

Abstract
Next-generation transcriptome sequencing is increasingly integrated with mass spectrometry to enhance MS-based protein and peptide identification. Recently, a breakthrough in transcriptome analysis was achieved with the development of ribosome profiling (ribo-seq). This technology is based on the deep sequencing of ribosome-protected mRNA fragments, thereby enabling the direct observation of in vivo protein synthesis at the transcript level. In order to explore the impact of a ribo-seq-derived protein sequence search space on MS/MS spectrum identification, we performed a comprehensive proteome study on a human cancer cell line, using both shotgun and N-terminal proteomics, next to ribosome profiling, which was used to delineate (alternative) translational reading-frames. By including protein-level evidence of sample-specific genetic variation and alternative translation, this strategy improved the identification score of 69 proteins and identified 22 new proteins in the shotgun experiment. Furthermore, we discovered 18 new alternative translation start sites in the N-terminal proteomics data and observed a correlation between the quantitative measures of ribo-seq and shotgun proteomics with a Pearson correlation coefficient ranging from 0.483 to 0.664. Overall, this study demonstrated the benefits of ribosome profiling for MS-based protein and peptide identification and we believe this approach could develop into a common practice for next-generation proteomics.
Keywords: proteogenomics, ribosome profiling, N-terminomics, bioinformatics, translation initiation
Introduction
A shotgun proteomics experiment typically involves the fractionation of a complex peptide mixture followed by LC-MS/MS analysis and the identification of peptides using one of several protein or peptide sequence database search tools [1–3]. N-terminal proteomics techniques such as N-terminal COFRADIC (combined fractional diagonal chromatography) expand on the results of a typical shotgun experiment by enriching for N-terminal peptides, thus revealing (alternative) translation start sites, while simultaneously measuring co-translational modifications of protein N-termini [4]. Protein reference databases only contain experimentally verified and/or predicted sequences and are therefore unlikely to contain a comprehensive representation of the actual protein content of a given sample. To resolve this shortcoming, recent efforts have been directed towards the combination of proteomics and next-generation transcriptome sequencing [5–8]. Proteogenomic approaches that delineate translation products based on mRNA sequencing data may improve protein identification in multiple ways. The transcriptome of a sample offers a more representative expression profile than could be obtained with a public database alone while at the same time reducing the search space through the elimination of unexpressed gene products [9]. The transcript data also contains useful information about sequence variations such as single nucleotide polymorphisms (SNP) or mutations and RNA splice and editing variants [9–11], which increases the chances of detecting new proteins or protein forms [12–14]. Despite the benefits of adding next-generation transcriptome sequencing to an MS-based proteomics experiment, there are still several improvements possible. Because of extensive translation regulation, the presence of a transcript does not necessarily imply the presence of the corresponding protein [15–17]. On top of that, several factors, including internal ribosome entry sites, the presence of multiple ORFs per transcript, non-AUG start codons and leaky scanning on top of ribosome frameshifting and stop codon readthrough hamper the prediction of the exact protein sequence(s) from a single transcript sequence [18–20]. Recently, a novel technique has been described that attempts to tackle these limitations: ribosome profiling [21]. Ribosome profiling, or ribo-seq, is based on the deep sequencing of ribosome-associated mRNA fragments, thus enabling the study of in vivo protein synthesis at the transcript level. In a ribo-seq experiment, eukaryotic translation is often halted using cycloheximide (CHX). The mRNA that is not protected by ribosomes after the translation halt is digested with nucleases and the monosome-mRNA complexes are isolated. Next, the protected mRNA sequences are separated from the ribosomes and converted into a DNA library, ready to be sequenced. The sequencing results in a genome-wide snapshot of the mRNA that enters the translation machinery. Additionally, (alternative) translation initiation sites can be studied with sub-codon to single-nucleotide precision through the use of antibiotics such as harringtonine (HARR) or lactimidomycin (LTM), which cause the ribosomes to halt at sites of translation initiation [22, 23]. When the exact translation start site is known, the ORF can be delineated, thus eliminating the need to translate the transcripts in three or six reading frames. The measurement of mRNA at the translation level, combined with the knowledge of the exact translation start sites, makes ribosome profiling an excellent choice for the creation of a custom protein sequence search space for MS/MS-based peptide identification [24]. It has to be noted that ribo-seq does not generate direct evidence of mature proteins or protein stability and that some non-coding transcripts do not result in a protein product, despite being associated with ribosomes [25–27]. However, MS-assisted validation may help to resolve both issues. Apart from canonical translation products, ribosome profiling also aids in the identification of unannotated truncated and N-terminally extended protein variants and the validation of these variants can come from matching N-terminal COFRADIC data [24, 28]. In this study we created a custom protein sequence database based on LTM ORF delineation for the HCT116 cell line, a widely used human colon cancer cell model, to serve as the search space for MS/MS spectra obtained by means of shotgun proteomics and N-terminal COFRADIC (Figure 1). Translation products derived from the ribosome profiling data of the HCT116 cells were combined with the public Swiss-Prot protein sequence database [29] to build an optimal protein search space for our proteomics data. The addition of ribo-seq data resulted in the identification of 22 new proteins, i.e. proteins that were not contained in the Swiss-Prot database, out of a total of 2,816 protein identifications in our shotgun proteomics experiment. On top of that, the inclusion of ribo-seq data improved the score of 69 proteins as a result of the discovery of proteins with a mutation, new isoforms and homologs and extended protein forms. Out of a total of 1,262 peptides, ribo-seq identified 18 extra N-termini in the COFRADIC experiment compared to Swiss-Prot alone, including 6 N-termini originating from extended protein forms with a near-cognate start site (i.e. the protein does not start with the canonical AUG codon). It needs to be noted that in the shotgun proteomics experiment 312 proteins were uniquely identified using the Swiss-Prot database, emphasizing the importance of proteomics techniques for the validation of next-generation transcriptome sequencing datasets. Finally, the correlation between the ribo-seq and shotgun proteomics data was calculated. Depending on the settings used, the Pearson correlation coefficient between the ribo-seq-derived normalized ribosome-protected fragments (RPF) counts and the normalized spectral counts of the shotgun experiment (i.e. emPAI [30] and NSAF [31] values) ranged from 0.483 to 0.664.
Figure 1. Proteogenomic strategy for the identification of proteins and peptides using a Swiss-Prot/ribo-seq-derived database.
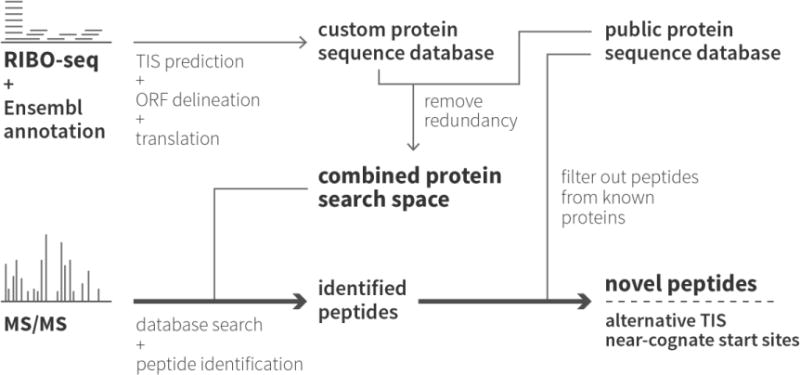
Ribo-seq was performed twice on the human colon cancer cell line HCT116, once with CHX to halt translation globally and once with LTM to stop translation specifically at translation initiation sites. After translation initiation site (TIS) prediction, the ribo-seq-derived ORFs were translated to create a custom protein sequence database. This database was then combined with the human Swiss-Prot protein sequence database. Proteome samples were prepared from the same HCT116 cells and analyzed using both shotgun proteomics and N-terminal COFRADIC. The proteins and peptides in these samples were then identified using the custom combined protein search space.
Material & Methods
Cell culture for proteomics
The HCT116 cell line was kindly provided by the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center (Baltimore, USA). Cells were cultivated in DMEM medium supplemented with 10% fetal bovine serum (HyClone, Thermo Fisher Scientific Inc.), 100 units/ml penicillin (Gibco, Life Technologies) and 100 μg/ml streptomycin (Gibco) in a humidified incubator at 37°C and 5% CO2. Prior to the proteomics experiments, the HCT116 cells were subjected to SILAC labeling [32] as part of another experiment that compared the wild type HCT116 cells to a double knockout line, which was differently labeled (manuscript in preparation). For the N-terminal COFRADIC analysis, cells were transferred to media containing 140 μM heavy (13C615N4) L-arginine (Cambridge Isotope Labs, Andover, MA, USA). For the shotgun proteome analysis, cells were cultured in medium supplemented with 140 μM medium heavy (13C6) L-arginine and 800 μM heavy (13C6) L-lysine. To achieve a complete incorporation of the labeled amino acids, cells were maintained in culture for at least 6 population doublings.
Cell culture and sample preparation for ribosome profiling
The HCT116 cells for the ribosome profiling experiments were cultivated in McCoy’s 5A (Modified) Medium (Gibco) supplemented with 10% fetal bovine serum, 2 mM alanyl-L-glutamine dipeptide (GlutaMAX, Gibco), 50 units/ml penicillin and 50 μg/ml steptamycin at 37°C and 5% CO2. Cultures at 80–90% confluence were treated with 50 μM LTM [33, 34] or 100 mg/ml CHX (Sigma, USA) at 37°C for 30 min. Subsequently, cells were washed with PBS, harvested by trypsin-EDTA, rinsed again with PBS and recovered by 5 min of centrifugation at 300 × g, all in the presence of CHX to maintain the polysomal state. Cell pellets were resuspended in ice-cold lysis buffer, formulated according to Guo et al. (2010) [35] (10 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 100 mM KCl, 1% Triton X-100, 2 mM dithiothreitol (DTT), 100 mg/ml CHX, 1 × complete and EDTA-free protease inhibitor cocktail (Roche)), at a concentration of 40 × 106 cells/ml. After 10 min of incubation on ice with periodic agitation, lysed samples were passed across QIAshredder spin columns (Qiagen) to shear the DNA. Subsequently, the flow-throughs were centrifuged for 10 min at 16,000 × g and 4°C. The recovered supernatant was aliquoted, snap-frozen in liquid nitrogen and stored at −80°C for subsequent ribosome footprint recovery and cDNA library generation.
Shotgun proteome analysis
4.2× 106 cells were lysed in 20 mM NH4HCO3 pH 7.9 by three rounds of freeze-thawing. Total protein concentration in cell extracts was measured using Biorad’s Protein Assay (Biorad Laboratories, Munich, Germany) and 2 mg protein material was used for downstream processing. Digestion was performed overnight using trypsin (Promega, Madison, WI, USA; enzyme/substrate, 1/50) after adding 0.5 M guanidinium hydrochloride and 2% ACN to aid in protein denaturation. Methionines were uniformly oxidized to methionine sulfoxides by adding 20 μl of 3% (w/v) H2O2 to 100 μl sample (equivalent to 500 μg proteins) for 30 min at 30°C. For chromatographic separation 100 μl peptide mixture was then immediately injected onto an RP-HPLC column (Zorbax® 300SB-C18 Narrow-bore, 2.1 mm internal diameter × 150 mm length, 5 μm particles, Agilent). Following 10 min of isocratic pumping with solvent A (10 mM ammonium acetate in water/ACN (98:2 v/v), pH 5.5), a gradient of 1% solvent B increase per minute (solvent B: 10 mM ammonium acetate in ACN/water (70:30 v/v), pH 5.5) was started. The column was then run at 100% solvent B for 5 min, switched to 100% solvent A and re-equilibrated for 20 min. The flow was kept constant at 80 μL/min using Agilent’s 1100 series capillary pump with the 100 μL/min flow controller. Fractions of 30 sec wide were collected from 20 to 80 min after sample injection. To reduce LC-MS/MS analysis time, fractions eluting 12 min apart were pooled, vacuum dried and re-dissolved in 20 μl 20 mM tris(2-carboxyethyl)phosphine (TCEP) in 2% acetonitrile.
N-terminal COFRADIC analysis
HCT116 cells were lysed in 50 mM HEPES pH 7.4, 100 mM NaCl and 0.8% CHAPS containing a cocktail of protease inhibitors (Roche) for 10 min on ice and centrifuged for 15 min at 16,000 g at 4°C. The protein sample was then subjected to N-terminal COFRADIC as described by Staes et al. (2011) [4].
LC-MS/MS analysis
The shotgun proteomics sample was subjected to LC-MS/MS analysis using an Ultimate 3000 RSLC nano HPLC (Dionex, Amsterdam, the Netherlands) in-line connected to an LTQ Orbitrap Velos (Thermo Fisher Scientific, Bremen, Germany). The sample mixture was loaded on a trapping column (made in-house, 100 μm id × 20 mm, 5 μm beads C18 Reprosil-HD, Dr. Maisch). After back flushing from the trapping column, the sample was loaded on a reverse-phase column (made in-house, 75 μm id × 150 mm, 5 μm beads C18 Reprosil-HD, Dr. Maisch). Peptides were loaded in solvent A′ (0.1% trifluoroacetic acid, 2% ACN) and separated with a linear gradient from 2% solvent A″ (0.1% formic acid) to 50% solvent B′ (0.1% formic acid and 80% ACN) at a flow rate of 300 nl/min followed by a wash reaching 100% solvent B′. The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the ten most abundant peaks in a given MS spectrum. Mascot Generic Files were created from the MS/MS data in each LC run using the Distiller software (version 2.3.2.0).
The N-terminal COFRADIC sample was analyzed on the LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) which was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the six most abundant peaks in a given MS spectrum.
All the MS data were converted using the PRIDE Converter [36] and are available through the PRIDE database [37] with the dataset identifier PXD000304 and DOI 10.6019/PXD000304 (http://www.ebi.ac.uk/pride/archive/login, PX reviewer account: username: review48267, password: TTewpyNH).
Peptide and protein identification and interpretation
The protein and peptide searches were performed against our custom database using X! Tandem Sledgehammer (2013.09.01.1) and OMSSA 2.1.9 in combination with the SearchGui (1.16.4) tool [38]. For the shotgun proteomics experiment, pyroglutamate formation of N-terminal glutamine, acetylation of N-termini (both at peptide level) and methionine oxidation to methionine-sulfoxide were selected as variable modifications. Heavy labelled arginine (13C6) and lysine (13C6) were selected as fixed modifications. Mass tolerance was set to 10 ppm on precursor ions and to 0.5 Da on fragment ions. The peptide charge was set to 2+, 3+, 4+. Trypsin was selected as the enzyme setting, one missed cleavage was allowed and cleavage was also allowed when arginine or lysine was followed by proline.
For the N-terminomics experiment, the generated MS/MS peak lists were searched with Mascot (version 2.3) [39]. Mass tolerance on precursor ions was set to 10 ppm (with Mascot’s C13 option set to 1) and to 0.5 Da on fragment ions. The peptide charge was set to 1+, 2+, 3+ and the instrument setting to ESI-TRAP. Methionine oxidation to methionine-sulfoxide, 13C2D3-acetylation on lysines and carbamidomethylation of cysteine were set as fixed modifications. Variable modifications were 13C2D3 acetylation of N-termini, acetylation of N-termini and pyroglutamate formation of N-terminal glutamine (all at peptide level). 13C615N4 L-arg was set as fixed modification. Endoproteinase semi-Arg-C/P (Arg-C specificity with arginine-proline cleavage allowed) was set as enzyme allowing for no missed cleavages.
Protein and peptide identification and data interpretation were done using the PeptideShaker algorithm (http://code.google.com/p/peptide-shaker, version 0.26.2), setting the FDR to 1% at all levels (peptide-to-spectrum matching, peptide and protein).
Ribosome profiling
100 μl of the clarified HCT116 cell lysate (equivalent to 4×106 cells) was used as input for ribosome footprinting. The A260 absorbance of the lysate was measured with Nanodrop (Thermo Scientific) and for each A260, 5 units of ARTseq Nuclease (Epicentre) were added to the samples. The nuclease digestion proceeded for 45 min at room temperature and was stopped by adding SUPERase. In Rnase Inhibitor (Life Technologies). Next, the ribosome protected fragments (RPFs) were isolated using Sephacryl S400 spin columns (GE Healthcare) according to the procedure described in ‘ARTseq Ribosome Profiling Kit, Mammalian’ (Epicentre). The RNA was extracted from the samples using acid 125 phenol : 24 chloroform : 1 isoamyl alcohol and precipitated overnight at −20°C by adding 2 μl glycogen, 1/10th volume of 5 M ammonium acetate and 1.5 volumes of 100% isopropyl alcohol. After centrifugation at 18,840 × g and 4°C for 20 min, the purified RNA pellet was resuspended in 10 μl nuclease free water.
Library preparation and sequencing
Libraries were created according to the guidelines described in the ARTseq Ribosome profiling Kit, Mammalian protocol (Epicentre). The RPFs were initially rRNA depleted using the Ribo-Zero Magnetic Kit (Human/Mouse/Rat, Epicentre), omitting the 50°C incubation step. Cleanup of the rRNA depletion reactions was performed through Zymo RNA Clean & Concentrator-5 kit (Zymo Research) using 200 μl binding buffer and 450 μl absolute ethanol. The samples were separated on a 15% urea-polyacrylamide gel and footprints of 26 to 34 nucleotides long were excised. RNA was extracted from the gel and precipitated. The pellet was resuspended in 20 μl nuclease-free water. Next, RPFs were end polished, 3′ adaptor ligated, reverse transcribed and PAGE purified. Five μl of circularized template DNA was used in the PCR reaction and amplification proceeded for 11 cycles. The libraries were purified with AMPure XP beads (Beckman Coulter) and their quality was assessed on a High Sensitivity DNA assay chip (Agilent technologies). The concentration of the libraries was measured with qPCR and they were single end sequenced on a Hiseq (Illumina) for 50 cycles. The ribo-seq libraries have been deposited in NCBI’s Gene Expression Omnibus [40] and are accessible through the GEO series accession number GSE58207 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58207).
Swiss-Prot/ribo-seq integrated database construction
The merged database was constructed using all human Swiss-Prot proteins (downloaded from http://www.uniprot.org, version 2014_03) and the translation products obtained from the ribosome profiling experiment (Figure 1). The ribo-seq-derived translation products were created from both the predicted (alternative) TIS genomic locations based on the LTM ribosome profiling information (according to Lee et al., 2012 [23]) and the corresponding mRNA sequences obtained from Ensembl (version 70) that displayed overall CHX ribosome protected fragment (RPF) coverage. After reconstructing the amino acid sequences, the Ensembl identifiers were mapped to Swiss-Prot identifiers (to safeguard uniformity) using the pBlast algorithm.
In order to remove redundancy introduced by the combination of the ribo-seq-derived translation products and the Swiss-Prot protein sequences, duplicated sequences were removed, retaining the custom sequence. Moreover, only the longest form of a series of gene translation products (N-terminal extended or canonical) was withheld in the combined database. The custom database contained 68,961 sequences as compared to the 20,264 proteins in UniProtKB-SwissProt version 2014_03. Extra information on the custom DB creation can be found in Menschaert et al (2013) [24].
Correlation analysis
Only the transcripts identified in both Swiss-Prot and the ribo-seq-derived translation products were selected for the correlation analysis. Ribo-seq measurements were expressed as the number of ribosomal footprints per CDS (RPF count), hereby correcting for a possible 3′UTR and 5′UTR bias as suggested by Ingolia et al. (2011) [22]. Two quantitative measures for protein abundance based on spectral counts (emPAI [30] and NSAF [31]) were calculated using the shotgun data. While the first method uses the number of peptides per protein normalized by the theoretical number of peptides, the so-called protein abundance index (PAI), the NSAF method takes both the protein length and the total number of identified MS/MS spectra in an experiment into account. For each dbTIS transcript for which quantitative ribo-seq and shotgun proteomics information was available a Pearson correlation coefficient was calculated between its normalized RPF count and its normalized spectral count. When more than one ribo-seq-derived transcript corresponded with a particular Swiss-Prot protein sequence, the one with the highest normalized RPF count was used. The different normalization and identification approaches were combined with the following additional transcript filtering settings: i) no extra cutoffs, ii) only dbTIS transcripts with a validated MS/MS-based identification (meaning that the spectral count value was higher than 2), iii) only dbTIS transcripts with a total RPF count ≥ 200, and iv) only dbTIS transcripts with both a validated MS identification and an RPF count ≥ 200. All correlation coefficients were computed using log-transformed RPF and emPAI/NSAF measures.
Results
A regular shotgun and an N-terminal COFRADIC proteomics experiment were performed on a HCT116 cell line to determine the effect of the addition of ribo-seq-derived translation products to the Swiss-Prot protein sequence database on MS/MS spectrum identification. The shotgun data were used for the overall assessment of protein expression, whereas the N-terminal COFRADIC data were specifically used for the validation of the ribo-seq-predicted translation initiation sites.
Shotgun proteomics
Using the combination of Swiss-Prot and the ribo-seq-derived database, we identified a total of 2,816 proteins in the HCT116 cells (Figure 2a). The majority of these proteins (2,482 or 88.1%) were identified in both Swiss-Prot and the custom database. The addition of the ribo-seq data to the protein search space led to 22 extra identifications, which would not have been picked up with just the Swiss-Prot database. Besides 9 previously unannotated protein products, these new identifications included 13 proteins with a mutation and three alternatively spliced isoforms. The inclusion of ribo-seq data also improved protein identification and score significance for 69 proteins since higher peptide coverage was obtained (Supplemental Figure 1 shows three examples). The proteins with an improved score coincided with mutation sites (52 proteins), alternatively spliced isoforms (14 proteins) and three N-terminal extensions. The ribo-seq experiment also missed 312 proteins, but these were still picked up thanks to the inclusion of Swiss-Prot in the search space. All the identified proteins and their respective annotations can be found in Supplemental Table 1. An approximate analysis of the turnover rate and half-lives of the 312 missed proteins using publically available datasets [41, 42] showed no significant difference between the missed and the other identified proteins (Wilcoxon rank-sum test, p > 0.05). A gene ontology enrichment analysis using the DAVID tool [43] revealed that several biological process ontologies involving protein transport and localization were significantly enriched in the 312 missed proteins, just as the corresponding cellular localization ontologies linked to the cytoskeleton, cytosol and non-membrane-bounded organelles (Supplemental Table 2).
Figure 2. Bar charts showing the number of protein and peptide identifications obtained from the shotgun proteomics and N-terminal COFRADIC experiments.
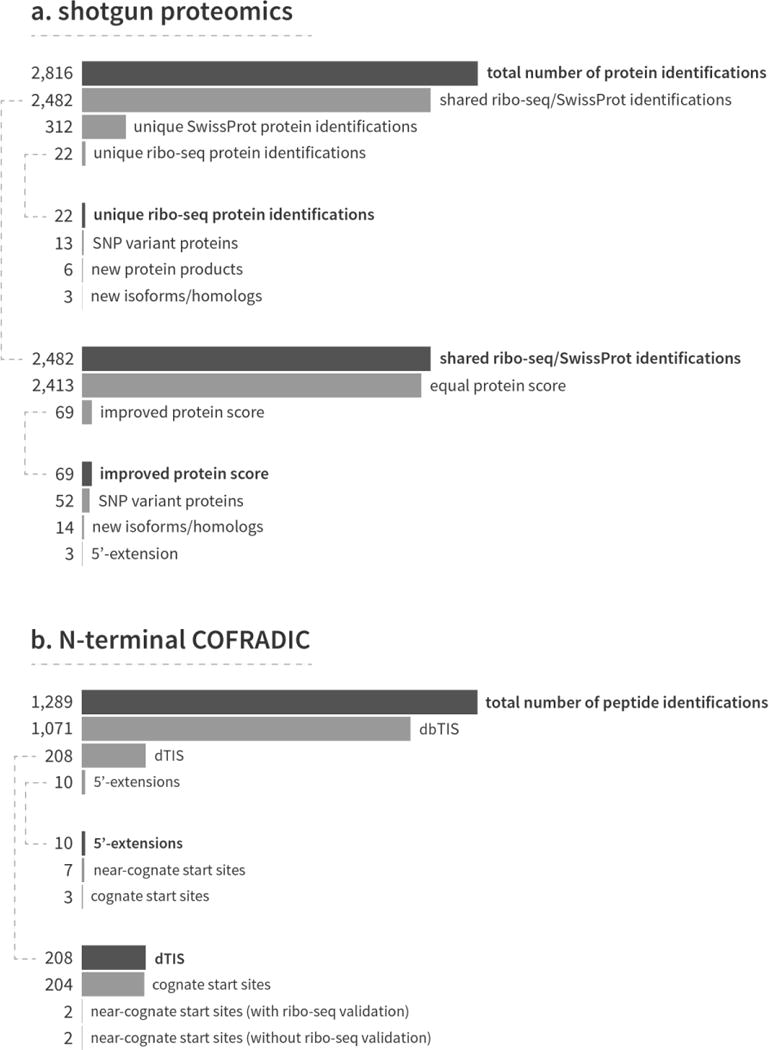
a) Shotgun proteomics. The custom combined protein sequence database resulted in the identification of 2,816 proteins. Most of these proteins (2,482 or 88.1%) were picked up by both databases independently, while 312 and 22 proteins were uniquely identified in the Swiss-Prot and ribo-seq databases respectively. The 22 unique ribo-seq identifications contained six new proteins, 13 proteins with a mutation site and 3 unannotated isoforms. The ribo-seq data also improved the protein identification and score of 69 proteins. b) N-terminal COFRADIC. Most of the 1,289 peptides that were found in the custom combined protein sequence database mapped to canonical, annotated N-termini (1,071 dbTIS peptides or 83.1%). Of the remaining N-termini, 208 started downstream of the canonical start site (beyond protein position 2), 9 mapped to a 5′-extension and one to an uORF. For both the up- and downstream start sites, we identified several near-cognate start sites.
N-terminal COFRADIC
In order to validate the TISs identified by the ribo-seq experiment and thus the corresponding N-terminal protein isoforms, positional proteomics in the form of N-terminal COFRADIC was applied to the HCT116 cells. After LC-MS/MS analysis and the subsequent combined database search, we identified 1,289 N-terminal peptides (Figure 2b). The greater part of these peptides mapped to canonical start sites (1,071 peptides or 83.1%), 208 peptides started downstream of the canonical start site (past protein position 2 in reference to Swiss-Prot), 9 peptides mapped to a 5′-extension and one to an uORF. Two examples of proteins with an N-terminal extension or truncation are given in Figure 3. Ribo-seq uniquely identified 18 peptides, which would have been missed when only searching Swiss-Prot. Both the N-terminal COFRADIC and ribo-seq experiment provided evidence of translation initiation at near-cognate start sites, which was also reported in previous COFRADIC and ribo-seq studies [22, 23]. A complete list of all identified N-terminal peptides is provided as Supplemental Table 1.
Figure 3. Depiction of two different N-termini that were predicted by ribo-seq and identified using N-terminal COFRADIC.
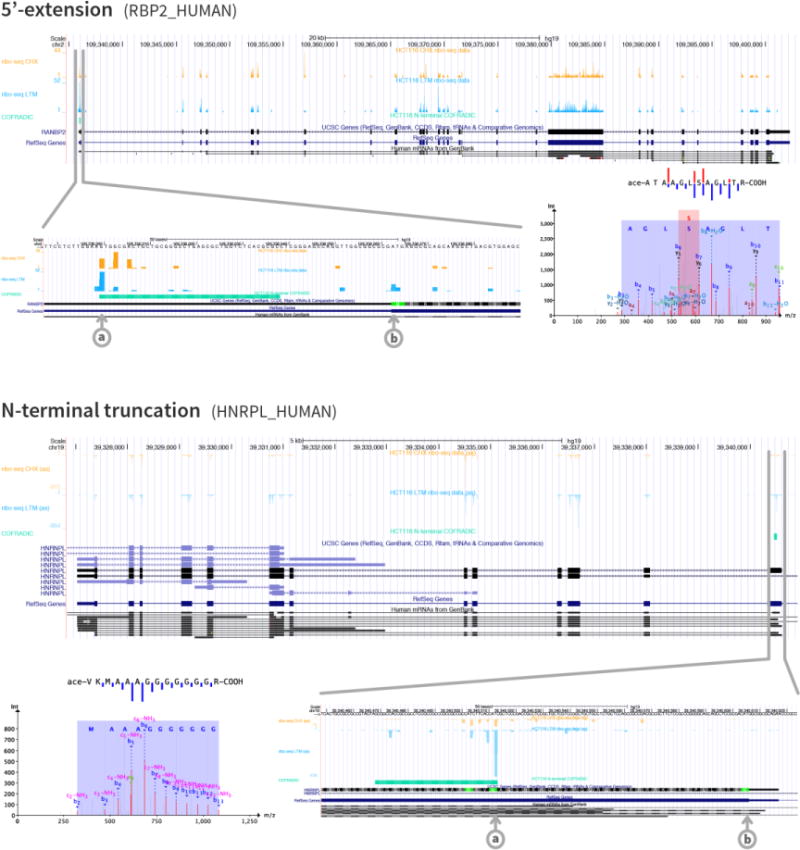
The figure shows a 5′-extension (Swiss-Prot entry name RBP2_HUMAN) and an N-terminal truncation (Swiss-Prot entry name HNRPL_HUMAN). The UCSC genome browser [57] was used to create the plots of the ribo-seq and N-terminal COFRADIC data and the different browser tracks are from top to bottom: CHX treatment data, LTM treatment data, N-terminal COFRADIC data, UCSC genes, RefSeq genes and human mRNA from GenBank. The different start sites (a: alternative start site, b: canonical start site) are clearly visible in the zoomed genome browser views, just as the three-nucleotide periodicity of the ribo-seq data, especially in the N-terminal truncation image. The MS/MS spectra and sequence fragmentations indicate the confidence and quality of the peptide identifications.
We compared the list of identified protein extensions starting at non-AUG start sites with the previously published list of non-AUG derived N-terminal extensions predicted by Ivanov et al. (2011) [44] and found matching evidence for one N-terminally extended protein (Swiss-Prot entry name HDGF_HUMAN; extension of 50 amino acids starting at GTG) out of 9 identified in our proteomics study.
Correlation analysis
We calculated a Pearson correlation coefficient to investigate the relation between the ribo-seq coverage and MS protein abundance measurements. Only transcripts for which quantitative information was available from both the ribo-seq and shotgun proteomics experiments were used in all the plots and calculations. The Pearson correlation values for the different normalization and identification approaches are listed in Table 1 and Figure 4a shows the correlation plots for the NSAF values, which were better correlated with the ribo-seq coverage than the emPAI values. The highest correlation (r2 = 0.664) was obtained when using only validated dbTIS transcripts with a total RPF count ≥ 200. The correlation coefficients were also calculated for the 312 protein identifications that were present in Swiss-Prot, but not in our ribo-seq-derived search space (Supplemental Figure 2). These 312 identifications were missing from the ribo-seq data because no TISs were identified in the LTM-treated cells, but, as there was coverage in the CHX-treated cells, the correlation could still be calculated. The Pearson correlation coefficients ranged from 0.464 to 0.713, depending on the protein selection and normalization procedure, and were similar for the proteins identified in both the Swiss-Prot and ribo-seq database.
Table 1. Pearson correlation coefficients between MS protein abundance and ribo-seq coverage.
MS protein identifications were performed with an FDR of 1% and protein abundances were calculated as emPAI and NSAF values. The correlation coefficients were computed for each of the following transcript filtering settings: i) all dbTIS transcripts without additional thresholds, ii) only transcripts with a validated MS identification (i.e. transcripts with a spectral count value > 2), iii) only dbTIS transcripts with a total RPF count ≥ 200 and iv) only dbTIS transcripts with both a validated MS/MS-based identification and an RPF count ≥ 200.
| i | ii | iii | iv | |
|---|---|---|---|---|
| 1% FDR | ||||
| emPAI | 0.488 | 0.498 | 0.483 | 0.518 |
| NSAF | 0.608 | 0.642 | 0.634 | 0.664 |
Figure 4.
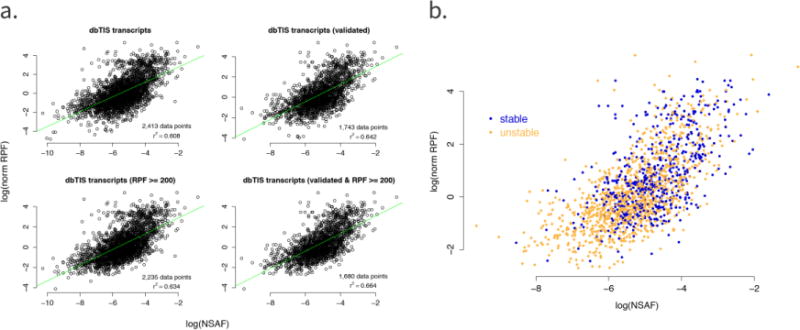
a) Correlation plots of protein abundance estimates based on NSAF values and RPF counts. Top left: all dbTIS transcripts; top right: dbTIS transcripts with a validated MS/MS-based identification (i.e. transcripts with a spectral count value > 2); bottom left: dbTIS transcripts with an RPF count ≥ 200; bottom right: dbTIS transcripts with both a validated MS identification and an RPF count ≥ 200. The regression line is shown in green. For each plot, the number of data points used (i.e. the number of dbTIS transcripts) as well as the corresponding Pearson correlation coefficient (r2) is shown. b) Correlation plot with the inclusion of stability data. Only dbTIS transcripts with both a validated MS/MS-based identification and an RPF count ≥ 200 were used (bottom right plot in Figure 4a). Instability indexes were determined with the ProtParam tool [45]: proteins with an instability index < 40 were classified as stable and are shown in blue, whereas proteins with an instability index ≥ 40 were classified as unstable and are shown in orange.
We also investigated the link between the correlation and the degree of protein stability. Figure 4b shows the correlation plot for validated dbTIS transcripts with an RPF ≥ 200 together with the instability indexes of the proteins. These indexes were obtained with the ExPASy ProtParam tool [45], where a protein with an instability index < 40 is predicted to be stable and a protein with an index ≥ 40 is considered unstable. The majority of unstable proteins were characterized by lower NSAF and RPF values than the stable proteins. As reported previously, protein stability is among the most significant factors governing the correlation between gene expression and protein abundance [11].
Discussion
The successful identification of proteins and peptides from MS/MS spectra depends on a number of factors. A state-of-the-art mass spectrometer that provides high resolution and mass accuracy is a vital element of a proteomics experiment. Solid experimental design and a robust identification pipeline are two other important factors. As even small changes in database search algorithms can lead to different identification results, combining several search engines, such as X!Tandem [2] and OMSSA[3], helps to increase the number of PSMs [46]. A more recent approach to improve the number of PSMs is based on the custom tailoring of the search space through the use of next-generation transcriptome sequencing [7, 24]. The new and improved protein identifications based on our ribo-seq-derived search space were a first indication of the success of our proteogenomics strategy. Especially the identification of N-terminally extended proteins would not have been possible when using only Swiss-Prot. The positive correlation between protein abundance (measured as NSAF and emPAI values) and the ribo-seq footprint coverage (measured as RPF counts) also justifies the usage of the described proteogenomics approach. It has been described before how NSAF gives a more accurate estimate of protein abundance than emPAI as it uses more information (e.g. fragment ion intensities and protein length) [47, 48]. This could explain why the NSAF values correlated better with the ribo-seq data. Interesting to note is that proteins with a lower stability index displayed both lower protein abundances as well as lower RPF counts than their more stable counterparts (Figure 4b). Several studies have reported correlation values between mRNA-seq coverage and protein abundance, ranging from 0.41–0.44 [49] to 0.51 [11] in mouse and between 0.42 and 0.43 in rat [14]. Nagaraj et al. (2011) published a Spearman’s correlation of 0.6 between FPKM-based transcript abundance and iBAQ-based protein abundance values for the human HeLa cell line [5]. The improved correlation observed in our study can be explained by the fact that, because it measures transcripts after they have entered the translation machinery, ribosome profiling is less affected by translation regulation. The ability of ribo-seq to take alternative translation events into account leads to a better delineation of ORFs, which could also improve the correlation. Another advantage of the ribo-seq-derived database was that it allowed us to identify translation initiation from non-AUG start sites at the protein level, for which only limited evidence is available so far [28, 50–52].
Without the addition of the Swiss-Prot database to our custom search space, a significant amount of proteins would have been missed (unique Swiss-Prot identifications in Figure 2). These proteins were missing from the ribo-seq-derived search space because no detectable LTM-signal could be observed. But since the CHX treatment resulted in coverage for these proteins, we could still calculate the correlation between protein abundance and RPF counts (Supplemental Figure 2). The abundance values and RPF counts, together with their correlation values, ruled out low abundance or coverage as a reason for the missed identifications. A suboptimal LTM treatment and/or TIS calling could help explain the lack of TIS recognition and the resulting absence of the corresponding proteins from the ribo-seq-derived search space. These results demonstrate the importance of reference databases and MS for the identification and validation of next-generation sequencing-derived translation products.
The combination of N-terminal COFRADIC and ribo-seq data identified a number of alternative TISs. Translation via these start sites produces protein isoforms with a different N-terminus if the new start site maintains the reading frame (e.g. the 5′-UTR extension in Figure 3). If the start site is not in the same reading frame, completely different proteins will be generated. The selection of upstream TISs can also lead to the creation of uORFs, which influence the downstream protein synthesis from the main ORF [53, 54]. Roughly half of all mammalian transcripts contain one or more upstream TISs, which are often associated with short ORFs [23]. In contrast to the previously reported frequent occurrence of uORFs in human and mouse ribosome profiling data [22, 23], we were able to identify only one N-terminal peptide of an upstream overlapping ORF in the PIDD gene (Supplemental Table 1). This limited evidence for uORF protein products could be attributed to several factors, such as a bias towards upstream (near-) cognate start site identification from ribosome profiling data [55] or the rapid degradation, small size and possibly low abundance of uORFs.
Conclusion
As sequencing techniques become more generally accessible, ribosome profiling has become [24, 27, 28, 50, 56] and will continue to be a valuable addition to MS-based protein and peptide identification, possibly taking over the role of mRNA sequencing for ORF delineation. The benefits of ribo-seq include the positive correlation between protein abundance and ribo-seq footprint coverage and the ability to predict TISs with single-nucleotide precision. Despite the advantages of ribo-seq, MS-based validation will remain indispensable, not only for the general identification of proteins (through shotgun proteomics), but also for the validation of ribo-seq-derived (alternative) TIS predictions (by means of N-terminomics techniques such as COFRADIC [4]). Furthermore, unlike ribo-seq or any other transcriptome sequencing technique, MS provides true in vivo evidence of proteins or peptides, while taking potential co- and post-translational modifications into account. We also found that both reference protein sequence databases and ribo-seq-derived search spaces can miss protein identifications and that the best results were obtained when these databases were combined. Overall, our results show the usefulness of a ribo-seq-based proteogenomics approach. The ultimate goal will now be the construction of an automated pipeline for the easy conversion of ribo-seq data into a custom protein sequence search space that incorporates both sequence variation information and TIS prediction, ready to be searched for protein identifications.
Supplementary Material
Abbreviations
- aTIS
alternative translation initiation site
- CDS
coding sequence
- CHX
cycloheximide
- COFRADIC
combined fractional diagonal chromatography
- dbTIS
database-annotated translation initiation site
- dTIS
downstream translation initiation site
- emPAI
exponentially modified protein abundance index
- HARR
harringtonine
- LTM
lactimidomycin
- NSAF
normalized spectral abundance factor
- PSM
peptide-to-spectrum match
- RPF
ribosome-protected fragment
- SNP
single nucleotide polymorphism
- TIS
translation initiation site
- uORF
upstream ORF
- UTR
untranslated region
References
- 1.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 2.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 3.Geer LY, Markey SP, Kowalak JA, Wagner L, et al. Open mass spectrometry search algorithm. Journal of proteome research. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 4.Staes A, Impens F, Van Damme P, Ruttens B, et al. Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nature protocols. 2011;6:1130–1141. doi: 10.1038/nprot.2011.355. [DOI] [PubMed] [Google Scholar]
- 5.Nagaraj N, Wisniewski JR, Geiger T, Cox J, et al. Deep proteome and transcriptome mapping of a human cancer cell line. Molecular systems biology. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S, Im H, Bairoch A, Cristofanilli M, et al. A chromosome-centric human proteome project (C-HPP) to characterize the sets of proteins encoded in chromosome 17. Journal of proteome research. 2013;12:45–57. doi: 10.1021/pr300985j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woo S, Cha SW, Merrihew G, He Y, et al. Proteogenomic database construction driven from large scale RNA-seq data. Journal of proteome research. 2014;13:21–28. doi: 10.1021/pr400294c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinto SM, Manda SS, Kim MS, Taylor K, et al. Functional annotation of proteome encoded by human chromosome 22. Journal of proteome research. 2014 doi: 10.1021/pr401169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Slebos RJ, Wang D, Halvey PJ, et al. Protein identification using customized protein sequence databases derived from RNA-Seq data. Journal of proteome research. 2012;11:1009–1017. doi: 10.1021/pr200766z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ning K, Nesvizhskii AI. The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: a preliminary assessment. BMC bioinformatics. 2010;11(Suppl 11):S14. doi: 10.1186/1471-2105-11-S11-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ning K, Fermin D, Nesvizhskii AI. Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-Seq gene expression data. Journal of proteome research. 2012;11:2261–2271. doi: 10.1021/pr201052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beck M, Schmidt A, Malmstroem J, Claassen M, et al. The quantitative proteome of a human cell line. Molecular systems biology. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Djebali S, Davis CA, Merkel A, Dobin A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low TY, van Heesch S, van den Toorn H, Giansanti P, et al. Quantitative and qualitative proteome characteristics extracted from in-depth integrated genomics and proteomics analysis. Cell reports. 2013;5:1469–1478. doi: 10.1016/j.celrep.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 15.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 16.Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Molecular cell. 2007;28:721–729. doi: 10.1016/j.molcel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Baek D, Villen J, Shin C, Camargo FD, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Touriol C, Bornes S, Bonnal S, Audigier S, et al. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biology of the cell/under the auspices of the European Cell Biology Organization. 2003;95:169–178. doi: 10.1016/s0248-4900(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 19.Michel AM, Choudhury KR, Firth AE, Ingolia NT, et al. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res. 2012;22:2219–2229. doi: 10.1101/gr.133249.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Namy O, Rousset JP, Napthine S, Brierley I. Reprogrammed genetic decoding in cellular gene expression. Molecular cell. 2004;13:157–168. doi: 10.1016/s1097-2765(04)00031-0. [DOI] [PubMed] [Google Scholar]
- 21.Ingolia NT. Genome-wide translational profiling by ribosome footprinting. Methods in enzymology. 2010;470:119–142. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- 22.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S, Liu B, Lee S, Huang SX, et al. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2424–2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menschaert G, Van Criekinge W, Notelaers T, Koch A, et al. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Molecular & cellular proteomics: MCP. 2013;12:1780–1790. doi: 10.1074/mcp.M113.027540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volders PJ, Helsens K, Wang X, Menten B, et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic acids research. 2013;41:D246–251. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. The EMBO journal. 2014 doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Damme P, Gawron D, Van Criekinge W, Menschaert G. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Molecular & cellular proteomics: MCP. 2014 doi: 10.1074/mcp.M113.036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishihama Y, Oda Y, Tabata T, Sato T, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Molecular & cellular proteomics: MCP. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Paoletti AC, Parmely TJ, Tomomori-Sato C, Sato S, et al. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18928–18933. doi: 10.1073/pnas.0606379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & cellular proteomics: MCP. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 33.Ju J, Lim SK, Jiang H, Seo JW, Shen B. Iso-migrastatin congeners from Streptomyces platensis and generation of a glutarimide polyketide library featuring the dorrigocin, lactimidomycin, migrastatin, and NK30424 scaffolds. Journal of the American Chemical Society. 2005;127:11930–11931. doi: 10.1021/ja053118u. [DOI] [PubMed] [Google Scholar]
- 34.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nature chemical biology. 2010;6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barsnes H, Vizcaino JA, Eidhammer I, Martens L. PRIDE Converter: making proteomics data-sharing easy. Nature biotechnology. 2009;27:598–599. doi: 10.1038/nbt0709-598. [DOI] [PubMed] [Google Scholar]
- 37.Martens L, Hermjakob H, Jones P, Adamski M, et al. PRIDE: the proteomics identifications database. Proteomics. 2005;5:3537–3545. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- 38.Vaudel M, Barsnes H, Berven FS, Sickmann A, Martens L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 2011;11:996–999. doi: 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- 39.Hirosawa M, Hoshida M, Ishikawa M, Toya T. MASCOT: multiple alignment system for protein sequences based on three-way dynamic programming. Computer applications in the biosciences: CABIOS. 1993;9:161–167. doi: 10.1093/bioinformatics/9.2.161. [DOI] [PubMed] [Google Scholar]
- 40.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doherty MK, Hammond DE, Clague MJ, Gaskell SJ, Beynon RJ. Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. Journal of proteome research. 2009;8:104–112. doi: 10.1021/pr800641v. [DOI] [PubMed] [Google Scholar]
- 42.Sandoval PC, Slentz DH, Pisitkun T, Saeed F, et al. Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. Journal of the American Society of Nephrology: JASN. 2013;24:1793–1805. doi: 10.1681/ASN.2013030279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ivanov IP, Firth AE, Michel AM, Atkins JF, Baranov PV. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic acids research. 2011;39:4220–4234. doi: 10.1093/nar/gkr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, et al. Protein identification and analysis tools in the ExPASy server. Methods in molecular biology. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 46.Searle BC, Turner M, Nesvizhskii AI. Improving sensitivity by probabilistically combining results from multiple MS/MS search methodologies. Journal of proteome research. 2008;7:245–253. doi: 10.1021/pr070540w. [DOI] [PubMed] [Google Scholar]
- 47.Colaert N, Vandekerckhove J, Gevaert K, Martens L. A comparison of MS2-based label-free quantitative proteomic techniques with regards to accuracy and precision. Proteomics. 2011;11:1110–1113. doi: 10.1002/pmic.201000521. [DOI] [PubMed] [Google Scholar]
- 48.McIlwain S, Mathews M, Bereman MS, Rubel EW, et al. Estimating relative abundances of proteins from shotgun proteomics data. BMC bioinformatics. 2012;13:308. doi: 10.1186/1471-2105-13-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwanhausser B, Busse D, Li N, Dittmar G, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 50.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, et al. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Branca RM, Orre LM, Johansson HJ, Granholm V, et al. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat Methods. 2014;11:59–62. doi: 10.1038/nmeth.2732. [DOI] [PubMed] [Google Scholar]
- 53.Wethmar K, Begay V, Smink JJ, Zaragoza K, et al. C/EBPbetaDeltauORF mice–a genetic model for uORF-mediated translational control in mammals. Genes & development. 2010;24:15–20. doi: 10.1101/gad.557910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medenbach J, Seiler M, Hentze MW. Translational control via protein-regulated upstream open reading frames. Cell. 2011;145:902–913. doi: 10.1016/j.cell.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Michel A, O’Connor P, Choudhury RK, Firth A, et al. EMBO Conference Series: Protein Synthesis and Translational Control. Heidelberg, Germany: 2013. [Google Scholar]
- 56.Vasquez JJ, Hon CC, Vanselow JT, Schlosser A, Siegel TN. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Nucleic acids research. 2014;42:3623–3637. doi: 10.1093/nar/gkt1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kent WJ, Sugnet CW, Furey TS, Roskin KM, et al. The human genome browser at UCSC. Genome research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.