

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is a relatively common inherited vascular disorder that was first described in 1864, and is notable for epistaxis, telangiectasia, and arterial venous malformations. While genetic tests are available, the diagnosis remains clinical, and is based on the Curacao criteria. Patients with HHT are at increased risk for both bleeding and clotting events. Because of these competing complications, hematologists are often faced with difficult clinical decisions. While the majority of management decisions revolve around bleeding complications, it is not infrequent for these patients to require anticoagulation for thrombosis. Any anticoagulation recommendations must take into account the bleeding risks associated with HHT. Recent reviews have found that HHT patients can be safely anticoagulated, with the most frequent complication being worsened epistaxis. Large clinical trials have shown that factor IIa and Xa inhibitors have less intracranial bleeding than warfarin, and basic coagulation research has provided a possible mechanism. This article describes the anticoagulation dilemma posed when a 62-year-old female patient with a history of bleeding events associated with HHT was diagnosed with a pulmonary embolism. The subsequent discussion focuses on the approach to anticoagulation in the HHT patient, and addresses the role of the new oral anticoagulants.
Keywords: Anticoagulation, Hereditary hemorrhagic telangiectasia, Hemorrhage, Thrombosis, Rivaroxaban, Apixaban, Dabigatran, Warfarin
Core tip: This article reviews an inherited disorder, hereditary hemorrhagic telangiectasia, in the context of a complicated clinical case. It highlights the problem of balancing the risks of bleeding and thrombosis, and raises the question of whether the new oral anticoagulants might provide safer therapy in such patients who need antithrombotic therapy.
CASE PRESENTATION
A 62-year-old female presented to our hematology clinic with previously diagnosed hereditary hemorrhagic telangiectasia (HHT). The patient had lifelong complications associated with this illness, including recurrent epistaxis and pulmonary arteriovenous malformations (AVMs). The patient had received multiple embolization procedures in the past to prevent complications from her pulmonary AVMs. The epistaxis was debilitating, with episodes occurring up to 5 times per day. Further medical history included the presence of a heterozygous prothrombin gene mutation (PTA20120G), discovered from a thrombophilia evaluation for recurrent superficial phlebitis 5 years prior. The patient’s more recent medical history included the development of a brain abscess 6 mo prior to presentation, which was treated with surgical evacuation. During her post-operative course, she developed a pulmonary embolism (PE) secondary to a deep venous thrombosis (DVT) in her left lower extremity. Initially, the patient was treated with the insertion of an inferior vena cava (IVC) filter, which was placed because of HHT-related bleeding concerns. The IVC filter ultimately developed extensive thrombosis, and the patient was initiated on warfarin therapy 3 mo prior to presentation. Since warfarin was initiated, she has had an increase in the severity of her epistaxis, but not the frequency. On physical exam, the patient had classic telangiectasia on the tongue and oral mucosa (Figure 1). The clinical question posed to our hematology clinic was how to proceed with anticoagulation for DVT/PE in the setting of a genetic thrombophilia and active bleeding in a patient with HHT.
Figure 1.
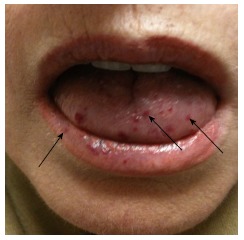
Arrows indicate telangiectatic lesions on tongue and lip of patient.
BACKGROUND
HHT, alternatively known as Osler-Weber-Rendu syndrome, is a relatively common autosomal dominant disorder, with an overall frequency of 1 per 5000 to 10000 individuals[1]. HTT was first described in 1864, but this account did not note a pattern of inheritance[2]. Subsequently, multiple case reports specifically included a familial component in their descriptions[3-5]. In 1909, Frederic Hanes officially used the phrase “hereditary hemorrhagic telangiectasia” for the first time[6].
As the name suggests, the disease is notable for autosomal dominant inheritance (hereditary), bleeding events (hemorrhagic), and visibly dilated blood vessels (telangiectasia). Additionally, most patients are affected by larger AVMs, commonly found in the pulmonary, hepatic, and cerebral vasculature[7]. Specific complications relate to the location of telangiectasia and AVMs, including, but not limited to, epistaxis, gastrointestinal bleeding, visible skin/mucosal manifestations, pulmonary arterial hypertension, pulmonary hemorrhage, and cerebral abscesses. Paradoxically, HHT patients are also burdened by a prothrombotic state due to elevated plasma levels of factor VIII[8]. The balance between hemorrhage and thrombosis is particularly difficult to manage in these patients, and was exemplified in our patient described above.
The underlying pathology in patients with HHT is a primary defect in the vascular wall. The pathophysiology leading to this defect is complex. Patients inherit a mutation in an autosomal dominant fashion; the penetrance of which is highly variable. The three major genes that have been identified are: ENG encoding endoglin (HHT type 1), ACVRL1 encoding activin receptor-like kinase (ALK-1) (HHT type 2), and SMAD4 encoding Smad4 (HHT in association with juvenile polyposis, JPHT)[9-11]. Over 80% of patients with HHT will have mutations in either the ENG or ACVRL1 gene, with the ENG gene accounting for the majority[12]. There is no common mutation in either the ENG or ACVRL1 genes, with over 470 mutations having been described in the ENG gene and 375 in the ACVRL1 gene[13]. Additionally, researchers have been studying two other gene mutations that can cause HHT: HHT3 and HHT4.
Animal models have shed light on how these mutations lead to vascular wall abnormalities. The mutated HHT genes described above encode proteins that alter signaling by the transforming growth factor-β superfamily[7]. It is suggested that endoglin, ALK-1, and Smad4 are all part of a common signaling pathway that is altered in HHT. Additionally, studies have shown that vascular endothelial growth factor is increased in HHT patients[14]. In the setting of HHT and an angiogenic stimulus, there is increased proliferation of endothelial cells, excessive vessel branching, and decreased recruitment of mural cells[7]. Ultimately, this process leads to the formation of telangiectases, which are focal dilatations of postcapillary venules. Once fully developed, these malformed vessels are dilated, convoluted, extend through the dermis, and have excessive layers of smooth muscle without elastic fibers[15,16]. These vessels lack capillaries and connect directly to dilated arterioles. AVMs are similar to telangiectases but have a direct connection between veins and arteries, and are thus much larger. These abnormal HHT blood vessels are prone to bleeding because of their inherently abnormal wall structure, as well as the presence of high perfusion pressures[7].
CLINICAL MANIFESTATIONS
Clinical diagnosis
The diagnosis of HHT remains clinical, although genetic testing has been increasingly utilized. The classic triad of epistaxis, telangiectases, and family history lacks sensitivity and specificity, thus diagnostic criteria were formally created, which are generally referred to as the Curacao criteria (Table 1)[17]. These criteria were recently validated in 263 patients who were screened for HHT and had first degree relatives available for genetic testing[18]. This analysis found that the positive predictive value for a definite clinical diagnosis was 100%, and a negative predictive value for an unlikely clinical diagnosis was 97.7%. Fifty-two study participants had a possible clinical diagnosis, of which 17 (32.7%) had an HHT-causing mutation. Therefore, the utility of genetic testing is most apparent in those with a possible clinical diagnosis. This lends itself to the application of a diagnostic algorithm that can be used to combine the clinical criteria with genetic testing (Figure 2).
Table 1.
The Curacao criteria for the diagnosis of hereditary hemorrhagic telangiectasia
| Criteria | Description | Percent manifestation |
| 1 Epistaxis | Spontaneous, recurrent | 90 |
| 2 Telangiectases | Multiple, at characteristic sites: | 80 |
| Lips | ||
| Oral cavity | ||
| Finger tips | ||
| Nose | ||
| 3 Visceral lesions | Gastrointestinal telangiectasia | 15-30 |
| Pulmonary AVMs | 50 | |
| Hepatic AVMs | 30-70 | |
| Cerebral AVMs | 10-20 | |
| Spinal AVMs | < 1 | |
| 4 Family history | Affected first degree relative | |
| Diagnosis of HHT: | ||
| Definite: 3-4 criteria | Possible: 2 criteria | Unlikely: 0-1 criterion |
HHT: Hereditary hemorrhagic telangiectasia; AVMs: Arteriovenous malformations.
Figure 2.
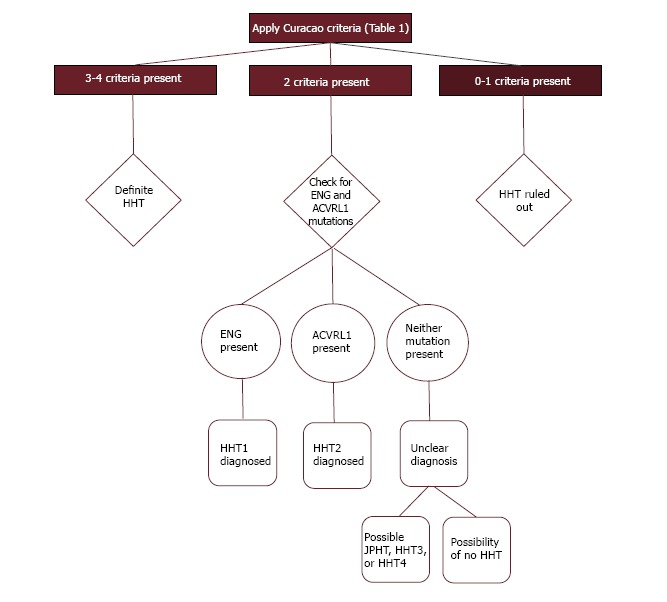
Diagnostic algorithm incorporating the Curacao criteria and genetic testing. HHT: Hereditary hemorrhagic telangiectasia.
Bleeding in HHT
Patients with HHT are at increased risk for both bleeding and thrombosis. Bleeding complications can arise from any location where telangiectases and AVMs are found. The most prevalent form of bleeding in HHT patients is epistaxis, which can be severe and recurrent. Mucosal telangiectases are very common, but their presence is mostly a cosmetic concern. Telangiectases in the gastrointestinal tract are an important source of chronic bleeding, and contribute to the common diagnosis of iron-deficiency anemia found in HHT patients. Perhaps the most important lesions in terms of both morbidity and mortality are pulmonary AVMs. These are present in roughly 50% of HHT patients, although the majority are asymptomatic[7]. The clinical implications of pulmonary AVMs can be divided into two categories: (1) Right-to-left shunting; and (2) Hemorrhage. Right-to-left shunting can result in severe complications, such as brain abscesses and ischemic strokes, as well as less dramatic events such as migraines and dyspnea[19]. Hemorrhagic complications from pulmonary AVMs can include hemoptysis as well as hemothorax. The consequences of pulmonary AVMs can be so severe that current recommendations include screening asymptomatic HHT patients with transthoracic contrast echocardiography (TTCE), or chest computed tomography if TTCE is not available[20]. Cerebral vascular malformations are less common than pulmonary AVMs, but can have devastating effects. Perhaps the most feared complication of HHT is intracranial hemorrhage (ICH), which is most commonly seen with cerebral arteriovenous fistulae (AVF). As vascular lesions in the brain decrease in size, ICH becomes less common. Therefore, AVFs have the greatest risk of ICH, AVMs have intermediate risk, and telangiectasia have the lowest risk. Current guidelines recommend screening for cerebral AVMs with magnetic resonance imaging[20]. One of the more prevalent, but usually asymptomatic, aspects of HHT is hepatic vascular malformations[21]. These can occur as hepatic AVMs (hepatic artery to hepatic vein), hepato-portal VMs (hepatic artery to portal vein), and porto-venous VMs (portal vein to hepatic vein). Lastly, patients rarely may have spinal AVMs that can lead to hemorrhage and subsequent paraplegia.
Thrombosis in HHT
Despite the presence of an overwhelming bleeding propensity, HHT patients also suffer from thrombotic complications. First, they may develop paradoxical thromboembolic stroke from pulmonary AVMs as described above. Second, it seems these patients may also possess an inherent prothrombotic state relating to disturbances in the regulation of coagulation at the endothelial surface. A recent study compared the presence of plasma proteins in HHT-affected adults without a history of thrombosis with non-HHT controls[8]. The researchers found statistically significant elevations in von Willebrand Factor and Factor VIII (FVIII) in HHT-affected adults. The researchers then evaluated the presence of elevated FVIII levels in the general HHT population and found that 87 of 125 (70%) individuals had measurements exceeding the upper limit of normal. An inverse correlation was also described between FVIII level and activated partial thromboplastin time. Furthermore, when compared to HHT individuals with no history of thrombus, HHT individuals who ultimately developed a thrombotic event were more likely to have an elevated FVIII level when it was initially measured at least 10 mo prior. To summarize the findings of this study, individuals with HHT have higher levels of FVIII than non-HHT controls, and the degree of FVIII elevation correlates with future thrombotic risk.
MANAGING THROMBOSIS IN HHT
Traditionally, physicians have been reluctant to treat HHT patients with either antiplatelet or anticoagulant therapy even if otherwise indicated. A recent survey found that 153 of 379 (40.4%) patients with HHT who received antiplatelet or anticoagulant therapy reported no change in epistaxis[22]. Furthermore, 86.9% of patients reported no hemorrhagic events other than epistaxis associated with antiplatelet and anticoagulant use. This survey supports the reasoning that HHT patients who have a strong indication for antiplatelet or anticoagulant use, should not have these agents withheld. Another study examining the use of antithrombotic agents in HHT patients arrived at a similar conclusion[23]. As in the Devlin study, this study found worsening of epistaxis to be the most common complication. There were no new or progressive cases of pulmonary or cerebral hemorrhage, likely because all patients in this study were pre-screened. These data again support the notion that anticoagulation should not be withheld from HHT patients with strong indications for its use. Although bleeding is the major complication associated with all anticoagulant medications, the rate of bleeding varies between agents, and may be of clinical importance in HHT patients.
Choice of anticoagulant agent
For the prior half century, warfarin has been the gold standard of oral anticoagulation. Within the past decade, new oral anticoagulants have been developed and evaluated in clinical trials. Specifically, these agents include the direct thrombin (factor IIa) inhibitor, dabigatran, and the factor Xa inhibitors, rivaroxaban and apixaban (Figure 3). Indications vary by agent, but all have been studied for stroke prevention in non-valvular atrial fibrillation and prevention and treatment of venous thromboembolism (VTE). Interestingly, dabigatran, as well as the factor Xa inhibitors, have been associated with reduced intracranial hemorrhage compared to warfarin in patients with non-valvular atrial fibrillation[24-26]. In studies examining the treatment of acute VTE, dabigatran and rivaroxaban had similar bleeding rates compared to warfarin, while apixaban had decreased bleeding relative to warfarin[27-30].
Figure 3.
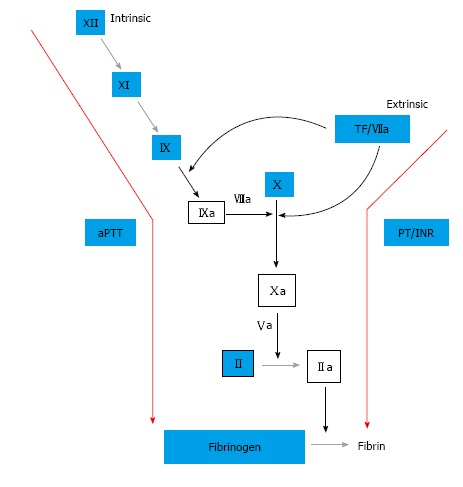
Coagulation cascade. Factors Xa and IIa (thrombin) are the principal targets for the new oral anticoagulants, while factors II, VII, IX, X, and protein C and S are decreased by the vitamin K antagonist, warfarin. Warfarin’s effect on the factor VIIa/TF complex is a possible cause of the increased intracranial bleeding seen in warfarin compared to the newer targeted agents.
The mechanism by which these new agents lead to similar or improved efficacy in relation to warfarin, while achieving less intracranial hemorrhage, has been evaluated. A recent study compared peak thrombin generation in the presence of both warfarin and dabigatran[31]. This study found that, while the mean lag times were equally prolonged in each group, the peak thrombin level was significantly decreased in the warfarin group. Furthermore, the authors found that in the presence of tissue factor (TF), the peak thrombin level increases. Lastly, it was shown that warfarin has a greater inhibitory effect on peak thrombin level in the presence of TF than dabigatran. When the mechanism of action of warfarin and dabigatran are compared, it is notable that one major difference is the suppressive effect warfarin has on formation of the TF-factor VIIa complex which initiates coagulation (Figure 3). Dabigatran, as well as the factor Xa inhibitors, lack this inhibitory effect since these agents blockade the coagulation cascade further downstream. The brain, in particular, has a rich endowment of tissue factor, thus the effect of warfarin is enhanced in the brain relative to dabigatran, which may explain the increased intracranial hemorrhage seen in patient’s taking warfarin vs the newer target-specific oral anticoagulants. Additionally, there is evidence that some of this effect may be due to reduced drug entry through the blood-brain barrier relative to warfarin[32].
Multiple studies have examined the efficacy of antithrombotic agents in the prevention of recurrent VTE after an initial course of anticoagulation. Both low-dose warfarin and low-dose aspirin have been shown to effectively reduce the risk of recurrent VTE, when compared to placebo, without increasing the risk of bleeding complications[33,34]. As for the new oral anticoagulants, dabigatran and rivaroxaban have been shown to effectively decrease the risk of recurrent VTE, but both increased the risk of clinically relevant bleeding when compared to placebo[35,36]. Notably, dabigatran had a lower risk of major or clinically relevant bleeding when compared to regular-dose warfarin (INR 2-3). Apixaban has been shown to have similar bleeding risk to aspirin when evaluated for stroke prevention in nonvalvular atrial fibrillation[37]. More recently, apixaban was compared at two doses (2.5 mg and 5 mg, twice daily) vs placebo in the extended treatment of VTE[38]. In this study, each dose was effective in reducing the risk for recurrent VTE relative to placebo. There was no increased risk of major or clinically relevant bleeding in either dose of apixaban vs placebo, or between the two doses.
Due to the inherent bleeding risk in HHT patients, any approach to anticoagulation that may decrease the risk of bleeding would be prudent. Through the use of either a factor IIa or Xa inhibitor, as opposed to warfarin for the acute treatment of VTE, the TF-VIIa interaction can be preserved, which may lead to decreased bleeding events, particularly in the brain. Based on the studies reviewed above, it would be reasonable to use either low-dose aspirin, low-dose warfarin, or apixaban to prevent recurrent VTE in a patient at risk for bleeding. Further research is necessary to fully describe these important differences between anticoagulant agents.
CONCLUSION
The patient presented with a difficult clinical scenario: new venous thromboembolism in the setting of a prothrombin gene mutation and bleeding complications secondary to HHT. The physicians who initially cared for our patient understood this dilemma and opted to place an IVC filter. Unfortunately, this filter thrombosed and the patient was initiated on warfarin. While on warfarin, the patient, like most patients studied, complained of worsening epistaxis, but no major bleeding events.
Our initial approach was to ensure she had been adequately screened for AVMs (pulmonary and cerebral) that could cause severe harm to our patient prior to the initiation of anticoagulation. The next management decision was to address the anticoagulation needs of our patient. Despite her bleeding risk, she had a strong indication for anticoagulation given progressive DVT and PE despite the presence of an IVC filter, as well as a prothrombotic state related to heterozygosity for the prothrombin gene mutation, immobilization, and recent surgery. Our primary options included: continuing warfarin (normal or low-dose); changing to low-dose aspirin; changing to a new oral anticoagulant, such as dabigatran, rivaroxaban, or apixaban; or discontinuing all anticoagulation. At the time of her appointment she had received 3 mo of anticoagulation with warfarin. Despite the role her recent surgery played in provoking the VTE, the presence of a thrombosed IVC filter and heterozygosity for prothrombin gene mutation indicated that she should receive long-term treatment. After 6 mo of anticoagulation, her IVC filter was retrieved as her thrombus burden had declined significantly in her IVC and lower extremities. We hoped retrieval would decrease her risk for recurrent VTE and allow us to eventually discontinue warfarin. Because her epistaxis was more symptomatic on therapeutic anticoagulation, we reduced her INR target range to 1.5-2. After 3 mo of warfarin therapy at a lower target INR, we discontinued anticoagulation completely. Unfortunately, 6 mo after her filter retrieval she developed recurrent right leg pain and a new iliofemoral DVT was identified. Subsequently, a new Celect IVC filter was placed. While recovering from this procedure, the patient developed abdominal pain and was ultimately found to have extensive thrombosis of the portal, superior and inferior mesenteric veins. Therapeutic anticoagulation with warfarin was resumed, and, after six months, her epistaxis became more severe, so her INR range was again reduced to 1.5-2. She has now been on low-dose warfarin for eighteen months and remains free of recurrent VTE or severe epistaxis. If a change to her anticoagulant regimen is warranted in the future, other options include low-dose aspirin and apixaban to prevent recurrent VTE, while minimizing the risk of bleeding.
HHT patients present hematologists with difficult clinical decisions due to the inherent bleeding and thrombotic complications associated with the disease. The majority of management decisions revolve around bleeding complications. When a thrombotic complication arises, anticoagulation recommendations must take into account the bleeding risks associated with HHT. Recent reviews have found that HHT patients can be safely anticoagulated, with the most frequent complication being worsened epistaxis. Patients should be aggressively screened for pulmonary and cerebral AVMs prior to initiating any anticoagulant. Large clinical trials have shown that factor IIa and Xa inhibitors have less intracranial bleeding than warfarin, and basic coagulation research has provided a possible mechanism. In light of this, there is an important role for the use of factor IIa and Xa inhibitors in HHT patients requiring acute anticoagulation. For long-term anticoagulation to prevent VTE recurrence, the agents associated with the lowest risk of bleeding relative to placebo are low-dose warfarin, low-dose aspirin, and apixaban. These agents will have an important role in the long-term prevention of VTE recurrence in patients with HHT.
Footnotes
P- Reviewer: Sabate M, Starke R S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: Dr. Dittus has no conflicts to report. Dr. Streiff has received research funding from Sanofi-Aventis, Bristol Myers Squibb and Portola, honoraria for CME lectures from Sanofi-Aventis and Ortho-McNeil, consulted for Sanofi-Aventis, Eisai, Daiichi-Sankyo, Boehringer-Ingelheim and Janssen HealthCare and has given expert witness testimony in various medical malpractice cases. Dr. Ansell serves as a consultant for Bristol Myers Squibb, Janssen, Boehringer Ingelheim, Pfizer, and Daiichi Sankyo Pharmaceuticals.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 24, 2014
First decision: November 27, 2014
Article in press: January 20, 2015
References
- 1.Sharathkumar AA, Shapiro A. Hereditary haemorrhagic telangiectasia. Haemophilia. 2008;14:1269–1280. doi: 10.1111/j.1365-2516.2008.01774.x. [DOI] [PubMed] [Google Scholar]
- 2.Sutton HG. Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular systeme. Med Mirror. 1864;1:769–781. [Google Scholar]
- 3.Rendu H. Epistaxis repetees chez un sujet porteur de petits angiomes cutanes et muqueux. Gaz Soc Hosp (Paris) 1896;68:1322–1323. [Google Scholar]
- 4.Osler W. On a family form of recurring epistaxis, associated with multiple telangiectases of the skin and mucous membranes. Bull Johns Hopkins Hosp. 1901;12:160–162. [Google Scholar]
- 5.Weber F. Multiple hereditary developmental angiomata (telangiectases) of the skin and mucous membranes associated with recurring haemorrhages. Lancet. 1907;2:160–162. [Google Scholar]
- 6.Hanes F. Multiple hereditary telangiectases causing hemorrhage (hereditary hemorrhagic telangiectasia) Bull Johns Hopkins Hosp. 1909;20:63–73. [Google Scholar]
- 7.Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24:203–219. doi: 10.1016/j.blre.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Shovlin CL, Sulaiman NL, Govani FS, Jackson JE, Begbie ME. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): association with venous thromboembolism. Thromb Haemost. 2007;98:1031–1039. [PubMed] [Google Scholar]
- 9.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 10.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 11.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 12.Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17:860–871. doi: 10.1038/ejhg.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.University of Utah, Department of Pathology. HHT Mutation Database. Available from: http: //www.arup.utah.edu/database/ENG/ENG_welcome.php.
- 14.Cirulli A, Liso A, D’Ovidio F, Mestice A, Pasculli G, Gallitelli M, Rizzi R, Specchia G, Sabbà C. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol. 2003;110:29–32. doi: 10.1159/000072411. [DOI] [PubMed] [Google Scholar]
- 15.Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918–924. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- 16.Braverman IM, Keh A, Jacobson BS. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Invest Dermatol. 1990;95:422–427. doi: 10.1111/1523-1747.ep12555569. [DOI] [PubMed] [Google Scholar]
- 17.Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am J Med Genet. 2000;91:66–67. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 18.van Gent MW, Velthuis S, Post MC, Snijder RJ, Westermann CJ, Letteboer TG, Mager JJ. Hereditary hemorrhagic telangiectasia: how accurate are the clinical criteria? Am J Med Genet A. 2013;161A:461–466. doi: 10.1002/ajmg.a.35715. [DOI] [PubMed] [Google Scholar]
- 19.Shovlin CL, Jackson JE, Bamford KB, Jenkins IH, Benjamin AR, Ramadan H, Kulinskaya E. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax. 2008;63:259–266. doi: 10.1136/thx.2007.087452. [DOI] [PubMed] [Google Scholar]
- 20.Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48:73–87. doi: 10.1136/jmg.2009.069013. [DOI] [PubMed] [Google Scholar]
- 21.Gincul R, Lesca G, Gelas-Dore B, Rollin N, Barthelet M, Dupuis-Girod S, Pilleul F, Giraud S, Plauchu H, Saurin JC. Evaluation of previously nonscreened hereditary hemorrhagic telangiectasia patients shows frequent liver involvement and early cardiac consequences. Hepatology. 2008;48:1570–1576. doi: 10.1002/hep.22514. [DOI] [PubMed] [Google Scholar]
- 22.Devlin HL, Hosman AE, Shovlin CL. Antiplatelet and anticoagulant agents in hereditary hemorrhagic telangiectasia. N Engl J Med. 2013;368:876–878. doi: 10.1056/NEJMc1213554. [DOI] [PubMed] [Google Scholar]
- 23.Edwards CP, Shehata N, Faughnan ME. Hereditary hemorrhagic telangiectasia patients can tolerate anticoagulation. Ann Hematol. 2012;91:1959–1968. doi: 10.1007/s00277-012-1553-8. [DOI] [PubMed] [Google Scholar]
- 24.Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, Pogue J, Reilly PA, Themeles E, Varrone J, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139–1151. doi: 10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 25.Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, Breithardt G, Halperin JL, Hankey GJ, Piccini JP, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883–891. doi: 10.1056/NEJMoa1009638. [DOI] [PubMed] [Google Scholar]
- 26.Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, Al-Khalidi HR, Ansell J, Atar D, Avezum A, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981–992. doi: 10.1056/NEJMoa1107039. [DOI] [PubMed] [Google Scholar]
- 27.Schulman S, Kearon C, Kakkar AK, Mismetti P, Schellong S, Eriksson H, Baanstra D, Schnee J, Goldhaber SZ. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361:2342–2352. doi: 10.1056/NEJMoa0906598. [DOI] [PubMed] [Google Scholar]
- 28.Bauersachs R, Berkowitz SD, Brenner B, Buller HR, Decousus H, Gallus AS, Lensing AW, Misselwitz F, Prins MH, Raskob GE, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499–2510. doi: 10.1056/NEJMoa1007903. [DOI] [PubMed] [Google Scholar]
- 29.Büller HR, Prins MH, Lensin AW, Decousus H, Jacobson BF, Minar E, Chlumsky J, Verhamme P, Wells P, Agnelli G, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012;366:1287–1297. doi: 10.1056/NEJMoa1113572. [DOI] [PubMed] [Google Scholar]
- 30.Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, Masiukiewicz U, Pak R, Thompson J, Raskob GE, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369:799–808. doi: 10.1056/NEJMoa1302507. [DOI] [PubMed] [Google Scholar]
- 31.Dale B, Eikelboom JW, Weitz JI, Young E, Paikin JS, Coppens M, Whitlock RP, Connolly SJ, Ginsberg JS, Hirsh J. Dabigatran attenuates thrombin generation to a lesser extent than warfarin: could this explain their differential effects on intracranial hemorrhage and myocardial infarction? J Thromb Thrombolysis. 2013;35:295–301. doi: 10.1007/s11239-012-0857-9. [DOI] [PubMed] [Google Scholar]
- 32.Ru San T, Chan MY, Wee Siong T, Kok Foo T, Kheng Siang N, Lee SH, Chi Keong C. Stroke prevention in atrial fibrillation: understanding the new oral anticoagulants dabigatran, rivaroxaban, and apixaban. Thrombosis. 2012;2012:108983. doi: 10.1155/2012/108983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ridker PM, Goldhaber SZ, Danielson E, Rosenberg Y, Eby CS, Deitcher SR, Cushman M, Moll S, Kessler CM, Elliott CG, et al. Long-term, low-intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N Engl J Med. 2003;348:1425–1434. doi: 10.1056/NEJMoa035029. [DOI] [PubMed] [Google Scholar]
- 34.Becattini C, Agnelli G, Schenone A, Eichinger S, Bucherini E, Silingardi M, Bianchi M, Moia M, Ageno W, Vandelli MR, et al. Aspirin for preventing the recurrence of venous thromboembolism. N Engl J Med. 2012;366:1959–1967. doi: 10.1056/NEJMoa1114238. [DOI] [PubMed] [Google Scholar]
- 35.Schulman S, Kearon C, Kakkar AK, Schellong S, Eriksson H, Baanstra D, Kvamme AM, Friedman J, Mismetti P, Goldhaber SZ. Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N Engl J Med. 2013;368:709–718. doi: 10.1056/NEJMoa1113697. [DOI] [PubMed] [Google Scholar]
- 36.Romualdi E, Donadini MP, Ageno W. Oral rivaroxaban after symptomatic venous thromboembolism: the continued treatment study (EINSTEIN-extension study) Expert Rev Cardiovasc Ther. 2011;9:841–844. doi: 10.1586/erc.11.62. [DOI] [PubMed] [Google Scholar]
- 37.Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, Flaker G, Avezum A, Hohnloser SH, Diaz R, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364:806–817. doi: 10.1056/NEJMoa1007432. [DOI] [PubMed] [Google Scholar]
- 38.Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, Porcari A, Raskob GE, Weitz JI. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368:699–708. doi: 10.1056/NEJMoa1207541. [DOI] [PubMed] [Google Scholar]