

Abstract
Genetic inactivation of the nuclear-encoded mitochondrial heart-muscle adenine nucleotide translocator-1 (ANT1), which exports mitochondrial ATP to the cytosol in both humans (ANT1−/−) and mice (Ant1−/−), results in lactic acidosis and mitochondrial cardiomyopathy and myopathy, the latter involving hyper-proliferation of mitochondria, induction of oxidative phosphorylation (OXPHOS) enzymes, increased reactive oxygen species (ROS), and excessive mtDNA damage. To understand these manifestations, we analyzed Ant1−/− mouse skeletal muscle for changes in gene expression using our custom 644 and 1087 gene MITOCHIP microarrays and for changes in the protein levels of key mitochondrial transcription factors. Thirty-four mRNAs were found to be up-regulated and 29 mRNAs down-regulated. Up-regulated mRNAs included the mitochondrial DNA (mtDNA) polypeptide and rRNA genes, selected nuclear-encoded OXPHOS genes, and stress-response genes including Mcl-1. Down-regulated mRNAs included glycolytic genes, pro-apoptotic genes, and c-Myc. The mitochondrial regulatory proteins Pgc-1α, Nrf-1, Tfam, and myogenin were up-regulated and could account for the induction of the OXPHOS and antioxidant enzymes. By contrast, c-Myc levels were reduced and might account for a reduction in apoptotic potential. Therefore, the Ant1−/− mouse skeletal muscle demonstrates that energy metabolism, antioxidant defenses, and apoptosis form an integrated metabolic network.
INTRODUCTION
Mitochondria play an important role in human health and disease as they regulate cellular energy production, reactive oxygen species (ROS) production, and apoptosis [1]. Mitochondria generate most of the cell’s energy by the process of OXPHOS. During OXPHOS, electrons are passed down the electron transport chain (ETC), encompassing respiratory complexes I–IV. As electrons traverse complexes I (NADH dehydrogenase), III, and IV (cytochrome c oxidase), protons are pumped out across the mitochondrial inner membrane creating an electrochemical gradient. The resulting combination membrane potential and pH gradient, the proton motor force, is then be used by the inner membrane proton-transporting ATP synthase (complex V) as a source of potential energy to drive the phosphorylation of ADP to ATP within the mitochondrial matrix. Matrix ATP is then exported out of the mitochondrion, in exchange for cytosolic ADP, by the adenine nucleotide translocators (ANT).
Humans have four ANT isoforms that are encoded by four different genes and are distributed in different tissue specific patterns. The human ANT1 gene is primarily expressed in the heart and in skeletal muscle [2]. Human ANT2 is weakly expressed or absent from most tissues, while human ANT3 is ubiquitously expressed [3]. The ANT1 and ANT3 isoforms have been proposed to export ATP from the mitochondrial matrix to the cytosol, while ANT2 has been proposed to be induced when mitochondrial energy production is impaired so that cytosolic ATP can be imported into the mitochondrion to sustain mitochondrial biogenesis and to maintain the membrane potential [4–6]. The fourth human ANT gene, ANT4, is expressed at highest levels in the testes and at lower levels in other organs including liver and brain [7]. Certain missense mutations in ANT1 have been shown to cause autosomal dominant progressive external opthalmoplegia (AdPEO) [8, 9], while null mutations in ANT1 cause autosomal recessive mitochondrial myopathy and cardiomyopathy [10].
Mice have three Ant isoforms encoded by three different genes. Ant1 is expressed primarily in the heart and skeletal muscle, like its human homolog, while Ant2 is expressed in virtually all tissues, but at very low levels in skeletal muscle [11, 12]. The murine equivalent to human ANT4, Ant4, has a highly restricted expression pattern, with mRNA only being detected in testes and germ cells [13, 14].
Mice in which the Ant1 gene has been insertionally inactivated develop an autosomal recessive (Ant1−/−) mitochondrial myopathy and cardiomyopathy [11] that is essentially identical to that observed in humans inheriting a homozygous null ANT1 mutation [10]. Systemic Ant1-deficient mice (Ant1−/−) are exercise intolerant in association with ragged red muscle fibers, massive proliferation of abnormal mitochondria, and marked elevation in muscle OXPHOS enzyme histochemistry. These mice also manifest metabolic abnormalities including elevated resting serum lactic acid and alanine and develop a progressive hypertropic cardiomyopathy [11]. The Ant1−/− mouse muscle and heart mitochondria also produce elevated levels of H2O2, exhibit evidence of chronic oxidative stress, and have increased numbers of mitochondrial DNA (mtDNA) rearrangements [15].
In addition to being the ATP/ADP translocase, the ANTs may be important regulators of the mitochondrial permeability transition pore (mtPTP) which in turn regulates apoptosis [16, 17]. While inactivation of the Ant1 and Ant2 genes in mouse liver markedly decreased mtPTP calcium activation and rendered the mtPTP insensitive to modulation by adenine nucleotide levels or atractyloside, loss of Ant1 and Ant2 did not eliminate the ability of the liver mitochondria to undergo permeability transition [18]. Proteomic analysis of the mouse liver mitochondrial inner membrane revealed peptides for Ant2, Ant1, and a third Ant, presumably Ant4, in the ratio of approximately 6:3:1 [19]. However, western blot analysis did not detect Ant4 mRNA in liver [13]. Therefore, Ant1 and/or Ant2 predominate in liver mitochondria and are important in regulating the liver mtPTP. Since over-expression of the human ANT1 or ANT3, but not ANT2, genes have been shown to induce apoptosis in cultured human cells [20, 21], we can conclude that Ant1 (ANT1) regulates the mtPTP and thus apoptosis in both human and mouse.
The hyper-proliferation of the mitochondria in the skeletal muscle of the Ant1−/− mice has been correlated with an up-regulation of OXPHOS gene expression by differential display reverse transcriptase-polymerase chain reaction (RT-PCR) analysis. This method identified 17 over-expressed genes in the mutant tissue Ant1−/− animals including components of the ETC as well as the muscle anti-apoptotic gene Mcl-1 [22].
To obtain a more detailed picture of the effect of Ant1 deficiency on muscle physiology, we compared the gene expression profiles in Ant1−/− skeletal muscle with those in wild-type muscle using our custom cDNA microarrays. These MITOCHIPs contain either 644 or 1087 genes relevant to energy metabolism, mitochondrial function, oxidative stress, and apoptosis. We found that in Ant1−/− skeletal muscle 34 genes were up-regulated including a number of OXPHOS genes while 29 genes were down-regulated including genes for glycolysis. This was associated with an increase in the protein levels of a number of transcriptions factors associated with induction of muscle oxidative metabolism including peroxisome proliferator-activated receptor γ (PPARγ)-coactivator-1α (PGC-1α, human; Pgc-1α, mouse), nuclear regulatory factor-1 (Nrf-1), mitochondrial transcription factor A (Tfam), and myogenin. The cellular Myelocytomatosis proto-oncogene gene and protein (c-Myc) were down-regulated, which may have reduced the potential for induction of apoptosis. These results imply that in Ant1−/− mouse skeletal muscle, experience energy starvation, oxidative stress, and apoptotic risk, and attempt to compensate through induction of OXPHOS and antioxidant defenses and the down-regulation of apopotosis.
MATERIALS AND METHODS
Assembly of MITOCHIP arrays
A majority of the genes represented on the MITOCHIP arrays were selected and obtained from the NIA 15K mouse cDNA clone set [23]. The complete gene lists for MITOCHIP v3 and v4 are provided in Supplemental Table I. In addition, the arrays also contained the differential display products isolated by Murdock et al [22] as well as all 13 protein-coding mtDNA genes and the 2 rRNA mtDNA genes that were amplified in-house. Other than the mtDNA genes, all the remaining cDNAs were amplified using the primers and PCR regimen described in NIA15K [23]. After amplification, the PCR products were purified using 96-well Qiagen PCR purification kits, and the dsDNA concentration was determined by fluorometry using the PicoGreen reagent (Invitrogen). The eluted pure PCR products were dried and reconstituted in 50% DMSO to a final DNA concentration of 75–100 ng/μl, and were then used for printing arrays. Arrays were printed using a GMS-412 arrayer, on poly-l-lysine coated glass slides. Poly-l-lysine coating, treatment and post-processing of slides were performed essentially as described by DeRisi et al [24], except that the succinic anhydride blocking step was omitted. The slides were prehybridized in a solution of 25% formamide, 5X SSC, 0.1% SDS and 1% BSA at 45°C for 45–60 min just before use, rinsed in water, dried by centrifugation at 500 rpm for 5 min and then used for hybridization.
Preparation of RNA
Total RNA was isolated from gastrocnemius muscles of wild type and mutant animals using Trizol reagent (Invitrogen) in accordance with the manufacturer’s protocol. The total RNA thus isolated was then treated with RNase-free DNase I (Roche) for 30′ at room temperature to rid the sample of any contaminating DNA. After DNase I treatment, the RNA was purified using RNeasy kits (Qiagen), quantitated by spectrophotometry and used for labeling.
Hybridization of MITOCHIP arrays
25–30 μg of total RNA were labeled with either Cy3-dCTP or Cy5-dCTP by reverse transcription for 2 hrs at 42°C, in a mix containing 1X Superscript II Buffer (Invitrogen), 10mM DTT, 3 μg Oligo-dT, 200μM each of dATP, dGTP and dTTP, 100μM dCTP and 100μM Cy3-dCTP or Cy-5 dCTP (Amersham) and 400 units Superscript II (Invitrogen), with the final reaction volume being 35 μl. After reverse transcription the labeled cDNA was rid of RNA denaturation with 17.5 μl of 1.0 N NaOH at 70°C for 10′, neutralized with 17.5 μl 1.0 N HCl, and purified using PCR purification columns (Qiagen). Equal amounts (picomoles of incorporated labeled nucleotide) of wild-type and mutant labeled cDNA were mixed, dried under vacuum, resuspended in 22 μl of hybridization buffer containing 25% formamide, 5X SSC, 0.1%SDS, 1 μg polyA RNA (Sigma) and 1μg mouse Cot1 DNA (Invitrogen) and applied to the arrays. The arrays were incubated for 12–15 hrs at 55°C, and then washed three times at 55°C with 2X SSC+0.1% SDS, three times with 1X SSC+0.1% SDS at room temperature, and three times with 0.1X SSC at room temperature. The incubations and washes were carried out in a GeneMachines HybStation (Genomic Solutions). After the final wash the slides were dried by rinsing with isopropanol followed by centrifugation at 500 rpm for 5 min and scanned immediately.
Scanning and processing of data
The arrays were scanned in an Affymetrix scanner and quantitated using the Imagene 4.1 software suite (BioDiscovery). The data was then exported into the GeneSight Lite module (BioDiscovery) and normalized such that the sum of all ratios was equal to one. The normalized data were then exported into Microsoft Excel where the lists were sorted to exclude spots that were flagged and were below background intensity in each channel. The ratios were then converted to Log2 to provide a symmetrical distribution around one for positive and negative values and average + or − changes of greater than 1.5 obtained for both versions of the MITOCHIP or 1.7 for one version of the MITOCHIP were reported.
Western Blots
Total protein was isolated from gastrocnemius muscles of 9–12 month old wild-type and Ant1−/− mice. One gastrocnemius muscle was homogenized in 2 ml of a buffer containing 50 mM Tris pH 7.2, 100 mg/ml Sucrose, 0.1% SDS and 1 tablet/50 ml Complete Protease Inhibitor cocktail (Roche). The homogenate was spun at 1000 g for 10 min and the supernatant was carefully aspirated. Protein concentrations were determined using Bradford’s reagent. μg of each protein sample were denatured in 1X (final conc) NuPage sample loading buffer (Invitrogen), run on 10% or 12% NuPage gels (Invitrogen), transferred to nitrocellulose membranes, stained with Ponceau S to ensure uniform loading and rinsed in 1X wash buffer (KPL). The blots were then blocked with 5% milk in 1X wash buffer for 1 hour, and incubated with primary antibody in 2% milk at 8–10°C overnight. The primary antibody was washed off with 5–8 washes over 30 min with 1X wash buffer, and the blots were incubated for 2 hours in 1X wash buffer containing the respective secondary antibody. The blots were washed 5–8 times over 30 min in 1X wash buffer, and developed using the ECL kit (Amersham) according to manufacturer’s protocols.
Antibodies and concentrations
Horse radish peroxidase (HRP)-conjugated secondary antibodies goat anti-mouse (GAM), goat anti-rabbit (GAR) and rabbit anti-goat (RAG) were obtained from Santa Cruz Biotechnology. Primary antibodies and concentrations used to detect the various proteins were as follows: PGC-1α detected using PGC-1(P-19) or PGC-1(K-15) primary antibody (Santa Cruz Biotechnology sc-5815 and sc-5816 respectively) at 1:500 dilution, with RAG secondary at 1:5000; Nrf1 detected using anti-NRF1 serum (kindly provided by Dr. R.C. Scarpulla) at 1:5000 dilution with GAR secondary at 1:5000; Tfam detected using anti-Tfam polyclonal serum (kindly provided by Dr. Gerald Shadel) at 1:10,000 with GAR secondary at 1:5000; mt-COI detected using monoclonal antibody 1D6 (Molecular Probes) at 100 ng/ml with GAM secondary at 1:10,000; myogenin detected using M-225 primary antibody (Santa Cruz Biotechnology sc-576) at 1:500 with GAR secondary at 1:5000; Myoglobin detected using M-19 primary antibody (Santa Cruz Biotechnology sc-8081) at 1:500 with RAG secondary at 1:5000; TnI-slow detected using TnI SS C-19 primary (Santa Cruz Biotechnology sc-8119) at 1:1000 with RAG secondary at 1:5000; TnI-fast detected using TnI FS C-19 primary (Santa Cruz Biotechnology sc-8120) at 1:1000 and RAG secondary at 1:5000; Myc was detected using monoclonal anti-c-myc 9E10 (Sigma M5546) at 1:5000 with GAM secondary at 1:5000.
RESULTS
Gene expression differences between skeletal muscles from Ant1−/− and wild type mice were studied using two versions of our mouse MITOCHIP, MITOCHIP vM3 containing 772 elements (representing 644 genes) and MITOCHIP vM4 containing 1152 elements (representing 1089 genes). Four experiments were performed with each version of the MITOCHIP. In each experiment, a pool of skeletal muscle RNA from at least 3 Ant1−/− mice was compared to a pool of skeletal muscle RNA from at least 3 wild-type mice. In all, tissues from 11 different mutant animals were compared with tissues from 11 different age-matched wild-type animals over the 8 experiments.
The differential fold-expression values for each gene from the four experiments using MITOCHIPv3 (Set 1) were averaged, as were those for the four experiments using MITOCHIP v4 (Set 2). Genes showing average expression differences of at least 1.5-fold in both Set 1 and Set 2 were considered to be significant. In instances where the gene was not represented in Set 1, an average differential expression of at least 1.7-fold in the Set 2 experiments was considered significant.
Genes up-regulated in Ant1−/− skeletal muscle
In all, 34 different mRNAs were found to be over-expressed in Ant1−/− skeletal muscle relative to wild type (Table I). These genes can be classified broadly into the following functional categories: (i) Genes encoding components of the mitochondrial OXPHOS machinery; (ii) Genes for other mitochondrial components; (iii) Genes whose products are enriched in slow-twitch (oxidative) muscle fibers; (iv) Genes for stress response, and (v) Miscellaneous genes. As can be seen from Table I, a number of the genes identified as up-regulated using the MITOCHIPs were also previously shown to be up-regulated by differential and northern analysis of mRNA from Ant1−/− skeletal muscle (18).
Table I.
Genes over-expressed in Ant1−/− skeletal muscle. NP = Not present in that version of the MITOCHIP.
| GENE | SET 1 | SET 2 |
|---|---|---|
| mtDNA OXPHOS subunits | ||
| mtDNA-ATPase 6 | 2.2 | 2.9 |
| mtDNA-ATPase 8 | 3.3 | 3.1 |
| mtDNA-COX 2* | 1.5 | 1.6 |
| mtDNA-COX 3* | 2.9 | 2.9 |
| mtDNA-Cyt b | 3.1 | 3.9 |
| mtDNA-NADH 1* | 1.8 | 2.6 |
| mtDNA-NADH 2* | 2.6 | 2.3 |
| mtDNA-NADH 3 | 2.5 | 1.6 |
| mtDNA-NADH 4* | 2.4 | 2.2 |
| mtDNA-NADH 4L* | 2.1 | 3.3 |
| mtDNA-NADH 5* | 1.9 | 2.9 |
| mtDNA-NADH 6* | 2.2 | 3.0 |
| DD69 (mtDNA_ND5)* | 2.2 | 1.7 |
| Nuclear OXPHOS subunits | ||
| NADH dehydrogenase (ubiquinone) Fe-S protein 4, 18kDa (NADH-coenzyme Q reductase) (Ndufs4)* | 1.9 | 1.9 |
| NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 5 (16kD) (Ndufb5) | 1.6 | 1.7 |
| NADH dehydrogenase (ubiquinone) 1, subcomplex unknown, 2 (14.5kD) (Ndufc2) | 1.6 | 1.5 |
| Cytochrome c oxidase, subunit VIIa 2 (Cox7a2) | 1.5 | 1.8 |
| Other Mitochondrial Components | ||
| Solute carrier family 40 (iron-regulated transporter), member 1 (Slc40a1) | 2.1 | 1.5 |
| Translocase of inner mitochondrial membrane 8 homolog b (Timm8b) | 1.6 | 1.7 |
| Mitochondrial Import Inner Membrane Translocase subunit TIM8 A (Deafness Dystonia Protein Homolog) | NP | 1.6 |
| mtDNA-12s rRNA | 3.6 | 3.5 |
| mtDNA-16s rRNA | 2.5 | 2.5 |
| Mitochondrial ribosomal protein L48 (Mrpl48)* | 2.7 | 1.7 |
| Inner membrane protein, mitochondrial (Immt, mitofilin) | 2.4 | 1.6 |
| Malate dehydrogenase, mitochondrial | 1.8 | 1.6 |
| Gene products enriched in Oxidative Muscle Fibers | ||
| Skeletal Muscle LIM protein (Fhl1) | 1.8 | 1.6 |
| ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 (Atp2a2) | 1.5 | 1.5 |
| Mus musculus ATPase, Na+/K+ transporting, beta 1 polypeptide (Atp1b1) | 1.5 | 1.6 |
| Mus musculus malate dehydrogenase, soluble (Mor2)* | 1.5 | 1.9 |
| Stress response | ||
| Microsomal glutathione S-transferase 1 (Mgst1) | 1.6 | 1.5 |
| Antigenic determinant of RecA protein (Kin 17)* | NP | 2.8 |
| Myeloid cell leukemia sequence 1 (Mcl1)* | 1.5 | 1.5 |
| Miscellaneous | ||
| Nrbf1 | NP | 2.7 |
| Ornithine decarboxylase antizyme 3 (Oaz3) | 1.7 | 2.7 |
| Homolog of Unc33/Collaspin reponse mediated protein 2 (Crmp2) | 3.1 | 3.5 |
Also identified as up-regulated by Murdock et al [22]
(i) OXPHOS Components
Sixteen of the 34 up-regulated genes encode subunits of the mitochondrial OXPHOS. Twelve of the 13 mRNAs encoded by the mtDNA were found to be strongly up-regulated in Ant1−/− muscle. Of the nuclear OXPHOS subunit mRNAs that are up-regulated, 3 belonged to complex I (Ndufs4, Ndufb5 and Ndufc2) while one belonged to complex IV (Cox7a2). Interestingly, Ndufs4 has been shown to be essential for the assembly of Complex I in humans [25], while Cox7a2 has been shown to be essential for assembly of Complex IV in yeast [26] and Dictyostelium [27]. The induction of mtDNA and nDNA OXPHOS proteins is consistent with the hyper-proliferation of mitochondrial and the increased complex II and IV staining in the Ant1−/− skeletal muscle [11, 15, 22].
One transcript encoded by the mtDNA, cytochrome c oxidase I (mt-CO1), yielded very poor signal on our arrays, though the reason for this is unclear. Since the mitochondrial DNA H-strand is transcribed as a polycistronic unit [28], it is unlikely that transcriptional differences can account for this discrepancy. However, certain mtDNA transcripts can have short half lifes [29] and thus the mt-CO1 mRNAs may have a differential stability relative to other mtDNA-encoded mRNAs. Indeed a previous study found that mt-CO1 mRNA levels were disproportionately regulated relative to other mtDNA and nDNA complex IV subunits in neurons [30].
To clarify the mt-CO1 mRNA levels, we used Real-Time PCR and found that the mt-COI mRNA was increased approximately 1.6-fold in Ant1−/− skeletal muscle (not shown). The levels of COI protein were also increased in Ant1−/− skeletal muscle (Fig. 1D). Hence, all of the mtDNA encoded OXPHOS genes were up-regulated in the Ant1−/− muscle.
Figure 1.
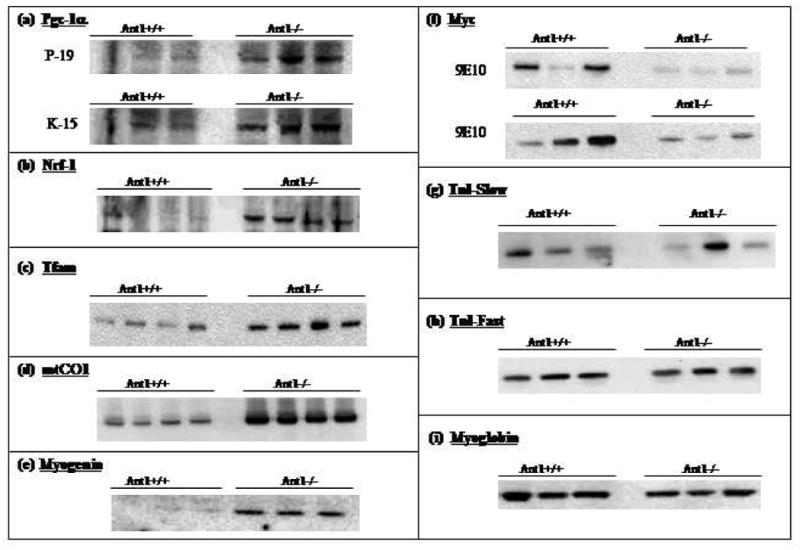
Proteins that regulate mitochondrial proliferation are over-expressed in Ant 1−/− skeletal muscle. Western blots showing levels in Ant1+/+ and Ant1−/− skeletal muscle of the following proteins: (a) Pgc-1α detected using antibody P-19 (amino terminus-specific) and K-15 (carboxy terminus-specific), (b) Nrf-1, (c) Tfam, (d) mtCO1, (e) Myogenin, (f) Myc (two different blots, both probed with antibody 9E10, (g) Tnni1, (h) Tnni2, and (i) Myoglobin.
(ii) Other Mitochondrial Components
mRNAs for three mitochondrial transporters were also up-regulated in Ant1−/− skeletal muscle. Slc40a1 (also called ferroportin) encodes a regulated iron transporter that appears to function mainly in the export of iron. Ferroportin over-expression has been shown to cause iron depletion in cultured cells [31] and mutations in ferroportin have been shown to cause diseases of iron overload in humans [32].
Timm8a and Timm8b encode components of the mitochondrial protein import machinery, essential for mitochondrial biogenesis. Mutations in Timm8a have been shown to cause deafness-dystonia-optic atrophy (Mohr-Tranebjaerg) syndrome in humans [33].
Five other mitochondrial components were over-represented in Ant1−/− skeletal muscle: three components of the mitochondrial ribosome [mtDNA 12S-rRNA (mt-rnr1), mtDNA 16S-rRNA (mt-rnr2) and mitochondrial ribosomal protein L52 (Mrpl48)], the TCA cycle enzyme malate dehydrogenase (Mor2), and mitofilin. Mitofilin is a mitochondrial inner membrane protein important in cristae structure, whose reduction leads to the collapse of the cristae network, increased membrane potential, and ROS production [34]. Mitofilin is also induced in brown adipose tissue when exposed to cold [35].
(iii) Oxidative Muscle Fiber-specific genes
The strong up-regulation of mitochondrial transcripts in Ant1−/− skeletal muscle indicates that the mutant muscle is attempting to compensate for the energy starvation by inducing OXPHOS. This notion is supported by the fact that the products of several over-expressed genes have also been shown to be enriched in slow-twitch or oxidative (Type I) muscle fibers. The skeletal muscle LIM domain protein (Fhl1), and cytoplasmic malate dehydrogenase (Mor2) have been shown to be over-expressed in soleus (an almost-exclusively oxidative, slow-twitch, muscle type) relative to the white quadriceps muscle (an almost exclusively glycolytic, fast-twitch, muscle type) [36]. Further, the Ca++ transporting ATPase subunit (Atp2a2) as well as the Na+/K+ transporting ATPase subunit (ATP1b1) have been shown to be enriched in oxidative, slow twitch, muscle fibers [37–39] relative to glycolytic, fast-twitch, muscle fibers.
(iv) Stress Response Genes
mRNAs encoding three different stress-response proteins were found to be over-expressed in Ant1−/− skeletal muscle. Of these, microsomal glutathione S-transferase mu (Mgst1) is a membrane bound protein that has been shown to have gluthione transferase and glutathione peroxidase activities [40], and to protect cells from oxidative stress [41, 42]. The other two stress response genes up-regulated in Ant1−/− skeletal muscle are Kin 17 and Mcl-1. The Kin 17 protein is induced in response to UV and ionizing radiation [43, 44] and has been shown to bind to curved DNA spanning stretches of illegitimate recombination [45]. Mcl-1 is an anti-apoptotic protein of the Bcl-2 family [46] and protects cells from apoptosis under a variety of conditions [47]. Mcl-1 has also been shown to be induced in response to oxidative stress [48].
(v) Miscellaneous
The three remaining genes that are up-regulated in Ant1−/− skeletal muscle (Nrbf1, Oaz3 and Crmp2) do not fit into any specific functional category but are interesting nevertheless. Nrbf1 was identified in a two-hybrid screen as a PPAR-α interacting protein. Nrbf1 was also shown to be able to interact with a variety of nuclear hormone receptors such as thyroid hormone receptor β (TRβ), retinoid acid receptor α (RARα), retinoid X receptor α (RXRα), and hepatocyte nuclear factor 4 (HNF4). Nrbf1 shares homology with the yeast protein MRF-1, a putative transcription factor regulating the expression of mitochondrial respiratory proteins [49]. Crmp2 (Collapsin mediated response protein 2) belongs to a family of molecules that are involved in axonal growth cone guidance [50], while Oaz3 encodes an ornithine decarboxylase antizyme [51].
Genes Down-regulated in Ant1−/− skeletal muscle
In all, mRNAs from 29 different genes were found to be down-regulated in Ant1−/− skeletal muscle relative to wild type (Table II). These genes encompass a variety of functions including metabolic enzymes, pro-apoptotic genes, signaling molecules, transporters, and others. Two classes of these genes (metabolic enzymes and apoptotic genes) are particularly interesting in the context of this study and will be discussed in more detail below.
Table II.
Genes under-expressed in Ant1−/− skeletal muscle. NP = Not present on that version of the MITOCHIP.
| GENE | SET 1 | SET 2 |
|---|---|---|
| Metabolic enzymes | ||
| Inducible 6-phosphofructo-2-kinase (Pfkfb3) | NP | −3.0 |
| Aldolase B | −8.2 | −2.7 |
| Protoporphyrinogen oxidase (Ppox) | −2.0 | −1.5 |
| Cytochrome P450 side chain cleavage enzyme 11a1 (Cyp11a1) | NP | −1.9 |
| Ornithine decarboxylase antizyme 2 (Oaz2) | −1.6 | −1.6 |
| Apoptosis effectors | ||
| Modulator of Apoptosis (Moap1) | NP | −1.8 |
| Bcl2-interacting killer-like (Biklk), mRNA | NP | −1.8 |
| Myelocytomatosis oncogene (Myc) | NP | −1.9 |
| RAD53 homolog (Chek2) | NP | −1.8 |
| Phospholipase A2, group VI (Pla2g6) | NP | −2.4 |
| Myc target genes | ||
| N-myc downstream regulated 1 (Ndr1) | NP | −2.2 |
| Ornithine decarboxylase, structural (Odc) | −3.2 | −2.3 |
| Transporters | ||
| Bumetanide-sensitive Sodium-(Potassium)-Chloride Cotransporter 1 (Slc12a2) | −1.6 | −1.5 |
| Solute carrier family 30 (zinc transporter), member 3 (Slc30a3) | −1.6 | −1.6 |
| Adenine nucleotide translocase-2 (Ant2) | −3.4 | −3.4 |
| Kinases | ||
| LIM motif-containing protein kinase 2 (Limk2) | NP | −1.7 |
| Casein kinase I alpha isoform (CSNK1A1) | NP | −1.9 |
| Casein kinase 1, epsilon (Csnk1e) | NP | −1.9 |
| Homeodomain interacting protein kinase 3 (Hipk3) | −2.0 | −1.7 |
| Mitogen activated protein kinase kinase kinase 12 (Map3k12) | NP | −1.9 |
| Rho-associated coiled-coil forming kinase 1 (Rock1) | NP | −1.7 |
| Signalling | ||
| Protein tyrosine phosphatase, non-receptor type 21 (Ptpn21) | NP | −2.0 |
| Mothers against Dpp homolog 1 (Madh1) | NP | −1.7 |
| Signal transducer and activator of transcription interacting protein 1 (Statip1) | NP | −1.7 |
| Activin receptor IIB (Acvr2b) | NP | −1.7 |
| Heat Shock Proteins | ||
| Crystallin, alpha C (Hsp 20) | NP | −1.8 |
| Heat shock protein, 86 kDa 1 (Hsp86-1) | NP | −1.7 |
| Other | ||
| Ubiquinol-cytochrome c reductase core protein 1 (Uqcrc1) | −1.6 | −1.6 |
| Microtubule-associated protein 4 (Mtap4) | NP | −1.9 |
NP: Not present on the array.
(i) Metabolic enzymes
Six metabolic enzyme genes were down-regulated in Ant1−/− skeletal muscle relative to wild type (Table II). Two of these are key glycolytic enzymes and thus genes, 6-phosphofructo 2-kinase (Pfkfb3) and Aldolase B (Aldob). Like mitochondrial proliferation, the down-regulation of these glycolytic genes may be indicative of the shift of the skeletal muscle in Ant1−/− animals to a more oxidative physiology. To explore this possibility further, we tabulated the relative expression levels of all the genes involved in glucose metabolism across our eight MITOCHIP experiments (Table III).
Table III.
Expression of glucose metabolism genes in Ant1−/− skeletal muscle.
| GENE | Mean Fold Expression |
|---|---|
| Glycolysis and Pyruvate Metabolism | |
| Mus musculus phosphofructokinase, muscle (Pfkm) | −1.1 |
| Mus musculus inducible 6-phosphofructo-2-kinase (Pfkfb3) | −2.9 |
| Mus musculus aldolase 1, A isoform (Aldoa) | −1.6 |
| Aldolase B (Aldob) | −3.4 |
| Mus musculus triosephosphate isomerase (Tpi1) | −1.5 |
| GAPDH | −1.3 |
| Mus musculus phosphoglycerate mutase 1 (Pgam1) | 1.0 |
| 3-Phosphoglycerate dehydrogenase | −1.3 |
| Mus musculus enolase 1, alpha non-neuron (Eno1) | −1.4 |
| Mus musculus pyruvate kinase 3 (Pk3) | −1.3 |
| Mus musculus lactate dehydrogenase 1, A chain (Ldh1) | −1.5 |
| Alanine aminotransferase | 1.2 |
| Alanine aminotransferase 2 (muscle specific) | −1.4 |
| Pentose Phosphate Pathway | |
| Mus musculus glucose-6-phosphate dehydrogenase X-linked (G6pdx) | 1.5 |
| Tricarboxylic acid cycle | |
| Isocitrate dehydrogenase 2 (NADP+), mitochondrial (Idh2) | 1.2 |
| Isocitrate dehydrogenase 3 (NAD+) alpha (Idh3a) | 1.2 |
| Aconitase 2, mitochondrial (Aco2) | 1.1 |
| ATP-specific Succinyl-CoA synthetase beta subunit | 1.2 |
| Succinate dehydrogenase (Ubiquinone) Flavoprotein subunit, Mitochondrial | 1.4 |
| SDHD gene_Small subunit of Cytochrome b of Succinate dehydrogenase | 1.5 |
| Fumarate hydratase (FH) | 1.4 |
| Malate dehydrogenase, mitochondrial (DD67) | 1.6 |
As can be seen from Table III, genes encoding glycolytic enzymes appear generally down-regulated in Ant1−/− skeletal muscle. In addition to Pfkfb3 and AldoB mentioned above, the genes for Aldolase I (Aldoa), Triosephosphate isomerase (Tpi1) and Enolase I (EnoI) were all down-regulated at least 1.4-fold. Also, Ldh1 (encoding lactate dehydrogenase A chain which converts pyruvate to lactate) and Aat2 (muscle specific alanine aminotransferase which converts pyruvate to alanine) were down-regulated by 1.5 and 1.4-fold, respectively.
The MITOCHIP results also suggest that the Ant2 gene may have been down-regulated, though the very low expression of this gene in skeletal muscle [11, 12] renders any estimate of extent of change immaterial. What is clear is that Ant2 was NOT up-regulated in the Ant1−/− muscle. This has been confirmed for both Ant2 mRNA in Northern blots and Ant2 protein in western blots (see Figure 3 c and d, respectively in [11]). The absence of induction of the Ant2 gene in the face of Ant1 deficiency initially seemed counter-institutive, since the simplest way to resolve the Ant1 deficiency would seem to have been induction of another Ant isoform that would complement the defect. Since the Ant2 gene is the only other systemic mouse Ant isoform, induction of this gene would have been the logical choice. One possible explanation for this anomaly could be that the mouse Ant2 protein has a similar function and properties as the human ANT2 protein. In contrast to ANT1 and ANT3, human ANT2 has been proposed to import cytosolic ATP into the mitochondria during periods of mitochondrial dysfunction, for example in hypoxia [5, 6]. If this is true for the mouse Ant2, then up-regulation of the Ant2 protein would not complement the Ant1 deficiency, but rather make the sarcoplasmic ATP deficiency worse. This would also explain the very low expression of Ant2 in skeletal muscle, where ATP would consistently need to be transported out of the mitochondrion to support muscle contraction and plasma membrane transport. Similarly, if Ant2 functions to import ATP into the mitochondria during hypoxia, then the gene should be induced during hypoxia, not hyperoxia as would be the situation in Ant1−/− skeletal muscle. Therefore, the lack of induction of Ant2 may reflect its converse function, relative to Ant1, rendering it both biochemically unable to complement Ant1 deficiency and inappropriately regulated to do so.
Table III also shows that mRNAs for several enzymes of the TCA cycle are moderately up-regulated in Ant1−/− skeletal muscle. This is consistent with the mitochondrial proliferation and associated induction of mitochondrial energy production in an effort to compensate for the cytosolic ATP deficiency.
Interestingly, G6pdx [glucose-6-phosphate dehydrogenase, which catalyzes the first and rate-limiting step of the Pentose Phosphate Pathway (PPP)] is over-expressed 1.5-fold in Ant1−/− skeletal muscle. The PPP provides much of the reducing power in the cell in the form of NADPH, and NADPH is important for maintaining reduced glutathione. Since flux through the PPP and the rates of NADPH production are directly proportional to glucose-6-phosphate dehydrogenase activity; then increased G6pdx expression may be is indicative of an increased demand for NADPH to manage oxidative stress.
The three remaining down-regulated genes were Ppox, Cyp11a1, and Odc1. The Ppox gene encodes the enzyme protoporphyrinogen oxidase, which catalyzes the penultimate reaction in heme biosynthesis. Mutations in the Ppox gene have been shown to cause variegated porphyria in humans [52]. The Cyp11a1 gene encodes the cytochrome P450 side chain cleaving enzyme that catalyzes the first step of cholesterol biosynthesis [53]. The Odc1 gene encodes ornithine decarboxylase, the key enzyme in the biosynthesis of polyamines which play central roles in cell proliferation and survival [54]. Down-regulation of Odc1 should cause the down-regulation of polyamine biosynthesis in Ant1−/− skeletal muscle.
(ii) Genes that regulate apoptosis
Several genes that encode pro-apoptotic genes were also down-regulated in Ant1−/− skeletal muscle including Myc, Modulator of apoptosis (Moap1), Bcl2-interacting killer (Bik), Chk2 checkpoint homolog (Chek2), and Phospholipase A2, group VI (Pla2g6). This suggests that the apoptotic threshold may be higher in the Ant1−/− skeletal muscle. The Myc oncogene regulates two complex and seemingly contradictory sets of regulatory pathways. While Myc over-expression or activation has been shown to be a key event in many cancers, Myc over-expression can also activate apoptotic pathways in a normal cell under conditions of serum deprivation or DNA damage [55]. Myc has been shown to promote cell death by suppressing the activity of the Cdk-inhibitor p21/Cip1 [56] and thus shifting the choice of cellular DNA damage response from cytostatic to apoptotic. The down-regulation of Myc in Ant1−/− skeletal muscle was also confirmed at the protein level by Western blotting (Fig. 1F).
The Moap1 gene was identified in a two-hybrid screen as encoding a protein that interacts with the apoptosis effector Bax. Moap1 has been shown to cause caspase-dependent apoptosis when over-expressed in cell culture [57, 58].
The Bik gene also encodes a potent death agonist. Over-expression of Bik causes caspase-dependent apoptosis in cell culture, and its apoptotic activity can be suppressed by co-over-expression of Bcl-2 or Bcl-XL [59]. The mouse Chek2, is required for p53 mediated DNA-damage response. Analyses of Chek2−/− mice have revealed that Chek2 is required for p53 mediated apoptosis in response to ionizing radiation. Thus, Chek2, like Myc, also appears to be involved in shifting the choice of the p53 pathway from cytostatic to apoptotic [60]. Finally, Pla2g6 has been shown to mediate Fas-induced cell death by causing the release of arachidonic acid. Pla2g6 may also be involved in regulating ion-channel activity and membrane phospholipids remodeling during apoptosis and inhibition of Pla2g6 has been shown to decrease Fas-induced cell death [61].
(iii) Other genes under-expressed in Ant1−/− muscle
As can be seen from Table II, several kinases, signaling molecules, transporters and other genes are down-regulated in Ant1−/− skeletal muscle. It is difficult to classify these genes further from a standpoint of metabolic pathways. However some of them do have common regulatory links. The genes for microtubule associated protein 4 (Mtap4) and heat shock protein 90 (Hsp86-1) (as well as Odc1 and several glucose metabolism genes) are known targets of Myc [62, 63]. Thus their down-regulation may be a consequence of the down-regulation of Myc in Ant1−/− skeletal muscle. Also, some of the down-regulated mRNAs may also be ones that are normally enriched in glycolytic muscle fibers, and their reduced expression may also be a result of the change in the metabolic nature of the muscle fibers in Ant1−/− animals.
Molecular basis of the mitochondrial proliferation in Ant1−/− skeletal muscle
Mitochondrial proliferation and changes in muscle fiber physiology and composition can be triggered by endurance training and exercise [64]. This process has been shown to be mediated by the PPARγ associated transcriptional coactivator-1α (PGC-1α[65, 66]. PGC-1α has also been shown, in ectopic expression studies, to be a master regulator of mitochondrial proliferation and muscle fiber-type switching [67] acting through regulation of transcription factors required for mitochondrial proliferation such as Nrf1 and Tfam [66].
Since we see marked mitochondrial proliferation as well as over-expression of several slow-twitch muscle fiber markers in Ant1−/− muscle, we examined whether these changes were also reflected the PGC-1α transcription pathway. As can be seen in Figure 1A–C, the levels of Pgc-1α Nrf1 and Tfam proteins were all elevated in Ant1−/− skeletal muscle, indicating that the mitochondrial proliferation observed in Ant1−/− skeletal muscle is mediated by the PGC-1αpathway. Interestingly, the levels of Pgc-1α mRNA appear to be slightly lower in Ant1−/− skeletal muscle relative to wild-type, as assayed by real-time PCR (not shown). However, Pgc-1α can be regulated by either transcriptional and/or post-transcriptional mechanisms [68–75].
To investigate whether the observed changes in Ant1−/− skeletal muscle were due to changes in fiber-type composition of the muscle (from fast-twitch to slow-twitch), or simply due to the metabolic nature of the muscle fibers, we examined the levels of myoglobin, Troponin I-slow (Tnni1; normally enriched in slow-twitch muscle fibers) and Troponin I-fast (Tnni2; normally enriched in fast-twitch muscle fibers) in Ant1−/− skeletal muscle. As can be seen from Figures 1G-I, the levels of Tnni1, Tnni2 and myoglobin are not significantly different in Ant1−/− skeletal muscle compared to wild-type. Immunohistochemical staining for different myosin heavy chain (MHC) isoforms also showed no significant change in the levels or distribution of the slow-twitch specific MHC isoforms in Ant1−/− skeletal muscle (A. Flierl and DC Wallace, unpublished results). While elevated expression of PGC-1α can induce a change in muscle-fiber type, it need not necessarily do so. Indeed, over-expression of one of the targets of PGC-1, myogenin, has been shown to promote increased oxidative metabolism in skeletal muscle fibers without effecting a complete change in muscle fiber type [76]. As can be seen in Figure 1E, levels of myogenin are significantly elevated in Ant1−/− skeletal muscle compared to wild-type. These results are all consistent with the idea that the skeletal muscle fibers in Ant1−/− animals adopt a more oxidative metabolism without changing the fiber-type composition of the muscle.
DISCUSSION
The skeletal muscle of Ant1−/− mice presents a fascinating regulatory conundrum from the standpoint of energy metabolism. On the one hand, the complete deficiency of Ant1 blocks virtually all transport of mitochondrial ATP into the cytosol, thus creating a severe energy deficiency within the nuclear-cytosol compartment of the muscle cells. As a result, there is little ATP for sarcoplasmic muscle contraction, ion transport, biogenesis, etc. causing severe exercise intolerance in the mice [11].
On the other hand, mitochondrial OXPHOS is fully functional and is even strongly up-regulated, as evidenced by the hyper-proliferation of skeletal muscle mitochondria and the strong induction of succinate dehydrogenase (complex II) and complex IV. Hence, the intra-mitochondrial ATP level would be expected to be near maximum. Indeed, the high resting serum lactate and alanine levels are indicative of a ‘stalled’ mitochondrial OXPHOS and TCA cycle. This would be the result if the mitochondrial matrix adenine nucleotides were maximally phosphorylated, thus depriving the ATP synthase of its ADP substrate. In the absence of ADP, the flux of protons through the ATP synthase proton channel would stop, and the mitochondrial membrane potential would remain hyper-polarized, blocking further proton pumping and stalling the ETC. Inhibition of the ETC, would also stall the TCA cycle, resulting in the accumulation of cytosolic pyruvate and NADH + H+, which would be converted to lactate and alanine [11].
With the mitochondrial ETC stalled, in the present of unlimited calories, the electron carriers would become fully reduced. Given a hyperoxic state, the excess electrons could readily be transferred directly to O2 to generate superoxide anion and the other ROS. The chronically elevated ROS would then damage the mitochondria and mtDNA in skeletal muscle and heart [15].
To investigate the effects of this metabolic conundrum on the molecular circuitry of energy metabolism, we employed the MITOCHIP to analyze changes in the transcript levels of over 1000 genes that are associated with energy metabolism, ROS biology, and apoptosis. These mRNA analyses were complemented by examining the protein levels of transcription factors known to regulate the expression of energy, antioxidant, and apoptotic genes. Our results revealed that the Ant1−/− skeletal muscle: (i) Is starved for energy and is attempting to compensate for this energy deficit by inducing oxidative metabolism while down regulating glycolysis; (ii) Is under oxidative stress and is attempting to combat this stress by up-regulating antioxidant defenses; (iii) Is inducing oxidative metabolism and antioxidant defenses through increased levels of nuclear (Pgc-1α, Nrf1, and myogenin) and mitochondrial (Tfam) DNA transcription factors; (iv) Is at increased risk for apoptosis and is compensating by up-regulating anti-apoptotic and down-regulating pro-apoptotic genes; and (v) Appears to be down-regulating the apoptotic pathways by decreased c-Myc.
The Metabolic Reorganization of Ant1−/− Mouse Skeletal Muscles
Previously, we had shown that as human myoblasts differentiate into myotubes, the expression of mtDNA and nDNA-encoded OXPHOS genes and of nDNA-encoded glycolytic genes change expression reciprocally. When myoblasts are differentiated in normoxic conditions, OXPHOS genes are up-regulated in myoblasts, down-regulated after commitment to differentiation, and up-regulated again in myotubes. ANT1 is not expressed in myoblasts but is progressively induced as myotubes are formed. By contrast, the glycolytic genes are down-regulated in myoblasts, up-regulated after commitment, and decline as myotubes are formed. If the myoblasts are differentiated into myotubes under hypoxic condition, however, the glycolytic gene expression levels remain high in the myotubes. As a consequence, by seven days after commitment the glycolytic mRNAs have increased six fold relative to OXPHOS transcripts [77].
While the ETC is stalled in the Ant1−/− mouse skeletal muscle, the cardiovascular system remains functional. Therefore, oxygen continues to be delivered to the muscle but can not be consumed by the mitochondria OXPHOS. Hence, the Ant1−/− muscle should become hyperoxic. In the hyperoxic state Ant1−/− muscle is attempting to compensate for the cytosolic ATP deficiency by up-regulating mitochondrial OXPHOS. However, this is futile due to the absence of Ant1, resulting in uncontrolled proliferation of the mitochondria [11]. All the mtDNA OXPHOS genes as well as four nuclear-encoded OXPHOS genes are significantly up-regulated in the Ant1 deficient muscle, with two of the nuclear encoded proteins, Ndufs4 and Cox7a2, being involved in the assembly of complex I and complex IV, respectively [25–27]. The fact that major up-regulated nuclear OXPHOS genes are involved in complex assembly suggests that most nuclear-encoded components of the ETC are not normally limiting in muscle and that the number of functional ETC complexes may be regulated at the level of complex assembly.
The up-regulation of several oxidative muscle fiber-specific marker genes including Fhl1, Mor2, Atp2a2, and Atp1b1 is also consistent the Ant1−/− muscle’s attempting to compensate for the energy deficiency by up-regulating oxidative metabolism. However, this is not associated with fiber type switching, since the myoglobin levels and troponin I and myosin heavy chain isoforms did not change. Hence, the primary response to the reduction in the sarcoplasmic ATP appears to be the up-regulation of mitochondrial OXPHOS.
Assuming that mouse and human muscle are under similar regulation, we would expect that the induction of mitochondrial OXPHOS should be associated with the reciprocal down-regulation of glycolysis, which we observed. Both the 6-phosphofructo-2-kinase (Pfkfb3) and aldolase B genes were strongly down-regulated, in parallel with the down regulation of several other glycolytic genes including the lactate dehydrogenase A (Ldh1) and muscle-specific alanine aminotransferase (Aat2) genes. Therefore, the “standard” muscle response to low sarcoplasmic ATP in the presence of normoxia is induction of OXPHOS and reduction of glycolysis.
The “standard” program for energy deficient muscle under normoxia proves to be exactly the wrong response when the cytosolic ATP deficiency is caused by Ant1 deficiency. The only source of ATP for the cytosol is then glycolysis. Since carbohydrates are not limiting, the resulting flux of carbohydrates through glycolysis is high. With OXPHOS and the TCA cycle stalled, pyruvate and NADH + H+ accumulate and are combined by lactate dehydrogenase to generate lactate. Since lactate is both the end product of glycolysis but also a substrate for OXPHOS, it is unclear if the Ant1−/− muscle would strive to export the excess lactate to control intra-cellular pH or import the lactate in a further attempt to bolster mitochondrial ATP production. Insight into this question might be obtained by analyzing the expression of the proton-linked monocarboxylate transporter (MCT) isoforms. Isoform MCT1 is expressed in Type I oxidative fibers and is thought to import lactate into muscle cells to be oxidized by OXPHOS, while isoform MCT4 is present in all muscles but at lower levels in oxidative muscle and is thought to export excess glycolytic lactate [78].
An alternative approach to increasing sarcoplasmic ATP would be to induce an alternative adenine nucleotide translocator, thus restoring the export of the excess mitochondrial ATP. Ant2 would be the logical choice, since it is expressed in virtually every tissue. However, Ant2 was NOT up-regulated [11]. This anomaly might be explained if Ant2 functions like the human ANT2 to import cytosolic ATP into the mitochondrion under anoxic conditions [5, 6]. If this is the case, then Ant2 should be induced under anoxic conditions and repressed under normoxic conditions, which is consistent with what we observed in the Ant1−/− mouse muscle.
Given the high rate of ROS production by the Ant1−/− muscle [15], we would expect the mitochondrial antioxidants defenses to be induced. While the steady state mRNA levels for the mitochondrial antioxidant genes were only mildly elevated, western blot analysis revealed that mitochondrial manganese superoxide dismutase (MnSOD) is induced six fold and glutathione peroxidase-1 is induced three fold in the Ant1−/− muscle [15]. In addition, the MITOCHIP revealed other stress-response genes were up-regulated in Ant1−/− skeletal muscle: Mgst1, G6pdx, and Kin17. Mgst1 is involved in protecting membranes and cells from lipid peroxidation damage, consistent with high oxidative stress. Glucose-6-phosphate dehydrogenase (G6pdx), the rate-limiting enzyme in the pentose phosphate pathway (PPP) and the primary source of NADPH, would be important in sustaining glutathione pathways to detoxify peroxides. The DNA damage caused by chronic oxidative stress [15], could be amelioated by the induction of the Kin17 DNA repair gene [43, 44].
Excessive oxidative stress and DNA damage make cells more prone to apoptosis. While the importance of the apoptotic pathway in muscle is still unclear, it has been implicated skeletal muscle denervation and remodeling, muscular dystrophy, and muscle aging [79–82]. H2O2 promotes apoptosis via activation of the MAP kinase and JNK pathways and subsequent inactivation of Mcl-1 [83], the muscle isoform of the Bcl-2 family [84]. Therefore, the up-regulation of Mcl1 indicates hat the Ant1−/− skeletal muscle is attempting to dampen the pro-apoptotic response. The down-regulation of the pro-apoptotic Bik, Moap1, Chek2, and Pla2g6 genes would further impede apoptosis. Finally, the down-regulation of the ornithine decarboxylase-1 gene (Odc1) indicates a down-regulation of polyamine biogenesis, which could also inhibit apoptosis by blocking the translocation of BAX into mitochondria [85].
Transcription Factor Regulation of the Energy deficiency in the Ant1−/− Muscle
Having established that the Ant1−/− muscle attempts to compensate for cytosolic ATP deficiency by up-regulating OXPHOS, and knowing that PGC-1α is implicated in the regulation of nDNA encoded mitochondrial genes, we examined the protein levels of Pgc-1α. Pgc-1α induces mitochondrial biogenesis in C2C12 cultured mouse muscle cells, acting through the nuclear respiratory factors 1 and 2 (NRF1 and 2) [66, 86] and Pgc-1α null mice show a reduction in MnSOD and Gpx1 [87]. Furthermore, transgenic mice over-expressing PGC-1α mRNA 16-fold exhibit a marked increase in exercise capacity and peak oxygen uptake; an increase in skeletal muscle oxidative metabolism proteins in including cytochrome c, various OXPHOS complex subunits, fatty acid oxidation enzymes; a 166% increase in mtDNA and up-regulation of Tfam transcription factor; and an induction of the antioxidant enzymes MnSOD, thioredoxin reductase-2, and catalase [75]. Since we observed a marked up-regulation of the Pgc-1α protein in the Ant1−/− skeletal muscle, we can infer that the up-regulation of mitochondrial OXPHOS and the increased antioxidant defenses in the Ant1−/− mice is a consequence of the increased levels of Pgc-1α protein. Up-regulation of Pgc-1α results in the up-regulation of the mtDNA transcription factor Tfam, which would increase mtDNA transcript levels. The muscle’s mitochondrial oxidative capacity can also be up-regulated by myogenin [76]. Similarly, up-regulation of Nrbf1, which interacts with PPARα as well as other transcription factors known to up-regulate mitochondrial gene expression, might also be a contributing factor [49].
Inhibition of apoptosis may have been achieved by down-regulation of c-Myc, which was seen by both the MITOCHIP and protein analyses. Over-expression of c-Myc has been shown to promote apoptosis in cultured cells under conditions of serum deprivation and oxidative stress [88, 89], proceeding via release of mitochondrial cytochrome c and activation of pro-apoptotic caspases [90]. c-Myc has also been shown to regulate some Nrf1 target genes and promote apoptosis by de-regulating genes involved in mitochondrial function [91]. In a normal Myc expression background, Nrf1 over-expression has been shown to trigger apoptosis in serum-deprived cells [91]. In the skeletal muscle of Ant1−/− mice Nrf1 levels and mitochondrial proliferation are increased, but c-Myc is down-regulated. Therefore, the down-regulation of c-Myc may be counteracting the pro-apoptotic tendency of increased Nrf1 levels. The down-regulation of c-Myc might also be important in the down-regulation of ornithine decarboxylase (Odc1), since Odc1 is a target gene of c-Myc. The down-regulation of Odc1 should diminish polyamine synthesis, inhibit the translocation of BAX into mitochondria, and repress apoptosis [85]. The inhibition of BAX translocation would also be augmented by the down-regulation of the BAX interacting Moap1 protein [57, 58].
Other c-Myc target genes that are down-regulated include Mtap4 and Hsp86-1. Myc can also regulate the expression of several glycolytic genes [63] which account for the down-regulation of Ldh1, and EnoI which are known to be positively regulated by Myc [62, 92]. However, Pfk1 and Gapd should also have been down-regulated but were not [63]. Overall, Myc down-regulation may have been the mechanism by which apoptosis was suppressed in the Ant1−/− skeletal muscle in the presence of high oxygen.
MITOCHIP Monitoring of Mitochondrial Function and for Drug Screening
Our mouse MITOCHIP is proving to be a powerful tool for analyzing the pathophysiology of mouse models of mitochondrial disease [93]. When complemented by analysis for key regulatory protein levels, the MITOCHIP and similar technologies are providing much needed readouts for identifying new mitochondrial therapeutic agents [94, 95].
Supplementary Material
Acknowledgments
This work was supported by grants NS41850, NS21328, and AG13154 awarded to DCW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERANCES
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li K, Warner CK, Hodge JA, Minoshima S, Kudoh J, Fukuyama R, Maekawa M, Shimizu Y, Shimizu N, Wallace DC. A human muscle adenine nucleotide translocator gene has four exons, is located on chromosome 4, and is differentially expressed. J Biol Chem. 1989;264:13998–14004. [PubMed] [Google Scholar]
- 3.Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem. 1992;267:14592–14597. [PubMed] [Google Scholar]
- 4.Torroni A, Stepien G, Hodge JA, Wallace DC. Neoplastic transformation is associated with coordinate induction of nuclear and cytoplasmic oxidative phosphorylation genes. J Biol Chem. 1990;265:20589–20593. [PubMed] [Google Scholar]
- 5.Chevrollier A, Loiseau D, Chabi B, Renier G, Douay O, Malthiery Y, Stepien G. ANT2 isoform required for cancer cell glycolysis. J Bioenerg Biomembr. 2005;37:307–316. doi: 10.1007/s10863-005-8642-5. [DOI] [PubMed] [Google Scholar]
- 6.Giraud S, Bonod-Bidaud C, Wesolowski-Louvel M, Stepien G. Expression of human ANT2 gene in highly proliferative cells: GRBOX, a new transcriptional element, is involved in the regulation of glycolytic ATP import into mitochondria. J Mol Biol. 1998;281:409–418. doi: 10.1006/jmbi.1998.1955. [DOI] [PubMed] [Google Scholar]
- 7.Dolce V, Scarcia P, Iacopetta D, Palmieri F. A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005;579:633–637. doi: 10.1016/j.febslet.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 8.Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, Keranen S, Peltonen L, Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 9.Napoli L, Bordoni A, Zeviani M, Hadjigeorgiou GM, Sciacco M, Tiranti V, Terentiou A, Moggio M, Papadimitriou A, Scarlato G, Comi GP. A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology. 2001;57:2295–2298. doi: 10.1212/wnl.57.12.2295. [DOI] [PubMed] [Google Scholar]
- 10.Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, Ferrero I, Zeviani M. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet. 2005;14:3079–3088. doi: 10.1093/hmg/ddi341. [DOI] [PubMed] [Google Scholar]
- 11.Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nature genetics. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- 12.Levy SE, Chen YS, Graham BH, Wallace DC. Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene. 2000;254:57–66. doi: 10.1016/s0378-1119(00)00252-3. [DOI] [PubMed] [Google Scholar]
- 13.Rodic N, Oka M, Hamazaki T, Murawski MR, Jorgensen M, Maatouk DM, Resnick JL, Li E, Terada N. DNA methylation is required for silencing of ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells. 2005;23:1314–1323. doi: 10.1634/stemcells.2005-0119. [DOI] [PubMed] [Google Scholar]
- 14.Brower JV, Rodic N, Seki T, Jorgensen M, Fliess N, Yachnis AT, McCarrey JR, Oh SP, Terada N. Evolutionarily conserved mammalian adenine nucleotide translocase 4 is essential for spermatogenesis. J Biol Chem. 2007;282:29658–29666. doi: 10.1074/jbc.M704386200. [DOI] [PubMed] [Google Scholar]
- 15.Esposito LA, Melov S, Panov A, Cottrell BA, Wallace DC. Mitochondrial disease in mouse results in increased oxidative stress. Proc Natl Acad Sci U S A. 1999;96:4820–4825. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halestrap AP, Brennerb C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 17.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 18.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Da Cruz S, Xenarios I, Langridge J, Vilbois F, Parone PA, Martinou JC. Proteomic analysis of the mouse liver mitochondrial inner membrane. J Biol Chem. 2003;278:41566–41571. doi: 10.1074/jbc.M304940200. [DOI] [PubMed] [Google Scholar]
- 20.Bauer MK, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol. 1999;147:1493–1502. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamora M, Granell M, Mampel T, Vinas O. Adenine nucleotide translocase 3 (ANT3) overexpression induces apoptosis in cultured cells. FEBS Lett. 2004;563:155–160. doi: 10.1016/S0014-5793(04)00293-5. [DOI] [PubMed] [Google Scholar]
- 22.Murdock DG, Boone BE, Esposito LA, Wallace DC. Up-regulation of nuclear and mitochondrial genes in the skeletal muscle of mice lacking the heart/muscle isoform of the adenine nucleotide translocator. J Biol Chem. 1999;274:14429–14433. doi: 10.1074/jbc.274.20.14429. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka TS, Jaradat SA, Lim MK, Kargul GJ, Wang X, Grahovac MJ, Pantano S, Sano Y, Piao Y, Nagaraja R, Doi H, Wood WH, 3rd, Becker KG, Ko MS. Genome-wide expression profiling of mid-gestation placenta and embryo using a 15,000 mouse developmental cDNA microarray. Proc Natl Acad Sci U S A. 2000;97:9127–9132. doi: 10.1073/pnas.97.16.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 25.Scacco S, Petruzzella V, Budde S, Vergari R, Tamborra R, Panelli D, van den Heuvel LP, Smeitink JA, Papa S. Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J Biol Chem. 2003;278:44161–44167. doi: 10.1074/jbc.M307615200. [DOI] [PubMed] [Google Scholar]
- 26.Calder KM, McEwen JE. Deletion of the COX7 gene in Saccharomyces cerevisiae reveals a role for cytochrome c oxidase subunit VII in assembly of remaining subunits. Mol Microbiol. 1991;5:1769–1777. doi: 10.1111/j.1365-2958.1991.tb01926.x. [DOI] [PubMed] [Google Scholar]
- 27.Sandona D, Bisson R. Inhibition of the synthesis of a cytochrome-c-oxidase subunit isoform by antisense RNA. Eur J Biochem. 1994;219:1053–1061. doi: 10.1111/j.1432-1033.1994.tb18588.x. [DOI] [PubMed] [Google Scholar]
- 28.Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. [DOI] [PubMed] [Google Scholar]
- 29.Cantatore P, Flagella Z, Fracasso F, Lezza AM, Gadaleta MN, de Montalvo A. Synthesis and turnover rates of four rat liver mitochondrial RNA species. FEBS Lett. 1987;213:144–148. doi: 10.1016/0014-5793(87)81480-1. [DOI] [PubMed] [Google Scholar]
- 30.Hevner RF, Wong-Riley MT. Mitochondrial and nuclear gene expression for cytochrome oxidase subunits are disproportionately regulated by functional activity in neurons. J Neurosci. 1993;13:1805–1819. doi: 10.1523/JNEUROSCI.13-05-01805.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 32.Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131–138. doi: 10.1016/j.bcmd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Koehler CM, Leuenberger D, Merchant S, Renold A, Junne T, Schatz G. Human deafness dystonia syndrome is a mitochondrial disease. Proc Natl Acad Sci U S A. 1999;96:2141–2146. doi: 10.1073/pnas.96.5.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell. 2005;16:1543–1554. doi: 10.1091/mbc.E04-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Navet R, Mathy G, Douette P, Dobson RL, Leprince P, De Pauw E, Sluse-Goffart C, Sluse FE. Mitoproteome plasticity of rat brown adipocytes in response to cold acclimation. J Proteome Res. 2007;6:25–33. doi: 10.1021/pr060064u. [DOI] [PubMed] [Google Scholar]
- 36.Campbell WG, Gordon SE, Carlson CJ, Pattison JS, Hamilton MT, Booth FW. Differential global gene expression in red and white skeletal muscle. Am J Physiol Cell Physiol. 2001;280:C763–768. doi: 10.1152/ajpcell.2001.280.4.C763. [DOI] [PubMed] [Google Scholar]
- 37.Mendler L, Szakonyi G, Zador E, Gorbe A, Dux L, Wuytack F. Expression of sarcoplasmic/endoplasmic reticulum Ca2+ ATPases in the rat extensor digitorum longus (EDL) muscle regenerating from notexin-induced necrosis. J Muscle Res Cell Motil. 1998;19:777–785. doi: 10.1023/a:1005499304147. [DOI] [PubMed] [Google Scholar]
- 38.Zador E, Szakonyi G, Racz G, Mendler L, Ver Heyen M, Lebacq J, Dux L, Wuytack F. Expression of the sarco/endoplasmic reticulum Ca(2+)-transport ATPase protein isoforms during regeneration from notexin-induced necrosis of rat soleus muscle. Acta Histochem. 1998;100:355–369. doi: 10.1016/S0065-1281(98)80033-0. [DOI] [PubMed] [Google Scholar]
- 39.Hundal HS, Marette A, Ramlal T, Liu Z, Klip A. Expression of beta subunit isoforms of the Na+,K(+)-ATPase is muscle type-specific. FEBS Lett. 1993;328:253–258. doi: 10.1016/0014-5793(93)80938-q. [DOI] [PubMed] [Google Scholar]
- 40.Morgenstern R, DePierre JW. Microsomal glutathione transferase. Purification in unactivated form and further characterization of the activation process, substrate specificity and amino acid composition. Eur J Biochem. 1983;134:591–597. doi: 10.1111/j.1432-1033.1983.tb07607.x. [DOI] [PubMed] [Google Scholar]
- 41.Mosialou E, Ekstrom G, Adang AE, Morgenstern R. Evidence that rat liver microsomal glutathione transferase is responsible for glutathione-dependent protection against lipid peroxidation. Biochem Pharmacol. 1993;45:1645–1651. doi: 10.1016/0006-2952(93)90305-g. [DOI] [PubMed] [Google Scholar]
- 42.Maeda A, Crabb JW, Palczewski K. Microsomal glutathione S-transferase 1 in the retinal pigment epithelium: protection against oxidative stress and a potential role in aging. Biochemistry. 2005;44:480–489. doi: 10.1021/bi048016f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biard DS, Miccoli L, Despras E, Frobert Y, Creminon C, Angulo JF. Ionizing radiation triggers chromatin-bound kin17 complex formation in human cells. J Biol Chem. 2002;277:19156–19165. doi: 10.1074/jbc.M200321200. [DOI] [PubMed] [Google Scholar]
- 44.Kannouche P, Pinon-Lataillade G, Tissier A, Chevalier-Lagente O, Sarasin A, Mezzina M, Angulo JF. The nuclear concentration of kin17, a mouse protein that binds to curved DNA, increases during cell proliferation and after UV irradiation. Carcinogenesis. 1998;19:781–789. doi: 10.1093/carcin/19.5.781. [DOI] [PubMed] [Google Scholar]
- 45.Mazin A, Milot E, Devoret R, Chartrand P. KIN17, a mouse nuclear protein, binds to bent DNA fragments that are found at illegitimate recombination junctions in mammalian cells. Mol Gen Genet. 1994;244:435–438. doi: 10.1007/BF00286696. [DOI] [PubMed] [Google Scholar]
- 46.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou P, Qian L, Kozopas KM, Craig RW. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630–643. [PubMed] [Google Scholar]
- 48.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a member of the BLC-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J Cell Physiol. 1996;166:523–536. doi: 10.1002/(SICI)1097-4652(199603)166:3<523::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 49.Masuda N, Yasumo H, Furusawa T, Tsukamoto T, Sadano H, Osumi T. Nuclear receptor binding factor-1 (NRBF-1), a protein interacting with a wide spectrum of nuclear hormone receptors. Gene. 1998;221:225–233. doi: 10.1016/s0378-1119(98)00461-2. [DOI] [PubMed] [Google Scholar]
- 50.Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995;376:509–514. doi: 10.1038/376509a0. [DOI] [PubMed] [Google Scholar]
- 51.Tokunaga F, Goto T, Koide T, Murakami Y, Hayashi S, Tamura T, Tanaka K, Ichihara A. ATP- and antizyme-dependent endoproteolysis of ornithine decarboxylase to oligopeptides by the 26 S proteasome. J Biol Chem. 1994;269:17382–17385. [PubMed] [Google Scholar]
- 52.Frank J, Christiano AM. Variegate porphyria: past, present and future. Skin Pharmacol Appl Skin Physiol. 1998;11:310–320. doi: 10.1159/000029854. [DOI] [PubMed] [Google Scholar]
- 53.Hu MC, Hsu HJ, Guo IC, Chung BC. Function of Cyp11a1 in animal models. Mol Cell Endocrinol. 2004;215:95–100. doi: 10.1016/j.mce.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 54.Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem J. 2003;376:1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 56.Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–734. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- 57.Tan KO, Tan KM, Chan SL, Yee KS, Bevort M, Ang KC, Yu VC. MAP-1, a novel proapoptotic protein containing a BH3-like motif that associates with Bax through its Bcl-2 homology domains. J Biol Chem. 2001;276:2802–2807. doi: 10.1074/jbc.M008955200. [DOI] [PubMed] [Google Scholar]
- 58.Sharpe JC, Arnoult D, Youle RJ. Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta. 2004;1644:107–113. doi: 10.1016/j.bbamcr.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 59.Hegde R, Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Blk, a BH3-containing mouse protein that interacts with Bcl-2 and Bcl-xL, is a potent death agonist. J Biol Chem. 1998;273:7783–7786. doi: 10.1074/jbc.273.14.7783. [DOI] [PubMed] [Google Scholar]
- 60.Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, Okada H, Sarkissian T, Wong JA, Sakai T, De Stanchina E, Bristow RG, Suda T, Lowe SW, Jeggo PA, Elledge SJ, Mak TW. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol. 2002;22:6521–6532. doi: 10.1128/MCB.22.18.6521-6532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taketo MM, Sonoshita M. Phospolipase A2 and apoptosis. Biochim Biophys Acta. 2002;1585:72–76. doi: 10.1016/s1388-1981(02)00326-8. [DOI] [PubMed] [Google Scholar]
- 62.Guo QM, Malek RL, Kim S, Chiao C, He M, Ruffy M, Sanka K, Lee NH, Dang CV, Liu ET. Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res. 2000;60:5922–5928. [PubMed] [Google Scholar]
- 63.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 64.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- 65.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. Faseb J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 66.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 67.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 68.Corton JC, Brown-Borg HM. Peroxisome proliferator-activated receptor gamma coactivator 1 in caloric restriction and other models of longevity. J Gerontol A Biol Sci Med Sci. 2005;60:1494–1509. doi: 10.1093/gerona/60.12.1494. [DOI] [PubMed] [Google Scholar]
- 69.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 70.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 71.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koo SH, Montminy M. In vino veritas: a tale of two sirt1s? Cell. 2006;127:1091–1093. doi: 10.1016/j.cell.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 73.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 75.Calvo JA, Daniels TG, Wang X, Paul A, Lin J, Spiegelman BM, Stevenson SC, Rangwala SM. Muscle-specific expression of PPAR{gamma}coactivator-1{alpha} improves exercise performance and increases peak oxygen uptake. J Appl Physiol. 2008 doi: 10.1152/japplphysiol.01231.2007. [DOI] [PubMed] [Google Scholar]
- 76.Hughes SM, Chi MM, Lowry OH, Gundersen K. Myogenin induces a shift of enzyme activity from glycolytic to oxidative metabolism in muscles of transgenic mice. J Cell Biol. 1999;145:633–642. doi: 10.1083/jcb.145.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Webster KA, Gunning P, Hardeman E, Wallace DC, Kedes L. Coordinate reciprocal trends in glycolytic and mitochondrial transcript accumulations during the in vitro differentiation of human myoblasts. J Cell Physiol. 1990;142:566–573. doi: 10.1002/jcp.1041420316. [DOI] [PubMed] [Google Scholar]
- 78.Juel C, Halestrap AP. Lactate transport in skeletal muscle - role and regulation of the monocarboxylate transporter. J Physiol. 1999;517(Pt 3):633–642. doi: 10.1111/j.1469-7793.1999.0633s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pollack M, Phaneuf S, Dirks A, Leeuwenburgh C. The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann N Y Acad Sci. 2002;959:93–107. doi: 10.1111/j.1749-6632.2002.tb02086.x. [DOI] [PubMed] [Google Scholar]
- 80.Alway SE, Degens H, Krishnamurthy G, Chaudhrai A. Denervation stimulates apoptosis but not Id2 expression in hindlimb muscles of aged rats. J Gerontol A Biol Sci Med Sci. 2003;58:687–697. doi: 10.1093/gerona/58.8.b687. [DOI] [PubMed] [Google Scholar]
- 81.Sandri M, Carraro U. Apoptosis of skeletal muscles during development and disease. Int J Biochem Cell Biol. 1999;31:1373–1390. doi: 10.1016/s1357-2725(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 82.Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nature genetics. 2003;35:367–371. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- 83.Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, Umezawa A, Ichijo H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem. 2002;277:43730–43734. doi: 10.1074/jbc.M207951200. [DOI] [PubMed] [Google Scholar]
- 84.Krajewski S, Bodrug S, Krajewska M, Shabaik A, Gascoyne R, Berean K, Reed JC. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am J Pathol. 1995;146:1309–1319. [PMC free article] [PubMed] [Google Scholar]
- 85.Yuan Q, Ray RM, Johnson LR. Polyamine depletion prevents camptothecin-induced apoptosis by inhibiting the release of cytochrome c. Am J Physiol Cell Physiol. 2002;282:C1290–1297. doi: 10.1152/ajpcell.00351.2001. [DOI] [PubMed] [Google Scholar]
- 86.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 87.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 88.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 89.Xu Y, Nguyen Q, Lo DC, Czaja MJ. c-myc-Dependent hepatoma cell apoptosis results from oxidative stress and not a deficiency of growth factors. J Cell Physiol. 1997;170:192–199. doi: 10.1002/(SICI)1097-4652(199702)170:2<192::AID-JCP11>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 90.Juin P, Hueber AO, Littlewood T, Evan G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999;13:1367–1381. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morrish F, Giedt C, Hockenbery D. c-MYC apoptotic function is mediated by NRF-1 target genes. Genes Dev. 2003;17:240–255. doi: 10.1101/gad.1032503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gu G, Deutch AY, Franklin J, Levy S, Wallace DC, Zhang J. Profiling genes related to mitochondrial function in mice treated with N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. 2003;308:197–205. doi: 10.1016/s0006-291x(03)01233-6. [DOI] [PubMed] [Google Scholar]
- 94.Smith JV, Burdick AJ, Golik P, Khan I, Wallace D, Luo Y. Anti-apoptotic properties of Ginkgo biloba extract EGb 761 in differentiated PC12 cells. Cell Mol Biol (Noisy-le-grand) 2002;48:699–707. [PubMed] [Google Scholar]
- 95.Wagner BK, Kitami T, Gilbert TJ, Peck D, Ramanathan A, Schreiber SL, Golub TR, Mootha VK. Large-scale chemical dissection of mitochondrial function. Nat Biotechnol. 2008 doi: 10.1038/nbt1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.