

Abstract
Alzheimer's disease (AD) has been associated with increased phosphorylation of the translation initiation factor 2α (eIF2α) at serine 51. Increased phosphorylation of eIF2α alters translational control and may thereby have adverse effects on synaptic plasticity, learning, and memory. To analyze if increased levels of p-eIF2α indeed promote AD-related neurocognitive impairments, we crossed 5xFAD transgenic mice with an eIF2α S51A knock-in line that expresses the nonphosphorylatable eIF2α variant eIF2α S51A. Behavioral assessment of the resulting mice revealed motor and cognitive deficits in 5xFAD mice that were, with the possible exception of locomotor hyperactivity, not restored by the eIF2α S51A allele. Telemetric intracranial EEG recordings revealed no measurable effects of the eIF2α S51A allele on 5xFAD-associated epileptic activity. Microarray-based transcriptome analyses showed clear transcriptional alterations in 5xFAD hippocampus that were not corrected by the eIF2α S51A allele. In contrast to prior studies, our immunoblot analyses did not reveal increased levels of p-eIF2α in the hippocampus of 5xFAD mice, suggesting that elevated p-eIF2α levels are not a universal feature of AD models. Collectively, our data indicate that 5xFAD-related pathologies do not necessarily require hyperphosphorylation of eIF2α to emerge; they also show that heterozygosity for the nonphosphorylatable eIF2α S51A allele has limited effects on 5xFAD-related disease manifestations.
1. Introduction
Alzheimer's disease (AD) is associated with progressive cognitive and neurological impairments. Synaptic dysfunction downstream of toxic amyloid species is thought to play a major role in altered brain function and cognitive impairments in AD [1].
A growing body of literature suggests that translational regulatory mechanisms are disrupted in AD [2–4]. More specifically, AD has been associated with elevated phosphorylation levels of the eukaryotic translation initiation factor 2α (eIF2α). Hyperphosphorylation of eIF2α has been observed in postmortem samples derived from subjects affected by AD [5–7]. Studies in AD mouse models that are based on rare familial mutations associated with high risk for AD (i.e., mice overexpressing mutant amyloid precursor protein and/or presenilin) have indicated that eIF2α hyperphosphorylation can be seen in some of these disease models as well [6–8]. eIF2α is phosphorylated in the context of cellular stress responses, such as the endoplasmic reticulum (ER) unfolded protein response (UPR); increased eIF2α phosphorylation then leads to a general inhibition of protein synthesis [9]. In addition, elevated eIF2α phosphorylation favors the translational expression of certain mRNAs, including activating transcription factor 4 (ATF4) [10].
De novo protein synthesis is well known to play import roles in the establishment of long-lasting synaptic plasticity and the formation of long-term memory [11, 12]. Hyperphosphorylation of eIF2α may interfere with protein synthesis-dependent forms of plasticity and memory formation by inhibiting de novo protein synthesis [13]. Additionally, eIF2α hyperphosphorylation may perturb synaptic plasticity and memory formation by suppressing cAMP response element binding protein- (CREB-) dependent gene expression via upregulation of ATF4 [13]. These considerations suggest that p-eIF2α-mediated translational and transcriptional effects may contribute to altered plasticity and cognitive dysfunction in AD. Indeed, it has been reported that the genetic removal of either of two different eIF2α kinases (PRKR-like endoplasmic reticulum kinase, Perk, and general control nonderepressible 2, Gcn2) restores plasticity and memory impairments in an APP/PS1 mouse model of AD [14].
We, here, set out to explore the role of eIF2α phosphorylation in Alzheimer's pathogenesis by crossing the 5xFAD mouse model [15] with an eIF2α S51A knock-in line [16], in which eIF2α cannot be phosphorylated on the mutant allele due to substitution of serine at residue 51 by alanine. While most mouse model studies have looked at kinases upstream of eIF2α [14, 17], we were interested in analyzing the effects of a nonphosphorylatable eIF2α S51A allele on disease progression in an AD mouse model. eIF2α phosphorylation globally inhibits protein synthesis and thereby attenuates the flow of new protein into the endoplasmic reticulum under conditions of ER stress [16]. Mouse embryonic fibroblasts (MEFs) derived from eIF2α S51A/S51A animals lack a global repression of protein synthesis, as well as an induction of UPR-inducible genes during ER stress [16]. As a consequence, viability under ER stress is much reduced in eIF2α S51A/S51A cells compared to wild-type control cells [16]. Homozygous eIF2α S51A mutant mice were found to die after birth, likely due to extended hypoglycemia caused by defective gluconeogenesis [16]; heterozygous eIF2α S51A mutants do not exhibit compromised viability. 5xFAD mice overexpress (under control of Thy1 promoter) both mutant human APP with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial Alzheimer's disease (FAD) mutations and human PSEN1 harboring two FAD mutations (M146L and L286V) [15]. These animals show early and progressive amyloid deposits (beginning at 2 months), gliotic and inflammatory changes, neuronal loss, and neurocognitive impairments [15].
2. Material and Methods
2.1. Animals
5xFAD mice (genetic background: B6/SJL) overexpress mutant forms of human APP (the Swedish mutation: K670N, M671L; the Florida mutation: I716V; the London mutation: V717I) and mutant PSEN1 (M146L, L286V) [15]. eIF2α +/S51A mice (genetic background: C57BL/6J) were generated as previously described [16]. 5xFAD mice were crossed with eIF2α +/S51A mice, yielding animals of four different genotypes: wild-type, 5xFAD, 5xFAD;eIF2α +/S51A, and eIF2α +/S51A. Our studies were performed using heterozygous eIF2α S51A mutant mice because homozygous eIF2α S51A mutants die shortly after birth [16] and are therefore not suitable to address the aims of the present paper.
One cohort of animals was generated for behavioral and neurological assessments. Tests were conducted in the following order/at the following age of the animals: open field (8 months, 11 months), wire hang test (10-11 months), tail suspension test (10-11 months), Morris water maze (12 months), and contextual fear conditioning (13-14 months). Ages of animals used for electrophysiological, biochemical, and gene expression analyses are provided in the respective sections below (see below). The present study was approved by “Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen” (Recklinghausen, Germany).
2.2. Tail Suspension Test
Assessment of pathological hindlimb clasping was performed using the tail suspension test. Hindlimb movements in the test were assigned to one of the four following categories: 1 = normal hindlimb movements, 2 = intermittent clasping of one hindlimb, 3 = intermittent clasping of both hindlimbs, and 4 = enduring clasping of both hindlimbs. Statistical analysis was performed by ordered logistic regression with the factors of 5xFAD genotype (5xFAD transgenes present versus absent) and eIF2α genotype (eIF2α +/S51A versus eIF2α +/+).
2.3. Wire Hang Test
Mice were placed on a wire cage lid, which was turned upside down and positioned about 25 cm above an empty cage. Latency to fall was recorded with a maximum duration of 10 min. Mice received one trial per day over a period of 3 days (latencies were averaged across these trials). Statistical analysis was performed using two-way ANOVA with the between-subjects factors 5xFAD genotype (5xFAD transgenes present versus absent) and eIF2α genotype (eIF2α +/S51A versus eIF2α +/+).
2.4. Open Field
To assess motor activity in a novel environment, mice were placed in an open field (27.5 cm × 27.5 cm × 20 cm) for 20 min on each of 3 consecutive days. The distance travelled was recorded using the EthoVision XT video tracking system (Noldus, Wageningen, Netherlands) and an average across all 3 sessions was calculated for each animal. Statistical analysis was performed using two-way ANOVA with the between-subjects factors 5xFAD genotype (5xFAD transgenes present versus absent) and eIF2α genotype (eIF2α +/S51A versus eIF2α +/+).
2.5. Morris Water Maze
Mice were trained on a hidden version of the Morris water maze (Ø 135 cm). In this task animals learned to find an escape platform (Ø 10 cm) hidden underneath the water surface in a constant location of the pool. Mice received 6 training trials per day for 7 consecutive days. Training trials were completed when the animal had reached the escape platform or when 60 s were elapsed, whichever came first. During training trials, animals were started from randomly alternating starting positions. If animals did not manage to get on the escape platform within 60 s, they were gently guided to the escape platform. There was a 15 s posttrial period on the escape platform. To test how well the animals had learned the position of the escape platform, we gave a probe trial at the end of training day 7. During the probe trial the platform was removed from the pool and the swim pattern was analyzed (with respect to quadrant occupancy and target crossings). After completion of the 7 days of hidden training, we gave one day of cued training (6 trials), during which the platform position was indicated by a visible cue. Swim patterns of mice were recorded using the EthoVision XT video tracking system (Noldus, Wageningen, Netherlands). Time spent in quadrants and target crossings were analyzed using the built-in features of the software. Further analyses were done based on the raw time-tagged xy-coordinates using Matlab (The Mathworks). Search strategies were classified according to parameters described in a previous study [18], originally based on [19]. Search strategies were defined by no more than two quantitative parameters that were chosen to reflect the unique abstract properties of a given strategy and that were not dependent on the specific pool dimensions used. Statistical analysis of the groups was done by three-way repeated-measures ANOVA with the between-subjects factors 5xFAD genotype (5xFAD transgenes present versus absent) and eIF2α genotype (eIF2α +/S51A versus eIF2α +/+) and one of the following within-subjects factors: training trials (for the analysis of the escape latency curves); quadrants (for the analysis of probe trial data, i.e., target crossings and quadrant occupancy, resp.).
2.6. Contextual Fear Conditioning
Mice were placed in a conditioning chamber (Med Associates, St. Albans, Vermont, USA) for 3 min and received foot shocks (0.75 mA, 2 s) after minutes 1 and 2. One day later, for testing, mice were again placed in the conditioning chamber for a period of 3 min. Freezing behavior and activity levels during the baseline (i.e., first minute on the training day) and the test were recorded and analysed using Video Fear Conditioning software (Med Associates, St. Albans, Vermont, USA). Due to differences in baseline activity levels between groups (data not shown), we do not report freezing scores but report activity suppression ratios instead, which were calculated as follows: activitytest/(activitybaseline + activitytest). Statistical comparison of the groups was performed by two-way ANOVA with the between-subjects factors 5xFAD genotype (5xFAD transgenes present versus absent) and eIF2α genotype (eIF2α +/S51A versus eIF2α +/+).
2.7. Radiotelemetric EEG Recordings
Mice (>10 months of age) were anesthetized by intraperitoneal injection containing ketamine (KetanestR, Parke-Davis/Pfizer, Germany)/xylazine (RompunR 2%, Bayer Vital, Germany) at 100/10 mg/kg. For measuring of the electroencephalogram (EEG), TL11M2-F20-EET 2-channel transmitters (technical specification: 3.9 g, 1.9 cc; Data Science International (DSI), USA) were implanted into a subcutaneous pouch on the back of the animals. The first channel was used to target the primary motor cortex region (M1). A differential epidural electrode was placed at the following stereotaxic coordinates: (M1-) lead bregma +1 mm, lateral of bregma 1.5 mm (left hemisphere). For deep brain recordings targeting the hippocampal CA1 region, the differential electrode of channel 2 was positioned as follows: (CA1-) lead, bregma −2 mm, lateral of bregma 1.5 mm (right hemisphere), dorsoventral (depth) 1.5 mm. For both channels, epidural reference electrodes were placed at bregma −6 mm, lateral of bregma 1 mm (left hemisphere), and bregma −6 mm, lateral of bregma 1 mm (right hemisphere). Electrodes were fixed at the neurocranium using glass ionomer cement (Kent Express, UK), and the scalp was closed using over and over sutures (Ethilon, 6-0). A detailed description of the implantation procedure is available in a previous publication [20]. For postoperative pain management carprofen (5 mg/kg; Rimadyl, Pfizer, Germany) was administered subcutaneously to the animals.
Following a 10-day recovery period, simultaneous video-EEG recordings from the motor cortex (M1) and the hippocampal CA1 region were performed for 48 h using Dataquest ART 4.2 software (DSI) at a sampling rate of 500 Hz with no a priori filter cut-off.
For further analyses, data were processed using Neuroscore 2.1 (DSI). Seizure analyses and calculations were performed using Neuroscore Spike Train Detector, that is, a seizure detection module. Seizure protocols contained total number of episodes and spikes, spike frequency, total spike train duration, and shortest and longest spike train duration.
2.8. Histology and Immunostaining
Mice (13 months old) were anaesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (7 mg/kg). For perfusion, the bloodstream was rinsed with sterile 0.9% sodium chloride and organs were fixed using 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS). Brains were dissected and postfixed in 4% PFA in PBS overnight at 4°C and dehydrated in 30% sucrose in PBS at 4°C. Coronal brain sections of 40 μm thickness were cut using a sliding microtome (Leica, Wetzlar, Germany) and were stored in cryoprotectant buffer (0.05 M phosphate buffer, 25% glycerol, and 25% ethylene glycol) at −20°C. Brain sections spaced 240 μm apart were transferred into Tris-buffered saline (TBS), washed twice for 5 min and incubated in 0.6% H2O2 in TBS for 30 min. Sections were then washed three times in TBS for 5 min. For blocking, sections were incubated for 30 min in TBS-plus (TBS, 0.1% Triton X-100, and 3% donkey serum). Next, sections were incubated with primary antibody (rabbit anti-choline acetyltransferase (ChAT), 1 : 100, Millipore, Darmstadt, Germany) in TBS-plus for 48 h at 4°C, washed twice in TBS for 5 min, and blocked in TBS-plus for 45 min. This was followed by a 1-hour incubation step (at room temperature) in TBS-plus containing biotin-conjugated secondary antibody (donkey anti-rabbit, 1 : 500, Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Sections were then washed three times in TBS for 5 min and incubated in 3,3′-diaminobenzidine (DAB) for 2 min using the DAB Substrate Kit (Roche, Mannheim, Germany). To stop the peroxidase reaction, sections were washed in tap water and TBS. Next, sections were mounted on glass slides, dehydrated in an ascending series of alcohol (twice 70%, once 96%, and twice 100% Ethanol), cleared in xylene, and covered with a coverslip. Stereological analysis of brain sections was performed using a bright field microscope Eclipse 90i (Nikon, Düsseldorf, Germany). Immunoreactive neurons within the medial septum (MS) and vertical limb of the diagonal band of Broca (VDB) were quantified using the software Stereo Investigator (MBF Bioscience, Magdeburg, Germany). Statistical analysis was performed via two-way ANOVA with the between-subjects factors 5xFAD genotype (5xFAD transgenes present versus absent) and eIF2α genotype (eIF2α +/S51A versus eIF2α +/+).
2.9. Immunoblot Analysis
Hippocampal samples were taken from 14- to 15-month-old mice after cervical dislocation and snap-frozen in liquid nitrogen. Each sample was homogenized in 250 μL lysis buffer containing RIPA buffer, Phospho-Stop (Roche, Mannheim, Germany), Protease-Inhibitor (Roche, Mannheim, Germany), 50 mM sodium fluoride, and 5 mM sodium orthovanadate and incubated on a rotator for 30 min at 4°C. Samples were centrifuged at 14000 rpm for 15 min at 4°C. Protein concentration was determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific, Bonn, Germany). If not stated otherwise, 15 μg protein was loaded on 10% tris glycine gels (APP, ChAT, eIF2α) or 16% tris tricine gels (CTF) for PAGE. Proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane and incubated in TBS with 10% Western Blocking Reagent (Roche, Mannheim, Germany) for 1 h. Incubation of PVDF membrane in primary antibody (rabbit anti-eIF2α, 1 : 2000, Cell Signaling, Danvers, MA, USA; rabbit anti-p-eIF2α, 1 : 2000, Cell Signaling, Danvers, MA, USA; rabbit anti-APP, 1 : 1000, Cell Signaling, Danvers, MA, USA; rabbit anti-CTF, 1 : 1000, Sigma-Aldrich, Taufkirchen, Germany; goat anti-ChAT, 1 : 1000, Millipore, Darmstadt, Germany; mouse anti-Aβ, clone 6E10, 1 : 1000, Covance, Princeton, NJ, USA; mouse anti-α-tubulin, 1 : 20000, Abcam, Cambridge, UK; mouse anti-β-actin, 1 : 5000, MP Biomedicals, Solon, OH, USA) in TBS with 5% Western Blocking Reagent was carried out overnight at 4°C. After multiple washing steps in TBS with 0.1% Tween-20, membranes were incubated in horseradish peroxidase-conjugated secondary antibody (donkey anti-rabbit IgG, 1 : 1000, Jackson ImmunoResearch Laboratories, West Grove, PA, USA; donkey anti-mouse IgG, 1 : 1000, Jackson ImmunoResearch Laboratories, West Grove, PA, USA; donkey anti-goat IgG, 1 : 10000, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) in TBS with 5% Western Blocking Reagent (for 1 h at room temperature or overnight at 4°C). Next, membranes were washed several times in TBS with 0.1% Tween-20. Immunosignals were detected using enhanced chemiluminescence (Amersham ECL Western Blotting Detection Reagents, GE Healthcare, Munich, Germany, or SuperSignal West Femto Maximum Sensitivity Substrate, Thermo Fisher Scientific, Bonn, Germany) and were quantified using a Chemidoc XRS imager (Bio-Rad, Munich, Germany). Densitometric analysis was performed using Image Lab software (Bio-Rad, Munich, Germany). Proteins were normalized to α-tubulin or β-actin, and phosphorylated proteins were normalized to their respective total proteins. eIF2α and p-eIF2α were detected on different blots together with the respective loading controls. Statistical analysis was accomplished by t-test or two-way ANOVA with the between-subjects factors 5xFAD genotype (5xFAD transgenes present versus absent) and eIF2α genotype (eIF2α +/S51A versus eIF2α +/+), as appropriate.
2.10. Aβ Enzyme-Linked Immunosorbent Assays (ELISA)
Hippocampal homogenates in RIPA buffer (as described above) were used for quantitative analysis of the two abundant species of amyloid β (Aβ), Aβ40 and Aβ42, using ELISA kits (Life Technologies, Carlsbad, CA, USA) according to manufacturer's instructions. Amounts of Aβ peptides were subsequently normalized to protein concentration of the respective sample.
2.11. RNA Extraction and Affymetrix Microarray Procedures
Microarray experiments were carried out using GeneChip Mouse Exon 1.0 ST v1 Expression Chip (Affymetrix). These exon arrays contain 6.553.600 probes with a coverage density of 4 probes for each exon of all known and predicted genes of the mouse genome. Mouse hippocampal tissue of 14- to 15-month-old mice was processed with the RNeasy Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RNA integrity was determined with Agilent's Bioanalyzer and only samples with integrity numbers between 9.2 and 10 were chosen for downstream applications. Arrays were washed and stained according to the manufacturer's recommendations. Labeled and purified cDNA was fragmented (5.5 μg) and subsequently hybridized to the arrays before scanning in a GeneChip 3000 7G scanner (Affymetrix). Normalization to the median of all samples, background correction as well as statistical analysis, was performed with GeneSpringGX software (Agilent technologies). An implemented GC-RMA algorithm was applied on all chips to summarize probe level information. Microarray data were analyzed using moderated t-tests. Benjamini Hochberg FDR was employed and comparisons with a P value <0.02 were considered statistically significant. Differentially regulated transcripts with a fold change (FC) greater than 1.6 were then subjected to hierarchical clustering analysis in order to visualize gene expression changes across groups. Array data are available in the GEO database under GSE50521. DAVID [21, 22] was used to carry out gene ontology enrichment analyses in the gene set differentially expressed between 5xFAD and wild-type controls. In addition, we used ingenuity pathway analysis to help define common features of genes differentially expressed in 5xFAD hippocampus.
2.12. Cell Culture
Confluent SHSY5Y wild-type cells in 6-well plates were treated with vehicle (ddH2O) for 15 min or with 2 mM dithiothreitol (DTT) for 15 or 60 min. After treatment cells were washed with PBS, detached with Trypsin/EDTA, and centrifuged at 500 ×g for 5 min at 4°C. Pelleted cells were resuspended in 100 μL lysis buffer (TBS, 1% Triton X-100, Protease-Inhibitor (Roche, Mannheim, Germany), and Phospho-Stop (Roche, Mannheim, Germany)) and incubated on ice for 30 min. Cell homogenates were centrifuged at 10000 ×g for 10 min at 4°C. The supernatant was used for immunoblot analysis.
3. Results
In order to test if increased eIF2α phosphorylation contributes to cognitive dysfunction and disease progression in 5xFAD mice, we crossed these mice with an eIF2α S51A knock-in line [16], in which eIF2α cannot be phosphorylated on one allele due to substitution of serine at residue 51 by alanine.
We performed immunoblot analyses to measure the abundance of p-eIF2α (i.e., eIF2α phosphorylated at serine 51), total eIF2α, APP, and APP cleavage products in lysates prepared from hippocampal tissue of 5xFAD, 5xFAD;eIF2α +/S51A, and eIF2α +/S51A animals as well as wild-type controls. Unexpectedly but in agreement with one other recent study [23], our analyses showed no significant effect of the 5xFAD transgenes on p-eIF2α levels (Figure 1(a); two-way ANOVA with between-subjects factors 5xFAD genotype and eIF2α genotype, effect of 5xFAD genotype, P = 0.15). Additional experiments showed the expected [24] increase in p-eIF2α abundance in cells treated with 2 mM DTT (see Supplementary Figure 1 in the Supplementary Material available online at http://dx.doi.org/10.1155/2015/825157; one-way ANOVA with treatment as between-subjects factor, P = 0.01), indicating that our experimental settings were suited, in principle, to detect eIF2α hyperphosphorylation. Consistent with previously published work [7], the 5xFAD transgenes affected total eIF2α levels with slightly increased eIF2α abundance in animals bearing the 5xFAD transgenes (Figure 1(a); two-way ANOVA with between-subjects factors 5xFAD genotype and eIF2α genotype, effect of 5xFAD genotype, P = 0.01). The eIF2α phosphorylation status did not differ significantly between heterozygous carriers of the eIF2α S51A mutation and animals carrying two wild-type eIF2α alleles (Figure 1(a); two-way ANOVA with between-subjects factors 5xFAD genotype and eIF2α genotype, effect of eIF2α genotype, P = 0.62). These biochemical findings suggest that an excessive eIF2α phosphorylation is not a universal feature of AD mouse models, such as 5xFAD mice, and they also indicate that the heterozygous eIF2α S51A mutation is not necessarily sufficient to suppress hippocampal eIF2α phosphorylation.
Figure 1.

The eIF2α S51A mutation had no measurable effects on eIF2α phosphorylation and APP processing. Shown are representative immunoblots of (a) p-eIF2α and total eIF2α, (b) human APP and total APP, and (c) α-CTF and β-CTF all prepared from hippocampal homogenates, along with the respective quantitative densitometric data (WT, n = 6 mice; 5xFAD, n = 7 mice; 5xFAD;eIF2α S51A, n = 6 mice; eIF2α S51A, n = 5 mice). (d) Concentrations of abundant Aβ-species, Aβ40 and Aβ42, in 5xFAD hippocampal homogenates were determined using ELISA (5xFAD, n = 7 mice; 5xFAD;eIF2α S51A, n = 6 mice). All data are presented as mean ± SEM. Data were analyzed using two-way ANOVAs with the between-subjects factors 5xFAD genotype/eIF2α genotype (effect of 5xFAD transgenes; effect of eIF2α genotype; 5xFAD × eIF2α interaction) and t-tests (5xFAD versus 5xFAD;eIF2α S51A) as appropriate. Statistically significant differences (P < 0.05) are denoted by bold font.
Further immunoblot analyses showed no clear effects of the eIF2α S51A mutation on the abundance of total APP (Figure 1(b); two-way ANOVA with between-subjects factors 5xFAD genotype and eIF2α genotype, effect of eIF2α genotype, P = 0.31), human APP (Figure 1(b); t-test, 5xFAD versus 5xFAD;eIF2α +/S51A, P = 0.97), and APP cleavage products (Figure 1(c); α-CTF, t-test, 5xFAD versus 5xFAD;eIF2α +/S51A, P = 0.66; β-CTF, t-test, 5xFAD versus 5xFAD; eIF2α +/S51A, P = 0.39). ELISA analyses showed no group differences regarding hippocampal Aβ40 and Aβ42 concentrations (Figure 1(d); Aβ40, t-test, 5xFAD versus 5xFAD;eIF2α +/S51A, P = 0.90; Aβ42, t-test, 5xFAD versus 5xFAD;eIF2α +/S51A, P = 0.30).
In line with these biochemistry results, our behavioral, electrophysiological, and gene expression analyses revealed limited effects of the eIF2α S51A allele on disease phenotypes present in 5xFAD mice, as outlined below.
In brief, the eIF2α S51A allele did not appear to ameliorate pathological hindlimb clasping in 5xFAD mice (Figure 2(a); ordered logistic regression, effect of 5xFAD transgenes, P < 0.0001; effect of eIF2α genotype P = 0.24; 5xFAD genotype × eIF2α genotype interaction, P = 0.081), nor was there any apparent effect on motor impairments as measured in the wire hang test (Figure 2(b); two-way ANOVA with between-subjects factors 5xFAD genotype and eIF2α genotype, effect of 5xFAD genotype, P < 0.0001; effect of eIF2α genotype, P = 0.56; 5xFAD × eIF2α interaction, P = 0.87). We analyzed learning and memory using a context fear conditioning paradigm (Figure 2(c)) and the Morris water maze (Figures 2(d)–2(g)). In context fear conditioning, 5xFAD mice showed higher activity suppression scores upon testing, indicative of associative learning impairments in these animals (Figure 2(c); two-way ANOVA with between-subjects factors 5xFAD genotype and eIF2α genotype, effect of 5xFAD genotype, P = 0.0003), that were not influenced by the eIF2α S51A allele in any obvious way (Figure 2(c); two-way ANOVA with between-subjects factors 5xFAD genotype and eIF2α genotype, effect of eIF2α genotype, P = 0.94; 5xFAD × eIF2α interaction, P = 0.87).
Figure 2.

The eIF2α S51A allele had limited effects on most neurological phenotypes but restored hyperactivity in 5xFAD mice. (a) Hindlimb clasping scores, as assessed in the tail suspension test (WT, n = 11 mice; 5xFAD, n = 12 mice; 5xFAD;eIF2α S51A, n = 9 mice; eIF2α S51A, n = 6 mice). (b) Latencies to fall in the context of a wire hang test (WT, n = 11 mice; 5xFAD, n = 13 mice; 5xFAD;eIF2α S51A, n = 11 mice; eIF2α S51A, n = 8 mice). (c) Activity suppression ratios in a context fear conditioning paradigm (WT, n = 11 mice; 5xFAD, n = 12 mice; 5xFAD;eIF2α S51A, n = 9 mice; eIF2α S51A, n = 6 mice). ((d)–(g)) Results of an assessment of spatial learning and memory in the Morris water maze (WT, n = 11 mice; 5xFAD, n = 12 mice; 5xFAD;eIF2α S51A, n = 9 mice; eIF2α S51A, n = 6 mice). (d) Escape latencies during training trials. (e) Quadrant occupancy and (f) target crossings measures obtained during a probe trial given after the completion of training day 7. For each genotype, bars represent (from left to right) target quadrant, opposite quadrant, adjacent right quadrant, and adjacent left quadrant. (g) Results of an extended swim path analysis: for each group, the proportion of animals in the respective search categories is plotted against training trial. ((h),(i)) Distance travelled in two open field experiments performed at either 8 months ((h); WT, n = 11 mice; 5xFAD, n = 13 mice; 5xFAD; eIF2α S51A, n = 11 mice; eIF2α S51A, n = 8 mice) or 11 months of age ((i); WT, n = 11 mice; 5xFAD, n = 13 mice; 5xFAD;eIF2α S51A, n = 11 mice; eIF2α S51A, n = 8 mice), respectively. Data were analyzed using two-way ANOVAs with the between-subjects factors 5xFAD genotype and eIF2α genotype ((a)–(c), (h), (i)) or using three-way ANOVAs with the between-subjects factors 5xFAD genotype and eIF2α genotype and the within-subjects factor trial (d) or quadrant ((e), (f)). Statistically significant differences (P < 0.05) are denoted by bold font. For additional information regarding the results of statistical analyses, see main text. Bar and line graphs show means ± SEM.
In the Morris water maze, 5xFAD animals showed substantially higher escape latencies during training trials than controls (Figure 2(d); three-way repeated-measures ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors and training trial as within-subjects factor, effect of 5xFAD transgenes, P < 0.0001; effect of eIF2α genotype, P = 0.76; 5xFAD × eIF2α interaction, P = 0.67), as well as a reduced number of target crossings during the probe trial given after completion of training (Figure 2(f); three-way repeated-measures ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors and quadrant as within-subjects factor, 5xFAD genotype × quadrant interaction, P = 0.03; eIF2α genotype × quadrant interaction, P = 0.89; eIF2α genotype × 5xFAD genotype × quadrant interaction, P = 0.58). An extended strategy analysis, in the context of which behaviors during the training trials were classified into increasingly hippocampus-dependent (directed search, focal search, and direct swimming), as well as primarily hippocampus-independent (chaining, scanning, random search, and thigmotaxis) search categories, revealed an excessive use of less hippocampus-dependent strategies in animals with the 5xFAD transgenes (Figure 2(g)). The eIF2α S51A allele had no obvious modulatory effect on any of these 5xFAD water maze phenotypes (Figures 2(d)–2(g)).
Our behavioral analyses did, however, reveal one neurobehavioral 5xFAD phenotype that appeared to be restored by the eIF2α S51A allele (Figures 2(h) and 2(i)). 5xFAD animals showed pronounced motor hyperactivity in an open field assay (two-way ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors, effect of 5xFAD genotype, P = 0.02 and 0.0003, resp.). 5xFAD-related hyperactivity appeared to be reduced in animals carrying the eIF2α S51A allele (two-way ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors, 5xFAD × eIF2α genotype, P = 0.06 for panel (h) and P = 0.07 for panel (i)).
The neurobiology underlying this possible eIF2α S51A-mediated rescue of 5xFAD-related hyperactivity remains to be elucidated. Here, we considered one possibility which is that the eIF2α S51A allele modifies the degeneration of the cholinergic system in 5xFAD animals, which may contribute to hyperactive behaviors in AD mouse models [25–30]. Initial stereological cell counting showed neither a significant effect of the 5xFAD transgenes (two-way ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors, effect of 5xFAD genotype, P = 0.74) nor an effect of the eIF2α S51A allele (two-way ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors, effect of eIF2α genotype, P = 0.32) on the number of ChAT-positive neurons in the basal forebrain (Supplementary Figure 2). Immunoblot analyses of ChAT abundance in the hippocampus, one of the target areas that basal forebrain cholinergic neurons project into, however, revealed lower ChAT levels in animals carrying the 5xFAD transgenes (Supplementary Figure 2; two-way ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors, effect of 5xFAD genotype, P = 0.002). The eIF2α S51A allele had no apparent effect on hippocampal ChAT protein abundance (two-way ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors, effect of eIF2α genotype, P = 0.49) and also showed no significant interaction with the 5xFAD genotype (two-way ANOVA with 5xFAD genotype and eIF2α genotype as between-subjects factors, 5xFAD × eIF2α genotype interaction, P = 0.78), indicating that the eIF2α S51A allele did not protect against the hippocampal ChAT loss in 5xFAD mice.
Network hyperexcitability and seizures are important features of animal models of AD [31, 32]. We performed electrocorticographic (M1) and deep intrahippocampal CA1 EEG recordings in 5xFAD animals crossed into the eIF2α S51A background (Figure 3). Qualitative and quantitative seizure analysis using Neuroscore Seizure Module (DSI) revealed that animals of all genotypes with the exception of wild-type animals exhibited seizure activity in the M1 recording, although ictal discharges were not seen in the deep, intrahippocampal CA1 recording (in none of the groups; not shown). Video analysis revealed that none of the cortical seizures was associated with motoric exacerbation. Thus, we observed predominantly nonconvulsive seizure activity in 5xFAD mice. eIF2α +/S51A mice showed nonconvulsive seizure activity as well and introduction of the eIF2α S51A mutation in 5xFAD mice did not appear to modify the epileptic phenotype in any apparent way.
Figure 3.

EEG recordings revealed nonconvulsive seizure activity in the motor cortex of animals carrying the 5xFAD transgenes and/or the eIF2α +/S51A allele. Radiotelemetric recordings from the primary motor cortex (M1) of wild-type, 5xFAD, 5xFAD;eIF2α +/S51A, and eIF2α +/S51A mice (WT, n = 4 mice; 5xFAD, n = 6 mice; 5xFAD;eIF2α S51A, n = 3 mice; eIF2α S51A, n = 4 mice). Wild-type mice did not exhibit seizure activity, whereas all other genotypes showed episodes of spike, polyspike, and spike-wave activity. Shown are example traces, as well as a quantification of the number of seizure episodes and the number of spikes, respectively. Bar graphs show mean ± SEM.
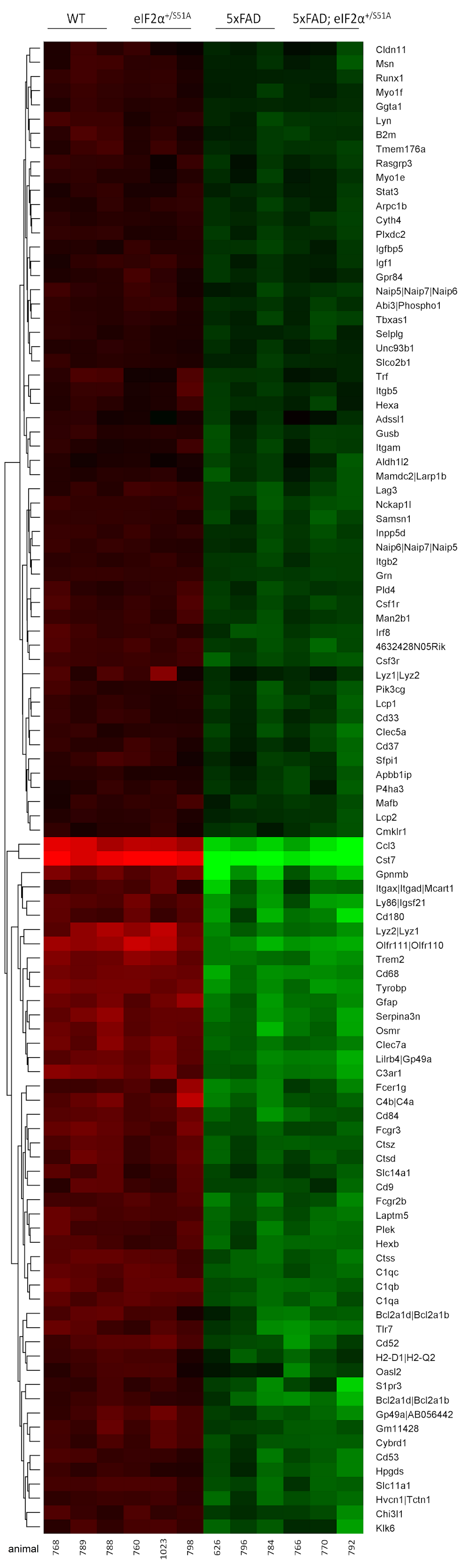
AD is associated with considerable transcriptional alterations in key brain areas [33–36]. Microarray analyses performed on hippocampal tissue of 5xFAD animals crossed into the eIF2α S51A background revealed a number of 5xFAD-related transcriptional alterations: statistical analysis revealed 106 genes differentially regulated between 5xFAD mice and wild-type controls (Supplementary Figure 3). Gene ontology analysis showed a substantial enrichment of immune-response related genes in this gene set (Supplementary Tables 1 and 2), which is in agreement with considerable inflammatory and immunological alterations observed in the context of cerebral amyloidosis. A comparison between 5xFAD and 5xFAD;eIF2α +/S51A animals revealed no differentially expressed genes, indicating that the eIF2α S51A mutation had no measurable effects on hippocampal transcriptional changes induced by the 5xFAD transgenes.
4. Discussion
In this study, we assessed the effects of a nonphosphorylatable eIF2α allele (eIF2α S51A) on disease progression in the 5xFAD mouse model of familial AD. While profound pathology was evident in 5xFAD mice, these abnormalities remained (mostly) unmodified by the heterozygous eIF2α S51A mutation. This was the case for a wide range of molecular (APP expression and processing; gene expression), electrophysiological (EEG), and neurobehavioral (motor impairments; learning and memory impairments) features of the model.
Currently, contradictory reports exist regarding the eIF2α phosphorylation status in 5xFAD mice. Elevated p-eIF2α levels in the brain of 5xFAD mice have been published previously [7, 17], but more recent studies found no elevated phosphorylation of eIF2α in 5xFAD mice [23], which is in agreement with our observation of unaltered p-eIF2α levels in the hippocampus of 5xFAD mice. We currently do not know the factors that might account for these discrepant findings. Possibilities that need to be formally addressed in future studies include differences in genetic background, on which the 5xFAD mutations were kept, differences in the brain areas examined, and age at assessment. The 5xFAD breeders used to generate the animals of the present study were obtained on a mixed B6/SJL genetic background (this is expected to result in background composition varying from animal to animal) and were crossed to eIF2α +/S51A mice on a C57BL/6J background. Other studies employed 5xFAD animals on a mixed B6/SJL genetic background (again, with background composition varying from animal to animal) [7] or on a C57BL/6 background [17]. Age may be another important variable in modifying the effect that the 5xFAD genotype has on eIF2α hyperphosphorylation. Younger (i.e., 3–6 months old) 5xFAD animals showed only modest or no measurable hyperphosphorylation of eIF2α or related kinases (Perk) despite the presence of notable plaque pathology at this age [7, 37], indicating that pronounced eIF2α hyperphosphorylation, if at all present, may be a feature of more advanced stages of cerebral amyloidosis. Finally, when analyzing eIF2α phosphorylation status, it is particularly important to normalize p-eIF2α abundance to total eIF2α protein abundance because the 5xFAD genotype may be associated with an increased abundance of total eIF2α ([7] and Figure 1 of the present study).
While we did not observe measurable eIF2α +/S51A-associated decrements in p-eIF2α abundance, others have reported moderate reductions in p-eIF2α abundance in eIF2α +/S51A mice [13, 23]. One possibility, again, is that this discrepancy is due to modulatory effects of genetic backgrounds that may have differed between these studies (C57BL/6J in [13]; nonstandardized, mixed B6/SJL backgrounds in the present study, as well as in [23]).
Ma et al. reported that conditional homozygous deletion of either of two different eIF2α kinases (Perk, Gcn2) in forebrain neurons improved spatial memory impairments in the APPswe/PSEN1dE9 mouse model of AD [14]. Deletion of either Perk or Gcn2 led to reduced p-eIF2α levels in this model [14], suggesting that these genetic manipulations were more effective in suppressing eIF2α phosphorylation than the heterozygous eIF2α S51A mutation used in the present study. Accordingly, limited effects on neurological impairments observed in our study could be related to a less effective suppression of p-eIF2α levels by the heterozygous eIF2α S51A mutation. Homozygous eIF2α S51A mutants could not be examined because of early postnatal lethality associated with this genotype [16]. In addition, it is possible that we would have been able to detect more pronounced rescue effects of the eIF2α S51A mutation on earlier stages of the disease process (i.e., in younger animals) in the model employed. Moreover, amyloid pathology progresses at a faster pace in 5xFAD animals (used in the present study) than in the model examined by Ma et al. (APPswe/PSEN1dE9 mice) and, hence, neurological impairments in the 5xFAD model may generally be less accessible to amelioration than those in APPswe/PSEN1dE9 mice.
One exception to the notion that the eIF2α +/S51A genotype did not influence 5xFAD phenotypes was the observation of restored motor hyperactivity in 5xFAD;eIF2α +/S51A mice. Elevated motor activity levels are a consistent feature of AD rodent models [38–41] and may also be observed in human individuals affected by the disorder [42]. Given the profound degeneration of basal forebrain cholinergic neurons in AD [43–46] and the role that basal forebrain cholinergic neurons play in the regulation of motor activity levels (ablation of basal forebrain cholinergic nuclei and anticholinergic pharmacological interventions increase motor activity in rats [30]), we asked whether the eIF2α S51A allele might restore hyperactivity by rescuing basal forebrain cholinergic neuron loss in 5xFAD mice. Our stereological analyses showed, however, no significant reduction of ChAT-immunoreactive neurons in the MS and VDB of 5xFAD mice, indicating that frank loss of cholinergic neurons was limited in the model at the age assessed. Nevertheless, immunoblot analyses were sensitive enough to pick up clear 5xFAD-related reductions in ChAT protein levels in one of the target areas of basal forebrain cholinergic nuclei, namely, the hippocampus, which is consistent with prior studies in transgenic AD mouse models [25–29]. 5xFAD and 5xFAD;eIF2α +/S51A mice did not differ significantly in their hippocampal ChAT protein levels, indicating that the eIF2α S51A allele did not rescue the aberrant cholinergic system of 5xFAD mice. Besides cholinergic dysfunction, AD is associated with perturbations in other neurotransmitter systems, such as the serotonergic system [47–53], and serotonergic alterations may potentially contribute to AD-associated locomotor hyperactivity [51, 54–56]. Future studies should further assess the possible mechanistic underpinnings of the eIF2α S51A effect on 5xFAD-related motor hyperactivity and assessments of the serotonergic system may provide one possible starting point for such studies.
Our finding that the eIF2α +/S51A allele had no effects on the APP abundance in 5xFAD mice is in line with a recent report that used a combination of genetic approaches and found no effect of p-eIF2α reductions on Aβ-dependent APP, as well as BACE1 levels [23], indicating that Aβ-dependent increases in APP and BACE1 levels do not involve eIF2α-dependent translational mechanisms.
In conclusion, our study revealed few effects of the eIF2α +/S51A genotype on disease progression in the 5xFAD mouse model of AD with the exception of a possible amelioration of 5xFAD-related motor hyperactivity. Our data indicate that eIF2α S51A heterozygosity is not necessarily sufficient to measurably suppress eIF2α phosphorylation in the 5xFAD mouse model employed. Future studies need to further elaborate on the specific conditions, under which a 5xFAD genotype may be associated with eIF2α hyperphosphorylation.
Supplementary Material
Supplementary Figure 1. Enhanced phosphorylation of eIF2α in SHSY5Y cells incubated with DTT. Shown are p-eIF2α and eIF2α immunoblots performed on lysates of SHSY5Y cells that had been treated with vehicle or 2mM DTT for 15 or 60 min (n = 2 samples per condition). Densitometric quantification revealed the expected increase in eIF2α phosphorylation in SHSY5Y cells incubated with DTT. P-values refer to results of posthoc Tukey tests.
Supplementary Figure 2. The cholinergic system was not involved in the eIF2α+/S51A-related restoration of hyperactivity in 5xFAD mice. (A) Shown are micrographs of ChAT-immunoreactive neurons in the basal forebrain of wild-type, 5xFAD, 5xFAD;eIF2α+/S51A and eIF2α+/S51A mice. Scale bar = 150 μm. Stereological quantification of ChAT-immunoreactive neurons in the MS and VDB of the basal forebrain demonstrated no significant differences between the four groups of mice (n = 3 mice per group). (B) Shown are immunoblots of ChAT from hippocampal homogenates of wild-type, 5xFAD, 5xFAD;eIF2α+/S51A and eIF2α+/S51A mice (n = 5-7 mice per group). Data were analyzed using two-way ANOVAs with the between-subjects factors 5xFAD genotype and eIF2α genotype. Statistically significant differences (p < 0.05) are denoted by bold font. Bar graphs show mean ± SEM.
Supplementary Figure 3. The eIF2αS51A allele had limited effects on transcriptional dysregulation in 5xFAD mice. Differentially regulated transcripts (Benjamini Hochberg FDR, p < 0.02) with a fold change higher than 1.6 were subjected to hierarchical clustering to visualize gene expression changes between groups (n = 3 mice per genotype).
Supplementary Table 1. A gene ontology analysis (DAVID) revealed a significant enrichment of immune-related transcripts in gene set differentially expressed in 5xFAD hippocampus.
Supplementary Table 2. Ingenuity pathway analysis also indicated a substantial enrichment of genes related to immune and inflammatory processes in the gene set differentially expressed in 5xFAD hippocampus.
{kind=link}
Acknowledgments
This work has been funded by the German Center for Neurodegenerative Diseases (DZNE) and by the Federal Institute for Drugs and Medical Devices (BfArM). The authors would like to thank the DZNE animal caretaker team for excellent technical support.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Palop J. J., Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nature Neuroscience. 2010;13(7):812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langstrom N. S., Anderson J. P., Lindroos H. G., Winbland B., Wallace W. C. Alzheimer's disease-associated reduction of polysomal mRNA translation. Molecular Brain Research. 1989;5(4):259–269. doi: 10.1016/0169-328X(89)90060-0. [DOI] [PubMed] [Google Scholar]
- 3.Gamliel A., Teicher C., Michaelson D. M., et al. Increased expression of presenilin 2 inhibits protein synthesis. Molecular and Cellular Neuroscience. 2002;19(1):111–124. doi: 10.1006/mcne.2001.1068. [DOI] [PubMed] [Google Scholar]
- 4.Ding Q., Markesbery W. R., Chen Q., Li F., Keller J. N. Ribosome dysfunction is an early event in Alzheimer's disease. Journal of Neuroscience. 2005;25(40):9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang R. C. C., Wong A. K. Y., Ng H.-K., Hugon J. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimer's disease. NeuroReport. 2002;13(18):2429–2432. doi: 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- 6.Kim H.-S., Choi Y., Shin K.-Y., et al. Swedish amyloid precursor protein mutation increases phosphorylation of eIF2αin vitro and in vivo . Journal of Neuroscience Research. 2007;85(7):1527–1537. doi: 10.1002/jnr.21267. [DOI] [PubMed] [Google Scholar]
- 7.O'Connor T., Sadleir K. R., Maus E., et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60(6):988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mouton-Liger F., Paquet C., Dumurgier J., et al. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochimica et Biophysica Acta. 2012;1822(6):885–896. doi: 10.1016/j.bbadis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Harding H. P., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 10.Blais J. D., Filipenko V., Bi M., et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Molecular and Cellular Biology. 2004;24(17):7469–7482. doi: 10.1128/mcb.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa-Mattioli M., Sossin W. S., Klann E., Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61(1):10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klann E., Dever T. E. Biochemical mechanisms for translational regulation in synaptic plasticity. Nature Reviews Neuroscience. 2004;5(12):931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- 13.Costa-Mattioli M., Gobert D., Harding H., et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2α kinase GCN2. Nature. 2005;436(7054):1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma T., Trinh M. A., Wexler A. J., et al. Suppression of eIF2α kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nature Neuroscience. 2013;16(9):1299–1305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oakley H., Cole S. L., Logan S., et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. Journal of Neuroscience. 2006;26(40):10129–10140. doi: 10.1523/jneurosci.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheuner D., Song B., McEwen E., et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular Cell. 2001;7(6):1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 17.Devi L., Ohno M. Deletion of the eIF2α Kinase GCN2 fails to rescue the memory decline associated with Alzheimer's disease. PLoS ONE. 2013;8(10) doi: 10.1371/journal.pone.0077335.e77335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garthe A., Behr J., Kempermann G. Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005464.e5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balschun D., Wolfer D. P., Gass P., et al. Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? Journal of Neuroscience. 2003;23(15):6304–6314. doi: 10.1523/JNEUROSCI.23-15-06304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiergräber M., Henry M., Hescheler J., Smyth N., Schneider T. Electrocorticographic and deep intracerebral EEG recording in mice using a telemetry system. Brain Research Protocols. 2005;14(3):154–164. doi: 10.1016/j.brainresprot.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Huang D. W., Sherman B. T., Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 22.Huang D. W., Sherman B. T., Lempicki R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadleir K. R., Eimer W. A., Kaufman R. J., Osten P., Vassar R. Genetic inhibition of phosphorylation of the translation initiation factor eIF2α does not block Aβ-dependent elevation of BACE1 and APP levels or reduce amyloid pathology in a mouse model of alzheimer's disease. PLoS ONE. 2014;9(7) doi: 10.1371/journal.pone.0101643.e101643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prostko C. R., Brostrom M. A., Brostrom C. O. Reversible phosphorylation of eukaryotic initiation factor 2α in response to endoplasmic reticular signaling. Molecular and Cellular Biochemistry. 1993;127-128:255–265. doi: 10.1007/bf01076776. [DOI] [PubMed] [Google Scholar]
- 25.Boncristiano S., Calhoun M. E., Kelly P. H., et al. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. The Journal of Neuroscience. 2002;22(8):3234–3243. doi: 10.1523/JNEUROSCI.22-08-03234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellucci A., Luccarini I., Scali C., et al. Cholinergic dysfunction, neuronal damage and axonal loss in TgCRND8 mice. Neurobiology of Disease. 2006;23(2):260–272. doi: 10.1016/j.nbd.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 27.Perez S. E., Dar S., Ikonomovic M. D., DeKosky S. T., Mufson E. J. Cholinergic forebrain degeneration in the APPswe/PS1DeltaE9 transgenic mouse. Neurobiology of Disease. 2007;28(1):3–15. doi: 10.1016/j.nbd.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christensen D. Z., Bayer T. A., Wirths O. Intracellular Aβ triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer's disease. Neurobiology of Aging. 2010;31(7):1153–1163. doi: 10.1016/j.neurobiolaging.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 29.Devi L., Ohno M. Phospho-eIF2α level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS ONE. 2010;5(9) doi: 10.1371/journal.pone.0012974.e12974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whishaw I. Q., O'Connor W. T., Dunnett S. B. Disruption of central cholinergic systems in the rat by basal forebrain lesions or atropine: effects on feeding, sensorimotor behaviour, locomotor activity and spatial navigation. Behavioural Brain Research. 1985;17(2):103–115. doi: 10.1016/0166-4328(85)90023-3. [DOI] [PubMed] [Google Scholar]
- 31.Palop J. J., Mucke L. Epilepsy and cognitive impairments in alzheimer disease. Archives of Neurology. 2009;66(4):435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verret L., Mann E. O., Hang G. B., et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149(3):708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loring J. F., Wen X., Lee J. M., Seilhamer J., Somogyi R. A gene expression profile of Alzheimer's disease. DNA and Cell Biology. 2001;20(11):683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- 34.Colangelo V., Schurr J., Ball M. J., Pelaez R. P., Bazan N. G., Lukiw W. J. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. Journal of Neuroscience Research. 2002;70(3):462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 35.Blalock E. M., Geddes J. W., Chen K. C., Porter N. M., Markesbery W. R., Landfield P. W. Incipient Alzheimer's disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang W. S., Dunckley T., Beach T. G., et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiological Genomics. 2008;33(2):240–256. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devi L., Alldred M. J., Ginsberg S. D., Ohno M. Sex- and brain region-specific acceleration of β-amyloidogenesis following behavioral stress in a mouse model of Alzheimer's disease. Molecular Brain. 2010;3(1, article 34) doi: 10.1186/1756-6606-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holcomb L., Gordon M. N., Mcgowan E., et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nature Medicine. 1998;4(1):97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 39.Dodart J.-C., Meziane H., Mathis C., Ungerer A., Bales K. R., Paul S. M. Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behavioral Neuroscience. 1999;113(5):982–990. doi: 10.1037//0735-7044.113.5.982. [DOI] [PubMed] [Google Scholar]
- 40.Holcomb L. A., Gordon M. N., Jantzen P., Hsiao K., Duff K., Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behavior Genetics. 1999;29(3):177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 41.Filali M., Lalonde R., Rivest S. Anomalies in social behaviors and exploratory activities in an APPswe/PS1 mouse model of Alzheimer's disease. Physiology & Behavior. 2011;104(5):880–885. doi: 10.1016/j.physbeh.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 42.White H. K., McConnell E. S., Bales C. W., Kuchibhatla M. A 6-month observational study of the relationship between weight loss and behavioral symptoms in institutionalized Alzheimer's disease subjects. Journal of the American Medical Directors Association. 2004;5(2):89–97. doi: 10.1016/S1525-8610(04)70061-4. [DOI] [PubMed] [Google Scholar]
- 43.Whitehouse P. J., Price D. L., Struble R. G., Clark A. W., Coyle J. T., Delon M. R. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 44.Vogels O. J. M., Broere C. A. J., ter Laak H. J., ten Donkelaar H. J., Nieuwenhuys R., Schulte B. P. M. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer's disease. Neurobiology of Aging. 1990;11(1):3–13. doi: 10.1016/0197-4580(90)90056-6. [DOI] [PubMed] [Google Scholar]
- 45.Jope R. S., Song L., Powers R. E. Cholinergic activation of phosphoinositide signaling is impaired in Alzheimer's disease brain. Neurobiology of Aging. 1997;18(1):111–120. doi: 10.1016/S0197-4580(96)00205-9. [DOI] [PubMed] [Google Scholar]
- 46.Cullen K. M., Halliday G. M. Neurofibrillary degeneration and cell loss in the nucleus basalis in comparison to cortical Alzheimer pathology. Neurobiology of Aging. 1998;19(4):297–306. doi: 10.1016/S0197-4580(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 47.Palmer A. M., Francis P. T., Benton J. S., et al. Presynaptic serotonergic dysfunction in patients with Alzheimer's disease. Journal of Neurochemistry. 1987;48(1):8–15. doi: 10.1111/j.1471-4159.1987.tb13120.x. [DOI] [PubMed] [Google Scholar]
- 48.Gottfries C. G. Disturbance of the 5-hydroxytryptamine metabolism in brains from patients with Alzheimer's dementia. Journal of Neural Transmission, Supplement. 1990;30:33–43. doi: 10.1007/978-3-7091-3345-3_4. [DOI] [PubMed] [Google Scholar]
- 49.Nazarali A. J., Reynolds G. P. Monoamine neurotransmitters and their metabolites in brain regions in Alzheimer's disease: a postmortem study. Cellular and Molecular Neurobiology. 1992;12(6):581–587. doi: 10.1007/bf00711237. [DOI] [PubMed] [Google Scholar]
- 50.Sparks D. L., Hunsaker J. C., III, Slevin J. T., DeKosky S. T., Kryscio R. J., Markesbery W. R. Monoaminergic and cholinergic synaptic markers in the nucleus basalis of Meynert (nbM): normal age-related changes and the effect of heart disease and Alzheimer's disease. Annals of Neurology. 1992;31(6):611–620. doi: 10.1002/ana.410310608. [DOI] [PubMed] [Google Scholar]
- 51.Garcia-Alloza M., Hirst W. D., Chen C. P. L.-H., Lasheras B., Francis P. T., Ramírez M. J. Differential involvement of 5-HT(1B/1D) and 5-HT6 receptors in cognitive and non-cognitive symptoms in Alzheimer's disease. Neuropsychopharmacology. 2004;29(2):410–416. doi: 10.1038/sj.npp.1300330. [DOI] [PubMed] [Google Scholar]
- 52.Cross A. J. Serotonin in Alzheimer-type dementia and other dementing illnesses. Annals of the New York Academy of Sciences. 1990;600:405–417. doi: 10.1111/j.1749-6632.1990.tb16897.x. [DOI] [PubMed] [Google Scholar]
- 53.Lai M. K. P., Tsang S. W. Y., Francis P. T., et al. Postmortem serotoninergic correlates of cognitive decline in Alzheimer's disease. NeuroReport. 2002;13(9):1175–1178. doi: 10.1097/00001756-200207020-00021. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Alloza M., Gil-Bea F. J., Diez-Ariza M., et al. Cholinergic-serotonergic imbalance contributes to cognitive and behavioral symptoms in Alzheimer's disease. Neuropsychologia. 2005;43(3):442–449. doi: 10.1016/j.neuropsychologia.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 55.Mintzer J., Brawman-Mintzer O., Mirski D. F., et al. Fenfluramine challenge test as a marker of serotonin activity in patients with Alzheimer's dementia and agitation. Biological Psychiatry. 1998;44(9):918–921. doi: 10.1016/s0006-3223(98)00004-3. [DOI] [PubMed] [Google Scholar]
- 56.Adriani W., Ognibene E., Heuland E., Ghirardi O., Caprioli A., Laviola G. Motor impulsivity in APP-SWE mice: a model of Alzheimer's disease. Behavioural Pharmacology. 2006;17(5-6):525–533. doi: 10.1097/00008877-200609000-00019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Enhanced phosphorylation of eIF2α in SHSY5Y cells incubated with DTT. Shown are p-eIF2α and eIF2α immunoblots performed on lysates of SHSY5Y cells that had been treated with vehicle or 2mM DTT for 15 or 60 min (n = 2 samples per condition). Densitometric quantification revealed the expected increase in eIF2α phosphorylation in SHSY5Y cells incubated with DTT. P-values refer to results of posthoc Tukey tests.
Supplementary Figure 2. The cholinergic system was not involved in the eIF2α+/S51A-related restoration of hyperactivity in 5xFAD mice. (A) Shown are micrographs of ChAT-immunoreactive neurons in the basal forebrain of wild-type, 5xFAD, 5xFAD;eIF2α+/S51A and eIF2α+/S51A mice. Scale bar = 150 μm. Stereological quantification of ChAT-immunoreactive neurons in the MS and VDB of the basal forebrain demonstrated no significant differences between the four groups of mice (n = 3 mice per group). (B) Shown are immunoblots of ChAT from hippocampal homogenates of wild-type, 5xFAD, 5xFAD;eIF2α+/S51A and eIF2α+/S51A mice (n = 5-7 mice per group). Data were analyzed using two-way ANOVAs with the between-subjects factors 5xFAD genotype and eIF2α genotype. Statistically significant differences (p < 0.05) are denoted by bold font. Bar graphs show mean ± SEM.
Supplementary Figure 3. The eIF2αS51A allele had limited effects on transcriptional dysregulation in 5xFAD mice. Differentially regulated transcripts (Benjamini Hochberg FDR, p < 0.02) with a fold change higher than 1.6 were subjected to hierarchical clustering to visualize gene expression changes between groups (n = 3 mice per genotype).
Supplementary Table 1. A gene ontology analysis (DAVID) revealed a significant enrichment of immune-related transcripts in gene set differentially expressed in 5xFAD hippocampus.
Supplementary Table 2. Ingenuity pathway analysis also indicated a substantial enrichment of genes related to immune and inflammatory processes in the gene set differentially expressed in 5xFAD hippocampus.