

Abstract
G-protein-coupled receptors (GPCRs) are important membrane proteins that mediate cellular signaling and represent primary targets for about one-third of currently marketed drugs. Recent x-ray crystallographic studies identified distinct conformations of GPCRs in the active and inactive states. An allosteric sodium ion was found bound to a highly conserved D2.50 residue in inactive GPCRs, whereas the D2.50 allosteric pocket became collapsed in active GPCR structures. However, the dynamic mechanisms underlying these observations remain elusive. In this study, we aimed to understand the mechanistic effects of sodium ion binding on dynamic activation of the M3 muscarinic GPCR through long-timescale accelerated molecular dynamics (aMD) simulations. Results showed that with the D2.50 residue deprotonated, the M3 receptor is bound by an allosteric sodium ion and confined mostly in the inactive state with remarkably reduced flexibility. In contrast, the D2.50-protonated receptor does not exhibit sodium ion binding to the D2.50 allosteric site and samples a significantly larger conformational space. The receptor activation is captured and characterized by large-scale structural rearrangements of the transmembrane helices via dynamic hydrogen bond and salt bridge interactions. The residue motions are highly correlated during receptor activation. Further network analysis revealed that the allosteric signaling between residue D2.50 and key residues in the intracellular, extracellular, and orthosteric pockets is significantly weakened upon sodium ion binding.
Introduction
Muscarinic acetylcholine receptors belong to the superfamily of G-protein-coupled receptors (GPCRs), which are important cellular signaling proteins and represent primary targets of about one-third of currently marketed drugs (1). M3, one of five subtypes of the muscarinic receptors, preferentially couples with the Gq/11 proteins and stimulates the metabolism of phospholipids and calcium release. The M3 receptor has been targeted for treating many human diseases, including cancer (2), diabetes (3), and obesity (4).
Like many other GPCRs, the M3 muscarinic receptor exhibits a certain level of basal activity even without binding any agonists (5). This suggests that the ligand-free receptor exists in an ensemble of different conformations. Binding of agonists and inverse agonists in the orthosteric site biases the receptor conformational equilibrium toward the active and inactive states, respectively. The receptor is also able to bind neutral antagonists that have no signaling effects but just block the receptor from binding other ligands, as well as partial agonists that induce only submaximal activity (5).
The x-ray structure of the M3 muscarinic receptor has been determined in an inactive state bound by the tiotropium (TTP) antagonist (6). The M3 receptor consists of seven transmembrane (TM) helices that are connected by six alternating extracellular and intracellular loops (ECL1–ECL3 and ICL1–ICL3). Compared with the inactive M2 receptor, the M3 receptor exhibits a highly conserved orthosteric site in the TM domain and inward displacement of the cytoplasmic end of the TM5 helix toward TM6 by ∼4 Å. Although the x-ray structure has provided important insights into the structural scaffold of the M3 receptor and atomistic receptor-antagonist interactions, the active structure of the M3 receptor is still lacking and the dynamic mechanisms of the receptor activation remain unclear.
To date, x-ray crystallographic studies have revealed active structures of three other GPCRs: 1) rhodopsin, as in an activated form of apo opsin (7,8) and in the active metarhodopsin II state (9,10); 2) the β2-adrenergic receptor (β2AR) (11,12); and 3) the M2 muscarinic receptor (13). These active structures are characterized by opening of the G-protein-coupling site through rearrangements of the TM5, TM6, and TM7 helices relative to the inactive configuration. The cytoplasmic end of TM6 is tilted outward by ∼10–14 Å when coupled to the G-protein (11) or G-protein-mimetic nanobody (12,13), and a smaller magnitude of ∼6–7 Å in the absence of the G-protein (7). Correspondingly, the R3.50-E6.30 salt bridge, identified as the ionic lock in many inactive GPCRs, is broken. Moreover, the Y5.58 and Y7.53 residues relocate their side chains toward each other and form close interactions in the intracellular pocket (7,8,11,12). The Ballesteros-Weinstein numbering scheme (14) is used for residues in the TM helices as LN.XX, where L is the one-letter residue name, N is the TM helix number, and XX is the residue number relative to the most conserved residue (assigned as 50) in the TM helix, with the number decreasing toward the N-terminus and increasing toward the C-terminus.
Binding of an allosteric sodium ion to the D2.50 residue has also been observed in the x-ray structures of several inactive GPCRs, including the human A2A adenosine receptor (A2AAR) (15), β1-adrenergic receptor (β1AR) (16), protease-activated receptor 1 (PAR1) (17), and δ-opioid receptor (δ-OR) (18). The D2.50 allosteric site for sodium ion binding is highly conserved among class A GPCRs (19). It forms a pocket that comprises the bound sodium ion and a cluster of structurally ordered water molecules in the inactive x-ray structures. GPCR activation requires collapse of the D2.50 allosteric pocket due to a structural rearrangement of the TM helices, notably the inward displacement of the NPxxY motif in TM7 (15,19,20).
Along with breakthroughs in GPCR structural studies, computational simulations have been performed to investigate the conformational dynamics (21–24) and ligand binding (20,25–27) of GPCRs. Using the specialized supercomputer Anton, deactivation of β2AR from the active x-ray structure upon removal of the G-protein or its mimetic nanobody was modeled through microsecond-timescale conventional molecular dynamics (cMD) simulations (21). Anton simulations of the M2 and M3 receptors captured binding of antagonist TTP to the extracellular vestibule, but not to the orthosteric site (6). The ligand-free receptors maintained the inactive x-ray conformation without a large structural change (28). The Google Exacycle cloud-computing platform was used to simulate β2AR for a total of 2.15 ms by combining short cMD runs (24). Markov state models revealed multiple activation pathways of β2AR, and the use of simulation-derived structures improved molecular docking of ligand molecules. Moreover, binding of allosteric modulators to the extracellular vestibule of the M2 receptor was recently captured in Anton simulations (25). The binding of a sodium ion to D2.50 and its allosteric effects on the GPCR dynamics were also investigated for A2AAR (20), the D2 dopaminergic receptor (26), and μ-OR (29). Particularly, simulations on A2AAR suggested that GPCR activation and sodium ion binding to the D2.50 allosteric site are mutually exclusive (20).
For the M3 muscarinic receptor, experimental mutation of D2.50 to asparagine showed increased binding affinity of the nitrogen mustard analogs of the acetylcholine and McN-A-343 agonists (30), suggesting that the D2.50N mutation biases the M3 receptor toward the active conformational state. Random mutagenesis of the M3 receptor also suggested a conformational link between the highly conserved D2.50, R3.50, and Y5.58 residues that is critical for receptor activation and G-protein coupling (31,32). Although the protonation state of D2.50 in the M3 receptor remains unknown, the pKa of D2.50 in β2AR was calculated from free-energy simulations, suggesting that the protonated state of D2.50 is favored upon receptor activation (33). Further long-timescale cMD simulations showed that the protonated D2.50 biases the conformation of β2AR toward more active-like states (33). These results are consistent with a previous experimental finding that both the basal activity and the level of agonist-induced activation for β2AR are greater at pH 5 than at pH 8 (34). Therefore, protonation of D2.50 appears to play an important role in GPCR activation. In addition, analysis of earlier Anton cMD simulations of the M2 and M3 muscarinic receptors (6) using the simulation trajectories provided by DE Shaw Research showed that sodium ion binding is absent when D2.50 is protonated (no sodium ion is found within 5 Å of the Cγ atom of D2.50). A similar finding was obtained when we analyzed our previous accelerated molecular dynamics (aMD) simulations of the M2 muscarinic receptor (28). More recent Anton cMD simulations of the M2 receptor captured sodium ion binding when the D2.50 residue was deprotonated (25). Thus, our goal in this work was to systematically examine how protonation of D2.50 affects sodium ion binding and determine the corresponding allosteric effects on activation of a class A GPCR, particularly the M3 muscarinic receptor.
aMD is a biomolecular, enhanced sampling simulation technique that often works by adding a nonnegative boost potential to the potential energy surface, effectively decreasing energy barriers and thus accelerating transitions between the low-energy states (35–37). aMD simulations on timescales of hundreds of nanoseconds have been shown to capture millisecond-timescale events in proteins (38), including activation of the M2 receptor (28,39).
In our previous study of the M2 muscarinic receptor, we demonstrated aMD on GPCR activation (28); here, we investigate the allosteric effects of sodium ion binding on activation of the M3 muscarinic receptor through extensive aMD simulations (total length of 6 μs). Based on the fact that the M3 receptor exhibits basal activity even without binding any agonists (5), we removed the cocrystalized ligand from the x-ray structure for enhanced sampling of the receptor’s different conformational states. A sodium ion is observed to bind the deprotonated, but not the protonated, D2.50 residue. Binding of the allosteric sodium ion leads to remarkably reduced flexibility in the D2.50-deprotonated M3 receptor that is mostly confined to the inactive state. In contrast, the D2.50-protonated M3 receptor undergoes activation, sampling a significantly larger conformational space.
Materials and Methods
aMD
aMD enhances the conformational sampling of functional biomolecules, often by adding a nonnegative boost potential to the potential energy surface when the system potential is lower than a reference energy (35–37):
| (1) |
where is the original potential, E is the reference energy, and is the modified potential. The boost potential is given by
| (2) |
where α is the acceleration factor. As the acceleration factor α decreases, the potential energy surface is flattened and biomolecular transitions between the low-energy states are increased.
Dual-boost aMD (37), which has been shown to provide sufficient sampling to capture activation of the M2 receptor (28), is adopted in this study. A dihedral bias potential is applied to all dihedral angles in the system and another total boost potential is applied to all individual atoms. The input parameters (Edihed, αdihed; Etotal, αtotal) are calculated as
| (3) |
where Natoms is the total number of atoms; Vdihed_avg and Vtotal_avg are the average dihedral and total potential energies calculated from short cMD simulations (e.g., 100 ns in this study), respectively; and λ and ν are adjustable acceleration parameters (previous studies suggested that proper acceleration is achieved with λ = 0.3 and ν = 0.2 for enhanced sampling of membrane proteins, e.g., activation of the M2 receptor (28,39)).
Simulations of the M3 muscarinic receptor
After the ligand was removed from the inactive x-ray structure (Protein Data Bank (PDB): 4DAJ), the M3 receptor was embedded in a palmitoyl-oleoyl-phosphatidyl-choline (POPC) lipid bilayer and solvated in an aqueous medium, with all atoms represented explicitly. The CHARMM27 parameter set was used for the protein (with CMAP terms included) (40,41), CHARMM36 for POPC lipids (42), and TIP3P model for water molecules (43). After a 100 ns cMD simulation, we performed two, five, and three independent 600 ns aMD simulations on the D2.50-deprotonated receptor in 0.15 M Na+ solution, the D2.50-protonated receptor in 0.15 M Na+ solution, and the D2.50-protonated receptor without Na+, respectively (Table 1). We performed all MD simulations using NAMD2.9 (44). Details regarding the system preparation and simulation protocols are provided in the Supporting Material.
Table 1.
List of cMD and aMD simulations performed on the M3 muscarinic receptor
| System | D2.50 | [Na+] | Simulation | Duration (ns) |
|---|---|---|---|---|
| 1 | deprotonated | 0.15 M | cMD | 100 |
| aMD | 600 × 2 | |||
| 2 | protonated | 0.15 M | cMD | 100 |
| aMD | 600 × 5 | |||
| 3 | protonated | – | cMD | 100 |
| aMD | 600 × 3 | |||
Simulation analysis
Residue cross correlations of the M3 receptor were calculated based on mutual information between all Cα atoms in the protein using the generalized correlation analysis approach developed by Lange and Grubmüller (45). A network analysis of the M3 receptor was carried out using the NetworkView plugin in VMD (46,47). A network graph with each protein residue treated as a node was constructed. Edges were added to the network by connecting pairs of in-contact nodes, which are defined as having any heavy atom within 5 Å for >75% of the simulation time. Each edge is weighted by the correlation values of the two end nodes () as . The length of a path between two distant nodes equals the sum of the edge weights along the path, i.e., . The shortest paths (D0) were computed between residue D2.50 and distant residues in the orthosteric ligand-binding site (W6.48 and N6.52), intracellular G-protein coupling site (E6.30, Y5.58, and Y7.53), and extracellular vestibule (F221ECL2, N6.58, and W7.35). The number of suboptimal paths (Nsop) was calculated within two distance limits (δ = 10 and 20) of the shortest paths.
Results
Sodium ion binding and activation of the M3 muscarinic receptor
Starting from the inactive x-ray structure of the M3 receptor with antagonist TTP removed (Fig. 1 A), residue D2.50 located in the known allosteric site (15) was either deprotonated or protonated for separate simulations of the receptor in 0.15 M Na+ solution. Residue D3.32 located in the orthosteric site was deprotonated in all of the simulations. In an initial 100 ns cMD simulation of the D2.50-deprotonated system at 310 K, one sodium ion entered the orthosteric site and then bound to the D2.50 allosteric site (Fig. S1 A), followed by a second sodium ion binding to the D3.32 orthosteric site (Fig. S1, A and C). In a different 100 ns cMD simulation of the D2.50-protonated system, no sodium ion bound to the D2.50 allosteric site, but one bound to the D3.32 orthosteric site (Fig. S1, B and D).
Figure 1.
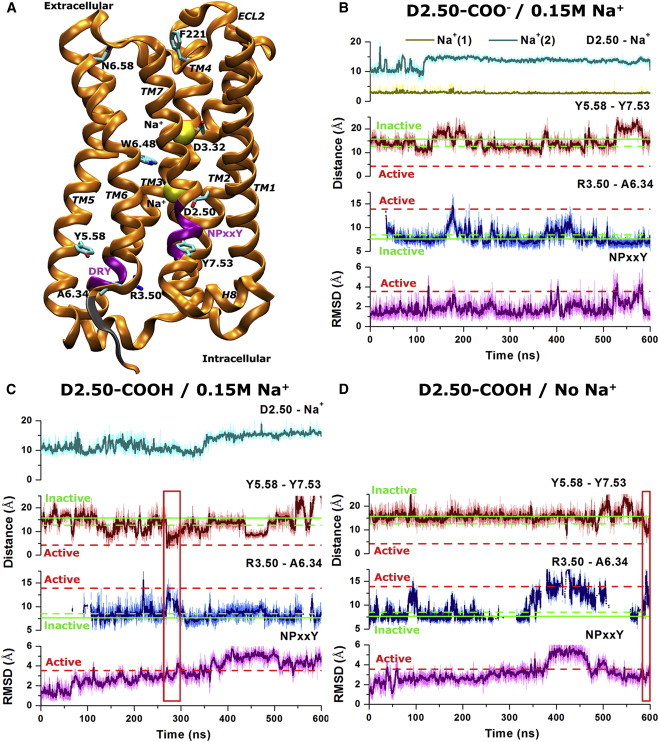
Activation and mechanistic effects of sodium ion binding of the M3 muscarinic receptor simulated via aMD. (A) Schematic representation of the M3 receptor (orange ribbons) with two sodium ions (yellow spheres) bound to the D3.32 orthosteric and D2.50 allosteric sites. Key residues, including D2.50, D3.32, R3.50, A6.34, W6.48, N6.58, Y5.58, Y7.53, and F221ECL2, are shown in sticks, and the two highly conserved DRY and NPxxY motifs are highlighted in purple. Three residues that are missing in the x-ray structure (K6.31−A6.33) are added and shown in gray. (B) In the D2.50-deprotonated receptor in 0.15 M Na+ solution, two sodium ions bind to the D2.50 allosteric and D3.32 orthosteric sites, respectively. (C) In the D2.50-protonated receptor in 0.15 M Na+ solution, only one sodium binds to the D3.32 orthosteric site. (D) A third system with D2.50 protonated is simulated in the absence of Na+. The distances of bound sodium ion(s) to the Cγ atom of D2.50 are plotted in (B) and (C). The corresponding distances between Y5.58 and Y7.53 (hydroxyl oxygens) and between R3.50 and A6.34 (Cα atoms) are plotted for each of the three simulated systems. Thick lines depict the running average over 1 ns (simulation frames were saved every 10 ps for the original). The Y5.58-Y7.53 and R3.50-A6.34 distances in the inactive x-ray structure of the M3 receptor (PDB: 4DAJ) are plotted in the solid green line and those of the inactive and active structures of the M2 receptor (PDB: 3UON and 4MQS, respectively) are plotted in dashed green and red lines, respectively. The R3.50-A6.34 distance is reported only when helical structure is formed up to residue A6.34 in the TM6 intracellular region (Fig. S3). The RMSD of the NPxxY motif between the active and inactive structures of the M2 receptor (3.6 Å) is also plotted in a dashed red line. Red rectangles highlight activation events of the M3 receptor in (C) and (D). To see this figure in color, go online.
Using the final structures of the cMD simulations, two and five independent 600 ns aMD simulations with randomized initial atomic velocities at 310 K were performed on the D2.50-deprotonated and protonated M3 receptor, respectively. In the D2.50-deprotonated system, the two sodium ions remain bound at the corresponding D2.50 allosteric and D3.32 orthosteric sites (Figs. 1 B and S2 A), whereas in the D2.50-protonated system, only one sodium ion remains bound to the D3.32 orthosteric site (Figs. 1 C and S2 B). In addition, three independent 600 ns aMD simulations were performed on the D2.50-protonated receptor in the absence of Na+ for comparison (Table 1).
During a 600 ns aMD simulation of the D2.50-deprotonated receptor, a sodium ion is bound to the D2.50 side chain at an average distance of 2.8 Å to the Cγ atom with slight fluctuations (0.4 Å standard deviation (SD); Fig. 1 B). The second sodium ion remains at an average distance of 2.7 Å to the Cγ atom of D3.32 with a 0.3 Å SD (Fig. S2 A). The distance between the hydroxyl oxygens of Y5.58 and Y7.53 stays above 8 Å, being similar to the Y5.58-Y7.53 distance observed in the inactive x-ray structures of the M2 and M3 receptors (12.6 Å and 15.6 Å, respectively) and significantly greater than that of the active M2 structure (4.2 Å). The distance between the Cα atoms of residues R3.50 and A6.34 in the cytoplasmic ends of TM3 and TM6 remains mostly at ∼8 Å and increases up to ∼13 Å during short periods of time near ∼180 ns and ∼420 ns. Note that the R3.50-A6.34 distance is reported only when helical structure is formed up to residue A6.34 in the TM6 intracellular region (Fig. S3), which has three residues (K6.31–A6.33) missing and adopts a random coil conformation in the x-ray structure (Fig. 1 A). For the NPxxY motif, the average root mean-square deviation (RMSD) of the M3 receptor relative to the inactive x-ray conformation is 2.3 Å, which is lower than the 3.6 Å RMSD calculated between the active and inactive x-ray structures of the M2 receptor. When the two independent 600 ns aMD simulations of the D2.50-deprotonated receptor in 0.15 M Na+ solution (1200 ns in total) are combined, calculations of the potential of mean force (PMF) show that the R3.50–A6.34 distance possesses an energy minimum at 7.0 Å (Fig. S4 A) and the Y5.58-Y7.53 hydrogen bond stays in the fully open conformation with a broad energy well at ∼12−16 Å (Fig. S4 C). Therefore, the D2.50-deprotonated M3 receptor that is bound by the allosteric sodium ion does not visit a conformation similar to the active M2 receptor.
In contrast, sodium ion binding to the D2.50 allosteric site is absent during a 600 ns aMD simulation of the D2.50-protonated M3 receptor (Fig. 1 C). The only sodium ion that enters the orthosteric site remains bound to the D3.32 side chain at 2.9 Å average distance with small fluctuations of 0.6 Å SD (Fig. S2 B). The side chains of Y5.58 and Y7.53 are able to move toward each other, forming close interaction briefly at 3.0 Å distance between the two hydroxyl oxygens during 260-300 ns of the aMD simulation (Fig. 1 C). Meanwhile, the distance between the Cα atoms of R3.50 and A6.34 increases from 7.5 Å to ∼14 Å due to outward tilting of the TM6 cytoplasmic end. Furthermore, the NPxxY motif moves inward with an ∼4 Å RMSD relative to the inactive x-ray conformation, due to rearrangement of the TM7 cytoplasmic end (Fig. 1 C). PMF calculations using a combined trajectory of the five independent 600 ns aMD simulations (3000 ns in total) show that the Y5.58-Y7.53 hydrogen bond visits the closed and water-bridged conformation with local energy wells centered at 3.0 Å and 4.5 Å, respectively (Fig. S4 D). Although the R3.50-A6.34 distance mostly adopts the closed conformation with the energy minimum found at 8.0 Å, it samples a fully open conformation with a second energy well centered at ∼15 Å (Fig. S4 B), which corresponds to outward tilting of the TM6 cytoplasmic end. Even without reweighting of the aMD simulations, the free energy of the fully open conformation of R3.50-A6.34 (active) is ∼1.5 kcal/mol greater than that of the closed (inactive) conformation, and the closed and water-bridged conformation of the Y5.58-Y7.53 hydrogen bond (active) is ∼3.0 kcal/mol greater than that of the fully open (inactive) conformation. These results suggest that the receptor active state with significantly higher free energies is less stable than the inactive state. Further PMF calculations of the distance between F221ECL2 and N6.58 in the extracellular vestibule show that in addition to the energy minimum at ∼13–14 Å, it visits a second energy well centered at ∼9 Å in the D2.50-protonated M3 receptor (Fig. S4 F), suggesting a narrowing of the extracellular mouth. These structural changes resemble those observed in activation of the M2 receptor (13,28), β2AR (11,12), and rhodopsin (7,8). Therefore, activation of the M3 receptor is captured in an aMD simulation of the D2.50-protonated form without sodium ion binding to the D2.50 allosteric site.
For further comparison, three independent 600 ns aMD simulations were also obtained on the D2.50-protonated M3 receptor in the absence of Na+. The results show that the distance between the hydroxyl oxygens of Y5.58 and Y7.53 decreases to 4.5 Å at ∼590 ns, accompanied by an increased distance between the Cα atoms of R3.50 and A6.34 to ∼17 Å (Fig. 1 D). Meanwhile, the RMSD of the NPxxY motif relative to the inactive x-ray conformation increases to ∼3 Å. The D2.50-protonated M3 receptor still undergoes activation in the absence of Na+, suggesting that activation of the M3 receptor is independent of sodium ion binding to the D3.32 orthosteric site.
Remarkably reduced flexibility of the M3 receptor upon sodium ion binding to D2.50
Here, we examined the mechanistic effects of sodium ion binding on the structural flexibility and dynamic motions of the M3 receptor. Calculations on the root mean-square fluctuations (RMSFs) of the receptor Cα atoms show that the D2.50-protonated form undergoes similar residue fluctuations in 0.15 M Na+ solution or in the absence of Na+, despite small differences observed in the TM1, TM2, and TM3 regions (Fig. 2 A). In comparison, the D2.50-deprotonated M3 receptor with sodium ion bound to the D2.50 allosteric site exhibits a remarkably reduced flexibility, by ∼30% on average, particularly in the TM3, TM4, TM5, TM6, and TM7 helices. Thus, the D2.50-deprotonated receptor is less likely to be activated because the structural rearrangement of these TM helices is highly involved in activation of class A GPCRs (11,13).
Figure 2.
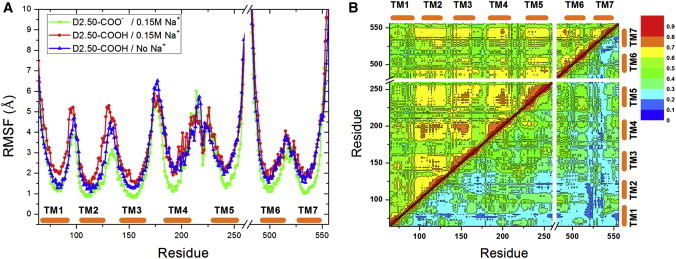
(A) The RMSFs of the M3 receptor were calculated by averaging the multiple independent 600 ns aMD simulations of the D2.50-deprotonated system in 0.15 M Na+ solution, the D2.50-protonated system in 0.15 M Na+ solution, and the D2.50-protonated system in the absence of Na+. (B) Generalized cross correlations of residue motions in the D2.50-deprotonated receptor (lower triangle) compared with the D2.50-protonated (upper triangle) in 0.15 M Na+ solution. Orange bars denote the seven TM helices (TM1–TM7). To see this figure in color, go online.
Fig. 2 B plots the generalized cross correlations of residue motions in the D2.50-deprotonated M3 receptor compared with the D2.50-protonated form, both of which were placed in the 0.15 M Na+ solution. In the D2.50-deprotonated form, although the residue motions in the extracellular region of TM3 and the ECL2 between the TM4 and TM5 extracellular ends exhibit correlations higher than 0.6, largely due to the presence of a disulphide bond connecting the two regions, the residue motions of other regions possess correlations lower than 0.6. Overall, the D2.50-protonated receptor undergoes increased correlated motions, with correlations between most TM helices being greater than 0.6 (Fig. 2 B). Notably, the TM2 helix, which is not stabilized by the allosteric sodium ion binding as in the D2.50-deprotonated system, exhibits higher correlations with the TM3, TM4, TM5, and TM7 helices. Moreover, correlations among the TM3, TM4, TM5, TM6, and TM7 helices are increased by ∼0.25 on average. The D2.50-protonated M3 receptor in the absence of Na+ shows similar increased residue correlations relative to the D2.50-deprotonated form with a sodium ion bound to the D2.50 allosteric site (Fig. S5). This is consistent with earlier findings that the apo M2 receptor (with D2.50 protonated) undergoes activation in the ligand-free form and exhibits significantly higher correlations of residue motions than in the antagonist-bound form, which maintains the inactive x-ray conformation (28). Correlated motions of the TM helices play a key role in activation and signaling of class A GPCRs.
Confined conformational space of the M3 receptor with sodium ion binding to D2.50
We further examined the conformational space sampled by the D2.50-deprotonated M3 receptor using the R3.50-A6.34 and Y5.58-Y7.53 distances as two reaction coordinates. With sodium ion bound to the D2.50 allosteric site, the M3 receptor mostly stays in the inactive state, with only transient visiting of an intermediate conformation (denoted IM (D2.50-COO−)), which exhibits an increased distance between R3.50 and A6.34 as shown in Fig. 3 A.
Figure 3.
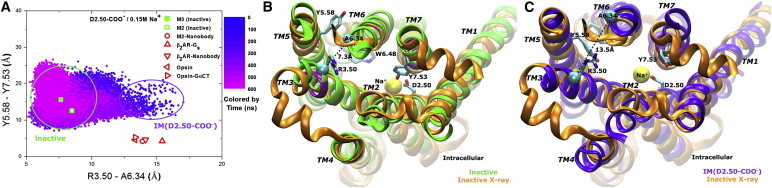
(A) Conformational space sampled by the D2.50-deprotonated M3 receptor during a 600 ns aMD simulation with sodium ion binding to the D2.50 allosteric site. Related GPCR x-ray structures, including the inactive M3 (PDB: 4DAJ), inactive M2 (PDB: 3UON), active M2 coupled by a G-protein-mimetic nanobody (PDB: 4MQS), active β2AR coupled by a Gs protein (PDB: 3SN6) or its mimetic nanobody (PDB: 3P0G), and opsin (active rhodopsin) with or without the C-terminal peptide of the Gα subunit (PDB: 3DQB and 3CAP, respectively) are marked for comparison. (B and C) The representative inactive (B, green) and intermediate IM (C2.50-COO−) (C, blue) conformations are compared with the x-ray structure (orange). Key residues, the DRY and NPxxY motifs, and sodium ions are shown similarly as described in Fig. 1A. To see this figure in color, go online.
A representative inactive conformation of the D2.50-deprotonated M3 receptor is depicted in Fig. 3 B. It largely overlaps with the inactive x-ray structure. The distance between R3.50 and A6.34 is 7.3 Å and the side chain of Y7.53 points toward Na+ in the D2.50 allosteric pocket. Marked structural changes are observed in the TM6 intracellular region, which forms an α helix in ∼60 ns of aMD simulation time (Fig. S3 A). Moreover, Y5.58 in the TM5 cytoplasmic end is able to reorient the side chain from the lipid-exposed surface to the interface between TM5 and TM6 helices in a slightly different inactive conformation (Fig. S6).
In the IM (D2.50-COO−) intermediate, the TM6 cytoplasmic end moves toward TM7, with the distance between R3.50 and A6.34 increasing to 13.5 Å (Fig. 3 C). Meanwhile, the NPxxY motif in TM7 tilts outward by ∼3 Å and Y7.53 flips the side chain from pointing toward Na+ in the D2.50 allosteric pocket to pointing toward the cytoplasmic solvent. This intermediate appears to result from the mobile nature of the TM6 cytoplasmic end identified in class A GPCRs (48). However, the presence of a sodium ion in the D2.50 allosteric pocket hinders inward movement of the NPxxY motif in TM7 (particularly reorientation of the Y7.53 side chain) and thus prohibits activation of the M3 receptor.
Activation of the M3 receptor without sodium ion binding to D2.50
In the absence of sodium ion binding to the D2.50 allosteric site, the D2.50-protonated M3 receptor samples a significantly larger conformational space compared with the D2.50-deprotonated form, since it is able to escape out of the inactive state and visit several distinct conformational states (Fig. 4 A). Of particular interest, the M3 receptor visits an active conformational state that resembles the x-ray structures of active GPCRs, including the M2 receptor (13,28), β2AR (11,12), and opsin (7,8).
Figure 4.
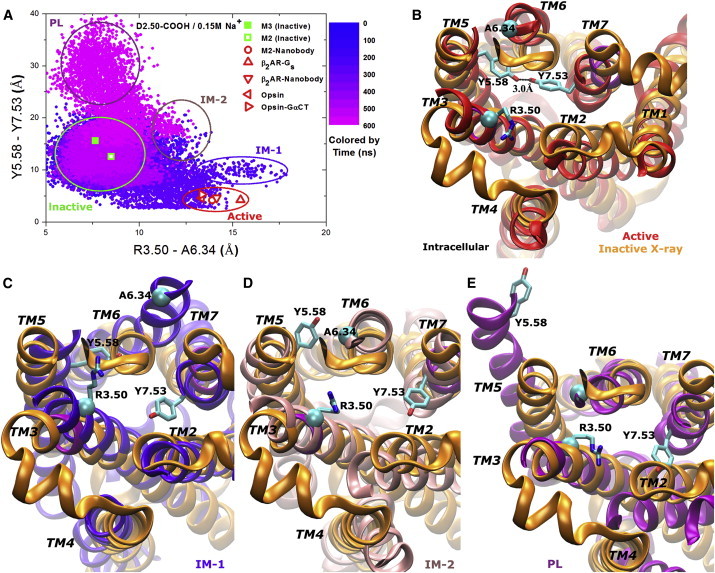
(A) Conformational space sampled by the D2.50-protonated M3 receptor in 0.15 M Na+ solution during a 600 ns aMD simulation. Related GPCR x-ray structures are marked similarly as in Fig. 3A. (B–E) Intracellular view of different conformers of the M3 receptor that might be relevant for GPCR phosphorylation are compared with the x-ray structure (orange): (B) active (red), (C) IM-1 intermediate (blue), (D) IM-2 intermediate (pink), and (E) PL (purple). To see this figure in color, go online.
During activation of the D2.50-protonated receptor in 0.15 M Na+ solution, the TM6 cytoplasmic end tilts outward by ∼6 Å (Fig. 4 B). Residue Y5.58 flips the side chain from the lipid-exposed surface to the interface between TM5 and TM6, and Y7.53 also reorients the side chain toward Y5.58 with ∼4 Å inward displacement of the NPxxY motif in TM7. Subsequently, the two tyrosines form a hydrogen bond at a 3.0 Å distance between the hydroxyl oxygens. An intermediate conformation that appears in the receptor activation pathway, IM-1, is shown in Fig. 4 C. In this conformation, Y7.53 flips the side chain from pointing toward the D2.50 allosteric site to pointing toward the cytoplasmic solvent as observed in the IM (D2.50-COO−) conformation. Moreover, Y5.58 slides its side chain into the space that is originally occupied by the TM6 cytoplasmic end, leading to a large outward tilting of the latter at a distance of ∼16 Å between R3.50 and A6.34 (Fig. 4 C). Additionally, the D2.50-protonated receptor visits a second intermediate, IM-2, which exhibits less outward tilting of the TM6 cytoplasmic end (∼12 Å distance between R3.50 and A6.34) compared with IM-1. In the IM-2 intermediate, the Y5.58 side chain remains on the lipid-exposed surface as observed in the x-ray structure, whereas Y7.53 reorients the side chain toward the cytoplasmic solvent as in IM-1 or IM (D2.50-COO−) (Fig. 4 D). Activation of the M3 receptor and both IM-1 and IM-2 intermediate conformations are also observed in an aMD simulation of the D2.50-protonated receptor in the absence of Na+, during which the Y5.58 and Y7.53 side chains form a water-bridged hydrogen-bonding interaction at a 4.5 Å distance between the two hydroxyl oxygens, and the TM6 cytoplasmic end tilts outward at a larger magnitude in the active conformation (∼14 Å) (Fig. S7).
During the last 50 ns of the aMD simulation of the D2.50-protonated receptor in 0.15 M Na+ solution, a distinct conformation is observed, with the Y5.58-Y7.53 distance increasing significantly to ∼30 Å (see Figs. 1 C and 4 A). This is largely due to the outward displacement of the mobile TM5 cytoplasmic end and flipping of the Y5.53 side chain away from the TM bundle (Fig. 4 E). Such a structural change can potentially lead to a large opening between the cytoplasmic ends of TM5 and TM6, and exposure of the third intracellular loop (ICL3). Since earlier site-directed mutagenesis experiments identified phosphorylation sites of GPCR in ICL3 (two clusters of Ser/Thr residues) (49,50), this conformation (denoted PL) may be relevant for coupling of GPCR with protein kinases for phosphorylation and arrestin binding. However, this strongly requires experimental validation, ideally with a high-resolution structure of arrestin-coupled GPCR. This observation is consistent with a previous finding regarding the M2 receptor, which also samples a similar conformational state during aMD simulations (28).
Network changes induced by sodium ion binding
To understand the effects of sodium ion binding on receptor allosteric signaling, we obtained dynamic networks for both the D2.50-protonated and D2.50-deprotonated M3 muscarinic receptor (see Materials and Methods). Previous studies on A2AAR and β2AR suggested that activation of GPCRs involves a collapse of the sodium-ion-binding pocket located at the D2.50 residue (19). Further simulations identified that protonation of the D2.50 residue shifts the conformation of the β2AR toward more active-like states (33). Therefore, we determined the shortest paths and number of suboptimal paths (Nsop) between residue D2.50 and distant residues in the orthosteric ligand-binding site (W6.48 and N6.52), the intracellular G-protein coupling site (E6.30, Y5.58, and Y7.53), and the extracellular vestibule (F221ECL2, N6.58, and W7.35) (Fig. 5). The shortest distances and Nsop values are summarized in Table 2.
Figure 5.
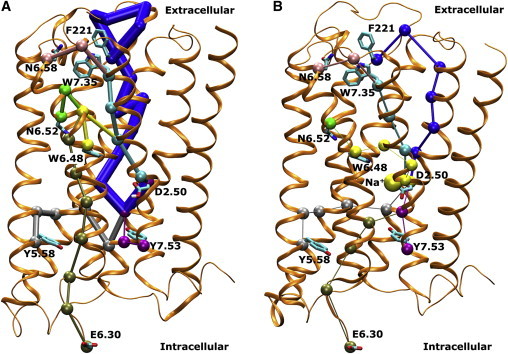
(A and B) The shortest paths between residue D2.50 and topographically distant residues obtained from network analysis of the (A) D2.50-protonated and (B) D2.50-deprotonated M3 receptor in 0.15 M Na+ solution. The analyzed signaling residues include W6.48 and N6.52 in the orthosteric ligand-binding site; E6.30, Y5.58, and Y7.53 in the intracellular G-protein-coupling site; and F221ECL2, N6.58, and W7.35 in the extracellular vestibule. The edge thickness is set proportional to the number of suboptimal paths (Nsop) obtained within the δ = 10 distance limit of the shortest paths as listed in Table 2. To see this figure in color, go online.
Table 2.
The shortest distance (D0) and number of suboptimal paths (Nsop) obtained from network analysis of the D2.50-deprotonated and D2.50-protonated M3 receptor between residue D2.50 and distant residues in the intracellular G-protein-coupling site, orthosteric ligand-binding site, and extracellular vestibule
| Residue | D2.50-protonated / 0.15 M Na+ |
D2.50-deprotonated / 0.15 M Na+ |
|||||
|---|---|---|---|---|---|---|---|
| D0 | Nsop (δ = 10) | Nsop (δ = 20) | D0 | Nsop (δ = 10) | Nsop (δ = 20) | ||
| Orthosteric site | W6.48 | 166 | 2 | 3 | 210 | 1 | 1 |
| N6.52 | 170 | 8 | 24 | 235 | 1 | 5 | |
| Intracellular | E6.30 | 340 | 5 | 28 | 349 | 2 | 2 |
| Y5.58 | 290 | 12 | 94 | 205 | 2 | 12 | |
| Y7.53 | 82 | 3 | 14 | 99 | 2 | 5 | |
| Extracellular | F221ECL2 | 638 | 24 | 150 | 276 | 5 | 15 |
| N6.58 | 161 | 9 | 33 | 259 | 4 | 17 | |
| W7.35 | 105 | 3 | 5 | 170 | 1 | 3 | |
The Nsop was calculated within two distance limits (δ = 10 and 20) of the shortest paths.
Overall, the shortest signaling distances between residue D2.50 and distant residues in the D2.50-deprotonated receptor bound by sodium ion are greater than those in the D2.50-protonated M3 receptor, except for interactions with the Y5.58 and F221ECL2 residues (Table 2). This suggests higher correlations between nodes along the shortest paths in the D2.50-protonated receptor. Furthermore, the D2.50-protonated receptor possesses a significantly larger number of degenerate suboptimal paths than the D2.50-deprotonated receptor. A similar trend was obtained for Nsop within two distance limits (δ = 10 and 20) of the shortest paths.
In addition, we compared the two D2.50-protonated and D2.50-deprotonated receptor forms regarding their network interactions involving key residues in the extracellular vestibule and orthosteric ligand-binding site (N6.58, W7.35, W6.48, and N6.52), and in the intracellular G-protein coupling site (E6.30, Y5.58, and Y7.53) (Table S1). Similar to the above findings, the D2.50-protonated receptor shows decreased distances and an increased number of suboptimal paths for signaling relative to the D2.50-deprotonated receptor. Therefore, the receptor exhibits stronger allosteric signaling during activation in the D2.50-protonated form. Such allosteric signaling is greatly weakened in the D2.50-deprotonated M3 receptor upon sodium ion binding.
Discussion
Comparison with activation of the M2 muscarinic receptor
The representative active conformations of the M3 receptor observed during aMD simulations of the D2.50-protonated form are compared with the x-ray structure of the active M2 receptor in Fig. S8. The orientation of the Y5.58 and Y7.53 side chains in the simulation-derived active M3 receptor is very similar to that of the active M2 x-ray structure, accompanied by a similar inward movement of the NPxxY motif in TM7. Relative to the inactive x-ray structures, the TM6 cytoplasmic end moves outward in both the active M3 and M2 receptors, although the displacement magnitude varies. In the active x-ray structure of the M2 receptor that is bound by agonist iperoxo and a G-protein mimetic nanobody, the TM6 cytoplasmic end tilts outward by 10.4 Å compared with the inactive antagonist-bound x-ray structure (13). In active conformations of the M3 receptor, the TM6 cytoplasmic end moves outward by ∼6 Å in aMD simulations of the D2.50-protonated receptor in 0.15 M Na+ solution and ∼14 Å in the absence of Na+. Different displacement magnitudes of the TM6 cytoplasmic end have also been observed in the active x-ray structures of opsin (∼6–7 Å) (7,8), β2AR coupled by the Gs protein (14 Å) (11), and a G-protein mimetic nanobody (11.4 Å) (12). These findings suggest that the TM6 cytoplasmic end is highly mobile in class A GPCRs. It is able to move outward across a wide range of magnitudes, from ∼6 Å to ∼14 Å. The presence of an intracellular G-protein or a G-protein mimetic nanobody is likely to stabilize GPCRs in an active conformation, with a larger outward displacement of the TM6 cytoplasmic end (e.g., x-ray structures of the active β2AR and M2 receptor) compared with one without the intracellular counterpart (e.g., opsin).
In addition, we compared the primary sequence of the rat M3 receptor studied here with the five subtypes of muscarinic receptors (Fig. S9). Notably, both the inactive and active x-ray structures have been determined for the human M2 receptor (13,51). Excluding the bulk of ICL3 that is missing in the x-ray structures, 81% of the protein residues are highly conserved between the M3 and M2 receptors. High sequence similarity is also identified between the M3 receptor and the M5 (93%), M1 (89%), and M4 (83%) subtypes. Such high similarity suggests a common structural scaffold for the seven TM helices, and similar activation mechanisms in the muscarinic receptors. This includes the characteristic conformational changes in the R3.50-E6.30 ionic lock and the Y5.58-Y7.53 hydrogen bond during activation, as well as binding of a sodium ion to the highly conserved D2.50 residue. However, 19% of the protein residues differ between the M2 and M3 receptors. These residues are mostly distributed on the receptor surface (Fig. S9 A). Notably, 10 and six residues are not conserved in the intracellular regions of the TM5 and TM6 helices, respectively. This is correlated with the high mobility of the TM5 and TM6 cytoplasmic ends, which have been crystalized in different configurations in the GPCR x-ray structures. For example, the TM5 cytoplasmic end in the inactive x-ray structure of the M3 receptor exhibits an ∼4 Å inward displacement compared with the inactive M2 structure, along with a distinct orientation of the Y5.58 side chain (6,51). Low conservation of residues in the TM6 cytoplasmic end could also contribute to its different displacement magnitude in the GPCR active conformations.
Sodium ion binding and the allosteric effects
Previous microsecond-timescale cMD simulations captured sodium ion binding to residue D2.50 in the D2 receptor and identified three low-energy sites along the binding pathway: the D3.32 orthosteric pocket, a cavity near W6.48, and the D2.50 allosteric pocket (26). Such sodium ion binding to the D2.50 allosteric site was also observed in cMD simulations of μ-OR (29). In addition, binding of sodium ion in the D2.50 allosteric pocket was determined in x-ray structures of the inactive A2AAR (15), β1AR (16), PAR1 (17), and δ-OR (18). It has been suggested that the muscarinic receptors possess a more open passage for sodium access compared with A2AAR and β1AR (19). In earlier simulations of the M2 muscarinic receptor, sodium ion was observed to bind to the deprotonated D2.50 residue (25), but this did not occur when residue D2.50 was protonated (6,28). In this study, we observed similar results in simulations of the M3 muscarinic receptor. In the D2.50-protonated M3 receptor, no sodium ion bound to the D2.50 allosteric site. In contrast, in the D2.50-deprotonated M3 receptor, one sodium ion diffused into the orthosteric site and then bound to the D2.50 allosteric site during a 100 ns cMD simulation. Therefore, sodium ion binding to the highly conserved D2.50 residue appears to a common characteristic of class A GPCRs.
Earlier 100-ns-timescale cMD simulations on A2AAR suggested that activation of A2AAR and sodium ion binding that greatly stabilizes the D2.50 allosteric pocket are mutually exclusive (20). Recent microsecond-timescale cMD simulations and free-energy calculations of β2AR identified that protonation of the D2.50 residue shifts the conformation of the β2AR toward more active-like states (33). Here, our extensive aMD simulations reveal that the D2.50-deprotonated M3 receptor with sodium ion bound stays in the inactive state, with only transient visiting of an intermediate conformation. In comparison, the D2.50-protonated M3 receptor samples a significantly larger conformational space, visiting an active state that resembles the active x-ray structures of the M2 receptor (13), β2AR (11,12), and opsin (7,8). Activation of the M3 receptor is characterized by outward tilting of the TM6 cytoplasmic end, formation of a close interaction between the Y5.58 and Y7.53 side chains, and inward displacement of the NPxxY motif in the TM7 intracellular region. Large correlations of residue motions and strong allosteric network interactions between distant regions in the receptor play important roles during activation of the M3 muscarinic receptor.
Conclusions
In this study, we investigated the mechanistic effects of sodium ion binding on activation of the M3 receptor through long-timescale aMD simulations. We performed aMD simulations (6 μs long in total) on the receptor, with different protonation of the D2.50 allosteric site and sodium ion concentration. During the aMD simulations, the formation of an α helix is observed in the intracellular region of the TM6 helix, which is missing three residues (K6.31−A6.33) and adopts a random coil conformation for residues A6.34−L6.37 in the x-ray structure. The TM6 intracellular region remained in a random coil conformation throughout our initial cMD simulations, illustrating the enhanced sampling power of aMD. Although aMD suffers from high energetic noise in simulations of large proteins such as GPCRs, and accurate reweighting to recover their original free-energy landscape remains challenging (39), our extensive aMD simulations provide sufficient conformational sampling of the M3 muscarinic receptor to reveal key conformational transitions. Activation is captured in the highly flexible D2.50-protonated M3 receptor, which exhibits large residue correlations and a strong allosteric network. When the D2.50 residue is deprotonated, the M3 receptor is bound by an allosteric sodium ion and is confined mostly in the inactive state, with remarkably reduced flexibility. The strength of network interactions among the D2.50 allosteric pocket, extracellular vestibule, orthosteric ligand-binding site, and intracellular G-protein coupling site is greatly weakened upon sodium ion binding to the D2.50 residue.
Author Contributions
Y.M. and J.A.M. designed research and wrote the manuscript. Y.M. performed research. Y.M., A.D.C., and J.A.M. analyzed data.
Acknowledgments
This work was supported by the National Science Foundation (grant MCB1020765), the National Institutes of Health (grant GM31749), the Howard Hughes Medical Institute, a UCSD Pharmacological Sciences Training Grant (A.D.C.), and the National Biomedical Computation Resource. Computing time was provided on the Gordon and Stampede supercomputers through the Extreme Science and Engineering Discovery Environment (awards TG-MCA93S013 and TG-MCB140011) and on the Hopper and Edison supercomputers through the National Energy Research Scientific Computing Center (project m1395).
Supporting Material
References
- 1.Kow R.L., Nathanson N.M. Structural biology: muscarinic receptors become crystal clear. Nature. 2012;482:480–481. doi: 10.1038/482480a. [DOI] [PubMed] [Google Scholar]
- 2.Spindel E.R. Muscarinic receptor agonists and antagonists: effects on cancer. Handb. Exp. Pharmacol. 2012;208:451–468. doi: 10.1007/978-3-642-23274-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruiz de Azua I., Scarselli M., Wess J. RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2010;107:7999–8004. doi: 10.1073/pnas.1003655107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weston-Green K., Huang X.F., Deng C. Effects of olanzapine on muscarinic M3 receptor binding density in the brain relates to weight gain, plasma insulin and metabolic hormone levels. Eur. Neuropsychopharmacol. 2012;22:364–373. doi: 10.1016/j.euroneuro.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Spalding T.A., Burstein E.S. Constitutive activity of muscarinic acetylcholine receptors. J. Recept. Signal Transduct. Res. 2006;26:61–85. doi: 10.1080/10799890600567349. [DOI] [PubMed] [Google Scholar]
- 6.Kruse A.C., Hu J., Kobilka B.K. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J.H., Scheerer P., Ernst O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 8.Scheerer P., Park J.H., Ernst O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 9.Deupi X., Edwards P., Standfuss J. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. USA. 2012;109:119–124. doi: 10.1073/pnas.1114089108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choe H.W., Kim Y.J., Ernst O.P. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 11.Rasmussen S.G.F., DeVree B.T., Kobilka B.K. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen S.G.F., Choi H.-J., Kobilka B.K. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kruse A.C., Ring A.M., Kobilka B.K. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballesteros J.A., Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Stuart C.S., editor. Methods in Neurosciences. Academic Press; New York: 1995. pp. 366–428. [Google Scholar]
- 15.Liu W., Chun E., Stevens R.C. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller-Gallacher J.L., Nehmé R., Tate C.G. The 2.1 Å resolution structure of cyanopindolol-bound β1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS ONE. 2014;9:e92727. doi: 10.1371/journal.pone.0092727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C., Srinivasan Y., Kobilka B.K. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fenalti G., Giguere P.M., Stevens R.C. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katritch V., Fenalti G., Stevens R.C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014;39:233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutiérrez-de-Terán H., Massink A., Stevens R.C. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure. 2013;21:2175–2185. doi: 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dror R.O., Arlow D.H., Shaw D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA. 2011;108:18684–18689. doi: 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niesen M.J.M., Bhattacharya S., Vaidehi N. The role of conformational ensembles in ligand recognition in G-protein coupled receptors. J. Am. Chem. Soc. 2011;133:13197–13204. doi: 10.1021/ja205313h. [DOI] [PubMed] [Google Scholar]
- 23.Provasi D., Artacho M.C., Filizola M. Ligand-induced modulation of the free-energy landscape of G protein-coupled receptors explored by adaptive biasing techniques. PLOS Comput. Biol. 2011;7:e1002193. doi: 10.1371/journal.pcbi.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohlhoff K.J., Shukla D., Pande V.S. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 2014;6:15–21. doi: 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dror R.O., Green H.F., Shaw D.E. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 26.Selent J., Sanz F., De Fabritiis G. Induced effects of sodium ions on dopaminergic G-protein coupled receptors. PLOS Comput. Biol. 2010;6:e1000884. doi: 10.1371/journal.pcbi.1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoichet B.K., Kobilka B.K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miao Y., Nichols S.E., McCammon J.A. Activation and dynamic network of the M2 muscarinic receptor. Proc. Natl. Acad. Sci. USA. 2013;110:10982–10987. doi: 10.1073/pnas.1309755110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan S., Vogel H., Filipek S. The role of water and sodium ions in the activation of the μ-opioid receptor. Angew. Chem. Int. Ed. Engl. 2013;52:10112–10115. doi: 10.1002/anie.201302244. [DOI] [PubMed] [Google Scholar]
- 30.Sagara Y., Mitsuya M., Mase T. Discovery of 2-aminothiazole-4-carboxamides, a novel class of muscarinic M(3) selective antagonists, through solution-phase parallel synthesis. Chem. Pharm. Bull. (Tokyo) 2005;53:437–440. doi: 10.1248/cpb.53.437. [DOI] [PubMed] [Google Scholar]
- 31.Li B., Nowak N.M., Wess J. Random mutagenesis of the M3 muscarinic acetylcholine receptor expressed in yeast: identification of second-site mutations that restore function to a coupling-deficient mutant M3 receptor. J. Biol. Chem. 2005;280:5664–5675. doi: 10.1074/jbc.M411623200. [DOI] [PubMed] [Google Scholar]
- 32.Hulme E.C. GPCR activation: a mutagenic spotlight on crystal structures. Trends Pharmacol. Sci. 2013;34:67–84. doi: 10.1016/j.tips.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Ranganathan A., Dror R.O., Carlsson J. Insights into the role of Asp79(2.50) in β2 adrenergic receptor activation from molecular dynamics simulations. Biochemistry. 2014;53:7283–7296. doi: 10.1021/bi5008723. [DOI] [PubMed] [Google Scholar]
- 34.Ghanouni P., Schambye H., Kobilka B.K. The effect of pH on beta(2) adrenoceptor function. Evidence for protonation-dependent activation. J. Biol. Chem. 2000;275:3121–3127. doi: 10.1074/jbc.275.5.3121. [DOI] [PubMed] [Google Scholar]
- 35.Markwick P.R.L., McCammon J.A. Studying functional dynamics in bio-molecules using accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2011;13:20053–20065. doi: 10.1039/c1cp22100k. [DOI] [PubMed] [Google Scholar]
- 36.Hamelberg D., Mongan J., McCammon J.A. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 37.Hamelberg D., de Oliveira C.A.F., McCammon J.A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007;127:155102. doi: 10.1063/1.2789432. [DOI] [PubMed] [Google Scholar]
- 38.Pierce L.C.T., Salomon-Ferrer R., Walker R.C. Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput. 2012;8:2997–3002. doi: 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao Y., Nichols S.E., McCammon J.A. Free energy landscape of G-protein coupled receptors, explored by accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2014;16:6398–6406. doi: 10.1039/c3cp53962h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 41.MacKerell A.D., Jr., Feig M., Brooks C.L., 3rd Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 42.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 44.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lange O.F., Grubmüller H. Generalized correlation for biomolecular dynamics. Proteins. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 46.Sethi A., Eargle J., Luthey-Schulten Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eargle J., Luthey-Schulten Z. NetworkView: 3D display and analysis of protein·RNA interaction networks. Bioinformatics. 2012;28:3000–3001. doi: 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deupi X., Standfuss J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr. Opin. Struct. Biol. 2011;21:541–551. doi: 10.1016/j.sbi.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Pals-Rylaarsdam R., Hosey M.M. Two homologous phosphorylation domains differentially contribute to desensitization and internalization of the m2 muscarinic acetylcholine receptor. J. Biol. Chem. 1997;272:14152–14158. doi: 10.1074/jbc.272.22.14152. [DOI] [PubMed] [Google Scholar]
- 50.Lee K.B., Ptasienski J.A., Hosey M.M. Acidic amino acids flanking phosphorylation sites in the M2 muscarinic receptor regulate receptor phosphorylation, internalization, and interaction with arrestins. J. Biol. Chem. 2000;275:35767–35777. doi: 10.1074/jbc.M002225200. [DOI] [PubMed] [Google Scholar]
- 51.Haga K., Kruse A.C., Kobayashi T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.