green L. 1991. Recombination in Escherichia coli and the definition of biological species. J Bacteriol 173:7257–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Reilly CE, Jaron P, Ochieng B, Nyaguara A, Tate JE, Parsons MB, Bopp CA, Williams KA, Vinje J, Blanton E, Wannemuehler KA, Vulule J, Laserson KF, Breiman RF, Feikin DR, Widdowson MA, Mintz E. 2012. Risk factors for death among children less than 5 years old hospitalized with diarrhea in rural western Kenya, 2005–2007: a cohort study. PLoS Med 9:e1001256. doi: 10.1371/journal.pmed.1001256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fullá N, Prado V, Durán C, Lagos R, Levine MM. 2005. Surveillance for antimicrobial resistance profiles among Shigella species isolated from a semirural community in the northern administrative area of Santiago, Chile. Am J Trop Med Hyg 72:851–854. [PubMed] [Google Scholar]
- 21.Prado V, Lagos R, Nataro JP, San Martin O, Arellano C, Wang JY, Borczyk AA, Levine MM. 1999. Population-based study of the incidence of Shigella diarrhea and causative serotypes in Santiago, Chile. Pediatr Infect Dis J 18:500–505. doi: 10.1097/00006454-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, Anson EL, Bolanos RA, Chou HH, Jordan CM, Halpern AL, Lonardi S, Beasley EM, Brandon RC, Chen L, Dunn PJ, Lai Z, Liang Y, Nusskern DR, Zhan M, Zhang Q, Zheng X, Rubin GM, Adams MD, Venter JC. 2000. A whole-genome assembly of Drosophila. Science 287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 23.Pop M, Phillippy A, Delcher AL, Salzberg SL. 2004. Comparative genome assembly. Brief Bioinform 5:237–248. doi: 10.1093/bib/5.3.237. [DOI] [PubMed] [Google Scholar]
- 24.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. 2009. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25:1968–1969. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai IJ, Otto TD, Berriman M. 2010. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol 11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otto TD, Sanders M, Berriman M, Newbold C. 2010. Iterative correction of reference nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 26:1704–1707. doi: 10.1093/bioinformatics/btq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angiuoli SV, Salzberg SL. 9 December 2010, posting date Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics doi: 10.1093/bioinformatics/btq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahl JW, Matalka MN, Rasko DA. 2012. Phylomark, a tool to identify conserved phylogenetic markers from whole-genome alignments. Appl Environ Microbiol 78:4884–4892. doi: 10.1128/AEM.00929-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Edgar RC. 2004. MUSCLE: a multiple sequence alignment with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rozen S, Skaletsky HJ. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386. [DOI] [PubMed] [Google Scholar]
- 37.Ochman H, Selander RK. 1984. Standard reference strains of Escherichia coli from natural populations. J Bacteriol 157:690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hazen TH, Sahl JW, Redman JC, Morris CR, Daugherty SC, Chibucos MC, Sengamalay NA, Fraser-Liggett CM, Steinsland H, Whittam TS, Whittam B, Manning SD, Rasko DA. 2012. Draft genome sequences of the diarrheagenic Escherichia coli collection. J Bacteriol 194:3026–3027. doi: 10.1128/JB.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hazen TH, Sahl JW, Fraser CM, Donnenberg MS, Scheutz F, Rasko DA. 15 July 2013, posting date Refining the pathovar paradigm via phylogenomics of the attaching and effacing Escherichia coli. Proc Natl Acad Sci U S A doi: 10.1073/pnas.1306836110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasko DA, Myers GS, Ravel J. 2005. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics 6:2. doi: 10.1186/1471-2105-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJ. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 44.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, Henderson IR, Sperandio V, Ravel J. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahl JW, Caporaso JG, Rasko DA, Keim P. 2014. The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. PeerJ 2:e332. doi: 10.7717/peerj.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O, Calteau A, Chiapello H, Clermont O, Cruveiller S, Danchin A, Diard M, Dossat C, Karoui ME, Frapy E, Garry L, Ghigo JM, Gilles AM, Johnson J, Le Bouguenec C, Lescat M, Mangenot S, Martinez-Jehanne V, Matic I, Nassif X, Oztas S, Petit MA, Pichon C, Rouy Z, Ruf CS, Schneider D, Tourret J, Vacherie B, Vallenet D, Medigue C, Rocha EP, Denamur E. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gordienko EN, Kazanov MD, Gelfand MS. 2013. Evolution of pan-genomes of Escherichia coli, Shigella spp., and Salmonella enterica. J Bacteriol 195:2786–2792. doi: 10.1128/JB.02285-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Day WA Jr, Fernandez RE, Maurelli AT. 2001. Pathoadaptive mutations that enhance virulence: genetic organization of the cadA regions of Shigella spp. Infect Immun 69:7471–7480. doi: 10.1128/IAI.69.12.7471-7480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Payne SM, Wyckoff EE, Murphy ER, Oglesby AG, Boulette ML, Davies NM. 2006. Iron and pathogenesis of Shigella: iron acquisition in the intracellular environment. Biometals 19:173–180. doi: 10.1007/s10534-005-4577-x. [DOI] [PubMed] [Google Scholar]
- 50.Hershberg R, Tang H, Petrov DA. 2007. Reduced selection leads to accelerated gene loss in Shigella. Genome Biol 8:R164. doi: 10.1186/gb-2007-8-8-r164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walters LL, Raterman EL, Grys TE, Welch RA. 2012. Atypical Shigella boydii 13 encodes virulence factors seen in attaching and effacing Escherichia coli. FEMS Microbiol Lett 328:20–25. doi: 10.1111/j.1574-6968.2011.02469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogura Y, Ooka T, Iguchi A, Toh H, Asadulghani M, Oshima K, Kodama T, Abe H, Nakayama K, Kurokawa K, Tobe T, Hattori M, Hayashi T. 2009. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc Natl Acad Sci U S A 106:17939–17944. doi: 10.1073/pnas.0903585106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sims GE, Kim SH. 2 May 2011, posting date Whole-genome phylogeny of Escherichia coli/Shigella group by feature frequency profiles (FFPs). Proc Natl Acad Sci U S A doi: 10.1073/pnas.1105168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiley EO. 1978. The evolutionary species concept reconsidered. Syst Zool 27(1):17–26. [Google Scholar]
- 55.Frankel G, Riley L, Giron JA, Valmassoi J, Friedmann A, Strockbine N, Falkow S, Schoolnik GK. 1990. Detection of Shigella in feces using DNA amplification. J Infect Dis 161:1252–1256. doi: 10.1093/infdis/161.6.1252. [DOI] [PubMed] [Google Scholar]
- 56.Farfán MJ, Garay TA, Prado CA, Filliol I, Ulloa MT, Toro CS. 2010. A new multiplex PCR for differential identification of Shigella flexneri and Shigella sonnei and detection of Shigella virulence determinants. Epidemiol Infect 138:525–533. doi: 10.1017/S0950268809990823. [DOI] [PubMed] [Google Scholar]
- 57.Vu DT, Sethabutr O, Von Seidlein L, Tran VT, Do GC, Bui TC, Le HT, Lee H, Houng HS, Hale TL, Clemens JD, Mason C, Dang DT. 2004. Detection of Shigella by a PCR assay targeting the ipaH gene suggests increased prevalence of shigellosis in Nha Trang, Vietnam. J Clin Microbiol 42:2031–2035. doi: 10.1128/JCM.42.5.2031-2035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lindsay B, Ochieng JB, Ikumapayi UN, Toure A, Ahmed D, Li S, Panchalingam S, Levine MM, Kotloff K, Rasko DA, Morris CR, Juma J, Fields BS, Dione M, Malle D, Becker SM, Houpt ER, Nataro JP, Sommerfelt H, Pop M, Oundo J, Antonio M, Hossain A, Tamboura B, Stine OC. 2013. Quantitative PCR for detection of Shigella improves ascertainment of Shigella burden in children with moderate-to-severe diarrhea in low-income countries. J Clin Microbiol 51:1740–1746. doi: 10.1128/JCM.02713-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moran NA. 2002. Microbial minimalism: genome reduction in bacterial pathogens. Cell 108:583–586. doi: 10.1016/S0092-8674(02)00665-7. [DOI] [PubMed] [Google Scholar]
- 60.Lan R, Reeves PR. 2002. Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect 4:1125–1132. doi: 10.1016/S1286-4579(02)01637-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.