

Abstract
Magic-angle spinning nuclear magnetic resonance is well suited for the study of membrane proteins in the nativelike lipid environment. However, the natural cellular membrane is invariably more complex than the proteoliposomes most often used for solid-state NMR (SSNMR) studies, and differences may affect the structure and dynamics of the proteins under examination. In this work we use SSNMR and other biochemical and biophysical methods to probe the structure of a seven-transmembrane helical photoreceptor, Anabaena sensory rhodopsin (ASR), prepared in the Escherichia coli inner membrane, and compare it to that in a bilayer formed by DMPC/DMPA lipids. We find that ASR is organized into trimers in both environments but forms two-dimensional crystal lattices of different symmetries. It favors hexagonal packing in liposomes, but may form a square lattice in the E. coli membrane. To examine possible changes in structure site-specifically, we perform two- and three-dimensional SSNMR experiments and analyze the differences in chemical shifts and peak intensities. Overall, this analysis reveals that the structure of ASR is largely conserved in the inner membrane of E. coli, with many of the important structural features of rhodopsins previously observed in ASR in proteoliposomes being preserved. Small, site-specific perturbations in protein structure that occur as a result of the membrane changes indicate that the protein can subtly adapt to its environment without large structural rearrangement.
Introduction
Membrane proteins comprise approximately a third of cellular proteins and perform many vital functions, for example, acting as transporters, receptors, and enzymes. Interaction with the membrane environment, e.g., lipids, other proteins, etc., is an important factor that often influences both the conformation and function of membrane proteins (1–3). However, lipid membranes are generally not suitable for structural studies by the primary structural methods of x-ray crystallography and solution NMR due to the need for three-dimensional crystals or rapid sample tumbling, respectively. Thus, most structural studies undertaken to date have involved purification of the protein of interest through removal from its native membrane, isolation and subsequent crystallization, or solubilization in suitable detergents.
Solid-state NMR (SSNMR) allows for the study of membrane proteins reconstituted into a lipid milieu that more closely mimics native cell membranes. Recent advances in SSNMR methodologies (4–8) have already resulted in several lipid-reconstituted membrane protein structures (9–14), and have paved the way for detailed investigations of structural perturbations caused by nonphysiological lipid mimetics. For example, comparison of structures of the bitopic oligomeric influenza A M2 proton channel in lipid bilayers, detergent micelles, and three-dimensional crystals reveals significant changes in helical packing (15,16). Furthermore, the trimeric oligomerization state of the polytopic α-helical Anabaena sensory rhodopsin in lipids (17) was found to be significantly different from the dimer found in three-dimensional crystals (18). In bacteriorhodopsin (BR), which has been well studied using a variety of methods, changes in the protein structure and oligomeric organization have also been observed between membrane mimetics (19–22).
While the lipid bilayer represents a good experimentally tractable model environment for structural and functional studies of membrane proteins, the natural cellular membrane is invariably more complex and includes a diverse array of lipids as well as peripheral and transmembrane proteins. The hydrophobic thickness and fluidity of the lipid bilayer, as well as interactions with other proteins present in the membrane, may affect the tilts and lengths of individual helices, interhelical packing, side-chain conformations, and protein dynamics, and, through these effects, influence the functional properties of proteins (1–3,16,23).
Several recent studies have demonstrated the feasibility of studying overexpressed peptides and proteins in the native Escherichia coli membrane using SSNMR, often enhanced by dynamic nuclear polarization (24–29). For example, as a result of high expression levels (70–80% of labeled protein), 12 of the 23 residues of the single-spanning transmembrane domain of LR11 could be assigned in E. coli membranes using a two-dimensional 13C-13C experiment (24). Studies of full-length M2 (26) in E. coli membranes have been used to validate the protein structure observed in synthetic liposomes, while in the outer membrane protein PagL (25) small structural perturbations were observed in some of the 13 detected residues in the loop and membrane-water interface regions, particularly at the level of amino-acid side chains.
Here, we extend these studies to polytopic α-helical proteins, and use multidimensional SSNMR to characterize the structure of a seven-transmembrane helical microbial photosensor Anabaena sensory rhodopsin (ASR) (30) in the membrane environment of E. coli, in which ASR is expected to partition into the inner membrane. ASR is a cyanobacterial photosensor that is believed to interact with a unique cytoplasmic soluble transducer (30), and may be responsible for chromatic adaptation in its host cell (30,31). Its three-dimensional structure has been solved by x-ray crystallography (18) and more recently by solid-state NMR (13). The wealth of structural and spectroscopic data available reveals a number of structural variations that are likely related to the lipid bilayer mimetic used in structural studies. For example, spectroscopic NMR assignments, which are available for 205 of the 229 residues of ASR reconstituted in a bilayer consisting of DMPC and DMPA lipids (referred to as “proteoliposomes” in the following) (32,33), reveal a single all-trans conformation of the retinal chromophore and of the retinal binding pocket in the dark state, but double (and structurally very similar) conformations for a number of residues located on the cytoplasmic side (32). In contrast, the all-trans and 13-cis conformations of retinal coexist in crystals (18), which results in double conformations for some of the residues in the vicinity of retinal (e.g., K210 and S86), but does not cause perturbations in the rest of the protein. We have pointed out above that the oligomeric organization of ASR is different in the lipid-reconstituted and crystalline states (13). Furthermore, there are also differences in the structure of some of the peripheral loops, as well as looser helical packing on the cytoplasmic side (13) of ASR in proteoliposomes. The latter observation is consistent with the intermediate timescale motions of helices (tens of nanoseconds) estimated from SSNMR relaxation measurements (34), as well as with the higher solvent accessibility of the cytoplasmic side of the protein, both in the dark state (33) and under illumination (35). Such increased conformational adaptability on the cytoplasmic side may be related to the function of ASR, especially to its ability to interact with its transducer.
The apparent sensitivity of the ASR structure to the surrounding environment and the available structural and spectroscopic SSNMR data make this protein an interesting target for the examination of the subtle effects of membrane composition. Here, we combine biochemical approaches, which allow us to enhance the relative NMR signal contribution from ASR, with the biophysical techniques of circular dichroism (CD) in the visible range, small-angle x-ray scattering (SAXS), Fourier transform infrared spectroscopy (FTIR), and SSNMR to probe the conformation of ASR in the E. coli inner membrane (IM) environment and compare it with the recently published data for ASR obtained in proteoliposomes (32,33). We show that ASR forms similarly structured trimers in both environments, which are arranged in two-dimensional crystals of different symmetry: a hexagonal bacteriorhodopsin-like lattice in the DMPC/DMPA proteoliposomes, versus a more loosely packed square two-dimensional lattice in the E. coli IM. Site-specific analysis of the chemical shifts obtained for ∼40% of ASR residues in the E. coli IM indicates a high degree of overall structural conservation. Many functionally important regions of ASR (the retinal binding pocket, the trimer interface, β-hairpin in the extracellular B-C loop) are unperturbed and represent very structurally stable elements. Outside of the conserved regions, small yet noticeable structural changes corresponding to chemical shift perturbations of up to 0.5 ppm for carbon and 1.0 ppm for nitrogen are observed in both transmembrane helices and the peripheral parts of the protein. Analysis of cross-peak intensities provides an approximate measure of the proteins’ mobility and indicates stabilization of the peripheral helix E′ on the cytoplasmic side, which may occur because of better matching between the hydrophobic thicknesses of ASR and the E. coli lipids compared to DMPC/DMPA, or due to the different lattice packing of trimers.
Materials and Methods
Materials
Common chemicals of reagent grade were purchased from either Fisher Scientific (Unionville, Ontario, Canada) or Sigma-Aldrich (Oakville, Ontario, Canada). Isotopically labeled compounds, such as 15NH4Cl and 13C6-glucose, were obtained from Cambridge Isotope Laboratories (Andover, MA). The Ni2+-NTA (nitrilotriacetic acid) agarose resin was purchased from Qiagen (Mississauga, Ontario, Canada). Lipids were purchased from Avanti Polar Lipids (Alabaster, AL).
Protein expression and purification
In Fig. 1 we briefly summarize our sample preparation strategies. ASR samples grown entirely in natural abundance, [U-15N], and [U-13C,15N]-labeled media are referred to as NA, UN, and UCN, respectively. Samples of ASR used in this study were: 1) isotopically labeled, solubilized, purified ASR reconstituted into liposomes, referred to as “proteoliposomes” or “PL-ASR” in the following; 2) ASR isolated in the E. coli membrane (EM), denoted “EM-ASR”; 3) samples of the E. coli membrane from noninduced cells subjected to isolation procedures identical to EM-ASR and used as a control for NMR assignments, referred to as “EM-NIC”; and 4) samples with reduced background signal (rb) in which cells are grown to the exponential phase in NA media and then resuspended at a high concentration in UN or UCN media before protein expression is induced, denoted as “rbUN” and “rbUCN”, respectively.
Figure 1.
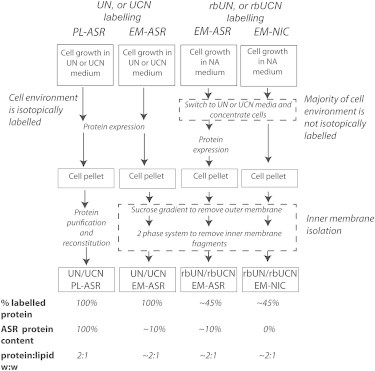
Summary of isotopic labeling, sample preparation strategies, and sample compositions. The terms NA, UN, and UCN denote natural abundance, uniformly 15N-labeled, and uniformly 13C,15N-labeled media or samples, respectively. A sample of purified ASR reconstituted in DMPC/DMPA lipids (PL-ASR, leftmost column) was used as reference. Two types of ASR samples expressed in the E. coli membrane (EM-ASR) were prepared. For the first type, ASR and background proteins were labeled at the same level, and outer membrane and inner membrane fractions not containing ASR were minimized using a sucrose gradient and His-tag binding (41,42) (second left column). To reduce NMR signal from the background proteins, cells were grown to the exponential phase in NA media, and then resuspended at a high concentration in labeled media before protein expression was induced (denoted as rbUN or rbUCN, second right column). An additional control sample was produced by following the expression and isolation procedures for the rbUCN EM-ASR sample, but without induction (EM-NIC, rightmost column). Sample compositions are given at the bottom of the figure and are explained in the Supporting Material.
Expression of ASR
For NA, UN, and UCN PL-ASR and EM-ASR samples, ASR was overexpressed in BL21-Codon-plus-RIL E. coli grown on M9 minimal medium at 30°C, using 4 g of glucose (NA or [U-13C] labeled for UCN labeling) and 1 g of NH4Cl (NA or 15NH4Cl for UN and UCN labeling) per liter of culture as the sole carbon and nitrogen sources, as described previously by Shi et al. (33). When cultures reached a target cell density of A600 = 0.4 OD, protein expression was induced through the addition of IPTG (isopropyl β-D-1-thiogalactopyranoside) to a concentration of 1 mM and retinal to a concentration of 7.5 μM. After ∼21 h, the cells were collected by centrifugation.
Expression of ASR with reduced isotopic labeling of background proteins
To reduce the background NMR signal from non-ASR cell components, isotopes can be strategically incorporated preferentially into the recombinant protein and coexpressed molecular components. This was achieved through allowing cells to grow to the exponential phase in unlabeled media (Fig. 1), before resuspending them in a lower volume of isotopically enriched media for protein expression (36). In this way, the majority of cellular components are produced in unlabeled media, and therefore do not contain isotopic labels, while the recombinant protein is produced in isotopically labeled media, and therefore contains a higher proportion of isotope labels.
To achieve this in our EM-ASR samples, cells were first grown to an OD600 of ∼0.7 in unlabeled LB media before being collected through gentle centrifugation (36). The cell pellet was then washed with minimal media salts before being resuspended in UN or UCN M9 minimal media at 4× their original concentration, resulting in a starting OD600 of ∼2.8. After ∼1 h of growth, expression was induced with IPTG at a concentration of 1 mM and retinal was added at a concentration of 7.5 μM. Approximately 4 h after induction the cells were collected by centrifugation. ASR was then isolated in the inner membrane as described below. Samples created from cells grown using this labeling method are denoted as “rbUN” and “rbUCN”.
Purification of ASR and reconstitution into liposomes
ASR was purified from E. coli cells as has been described previously by Shi et al. (33). Briefly, cell pellets were pretreated with lysozyme (12 mg/L of culture) and DNase I (600 units per liter of culture) and then broken by sonication. The membrane fraction was then solubilized in 1% DDM (n-dodecyl β-d-maltoside) at 4°C, and purified following the batch procedure described in the Qiagen Ni2+-NTA resin manual. Approximately 5 mg of ASR was purified from 1 L of culture. The molar amount of ASR was determined by the absorbance of opsin-bound retinal, using the extinction coefficient of 48,000 M−1 cm−1 (37). To reconstitute ASR into liposomes, the purified proteins were buffer-exchanged using an Amicon Ultra-15 10 K centrifugal filter (Millipore) into pH 8.0 buffer (5 mM Tris, 10 mM NaCl, 0.05% DDM), and concentrated to ∼1 mg/mL. The protein was then mixed with lipids, which were prepared by hydrating dried DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) and DMPA (1,2-dimyristoyl-sn-glycero-3-phosphate) premixed at a 9:1 ratio (w/w), at a protein/lipid ratio of 2:1 (w/w) and incubated for 6 h at 4°C before Bio-Beads SM (Bio-Rad Laboratories, Hercules, CA) were added for detergent removal. The functionality and protein/lipid ratios of proteoliposomes have been tested using visible and FTIR spectroscopy, as described previously in Shi et al. (38,39).
Isolation of ASR in the E. coli membrane
To facilitate the study of ASR in the E. coli membrane, it is desirable to remove as much background signal originating from areas of the cell that do not contain ASR, as possible. This is achieved both through physically separating fragments of the membrane containing ASR, and through isotopic labeling strategies that limit isotopic incorporation into background proteins. To physically remove background proteins and other material from the sample while maintaining the inner membrane environment around ASR, several steps were taken. To begin, cell pellets were pretreated with lysozyme (12 mg/L of culture) and DNase I (600 units/L culture) before being broken by sonication. Low-speed centrifugation was used to remove unbroken cells, and the supernatant was pelleted to remove soluble debris. The membrane pellet was then resuspended and the inner and outer membranes were separated by sucrose gradient (40). The raw membranes were first applied on top of a two-step sucrose gradient composed of 55% sucrose and 9% sucrose (all sucrose percentages are reported as w/w). These gradients were centrifuged for 2.5 h at 210,000×g in a swinging bucket rotor and the membrane fraction was collected from the top of the 55% sucrose layer. The membrane fraction was then diluted by at least 3× and subjected to a six-step sucrose gradient that was composed of discrete (from top to bottom) 30, 35, 40, 45, 50, and 55% sucrose fractions. The gradients were centrifuged for 15 h at 210,000×g and the IM fraction was collected from the top of the 40% sucrose layer. Both sucrose gradients were prepared in buffer containing 50 mM TEA, 1 mM EDTA, and 1 mM DTT at pH 7.5.
The IM fraction was then resuspended in buffer containing 5 mM Tris (pH 8) before being added to a two-phase affinity system (41,42) (10 mM Tris at pH 8, 0.3 M NaCl, 6.45% (w/w) PEG 3350, 8.45% (w/w) dextran, and 320 μL of Ni-NTA-agarose slurry per mL of system), which utilizes the C-terminal 6×His-tag on recombinant ASR to bind ASR-containing membrane fragments. After incubation with gentle mixing in the two-phase system for ∼30 min, the phases were separated by low-speed centrifugation (1600×g) for 5 min. The top phase, which contains IM vesicles not containing ASR, was then collected with a Pasteur pipette and replaced with an equal volume of buffer (6.45% (w/w) PEG 3350, 10 mM Tris, and 40 mM imidazole). The previous two steps were repeated until the absorption spectra showed a lack of protein in the top phase. Membranes were then eluted from the affinity beads in the bottom phase by incubation in 500 mM imidazole for 15 min, before low-speed centrifugation (1000×g) was used to sediment the affinity beads.
Because we are likely to still see a strong presence of background proteins in our SSNMR spectrum, a control sample was produced in which all steps of protein expression and IM-isolation were identical to EM-ASR preparations, including rbUCN labeling, but without the induction of ASR expression. As a result of this method, only the non-ASR proteins seen in rbUN- or rbUCN-labeled samples will be seen in the samples created from noninduced cells (EM-NIC), and a direct comparison of the spectra obtained on EM-NIC with the spectra obtained on EM-ASR samples can be used to confirm the ASR peaks in the two- and three-dimensional NMR spectra.
SSNMR spectroscopy and data analysis
All experiments on PL-ASR and EM-NIC, as well as initial one- and two-dimensional NMR experiments on EM-ASR, were performed on an Avance III spectrometer (Bruker Biospin, Billerica, MA) operating at 800.230 MHz using a 3.2-mm 1H-13C-15N E-FREE magic-angle spinning probe. All pulse sequences and their descriptions are given in Fig. S1 and the related discussion in the Supporting Material. Samples were packed into 3.2-mm thin-wall rotors (Bruker Biospin). All experiments at 800 MHz were performed at a spinning rate of 14.3 kHz, and at an effective temperature of 5°C, with a recycle delay of 1.7 s. Typical π/2 pulses were 2.5 μs for 1H, 4 μs for 13C, and 7 μs for 15N.
All three-dimensional experiments on EM-ASR were performed on a 900 MHz Avance III NMR spectrometer (Bruker Biospin) equipped with a low-E triple resonance 1H-13C-15N 3.2 mm magic-angle spinning probe designed and built at the National High Magnetic Field Laboratory (Tallahassee, FL) (43). All experiments at 900 MHz were performed with spinning at 15 kHz and at an effective temperature of 5°C with recycle delays of 1 or 1.2 s. The high sensitivity and fast recycle delays afforded by the 900-MHz probehead were critical for the detection of small amounts of ASR. Typical π/2 pulses were 2.5 μs for 1H, 3.5 μs for 13C, and 5.6 μs for 15N. Additional experimental details are given in the Supporting Material. Chemical shifts were referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid using the 13C adamantane down-field peak resonating at 40.48 ppm as a secondary external standard (44). Data processing was performed using the software NMRPIPE (45). Noise analysis and peak picking were performed in the CARA (46) environment.
CD measurements
CD spectra were recorded on a model No. J-810 spectropolarimeter (JASCO, Easton, MD). The samples were scanned at room temperature over the 350–700 nm range at a rate of 100 nm/min. Spectra taken on buffers were used for baseline correction. UV-visible spectra were collected simultaneously with the CD spectra. To collect the CD spectra of ASR in the proteoliposome and IM environment, the samples were suspended in 70% glycerol (v/v) to reduce light scattering via refraction index matching (47). The spectra were collected using a 0.1-cm path-length quartz microcell.
SAXS measurements
Synchrotron SAXS spectra, measured at the Stanford Synchrotron Radiation Lightsource (SLAC National Accelerator Laboratory, Menlo Park, CA), were obtained as has been described previously in Kuang et al. (48). Briefly, the membranes were sealed in quartz capillaries (diameter ∼1.5 mm; Hilgenberg, Malsfeld, Germany) and irradiated for ∼5 s with incident synchrotron x-rays (from an eight-pole wiggler) that were monochromatized (λ = 1.37776 Å) and focused using a cylindrical mirror. The scattered radiation was collected using a model No. MX225-HE detector (Rayonix, Evanston, IL), with each sample being measured six times. Final spectra are averages of these measurements. For all measurements, no radiation damage was observed. The two-dimensional SAXS powder patterns were integrated using the software FIT2D (www.esrf.eu/computing/scientific/FIT2D/), and the sample-to-detector distance was calibrated using silver behenate as a standard. Data were fitted by the nonlinear least-squares method using the software IGOR PRO 6 (WaveMetrics, Lake Oswego, OR).
Results and Discussion
Sample optimization and the state of ASR in E. coli membranes
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of the cell pellet (Fig. S2) indicates that, despite overexpression, the ASR content of whole cells is low, with only a small amount of ASR being detected after the cells have been induced. The gels show the strong presence of a ∼37 kDa protein, which likely corresponds to the major outer membrane proteins OmpA and OmpF (49–51), along with many other background proteins. This indicates that ASR would not be the dominant component in the sample and therefore additional measures are necessary in order to facilitate studies of ASR in the E. coli inner membrane. Our goal was to create a sample of ASR in which the E. coli membrane environment is maintained, while fewer background proteins and other molecules from the E. coli cells are present. Although the gels show a significant decrease in the amount of background proteins after the sucrose gradient, there is still a strong presence of the ∼37 kDa protein, which we have experimentally identified as a mixture of OmpF and OmpA (see details in the Supporting Material).
To more accurately monitor the inner membrane isolation process and the protein/lipid ratios of samples, FTIR was performed after each step (Fig. 2 A). The Amide I band is sensitive to protein secondary structure and can be used to monitor the relative amounts of α-helical, β-strand, and random coil secondary structures in the samples. Narrowing of this band around the frequencies corresponding to α-helical structure indicates that the relative amount of β-barrel proteins is indeed being reduced during the purification. However, the greater width of the final EM-ASR peak as compared to the PL-ASR peak points to the presence of other proteins in the final sample.
Figure 2.
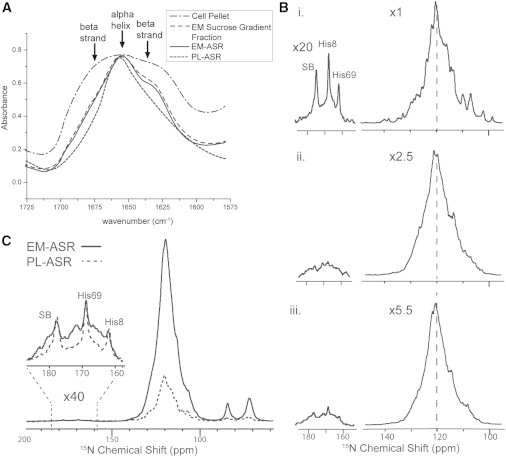
(A) FTIR spectra of NA EM-ASR throughout the inner membrane isolation process. By observing the narrowing of the Amide I peak and the reduction of its β-shoulder, the reduction of outer membrane content (β-barrel proteins) can be monitored. (B) One-dimensional 15N spectra of (i) UCN PL-ASR, (ii) UN EM-ASR, and (iii) rbUN EM-ASR. The relative increase in UN-labeled α-helical proteins in rbUN EM-ASR is evident from the relative increase in the downshifted maximum of the spectra (dashed line). All spectra are scaled to approximately match intensities, with scaling factors, and corrected for the number of scans taken in each experiment (noted to the left). The main peak is presented with no window function, whereas 60 Hz of line broadening is applied to the regions of the three resolved peaks. (C) One-dimensional 15N spectra of UCN PL-ASR and rbUN EM-ASR with the entire spectra processed with the stronger exponential window function of 60 Hz, and scaled such that the three resolved peaks (SB, His8, and His69) approximately match in intensity, showing that ASR accounts for only ∼1/4 of the labeled protein content in EM-ASR.
To evaluate the effectiveness of rbUN and rbUCN labeling, we compare UN EM-ASR and rbUN EM-ASR samples. The one-dimensional 15N spectrum of UN EM-ASR (Fig. 2 B) has an overall reduced intensity (∼40%) compared to that of PL-ASR, likely due to the increased lipid and water content of EM-ASR. In addition, it shows a significantly shifted and broadened maximum as compared to PL-ASR as well as a secondary maximum peak that aligns with the maximum of the 15N spectrum of PL-ASR. The first maximum peak is upshifted by ∼1 ppm relative to the PL-ASR maximum, and the bulk centroid of the spectrum is upshifted by much more, indicating the presence of isotopically labeled proteins with high β-strand content (52). Resolved residues seen in the one-dimensional 15N spectrum of PL-ASR (His8, His69, and Schiff base (SB)) cannot be seen in UN EM-ASR, even after long acquisition, indicating a small relative amount of isotopically labeled ASR in the sample.
The spectrum of EM-ASR with a reduced amount of isotopic labeling of background proteins (rbUN EM-ASR) shows a further decrease in overall signal intensity due to the increased presence of unlabeled protein. However, a noticeable relative increase in the ASR backbone peak, when compared to UN EM-ASR, is evident and some unique ASR residues can be resolved. In particular, the peaks at 177.2, 169.1, and 162.4 ppm correspond to the SB nitrogen of Lys210, His8, and His69 Nδ1 resonances, respectively. All three peaks appear to be unshifted in the EM-ASR sample, indicating that the retinal remains in an all-trans conformation and the conformations and protonation states of the His8 and His69 side chains are unaltered in the E. coli IM. Through comparisons of the intensity of these peaks in rbUN EM-ASR and PL-ASR (Fig. 2 C), we conclude that our rbUN and rbUCN EM-ASR NMR samples contain ∼1 mg (800 MHz, 3.2 mm thin wall rotor; Bruker) and 1.6 mg (900 MHz, thin wall rotor; Revolution NMR, Fort Collins, CO) of ASR, which comprises only ∼1/4 of the total isotopically labeled protein content.
The relative content of UCN labeled α-helical proteins in the rbUCN EM-ASR and EM-NIC samples was further analyzed using FTIR through observation of the Amide I band, which is dominated by CO stretching and is therefore sensitive to 13C labeling, and the Amide II band, which is sensitive to both 13C and 15N labeling. Isotopic labeling causes a downshift of the vibrational frequency of the amino acids containing the heavier atoms by ∼20–50 cm−1, and therefore the peaks representing the 13C and/or 15N-labeled proteins are downshifted from the frequencies observed in unlabeled proteins. As seen in Fig. 3 A, even in the cell pellet spectrum, the Amide II band is clearly split into two peaks representing isotopically labeled and natural abundance proteins. The Amide II peak representing isotopically labeled proteins shows a relative increase in the EM-ASR spectrum, indicating the removal of unlabeled proteins. Amide I peaks representing the NA α-helical and β-sheet content are present between ∼1658–1647 cm−1 and ∼1638–1632 cm−1, respectively, with their isotopically labeled counterparts being significantly downshifted by ∼40 cm−1 from these wavelengths. Although all four peaks have similar intensities in the cell pellet spectrum, the increase in the relative content of isotopically labeled α-helical proteins is evident in the EM-ASR spectrum.
Figure 3.
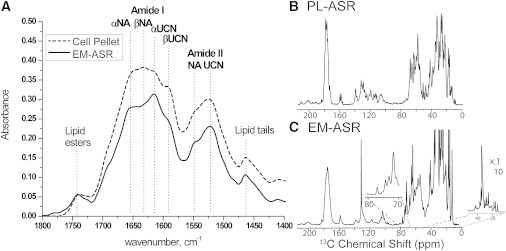
(A) FTIR spectra of rbUCN ASR before (cell pellet) and after (EM-ASR) the IM isolation process. (Dashed lines) Peaks corresponding to the natural abundance (NA) α-helical and β-strand (αNA and βNA) peaks as well as their isotopically labeled counterparts (αUCN and βUCN). A noticeable increase in the UCN α-helical peak in EM-ASR as compared to the cell pellet is observed. One-dimensional 13C NMR spectra of (B) UCN PL-ASR and (C) rbUCN EM-ASR. All NMR spectra are scaled to approximately match intensities.
The 13C spectrum of EM-ASR (Fig. 3 C) displays an amount of fine resolution in the Cα region similar to that of PL-ASR (Fig. 3 B). Being heterogeneous, EM-ASR samples show a number of additional resonances that are not observed in the PL-ASR sample. Notably, three intense peaks located at 132.0, 34.7, and 32.9 ppm likely originate from the fatty acyl chains of lipids in the sample. Additionally, four well-resolved peaks can be observed in the spectral region of 70–80 ppm (Fig. 3 C, inset) that are not seen in PL-ASR but have been reported before in solution NMR spectra of BR (53) and in SSNMR spectra of green proteorhodopsin (39). These signals have been attributed to sugar moieties or carbohydrates that are tightly associated and copurify with green proteorhodopsin or to glycolipids of BR.
We used absorption spectroscopy, CD spectroscopy in the visible range, and SAXS to further evaluate the functionality, oligomeric state, and possible two-dimensional crystallinity of ASR in the E. coli membrane. As can be seen in Fig. 4 B, although a strong background is present, the absorption spectrum of EM-ASR can be observed with a maximum at ∼540 nm, the wavelength at which ASR absorbs in the PL-ASR sample. Such an absorption peak is not observed in the EM-NIC sample (Fig. S3), confirming this spectral range to be ASR-specific. This also indicates that ASR remains correctly folded in EM-ASR. The appearance of a bilobal shape in the visible CD spectra of oligomers formed by microbial rhodopsins is related to the excitonic coupling of retinal chromophores of the monomers, with the exact shape of the spectra depending on the relative orientation of retinals, as well as additional contributions from aromatic side chains (54–56). CD spectra of ASR solubilized in DDM and in proteoliposomes show a bilobal shape that corresponds to a trimeric arrangement of ASR monomers, with the intermonomer interface being formed between helix B of one monomer, and helices E and D of another (17). In Fig. 4 A, we compare the CD spectra measured in both PL-ASR (as published previously by Wang et al. (17)) and EM-ASR. Although there is a strong membrane background signal at shorter wavelengths in EM-ASR, similar bilobal shapes are clearly visible at higher wavelengths in both samples, pointing to the presence of ASR trimers in E. coli membranes. The conservation of the trimeric structure is further supported by the analysis of SSNMR chemical shifts, as discussed below.
Figure 4.
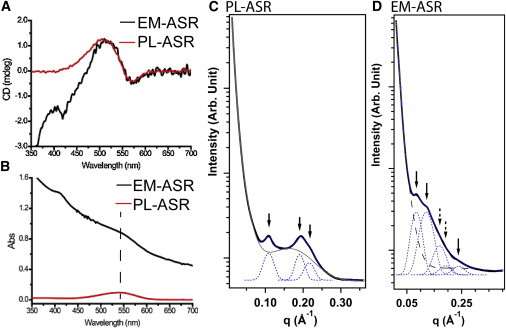
(A) Visible range CD and (B) absorption spectra of PL-ASR and EM-ASR. In (A), the bilobal shape is clearly present in both PL-ASR and EM-ASR, indicating the presence of trimers. SAXS spectra of (C) PL-ASR and (D) EM-ASR. Bragg diffraction peaks in the SAXS spectrum of PL-ASR indicate the presence of an ordered two-dimensional hexagonal lattice, while the peaks present in the EM-ASR spectrum indicate the presence of a two-dimensional tetragonal lattice. See text for details. To see this figure in color, go online
Given the high protein/lipid in our SSNMR samples, it is possible that ASR may form a two-dimensional lattice in PL-ASR and/or EM-ASR. Under native conditions, BR trimers assemble into a two-dimensional hexagonal lattice called the purple membrane (57). The synchrotron SAXS spectrum of PL-ASR shows a series of scattering peaks (marked by black arrows in Fig. 4 C). These peaks can be fit nicely to reveal a liposome form-factor background (gray trace) (58,59) and a series of scattering peaks (blue dotted traces) centered at 0.109, 0.190, and 0.218 Å−1, respectively. There correlations can be indexed as the q10, q11, and q20 scatterings of a two-dimensional hexagonal lattice with a lattice parameter of ∼66.4 Å, reminiscent of the purple membrane structure of BR, which has a lattice parameter of 62.7 Å (60). The width of these scattering peaks suggests a relatively small crystalline domain size, i.e., ∼500 Å as estimated using Sherrer’s equation (61).
In contrast, the synchrotron SAXS spectrum of EM-ASR samples, shown in Fig. 4 D, reveals a completely different two-dimensional lattice arrangement. There are two relatively strong scattering peaks centered at 0.085 and 0.120 Å−1 and a weak, albeit discernible, peak at 0.240 Å−1 (marked by solid black arrows), with position relationships of 1: √2: √8. Using a two-dimensional tetragonally packed lattice model, the scattering data can be fit to reveal two additional hidden peaks (marked by dotted arrows) centered at 0.169 and 0.191 Å−1, respectively. These five peaks are indexed as the q10, q11, q20, q21, and q22 of an in-membrane tetragonal membrane protein lattice with a lattice parameter of ∼74 Å. Because these peaks are not visible in the SAXS spectra that we have obtained on another retinal-binding protein (green proteorhodopsin) produced in a similar membrane environment (Fig. S3), we can conclude that they do not originate from background proteins and indicate that ASR is arranged in a tetragonal lattice in the E. coli membrane environment. The observed scatterings are too weak to extract meaningful estimates of the domain size, but their weak and diffusive nature suggests a smaller domain size than that observed in PL-ASR.
Two- and three-dimensional SSNMR spectra of ASR in cellular membranes
Chemical shifts are exquisitely sensitive to alterations in the chemical and structural environment, and can be used to detect variations in the structure of ASR in the E. coli inner membrane. Fig. 5 compares the two-dimensional NCA spectra measured in PL-ASR, rbUCN EM-ASR, and rbUCN EM-NIC. The two-dimensional NCA spectrum of PL-ASR is generally well resolved (Fig. 5 A), with linewidths of ∼0.5 ppm in the direct 13C and ∼1 ppm in the indirect (truncated) 15N dimensions. The linewidths of resolved ASR peaks in EM-ASR are comparable to those seen in PL-ASR, overall indicating that the protein remains structurally homogeneous even in the heterogeneous environment of the cell membrane. We note that the cross peaks, which correspond to other background proteins, are similarly narrow and are downshifted in the 13C dimension and upshifted in the 15N dimension, confirming the presence of a large amount of proteins with β-strand secondary structure. This suggests that much of the lack of resolution in the center of the spectra is caused by the overlap of many well-resolved peaks, as opposed to the presence of broad, unspecific peaks.
Figure 5.
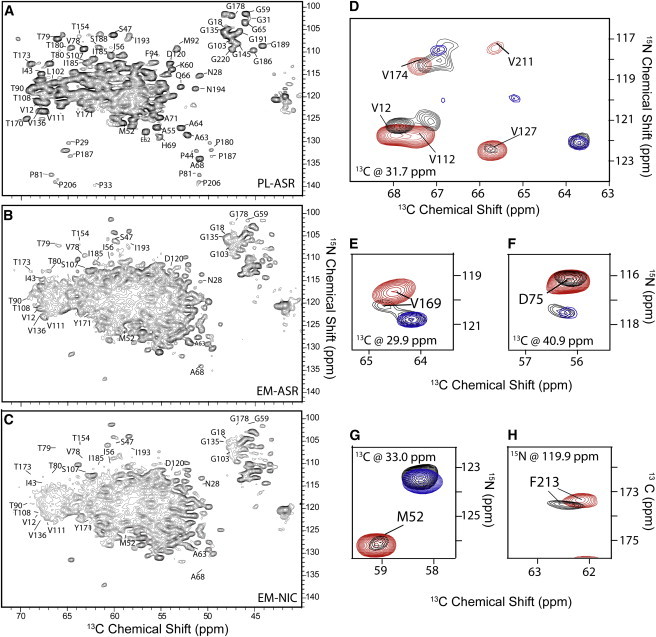
NCA SSNMR spectra of (A) PL-ASR, (B) rbUCN EM-ASR, and (C) rbUCN EM-NIC. All peaks are labeled according to their assignments in PL-ASR. All spectra were recorded on an 800 MHz spectrometer and processed identically using 15 Hz of Lorentzian line narrowing and 30 Hz of Gaussian line broadening in the indirect dimension, and 30 Hz of Lorentzian line narrowing and 60 Hz of Gaussian line broadening in the direct dimension. The spectrum in (A) was collected with 32 scans, while spectra (B) and (C) were both collected with 320 scans. In (A), the lowest contour is cut at 10×σ, while in (B) and (C) the lowest contour level is cut at 5×σ. Representative overlays of two-dimensional planes of (D–G) three-dimensional NCACB and (H) CANCO experiments performed on PL-ASR (red), EM-ASR (black), and EM-NIC (blue). All peaks are labeled according to the assignments. PL-ASR and EM-NIC spectra were recorded on an 800 MHz spectrometer while EM-ASR spectra were recorded on a 900 MHz spectrometer. Additional planes are shown in Fig. S4. To see this figure in color, go online.
Overall, a total of 16 ASR peaks could be identified in the two-dimensional NCA EM-ASR spectra (Fig. 5 B) using the known resonance assignments obtained from proteoliposome samples, and through a comparison with the spectra collected on EM-NIC (Fig. 5 C). Due to the reduced amount of ASR in the EM-ASR sample, many of the lower-intensity resolved peaks in the PL-ASR spectrum that do not overlap with background peaks, most notably in the proline region, are not visible in the EM-ASR spectra. Isolated residues with unchanged (conserved) chemical shifts are found in both the loop regions (A-B, B-C, C-D, E-F, and F-G) and in the intramembrane regions of helices B, C, D, and F. In agreement with the previous observation of the unchanged chemical shift of the SB 15N, signals of residues S47 and T79, which are part of the retinal binding pocket, are also not shifted. Additionally, the chemical shifts of M52 and I56, which are found on the extracellular side of helix B and are involved in the intermonomer contacts of the trimer, are nearly identical to those detected in PL-ASR, indicating that the monomer-monomer interface of the trimer is preserved in the E. coli inner membrane.
Due to the relatively large size of ASR, as well as the spectral overlap created by background proteins, three-dimensional spectroscopy is required for more detailed site-specific analysis. To extend our assignments, we have performed three-dimensional CANCO and NCACB experiments on PL-ASR, rbUCN EM-ASR, and rbUCN EM-NIC samples. CANCO experiments were performed to obtain backbone assignments, whereas three-dimensional NCACB experiments provide information on side-chain atoms and help validate the identification of shifted residues from the CANCO data in cases of overlapping peaks or where there are multiple assignments possible.
In Fig. 5, D–H, we show representative two-dimensional planes extracted from the three-dimensional NCACB and CANCO experiments recorded on the PL-ASR, rbUCN EM-ASR, and rbUCN EM-NIC samples. The two-dimensional plane of PL-ASR shown in Fig. 5 D contains five cross peaks corresponding to valine correlations. Four of them, although reduced in intensity due to the lower amount of ASR, are also clearly visible in the spectrum of EM-ASR, while V211 is below detection in this spectrum. We additionally show several conserved peaks of interest, such as that of D75, which is situated in the retinal binding pocket (Fig. 5 F), and M52, which is involved in the monomer-monomer interface (Fig. 5 G), as well as examples of peaks that are shifted in EM-ASR (F213, V169). Additional planes are shown in Fig. S4.
Structural evaluation of ASR in the E. coli inner membrane
With combined data from two- and three-dimensional experiments on PL-ASR, rbUCN EM-ASR, and rbUCN EM-NIC, it is found that N, CA, and in some cases CB resonances belonging to 82 of the 205 residues previously assigned in PL-ASR (32,33), have been confidently reassigned in EM-ASR. Additional assignments for CO resonances in 29 residues have also been obtained (summarized in Table S1 in the Supporting Material). Although several residues that are visible in PL-ASR are obscured by background peaks in EM-ASR, the majority of unassigned residues are missing due to insufficient sensitivity. Based on the signal-to-noise ratios (SNRs) of these peaks in the PL-ASR spectra, their absence in the EM-ASR spectra does not indicate that these residues undergo significantly increased motions. To verify that our spectra do not contain resolved peaks belonging to ASR that we are unable to assign due to large changes in chemical shifts, we have confirmed that any unassigned, resolved peaks found in the EM-ASR spectra are also found in the EM-NIC spectra, and thus correspond to the background proteins.
The observable ASR residues are evenly distributed throughout the protein, found in loops, membrane-water interface regions, and transmembrane regions alike, thus providing a site-specific view of conformational perturbations in ASR induced by the environment of the inner membrane of E. coli. In Fig. 6 we show residue-specific changes in chemical shifts for N, C′, Cα, and Cβ atoms, as well as changes in peak intensities. The largest perturbations range from up to a ppm in the 15N dimension to up to one-half a ppm in the 13C dimension, with most of them occurring in helices B and D and in the extracellular B-C loop (Fig. 7 A). Chemical shift perturbations of this magnitude do not correspond to large changes in the secondary structure of the protein, and we can thus conclude that the structure of ASR is largely preserved in the E. coli inner membrane. Additionally, noticeable changes in cross-peak intensities ranging from 2 to 3× are observed in the E-F and C-D loops.
Figure 6.
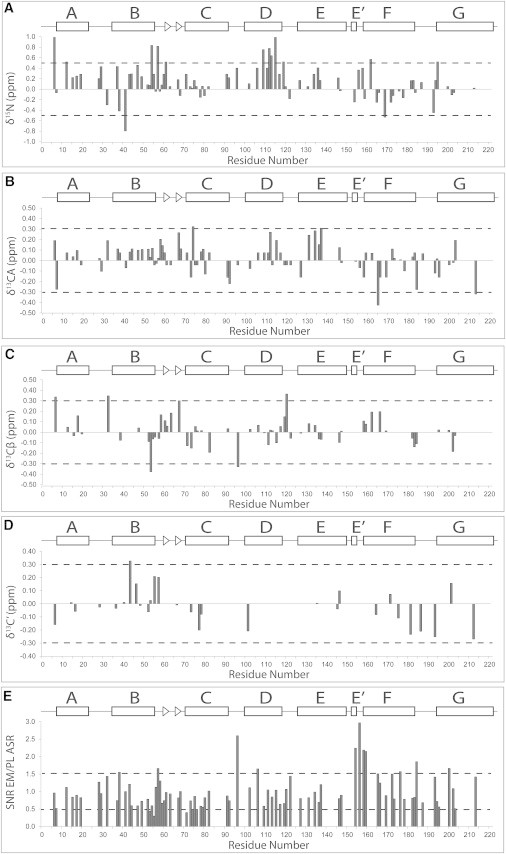
(A–D) Site-specific chemical shift perturbations and (E) SNR comparisons between EM-ASR and PL-ASR. For N and CA shifts and for the relative SNR, EM/PL data is combined from NCA, CANCO, and NCACB experiments. The relative SNR EM/PL is calculated with consideration for the number of scans performed on each sample and the relative amounts of UCN ASR present in each sample.
Figure 7.
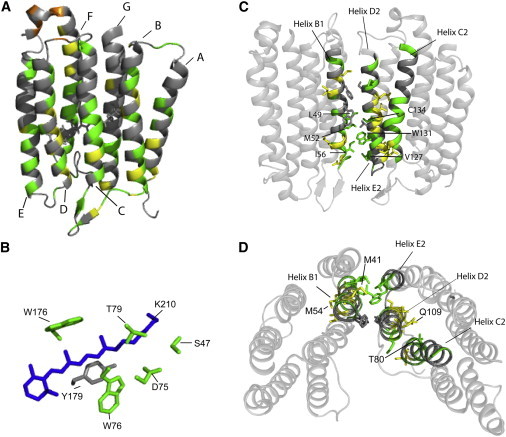
(A) Structural model of a monomer of trimeric ASR (Protein Data Bank: 2M3G). In all figures: (green) unperturbed residues, (yellow) residues experiencing significant changes in chemical shift, and (orange) residues with significantly enhanced signal intensity. (B) Closeup of the retinal pocket, showing the side chains of selected unperturbed residues (green) that form the pocket. (Blue) Retinal molecule. (C) Side view and (D) view from the extracellular side of the interaction interface (helices B1, E2, and D2) between two monomers and helix C2, which is tightly packed against many of the perturbed residues in helix D2. The side chains of perturbed residues and residues involved in the interaction interface are shown. In (C), unperturbed residues involved in the interaction interface are labeled, while in (D) several of the most perturbed residues and residues that pack tightly against these residues are labeled. In (A) and (C), the cytoplasmic side is on top. To see this figure in color, go online.
Many of the important structural features of rhodopsins previously observed for PL-ASR remain well conserved in EM-ASR. For example, in line with the aforementioned observations for unshifted peaks of the 15N SB and for S47, D75, and T79 (located in the retinal binding pocket), two additional residues interacting with the retinal (W76 and W176) are resolved in the three-dimensional spectra, and display no significant changes in chemical shifts (Fig. 7 B), confirming that the all-trans conformation of retinal and the structure of the retinal binding pocket remains unchanged.
Three-dimensional spectroscopy also allows for further expansion of our view of the intermonomer interface within the trimer, and confirms that tight protein-packing limits the structural flexibility. The interface is comprised of interacting residues on helix B1 of one monomer (F42, W46, L49, M52, and I56) and helix D2 (S107 and T114) and E2 (V127, W131, and C134) of a second monomer. Although the most perturbed residues are found in helices B and D, they are not involved in the intermonomer interface. In addition to M52 and I56, which are located in the extracellular half of helix B and observed in the two-dimensional NCA spectrum, V127, W131, and C134 on the extracellular half of helix E, and an additional residue, L49, in the middle of helix B, do not experience significant chemical shift changes. Thus, the structurally conserved parts of helices B and E involved in the intermonomer interface extend at least to the entire extracellular half (Fig. 7 C).
M41 on the cytoplasmic side and M54 on the extracellular side are among the most perturbed residues in helix B. Likewise, there are noticeable (>0.5 ppm) perturbations observed for residues Q109, V112, S115, and D120 of helix D. All of the mentioned residues face either the interior of the trimer, neighboring helices within the same monomer, or lipids, and do not directly affect the monomer-monomer interface (Fig. 7 D). Specifically, M41, whose nitrogen chemical shift is perturbed by ∼0.8 ppm, faces the lipid-exposed pocket at the interface formed by helices A and B of one monomer, and helices C and D of the neighboring monomer. It is thus possible that the shift in the M41 peak is caused by a change of lipids in this pocket in EM-ASR. Additionally, S115 is tightly packed against G135 of helix E, and Q109 (chemical shift of the backbone nitrogen perturbed by ∼0.7 ppm) is closely packed against T80 and P81 of helix C, which are unshifted themselves, indicating that the chemical shift changes observed in backbone atoms do not correspond to large changes in the conformation of amino-acid side chains. The lateral pressure exerted on ASR may differ between the tetragonal lattice of EM-ASR and the hexagonal lattice of PL-ASR, which could explain the changes in chemical shift observed in certain residues of helices B and D. Particularly, Q109 is known to participate in a strong interhelical H-bond (similar to D115 of BR) (62) and may serve as a sensitive indicator of changes in helical packing.
The B-C loop is well structured in many rhodopsins, and appears to be a common structural element that is often conserved in lipidic, detergent, and crystalline environments (39,63–67). In ASR the B-C loop is disordered in the x-ray structure (18), but gains partial β-structure in proteoliposomes (13). It is composed of two short antiparallel strands, V61-A63 and Q66-A68, and is subjected to collective motions as indicated by transverse relaxation measurements (34). While there are some nonnegligible chemical shift perturbations detected for the nitrogen of M54 in the immediate vicinity of the loop, for D57 in its flexible part, and for V61, the structure of the loop is largely unaffected by changes in the environment.
The E-F loop, which contains the peripheral helix E′, is another structural element commonly found in several other rhodopsins in the lipid environment (39,68–70), but which exhibits different behaviors in detergent micelles and crystals. It is natively helical in BR in purple membrane samples (20), but either not visible or has large B-factors in high-resolution x-ray structures (65,71,72) and displays multiple conformers in samples prepared in detergent micelles (68) or nanodisks (19). While the chemical shifts of residues detected in the E-F loop of ASR (T154, T156, S158, and S159) remain largely unchanged in the E. coli membrane, there is a noticeable increase in the relative cross-peak intensities for these residues (Figs. 6 E and 7 A), indicating partial immobilization of this loop in the E. coli membrane. This increase in intensity, along with the signal increase for K96 in the cytoplasmic C-D loop, may indicate a more symmetric position of ASR in the E. coli inner membrane. Our previous measurements indicated that the cytoplasmic side of ASR is more exposed in the DMPC/DMPA bilayer (33). This asymmetry could be caused by a mismatch in the hydrophobic dimensions of ASR and the PL environment. While DMPC and DMPA both contain fatty acyl chains with a 14:0 composition, the E. coli membrane contains mostly PE C16:0/cyC17:0, and C14:0, and the longer C16:1, C16:0, cyC17:0, and C18:1 constitute almost all of the fatty acyl substituents (73,74), and thus will have a larger hydrophobic thickness.
Alternatively, the stabilization of the E-F loop in EM-ASR could be due to the removal of steric clashes between E-F loops of adjacent monomers in the hexagonal lattice. It has been shown that the E-F loops of adjacent trimers in the PM of BR can create steric conflicts under illumination (75,76). ASR, which is somewhat more cytoplasmically open (13,33,35) than BR, could produce such a steric clash in the PM-like lattice of PL-ASR, while in the more loosely packed tetragonal lattice of EM-ASR, this conflict is removed, resulting in a different dynamic behavior of the E-F loop.
Conclusions
We have examined the conformation of a seven-transmembrane helical microbial photoreceptor, ASR, in the environment of the E. coli inner membrane and compared it to that in a bilayer formed by DMPC/DMPA lipids. In both environments ASR forms trimers, as confirmed by visible CD spectroscopy and site-specific comparison of SSNMR chemical shifts of residues forming the intermonomer interface. SAXS measurements show that ASR reconstituted in DMPC/DMPA lipids forms small, two-dimensional crystalline domains of hexagonal symmetry (typical size ∼500 Å) with a lattice parameter of ∼66.4 Å. While a two-dimensional lattice is also found in the E. coli inner membrane, the domains are likely smaller in size, and packing is of tetragonal symmetry and is looser, with a lattice parameter of ∼74 Å.
Although the outer surfaces of ASR trimers in EM-ASR are more loosely packed and therefore more susceptible to the effects of the lipid environment, a comparison of solid-state NMR chemical shifts reveals that the overall structure of the protein in the E. coli inner membrane remains similar to that in DMPC/DMPA lipids, and is not significantly influenced by different crystal packing or by interactions with lipids and other proteins present in the E. coli inner membrane. Many functionally important structural motifs are conserved between the two environments. In particular, the retinal remains in the all-trans conformation in the dark state in the E. coli membrane and, in full agreement with this, the conformation of the retinal-binding pocket is unperturbed.
While the intermonomer interface is structurally well conserved between the two environments, there are small but noticeable chemical shift perturbations detected in two of the helices, B and D, all occurring on the sides facing either the interior of a monomer or the lipid bilayer. This may be the consequence of different lateral pressure profiles experienced by ASR in the two systems studied. Additionally, the large, threefold increase in the relative cross-peak intensities for residues in the cytoplasmic C-D and E-F loops indicate the possibility of a relative shift in the position of the trimer in the E. coli membrane bilayer as compared to that in proteoliposomes, or a removal of steric conflict between EF loops of adjacent trimers.
In contrast to small peptides and single-spanning membrane proteins, the interprotein interactions in and between the ASR α-helical bundles likely minimize the effect of the environment on the protein structure. Data on the adaptability of membrane protein structures to their environment are of fundamental importance for understanding protein function, and for the validation of their high-resolution structures in cell membrane mimetics.
Author Contributions
M.E.W., R.M., and E.R. prepared all samples and performed biochemical characterization; M.E.W. and L.S.B. carried out FTIR experiments and data analysis; S.W. and M.E.W. performed CD experiments; Y.J. and H.L. conducted SAXS measurements and data analysis; P.L.G. built the SSNMR probe used in the study; M.E.W. and I.H. collected SSNMR data; M.E.W. and V.L. analyzed SSNMR data; L.S.B. and V.L. designed the study; and M.E.W., L.S.B., and V.L. wrote the article. All authors discussed the results of the study.
Acknowledgments
We thank Drs. Dyanne Brewer and Armen Charchoglyan of the University of Guelph Advanced Analysis Centre Mass Spectrometry Facility for their help with gel digestion, and Mr. Brian Bryksa for assistance with CD measurements.
This research was supported by the Natural Sciences and Engineering Research Council of Canada (Discovery Grants to V.L. and L.S.B.), the National Science Foundation (under grant No. DMR 1410825 to H.L.), the Canada Foundation for Innovation, and the Ontario Ministry of Research and Innovation. Part of the work has been performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement No. DMR-1157490 and the State of Florida, and at the NMR Facility of the University of Guelph Advanced Analysis Centre (supported in part by the University of Guelph). SAXS experiments were performed at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences under contract No. DE-AC02-76SF00515. V.L. was a Tier II Canada Research Chair in Biophysics. M.E.W. and E.R. are recipients of a Natural Sciences and Engineering Research Council of Canada Postgraduate Fellowship, and R.M. is a recipient of an Ontario Graduate Scholarship.
Footnotes
Shenlin Wang’s present address is Beijing NMR Center, Peking University, Beijing, China.
Contributor Information
Leonid S. Brown, Email: lebrown@uoguelph.ca.
Vladimir Ladizhansky, Email: vladizha@uoguelph.ca.
Supporting Material
Supporting Citations
References (77–82) appear in the Supporting Material.
References
- 1.Andersen O.S., Koeppe R.E., 2nd Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- 2.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Lee A. Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 4.Tang M., Comellas G., Rienstra C.M. Advanced solid-state NMR approaches for structure determination of membrane proteins and amyloid fibrils. Acc. Chem. Res. 2013;46:2080–2088. doi: 10.1021/ar4000168. [DOI] [PubMed] [Google Scholar]
- 5.Lewandowski J.R. Advances in solid-state relaxation methodology for probing site-specific protein dynamics. Acc. Chem. Res. 2013;46:2018–2027. doi: 10.1021/ar300334g. [DOI] [PubMed] [Google Scholar]
- 6.Murray D.T., Das N., Cross T.A. Solid state NMR strategy for characterizing native membrane protein structures. Acc. Chem. Res. 2013;46:2172–2181. doi: 10.1021/ar3003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Opella S.J. Structure determination of membrane proteins by nuclear magnetic resonance spectroscopy. Annu. Rev. Anal. Chem. 2013;6:305–328. doi: 10.1146/annurev-anchem-062012-092631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladizhansky V. Recent advances in magic-angle spinning solid-state NMR of proteins. Isr. J. Chem. 2014;54:86–103. doi: 10.1016/j.pnmrs.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Cady S.D., Mishanina T.V., Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J. Mol. Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma M., Yi M., Cross T.A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shahid S.A., Bardiaux B., Linke D. Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat. Methods. 2012;9:1212–1217. doi: 10.1038/nmeth.2248. [DOI] [PubMed] [Google Scholar]
- 12.Park S.H., Das B.B., Opella S.J. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S., Munro R.A., Ladizhansky V. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat. Methods. 2013;10:1007–1012. doi: 10.1038/nmeth.2635. [DOI] [PubMed] [Google Scholar]
- 14.Wang S., Ladizhansky V. Recent advances in magic angle spinning solid state NMR of membrane proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2014;82:1–26. doi: 10.1016/j.pnmrs.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 15.Cross T.A., Sharma M., Zhou H.-X. Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cross T.A., Dong H., Zhou H.-X. M2 protein from influenza A: from multiple structures to biophysical and functional insights. Curr. Opin. Virol. 2012;2:128–133. doi: 10.1016/j.coviro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S., Munro R.A., Ladizhansky V. Paramagnetic relaxation enhancement reveals oligomerization interface of a membrane protein. J. Am. Chem. Soc. 2012;134:16995–16998. doi: 10.1021/ja308310z. [DOI] [PubMed] [Google Scholar]
- 18.Vogeley L., Sineshchekov O.A., Luecke H. Anabaena sensory rhodopsin: a photochromic color sensor at 2.0 Å. Science. 2004;306:1390–1393. doi: 10.1126/science.1103943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etzkorn M., Raschle T., Wagner G. Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: biophysical properties and NMR accessibility. Structure. 2013;21:394–401. doi: 10.1016/j.str.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higman V.A., Varga K., Watts A. The conformation of bacteriorhodopsin loops in purple membranes resolved by solid-state MAS NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2011;50:8432–8435. doi: 10.1002/anie.201100730. [DOI] [PubMed] [Google Scholar]
- 21.Dencher N.A., Heyn M.P. Formation and properties of bacteriorhodopsin monomers in the non-ionic detergents octyl-β-D-glucoside and Triton X-100. FEBS Lett. 1978;96:322–326. doi: 10.1016/0014-5793(78)80427-x. [DOI] [PubMed] [Google Scholar]
- 22.Heyn M.P., Cherry R.J., Dencher N.A. Lipid-protein interactions in bacteriorhodopsin-dimyristoylphosphatidylcholine vesicles. Biochemistry. 1981;20:840–849. doi: 10.1021/bi00507a029. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H.-X., Cross T.A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu R., Wang X., Tian F. In situ structural characterization of a recombinant protein in native Escherichia coli membranes with solid-state magic-angle-spinning NMR. J. Am. Chem. Soc. 2011;133:12370–12373. doi: 10.1021/ja204062v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renault M., Tommassen-van Boxtel R., Baldus M. Cellular solid-state nuclear magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA. 2012;109:4863–4868. doi: 10.1073/pnas.1116478109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao Y., Qin H., Cross T.A. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew. Chem. Int. Ed. Engl. 2012;51:8383–8386. doi: 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renault M., Pawsey S., Baldus M. Solid-state NMR spectroscopy on cellular preparations enhanced by dynamic nuclear polarization. Angew. Chem. Int. Ed. Engl. 2012;51:2998–3001. doi: 10.1002/anie.201105984. [DOI] [PubMed] [Google Scholar]
- 28.Jacso T., Franks W.T., Reif B. Characterization of membrane proteins in isolated native cellular membranes by dynamic nuclear polarization solid-state NMR spectroscopy without purification and reconstitution. Angew. Chem. Int. Ed. Engl. 2012;51:432–435. doi: 10.1002/anie.201104987. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto K., Caporini M.A., Ramamoorthy A. Cellular solid-state NMR investigation of a membrane protein using dynamic nuclear polarization. Biochim. Biophys. Acta. 2014;1848:342–349. doi: 10.1016/j.bbamem.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung K.H., Trivedi V.D., Spudich J.L. Demonstration of a sensory rhodopsin in eubacteria. Mol. Microbiol. 2003;47:1513–1522. doi: 10.1046/j.1365-2958.2003.03395.x. [DOI] [PubMed] [Google Scholar]
- 31.Sineshchekov O.A., Trivedi V.D., Spudich J.L. Photochromicity of Anabaena sensory rhodopsin, an atypical microbial receptor with a cis-retinal light-adapted form. J. Biol. Chem. 2005;280:14663–14668. doi: 10.1074/jbc.M501416200. [DOI] [PubMed] [Google Scholar]
- 32.Wang S., Shi L., Ladizhansky V. Solid-state NMR 13C and 15N resonance assignments of a seven-transmembrane helical protein Anabaena sensory rhodopsin. Biomol. NMR Assign. 2013;7:253–256. doi: 10.1007/s12104-012-9421-y. [DOI] [PubMed] [Google Scholar]
- 33.Shi L., Kawamura I., Ladizhansky V. Conformation of a seven-helical transmembrane photosensor in the lipid environment. Angew. Chem. Int. Ed. 2011;50:1302–1305. doi: 10.1002/anie.201004422. [DOI] [PubMed] [Google Scholar]
- 34.Good D.B., Wang S., Ladizhansky V. Conformational dynamics of a seven transmembrane helical protein Anabaena sensory rhodopsin probed by solid-state NMR. J. Am. Chem. Soc. 2014;136:2833–2842. doi: 10.1021/ja411633w. [DOI] [PubMed] [Google Scholar]
- 35.Wang S., Shi L., Ladizhansky V. Site-specific solid-state NMR detection of hydrogen-deuterium exchange reveals conformational changes in a 7-helical transmembrane protein. Biophys. J. 2011;101:L23–L25. doi: 10.1016/j.bpj.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marley J., Lu M., Bracken C. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 37.Wada Y., Kawanabe A., Ohtani H. Quantum yields for the light adaptations in Anabaena sensory rhodopsin and bacteriorhodopsin. Chem. Phys. Lett. 2008;453:105–108. [Google Scholar]
- 38.Shi L., Yoon S.R., Brown L.S. Cytoplasmic shuttling of protons in Anabaena sensory rhodopsin: implications for signaling mechanism. J. Mol. Biol. 2006;358:686–700. doi: 10.1016/j.jmb.2006.02.036. [DOI] [PubMed] [Google Scholar]
- 39.Shi L., Ahmed M.A., Ladizhansky V. Three-dimensional solid-state NMR study of a seven-helical integral membrane proton pump-structural insights. J. Mol. Biol. 2009;386:1078–1093. doi: 10.1016/j.jmb.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Wagner S., Baars L., de Gier J.-W. Consequences of membrane protein overexpression in Escherichia coli. Mol. Cell. Proteomics. 2007;6:1527–1550. doi: 10.1074/mcp.M600431-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Barinaga-Rementeria Ramírez I., Abedinpour P., Jergil B. Purification of caveolae by affinity two-phase partitioning using biotinylated antibodies and NeutrAvidin-dextran. Anal. Biochem. 2004;331:17–26. doi: 10.1016/j.ab.2004.04.044. [DOI] [PubMed] [Google Scholar]
- 42.Everberg H., Clough J., Ramírez I.B. Isolation of Escherichia coli inner membranes by metal affinity two-phase partitioning. J. Chromatogr. A. 2006;1118:244–252. doi: 10.1016/j.chroma.2006.03.123. [DOI] [PubMed] [Google Scholar]
- 43.Gor’kov P.L., Chekmenev E.Y., Brey W.W. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J. Magn. Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 44.Morcombe C.R., Zilm K.W. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 45.Delaglio F., Grzesiek S., Bax A. NMRPIPE: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 46.Keller R. 1st Ed. Cantina; Goldau, Switzerland: 2004. The Computer Aided Resonance Assignment Tutorial. [Google Scholar]
- 47.Sasaki T., Kubo M., Demura M. Halorhodopsin from Natronomonas pharaonis forms a trimer even in the presence of a detergent, dodecyl-β-D-maltoside. Photochem. Photobiol. 2009;85:130–136. doi: 10.1111/j.1751-1097.2008.00406.x. [DOI] [PubMed] [Google Scholar]
- 48.Kuang L., Fernandes D.A., Liang H. “Frozen” block copolymer nanomembranes with light-driven proton pumping performance. ACS Nano. 2014;8:537–545. doi: 10.1021/nn4059852. [DOI] [PubMed] [Google Scholar]
- 49.Sugawara E., Nikaido H. OmpA protein of Escherichia coli outer membrane occurs in open and closed channel forms. J. Biol. Chem. 1994;269:17981–17987. [PubMed] [Google Scholar]
- 50.DiRienzo J.M., Nakamura K., Inouye M. The outer membrane proteins of Gram-negative bacteria: biosynthesis, assembly, and functions. Annu. Rev. Biochem. 1978;47:481–532. doi: 10.1146/annurev.bi.47.070178.002405. [DOI] [PubMed] [Google Scholar]
- 51.Molloy M.P., Herbert B.R., Gooley A.A. Proteomic analysis of the Escherichia coli outer membrane. Eur. J. Biochem. 2000;267:2871–2881. doi: 10.1046/j.1432-1327.2000.01296.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y., Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patzelt H., Ulrich A.S., Oesterhelt D. Towards structural investigations on isotope labeled native bacteriorhodopsin in detergent micelles by solution-state NMR spectroscopy. J. Biomol. NMR. 1997;10:95–106. [Google Scholar]
- 54.Cherry R.J., Heyn M.P., Oesterhelt D. Rotational diffusion and exciton coupling of bacteriorhodopsin in the cell membrane of Halobacterium halobium. FEBS Lett. 1977;78:25–30. doi: 10.1016/0014-5793(77)80265-2. [DOI] [PubMed] [Google Scholar]
- 55.Pescitelli G., Woody R.W. The exciton origin of the visible circular dichroism spectrum of bacteriorhodopsin. J. Phys. Chem. B. 2012;116:6751–6763. doi: 10.1021/jp212166k. [DOI] [PubMed] [Google Scholar]
- 56.Ebrey T.G., Becher B., Honig B. Exciton interactions and chromophore orientation in the purple membrane. J. Mol. Biol. 1977;112:377–397. doi: 10.1016/s0022-2836(77)80188-5. [DOI] [PubMed] [Google Scholar]
- 57.Blaurock A.E., Stoeckenius W. Structure of the purple membrane. Nat. New Biol. 1971;233:152–155. doi: 10.1038/newbio233152a0. [DOI] [PubMed] [Google Scholar]
- 58.Yang L., Gordon V.D., Wong G.C.L. Synthetic antimicrobial oligomers induce a composition-dependent topological transition in membranes. J. Am. Chem. Soc. 2007;129:12141–12147. doi: 10.1021/ja072310o. [DOI] [PubMed] [Google Scholar]
- 59.Liang H., Whited G., Stucky G.D. The directed cooperative assembly of proteorhodopsin into 2D and 3D polarized arrays. Proc. Natl. Acad. Sci. USA. 2007;104:8212–8217. doi: 10.1073/pnas.0702336104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Unwin P.N.T., Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J. Mol. Biol. 1975;94:425–440. doi: 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- 61.Patterson A. The Scherrer formula for x-ray particle size determination. Phys. Rev. 1939;56:978–982. [Google Scholar]
- 62.Perálvarez-Marín A., Lórenz-Fonfría V.A., Padrós E. Inter-helical hydrogen bonds are essential elements for intra-protein signal transduction: the role of Asp115 in bacteriorhodopsin transport function. J. Mol. Biol. 2007;368:666–676. doi: 10.1016/j.jmb.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 63.Gautier A., Mott H.R., Nietlispach D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat. Struct. Mol. Biol. 2010;17:768–774. doi: 10.1038/nsmb.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reckel S., Gottstein D., Dötsch V. Solution NMR structure of proteorhodopsin. Angew. Chem. Int. Ed. Engl. 2011;50:11942–11946. doi: 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luecke H., Schobert B., Lanyi J.K. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 66.Ganapathy S., Naito A., McDowell C.A. Paramagnetic doping as an aid in obtaining high-resolution 13C NMR spectra of biomolecules in the solid state. J. Am. Chem. Soc. 1981;103:6011–6015. [Google Scholar]
- 67.Royant A., Nollert P., Navarro J. X-ray structure of sensory rhodopsin II at 2.1-Å resolution. Proc. Natl. Acad. Sci. USA. 2001;98:10131–10136. doi: 10.1073/pnas.181203898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schubert M., Kolbe M., Schmieder P. Heteronuclear multidimensional NMR spectroscopy of solubilized membrane proteins: resonance assignment of native bacteriorhodopsin. ChemBioChem. 2002;3:1019–1023. doi: 10.1002/1439-7633(20021004)3:10<1019::AID-CBIC1019>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 69.Etzkorn M., Martell S., Baldus M. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Eng. 2007;46:459–462. doi: 10.1002/anie.200602139. [DOI] [PubMed] [Google Scholar]
- 70.Kimura Y., Vassylyev D.G., Fujiyoshi Y. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature. 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 71.Essen L., Siegert R., Oesterhelt D. Lipid patches in membrane protein oligomers: crystal structure of the bacteriorhodopsin-lipid complex. Proc. Natl. Acad. Sci. USA. 1998;95:11673–11678. doi: 10.1073/pnas.95.20.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Belrhali H., Nollert P., Pebay-Peyroula E. Protein, lipid and water organization in bacteriorhodopsin crystals: a molecular view of the purple membrane at 1.9 Å resolution. Structure. 1999;7:909–917. doi: 10.1016/s0969-2126(99)80118-x. [DOI] [PubMed] [Google Scholar]
- 73.Oursel D., Loutelier-Bourhis C., Lange C.M. Lipid composition of membranes of Escherichia coli by liquid chromatography/tandem mass spectrometry using negative electrospray ionization. Rapid Commun. Mass Spectrom. 2007;21:1721–1728. doi: 10.1002/rcm.3013. [DOI] [PubMed] [Google Scholar]
- 74.Gidden J., Denson J., Lay J.O. Lipid compositions in Escherichia coli and Bacillus subtilis during growth as determined by MALDI-TOF and TOF/TOF mass spectrometry. Int. J. Mass Spectrom. 2009;283:178–184. doi: 10.1016/j.ijms.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vonck J. Structure of the bacteriorhodopsin mutant F219L N intermediate revealed by electron crystallography. EMBO J. 2000;19:2152–2160. doi: 10.1093/emboj/19.10.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shibata M., Yamashita H., Ando T. High-speed atomic force microscopy shows dynamic molecular processes in photoactivated bacteriorhodopsin. Nat. Nanotechnol. 2010;5:208–212. doi: 10.1038/nnano.2010.7. [DOI] [PubMed] [Google Scholar]
- 77.Pines A., Gibby M.G., Waugh J.S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973;59:569–590. [Google Scholar]
- 78.Hartmann S.R., Hahn E.L. Nuclear double resonance in the rotating frame. Phys. Rev. 1962;128:2042–2053. [Google Scholar]
- 79.Baldus M., Petkova A.T., Griffin R.G. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Phys. 1998;95:1197–1207. [Google Scholar]
- 80.Verel R., Ernst M., Meier B.H. Adiabatic dipolar recoupling in solid-state NMR: the DREAM scheme. J. Magn. Reson. 2001;150:81–99. doi: 10.1006/jmre.2001.2310. [DOI] [PubMed] [Google Scholar]
- 81.Hohwy M., Jakobsen H.J., Nielsen N.C. Broadband dipolar recoupling in the nuclear magnetic resonance of rotating solids: a compensated C7 pulse sequence. J. Chem. Phys. 1998;108:2686–2694. [Google Scholar]
- 82.Fung B.M., Khitrin A.K., Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.