

Abstract
Traumatic brain injury (TBI) causes microglial activation and related neurotoxicity that contributes to chronic neurodegeneration and loss of neurological function. Selective activation of metabotropic glutamate receptor 5 (mGluR5) by the orthosteric agonist (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), is neuroprotective in experimental models of TBI, and has potent anti-inflammatory effects in vitro. However, the therapeutic potential of CHPG is limited due to its relatively weak potency and brain permeability. Highly potent, selective and brain penetrant mGluR5 positive allosteric modulators (PAMs) have been developed and show promise as therapeutic agents. We evaluated the therapeutic potential of a novel mGluR5 PAM, VU0360172, after controlled cortical impact (CCI) in mice. Vehicle, VU0360172, or VU0360172 plus mGluR5 antagonist (MTEP), were administered systemically to CCI mice at 3 h post-injury; lesion volume, hippocampal neurodegeneration, microglial activation, and functional recovery were assessed through 28 days post-injury. Anti-inflammatory effects of VU0360172 were also examined in vitro using BV2 and primary microglia. VU0360172 treatment significantly reduced the lesion, attenuated hippocampal neurodegeneration, and improved motor function recovery after CCI. Effects were mediated by mGluR5 as co-administration of MTEP blocked the protective effects of VU0360172. VU0360172 significantly reduced CD68 and NOX2 expression in activated microglia in the cortex at 28 days post-injury, and also suppressed pro-inflammatory signaling pathways in BV2 and primary microglia. In addition, VU0360172 treatment shifted the balance between M1/M2 microglial activation states towards an M2 pro-repair phenotype. This study demonstrates that VU0360172 confers neuroprotection after experimental TBI, and suggests that mGluR5 PAMs may be promising therapeutic agents for head injury.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-014-0298-6) contains supplementary material, which is available to authorized users.
Keywords: Traumatic brain injury, Metabotropic glutamate receptor 5, Positive allosteric modulator, Neuroprotection, Functional recovery, Microglial activation
Introduction
Allosteric modulators target a site on receptors separate from the orthosteric binding site of the endogenous ligand, and can either potentiate (positive allosteric modulator; PAM) or inhibit (negative allosteric modulator; NAM) receptor function [1]. For some G-protein coupled receptors (GPCRs), allosteric modulators can provide greater subtype selectivity than orthosteric ligands. Further, allosteric modulators can display functional selectivity by targeting downstream signaling pathways critical for therapeutic efficacy without modulating signaling pathways that may lead to adverse side effects [2].
Metabotropic glutamate receptors (mGluRs) are GPCRs that are promising drug targets for psychiatric and neurodegenerative diseases [3], as well as CNS injury [4]. mGluRs are divided into three subgroups based on agonist binding, signal transduction pathways, and sequence homology [3]. Group I mGluRs include mGluR1 and mGluR5, which are coupled to Gq/11 and mediate IP3/Ca2+ signal transduction. In neurons, group I mGluRs are localized post-synaptically where they regulate cell excitability and post-synaptic efficacy [3]. Although mGluR1 and mGluR5 share certain common signal transduction mechanisms, they show remarkably different profiles in CNS injury [4]. Agonists of mGluR1 exacerbate necrotic cell death [5], whereas selective antagonists of mGluR1 are neuroprotective in vitro and in vivo [6–8]. In contrast, activation of mGluR5 inhibits caspase-dependent apoptosis and is neuroprotective in neuronal cell death models in vitro [5, 9–11] and in multiple CNS injury models [12–17].
mGluRs are also expressed on glia (astrocytes, microglia and oligodendrocytes) and are activated under normal physiological as well as pathophysiological conditions [18]. We have shown that mGluR5 is expressed in microglia [19], and that a selective mGluR5 orthosteric agonist, (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), reduced microglial activation and the associated release of pro-inflammatory mediators following stimulation with either lipopolysaccharide (LPS) or interferon-γ (IFNγ) [19, 20]. CHPG treatment also reduced NADPH oxidase (NOX2) activity levels and abolished the neurotoxic potential of activated microglia in microglia/neuron co-culture models. The protective effects of CHPG were blocked by mGluR5 knockout or by addition of the mGluR5 antagonist, MTEP, demonstrating a selective mGluR5-mediated neuroprotective effect [19, 20]. Furthermore, we demonstrated that siRNAs directed against either of the two membrane subunits of NOX2 (p22phox or gp91phox) abolished CHPG’s anti-inflammatory effects, which indicated that its protective actions are mediated, in part, by inhibition of NOX2 in microglia [20].
Following experimental traumatic brain injury (TBI) we found that acute central administration of CHPG significantly limited post-traumatic sensorimotor and cognitive deficits, reduced lesion volumes, and attenuated post-traumatic microglial activation [16]. Further, we demonstrated persistent microglial activation in the TBI cortex through 1 year post-injury that was associated with progressive lesion expansion and hippocampal neurodegeneration [21]. Remarkably, a markedly delayed, single dose central administration of CHPG at 1 month post-TBI, reduced microglial activation, limited lesion progression, decreased hippocampal neurodegeneration and white matter loss, and significantly improved motor and cognitive recovery after experimental TBI [13]. These data indicate that targeting of mGluR5, even as late as 1 month post-injury, is highly neuroprotective after TBI. However, CHPG has a number of potential problems as a therapeutic agent. These include limited selectivity, poor penetration through the blood-brain barrier, and rapid desensitization of the mGluR5 receptor [22].
In recent years a number of mGluR5 PAMs have been developed and investigated as potential treatment for CNS conditions ranging from schizophrenia and anxiety disorders [23–26] to learning and cognition-related problems [27–30]. We demonstrated recently that selected first-generation mGluR5 PAMs significantly attenuated both LPS- and IFNγ-induced nitric oxide (NO) and tumor necrosis factor-α (TNFα) release in cultured microglia [31], with higher potency compared to the orthosteric agonist, CHPG. These data support the use of mGluR5 PAMs as anti-inflammatory based neuroprotective agents.
Here, we evaluated the therapeutic potential of a novel mGluR5 PAM, VU0360172, in the controlled cortical impact (CCI) model in mice, and hypothesized that VU0360172 would confer neuroprotection after TBI by modulating post-traumatic neuroinflammation. VU0360172 (N-Cyclobutyl-6-[2-(3-fluorophenyl)ethynyl]-3-pyridinecarboxamide hydrochloride) is a highly potent mGluR5 PAM (EC50 = 16 nM; Ki = 195 nM) that has no significant effect on the agonist response of mGluRs 1, 3, or 4 [32]. VU0360172 is an aqueous soluble and orally bioavailable drug that penetrates the CNS and dose-dependently reduces amphetamine-induced hyper-locomotion in a rodent model of antipsychotic activity [32]. We administered VU0360172 systemically after CCI and evaluated functional recovery through 28 days post-injury, as well as lesion volume, hippocampal neurodegeneration and microglial activation. The potential anti-inflammatory effects of VU0360172 treatment were evaluated in vitro using BV2 and primary mouse microglia.
Methods
Controlled Cortical Impact
All surgical procedures were carried out in accordance with protocols approved by University of Maryland School of Medicine Institutional Animal Care and Use Committee (IACUC). Our custom-designed CCI TBI device [33] consists of a microprocessor-controlled pneumatic impactor with a 3.5 mm diameter tip. Adult male C57Bl/6 mice (3 months old, 22–25 g; Taconic Farms, Rensselaer, NY) were anesthetized with isoflurane evaporated in a gas mixture containing 70 % N2O and 30 % O2 and administered through a nose mask (induction at 4 % and maintenance at 2 %). Depth of anesthesia was assessed by monitoring respiration rate and pedal withdrawal reflexes. Mice were placed on a heated pad, and core body temperature was maintained at 37 °C. The head was mounted in a stereotaxic frame, and the surgical site was clipped and cleaned with Nolvasan and ethanol scrubs. A 10-mm midline incision was made over the skull, the skin and fascia were reflected, and a 4-mm craniotomy was made on the central aspect of the left parietal bone. The impounder tip of the injury device was then extended to its full stroke distance (44 mm), positioned to the surface of the exposed dura, and reset to impact the cortical surface. Moderate-level CCI was induced using an impactor velocity of 6 m/s and deformation depth of 2 mm. After injury, the incision was closed with interrupted 6-0 silk sutures, anesthesia was terminated, and the animal was placed into a heated cage to maintain normal core temperature for 45 min post-injury. All animals were monitored carefully for at least 4 h after surgery and then daily. Sham animals underwent the same procedure as injured mice except for the impact.
Drug Treatments
At 3 h and 24 h post-injury sham-injured and CCI mice received an intraperitoneal (i.p.) injection of the mGluR5 PAM, VU0360172 {N-cyclobutyl-6-[2-(3-fluorophenyl)ethynyl]-3-pyridinecarboxamide hydrochloride; 50 mg/kg in saline + 10 % Tween-80} or equal volume vehicle (saline + 10 % Tween-80). A 5 mg/ml stock of VU0360172 was prepared and injected intraperitoneally at 10 ml/kg resulting in a final dose of 50 mg/kg per animal. Another group of TBI mice received the selective mGluR5 antagonist 3-[(2-Methyl- 4-thiazolyl)ethynyl]pyridine (MTEP; 10 mg/kg in saline) by intraperitoneal injection 15 min prior to VU0360172 treatment. Drug dosages were based upon prior in vivo pharmacological studies for VU0360172 in rodents [32], and MTEP administration following CCI in mice [13]. Sham-injured (craniotomy only) vehicle- and VU0360172-treated mice were included as controls in the study. The following n were included for each group: sham (vehicle) = 9, sham (VU0360172) = 8, TBI (vehicle) = 14, TBI (VU0360172), TBI (MTEP + VU0360172) = 8.
Motor Function
Fine motor co-ordination was assessed at 1, 3, 7, 14, 21 and 28 days post-injury using the beam walk task, as previously described [34]. Briefly, mice were trained for 3 days to cross a narrow wooden beam (6 mm wide and 120 cm in length), which was suspended 30 cm above a 60 mm thick foam rubber pad. The number of foot-faults for the right hindlimb was recorded over 50 steps on each day of testing by an investigator blinded to treatment groups. A basal level of competence at this task was established prior to sham-injury or CCI, with an acceptance level of <10 faults per 50 steps.
Stereological assessment of lesion volume and hippocampal neurodegeneration
At 28 days post-injury mice were anesthetized (>100 mg/kg Euthasol, i.p.) and transcardially perfused with 100 ml of 0.9 % saline followed by 300 ml 4 % paraformaldehyde. The brains were carefully removed and post-fixed in 4 % paraformaldehyde overnight and then cryoprotected in 30 % sucrose. Coronal sections were cut (3 × 60 μm followed by 3 × 20 μm sections) and serially collected throughout the injured brain, starting at +1.78 mm from the bregma. Sections were mounted onto glass slides for immunohistochemical analysis. Cresyl violet stained 60 μm coronal sections were used for neuronal loss and lesion volume analysis. TBI lesion volume was quantified based on the Cavalieri method of stereology as described previously [35]. Briefly, out of a total of 96 60 μm sections, every 8th section was analyzed beginning from a random start point (i.e., random selection of a 60 μm section from numbers 1 to 6), and a total of 12 sections were analyzed. Using the Cavalieri estimator with a grid spacing of 100 μm the missing tissue on the injured hemisphere was outlined using Stereoinvestigator software (MBF Biosciences, Williston, VT) to quantify the lesion size. To quantify hippocampal neuronal cell loss, the optical fractionator method of stereology was employed as previously described [35]. Briefly, every fourth 60 μm section between –1.22 mm and –2.54 mm from bregma was analyzed beginning from a random start point, (which was the section where different hippocampal sub-regions were distinctly visible). A total of 5 sections were analyzed. The optical dissector had a size of 50 μm × 50 μm in the x and y-axis, respectively, with a height of 10 μm and guard-zone of 4 μm from the top of the section. The sampled region for each hippocampal sub-region was demarcated in the injured hemisphere and cresyl-violet neuronal cell bodies were counted using Stereoinvestigator software. For the Cornu Ammonis (CA) 1 (CA1) and CA2/3 sub-regions a grid spacing of 75 μm in the x-axis and 100 μm in the y-axis was used, resulting in an area fraction of 1/12. For the dentate gyrus sub-region, a grid spacing of 175 μm in the x-axis and 100 μm in the y-axis was used, resulting in an area fraction of 1/28. The volume of each hippocampal sub-region was measured using the Cavalieri estimator method with a grid spacing of 50 μm. The number of surviving neurons in each hippocampal sub-region was divided by the volume of the region of interest to obtain the cellular density expressed in counts/mm3. According to best stereological practice all stereological probes were optimized to obtain a Gunderson coefficient of error (CE, m = 1) value of less than 0.10 for the TBI tissue.
Immunofluorescence Staining and Image Analysis
Immunofluorescence staining was performed on 20 μm frozen coronal sections using standard techniques. Briefly, sections were washed with 1× PBS (3 times), blocked for 1 h with 5 % donkey serum containing 0.3 % Triton X-100, and incubated overnight at 4 °C with the appropriate primary antibodies made up in 2 % donkey serum containing 0.3 % Triton X-100. For double immunofluorescence staining, sections were incubated with the following combinations of antibodies: goat anti-arginase 1 (N-20) (1:300; #SC-18351, Santa Cruz Biotechnology, Santa Cruz, CA) and rat anti-mouse CD11b [OX-42] (1:200; #MCA74GA, AbD Serotec, Raleigh, NC); rabbit anti-iNOS (1:500; #482728, EMD Millipore, Billerica, MA) and rat mouse anti-CD11b [OX-42] (1:200); mouse anti-gp91phox (1:400; #611415, BD Transduction Labs, Franklin Gardens, NJ) and rabbit anti-iba1 (1/300; #019-19741, Wako Chemicals, Richmond, VA). For CD68 immunofluorescnece staining, sections were incubated with rat anti-CD68 [ED1] (1:200; #MCA1957GA, AbD Serotec). Sections were then washed with 1× PBS (3 times) and incubated for 1 h with the appropriate Alexa Fluor conjugated secondary antibodies (Life Technologies, Grand Island, NY). Sections were washed with 1× PBS (3 times) and counterstained with 4′, 6-diamidino-2-phenylindole (1 mg/mL; Sigma-Alrich, St. Louis, MO), air-dried and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). In each treatment group, a total of 35–45 sections (n = 5 animals per group) were examined using a Nikon Eclipse Ti microscope (Nikon Instruments, Melville, NY). Image analysis was performed using the NIS-Element AR analysis software (Nikon Instruments) to determine the degree of overlap between arginase-1, iNOS or gp91phox immunoreative cells and CD11b- or Iba1-expressing cells. Results were expressed as Mander’s overlap coefficients. The same software was used to measure CD68 fluorescence intensity, and results were expressed as CD68 mean signal intensity.
In Vitro Studies
BV2 microglia (murine microglial cell line) were grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10 % fetal equine serum (Hyclone, Logan, UT), and 1 % penicillin and streptomycin (Invitrogen) at 37 °C with 5 % CO2. BV2 microglia were seeded at 2 × 105 cells/well in a 96-well plate for 24 h. Primary cortical microglia were obtained from postnatal day 1 C57Bl/6 mouse pups and cultured as previously described [20]. Briefly, the whole brain was dissected carefully and homogenized in L15 medium (Invitrogen). Mixed glial cultures were incubated for 8–10 days at 37 °C and 5 % CO2 in DMEM supplemented with 10 % fetal calf serum (Hyclone), 1 % l-glutamine (Invitrogen), 1 % sodium pyruvate (Invitrogen), and 1 % penicillin and streptomycin (Invitrogen). Mixed glial cultures were shaken for 1 h at 100 rpm and 37 °C, and detached microglia were collected and seeded at 4 × 105 cells per well in a 96-well plate in DMEM containing 10 % fetal calf serum. Cells were pre-treated with increasing concentrations of mGluR5 PAM, VU0360172 (1–50 μM), or mGluR5 orthosteric agonist, CHPG (2,000 μM; Tocris Bioscience, Ellisville, MO), for 1 h prior to stimulation with either lipopolysaccharide (LPS; 50 ng/mL for BV2s or 10 ng/ml for primary microglia, Sigma-Aldrich) or interferon-γ (IFNγ; 0.2 ng/ml for BV2s or 20 ng/ml for primary microglia; R&D Systems, Minneapolis, MN). At 24 h later, microglial activation was assessed by measurement of nitric oxide (NO) and tumor necrosis factor-α (TNFα) release into the culture medium. NO release was assayed using a Greiss reagent assay (#G7921; Invitrogen), according to the manufacturer’s instructions. NO concentrations were calculated using standard curves generated from a nitrite stock, and results were expressed in micromoles. TNFα release was assayed using a sandwich enzyme-linked immunosorbent assay for mouse TNFα (#DY410, R&D Systems), according to the manufacturer’s instructions. Cytokine concentrations were calculated using standard curves generated from recombinant mouse TNFα, and results were expressed in picograms per milliliter. In addition, cell viability was assayed using the CytoTox96 NonRadioactive Cytotoxicity Assay (#G1780, Promega, Madison, WI), according to the manufacturer’s instructions. In each assay, absorbance was measured using a Synergy HT Multi-Mode Microplate Reader (Biotek, Winooski, VT). For Western blot analysis, BV2 microglia were seeded at 2 × 106 cells/well in a 24-well plate for 24 h. Cells were pre-treated with 20 μM VU0360172 for 1 h prior to stimulation with 50 ng/ml LPS; 24 h later, protein was extracted using RIPA buffer, equalized, and loaded onto 5–20 % gradient gels for SDS PAGE (Bio-Rad; Hercules, CA). Proteins were transferred onto nitrocellulose membranes, and then blocked overnight in 5 % milk in 1× PBS containing 0.05 % Tween-20 (PBS-T). The membrane was incubated in rabbit anti-iNOS (1:300; #482728, EMD Millipore), goat anti-arginase 1 (N-20) (1:200; #SC-18351, Santa Cruz Biotechnology) or rabbit anti-Ym1 (1:1,000; 01404, Stem Cell Technologies, Vancouver, BC), β-actin (1:2,000; Sigma) for 2 h at room temperature, then washed three times in PBS-T, and incubated in appropriate HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. Membranes were washed three times in PBS-T, and proteins were visualized using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Rockford, IL). Chemiluminescence was captured ChemiDoc™ XRS+ System (Bio-Rad), and protein bands were quantified by densitometric analysis using BioRad Molecular Imaging Software. The data presented reflects the intensity of target protein band normalized based on the intensity of the endogenous control for each sample (expressed in arbitrary units).
Statistical Analysis
Quantitative data were expressed as mean ± standard errors of the mean (S.E.M.). Stereological and behavioral analyses were performed by an investigator blinded to treatment group. Motor function recovery was analyzed by one-way repeated measures ANOVA to determine the interactions of post-injury trial and groups, followed by post-hoc adjustments using a Tukey’s multiple comparison test. Lesion volume, hippocampal neuronal cell loss, TNFα, NO, and viability assays in microglia were analyzed by one-way ANOVA, followed by post-hoc adjustments using the Bonferonni multiple comparisons test. Western blots were analyzed by two-way ANOVA (drug treatment x stimulation), followed by post-hoc adjustments using the Bonferonni multiple comparisons test. Colocalization of immunolabled proteins in the injured cortex was analyzed using Mander’s overlap coefficient and unpaired t-tests. Statistical analysis was performed using the GraphPad Prism Program, Version 3.02 for Windows (GraphPad Software, San Diego, CA). A P < 0.05 was considered statistically significant.
Results
VU0360172 Improves Motor Function Recovery after TBI
To evaluate the neuroprotective effects of the novel mGluR5 PAM we administered VU0360172 systemically (50 mg/kg, i.p.) to moderate-level CCI mice at 3 h and 24 h post-injury, and compared them to vehicle (saline + 10 % Tween-80)-treated CCI mice. We also administered the mGluR5 antagonist, MTEP (10 mg/kg, i.p.), 15 min prior to VU0360172 treatment to another cohort of CCI mice to determine if protective effects were specifically mediated by mGluR5 receptor stimulation. Long-term motor and cognitive function recovery after CCI was assessed using beam walk and Morris water maze tests, respectively. Functional assessment of fine motor coordination was assessed on days 1, 3, 7, 14, 21 and 28 post-injury using a well-characterized beam walk test [36]. The interaction of ‘post-injury day X group’ in the beam walk test was statistically significant [F(20, 225) = 2.990, P < 0.0001]. CCI induced significant sensorimotor impairments at all time points when compared to sham-injured vehicle-treated mice (P < 0.001 vs vehicle; Fig. 1). VU0360172-treated CCI mice had significantly improved sensorimotor performance in this test on day 28 (P < 0.05) when compared to vehicle-treated CCI mice. In contrast, CCI mice that were co-administered VU0360172 + MTEP had similar performance to the vehicle-treated CCI mice. We also assessed spatial learning on days 14–17 post-injury using the acquisition phase of the Morris water maze test [34]. However, VU0360172 treatment failed to improve spatial learning performance after CCI (data not shown).
Fig. 1.
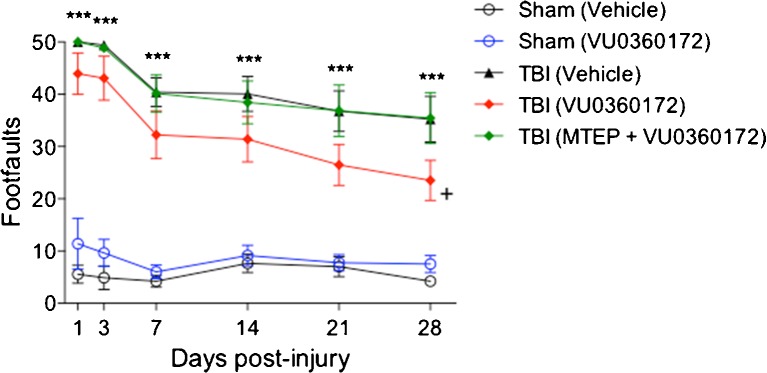
Systemic administration of VU0360172 improves motor function recovery after Traumatic brain injury (TBI). Fine motor coordination deficits were quantified using the beam walk test. Controlled cortical impact (CCI) induced significant sensorimotor impairments in vehicle-treated mice at all time points when compared to sham-injured vehicle-treated group (*** P < 0.001). VU0360172-treated CCI mice had significantly improved sensorimotor performance on day 28 (+ P < 0.05) when compared to vehicle-treated CCI mice. In contrast, CCI mice that were co-administered MTEP + VU0360172 had similar performance to the vehicle-treated CCI mice. Analysis by 2-way repeated measures ANOVA, followed by post-hoc adjustments using a Tukey’s multiple comparisons test. Mean ± standard error of the mean (S.E.M.) [sham (vehicle) = 9, sham (VU0360172) = 8, TBI (vehicle) = 14, TBI (VU0360172), TBI (MTEP + VU0360172) = 8]
VU0360172 Treatment Reduces Cortical Tissue Loss and Hippocampal Neurodegeneration After TBI
Following completion of the behavioral studies all animals were euthanized at 28 days post-injury and processed for histological analysis. We assessed TBI lesion volumes in cresyl violet stained coronal sections from randomly selected sub-groups of CCI mice in each of the vehicle- (n = 8), VU0360172- (n = 8), and MTEP+VU0360172- (n = 7) treated groups. Representative images of CCI tissue from each treatment group are presented in Fig. 2a. Stereological analysis revealed that CCI resulted in a large lesion in vehicle-treated mice (5.34 ± 0.43 mm3; Fig. 2b), and that the CCI lesion was reduced significantly in VU0360172-treated mice (3.57 ± 0.43 mm3; P < 0.05). CCI mice that received the combined treatment of MTEP and VU0360172 had similar lesions (5.59 ± 0.69 mm3) to the vehicle-treated CCI mice. We then assessed post-traumatic neuronal loss in the CA1, CA2/3 and dentate gyrus sub-regions of the hippocampus in sham-injured and CCI mice using stereological techniques. Representative high-powered images from the CA1, CA2/3 and dentate gyrus of vehicle-, VU0360172-, and MTEP+VU0360172-treated CCI groups are presented in Fig. 3a. CCI caused a significant loss of CA1 (P < 0.001), CA2/3 (P < 0.001), and dentate gyrus (P < 0.01) neurons in the vehicle-treated CCI group when compared to sham-injured vehicle-treated controls (Fig. 3b). VU0360172 treatment resulted in robust neuroprotection in the hippocampus, such that neuronal densities in the CA1 (P < 0.05) and the CA2/3 (P < 0.01) were increased significantly in the VU0360172-treated CCI group when compared to the vehicle-treated CCI group. Furthermore, co-administration of MTEP abrogated the neuroprotective effects of VU0360172 treatment, and there was a significant difference between the VU0360172-treated and MTEP+VU0360172-treated CCI groups in each sub-region [CA1 (P < 0.001), CA2/3 (P < 0.001), and dentate gyrus (P < 0.01)]. There was no significant difference in neuronal densities between the sham-injured vehicle-treated and the sham-injured VU0360172-treated groups. Thus, VU0360172 treatment resulted in a significantly reduced cortical lesion size and hippocampal neurodegeneration after injury. Moreover, our data demonstrate that the therapeutic actions of VU0360172 were mediated via selective mGluR5 receptor stimulation as co-administration of the mGluR5 antagonist, MTEP, blocked the neuroprotective effects VU0360172 treatment.
Fig. 2.

a, b VU0360172 treatment reduces TBI-induced lesion volume at 28 days post-injury. a TBI lesion volume was assessed by stereological methods in a subset of vehicle-, VU0360172, and MTEP + VU0360172-treated CCI mice at 28 days post-injury, and representative images of each group are shown. B The VU0360172-treated CCI group had significantly reduced lesion volumes (* P < 0.05) when compared to vehicle-treated CCI group. The vehicle- and MTEP + VU0360172-treated CCI groups had comparable lesion volumes. Analysis by 1-way ANOVA, followed by post-hoc adjustments using a Bonferonni multiple comparisons test. Mean ± S.E.M. [TBI (vehicle) = 8, TBI (VU0360172) = 8, TBI (MTEP + VU0360172) = 7]
Fig. 3.

a, b VU0360172 treatment reduces hippocampal neurodegeneration at 28 days post-injury. a Neuronal loss in the CA1, CA2/3 and dentate gyrus subfields of the hippocampus was assessed by stereological methods in vehicle-, VU0360172-, and MTEP + VU0360172-treated CCI mice at 28 days post-injury, and representative images of each group are shown. b CCI induced significant loss of CA1 (*** P < 0.001), CA2/3 (*** P < 0.001), and dentate gyrus (** P < 0.01) neurons in the vehicle-treated CCI group when compared to sham-injured vehicle-treated group. The VU0360172-treated CCI group had significantly reduced neuronal loss in the CA1 (+ P < 0.05) and the CA2/3 (++ P < 0.01) when compared to the vehicle-treated CCI group. There was a significant difference between the VU0360172-treated and MTEP + VU0360172-treated CCI groups in each sub-region [CA1 (^^^ P < 0.001), CA2/3 (^^^ P < 0.001), and dentate gyrus (^^ P < 0.01)]. Analysis by one-way ANOVA, followed by post-hoc adjustments using a Bonferonni multiple comparisons test. Mean ± S.E.M. [TBI (vehicle) = 8, TBI (VU0360172) = 8, TBI (MTEP + VU0360172) = 7]. Bar 25 μm
VU0360172 is Anti-Inflammatory and Polarizes Microglia Towards an M2 Phenotype In Vitro
Previously, we demonstrated that selective activation of microglial mGluR5 receptors by CHPG had strong anti-inflammatory effects and attenuated microglial activation in vitro and in vivo [13, 19, 16, 20]. To test whether VU0360172 has similar anti-inflammatory properties, we pre-treated cultured BV2 microglia for 1 h with increasing concentrations of VU0360172 (1–50 μM) and then stimulated the cells with/without LPS (50 ng/ml) or IFNγ (0.2 ng/ml) for a further 24 h. We included a CHPG treatment group (2,000 μM) as a positive control, and measured TNFα and NO release in the medium to assess microglial activation, as well as LDH release to assess cell cytotoxicity. VU0360172 pre-treatment significantly reduced LPS-stimulated TNFα production in a dose-dependent manner starting at 1 μM (P < 0.001; Fig. 4a), and significantly reduced NO production at 20 μM (P < 0.01; Fig. 4a). Similarly, VU0360172 pre-treatment significantly reduced IFNγ-stimulated TNFα production starting at 20 μM (P < 0.001; Fig. 4b), and significantly reduced NO production at 20 μM (P < 0.01; Fig. 4a). CHPG treatment also significantly reduced TNFα and NO release in both stimulation models (P < 0.001), whereas VU0360172 treatment alone did not produce either pro-inflammatory mediator. VU0360172 at a concentration of 50 μM was cytotoxic to BV2 microglia, but not at lower concentrations at which significant anti-inflammatory effects were observed (data not shown). To confirm the anti-inflammatory effects of VU0360172 in primary microglia we repeated these studies using microglia isolated from p1 C57Bl/6 mouse pups. We pre-treated primary microglia for 1 h with 20 μM VU0360172 and stimulated the cells with/without LPS (10 ng/ml) or IFNγ (20 ng/ml) for a further 24 h. VU0360172 treatment significantly reduced TNFα and NO release in response to LPS stimulation (P < 0.001; Fig. 4c), and in response to IFNγ stimulation (P < 0.001; Fig. 4d), thereby confirming significant anti-inflammatory properties of the mGluR5 PAM, VU0360172, in primary microglia.
Fig. 4.

a–d VU0360172 attenuates microglial activation in vitro. The anti-inflammatory effects of VU0360172 were assessed using lipopolysaccharide (LPS) or interferon-γ (IFNγ) models of microglial activation in BV2 and primary microglia. a LPS stimulation resulted in a significant increase in TNFα release and nitrite production (*** P < 0.001 vs control for both) in BV2 microglia. VU0360172 treatment significantly reduced LPS-stimulated TNFα release starting at 1 μM (+++ P < 0.001), and LPS-stimulated nitrite production at 20 μM (++ P < 0.01). Similarly, CHPG treatment (positive control) significantly reduced the release of both pro-inflammatory mediators (+++ P < 0.001 vs LPS), whereas VU0360172 treatment alone did not produce TNFα or nitrite. b IFNγ stimulation resulted in a significant increase in TNFα release and nitrite production (*** P < 0.001 vs control for both) in BV2 microglia. VU0360172 treatment significantly reduced IFNγ-stimulated TNFα release starting at 20 μM (+++ P < 0.001), and IFNγ-stimulated nitrite production at 20 μM (++ P < 0.01). Similarly, CHPG treatment (positive control) significantly reduced the release of both pro-inflammatory mediators (+++ P < 0.001 vs IFNγ). Analysis by one-way ANOVA, followed by post-hoc adjustments using a Bonferonni multiple comparisons test. Mean ± S.E.M. (n = 6/treatment). c LPS stimulation resulted in a significant increase in TNFα release and nitrite production (*** P < 0.001 vs control for both) in primary microglia; 20 μM VU0360172 significantly reduced LPS-stimulated TNFα release and nitrite production (+++ P < 0.001), whereas VU0360172 treatment alone had no effect. d IFNγ stimulation resulted in a significant increase in TNFα release and nitrite production (*** P < 0.001 vs control for both) in primary microglia; 20 μM VU0360172 significantly reduced IFNγ-stimulated TNFα release and nitrite production (+++ P < 0.001), whereas VU0360172 treatment alone had no effect. Analysis by 2-way ANOVA, followed by post-hoc adjustments using a Bonferonni multiple comparisons test. Mean ± S.E.M. (n = 6/treatment)
Microglia can be polarized towards an M1- or M2-activation phenotype depending on the stimuli in their local microenvironment [37]. We chose to evaluate polarization in BV2 microglia in order to determine whether VU0360172 treatment could alter microglial polarization following LPS stimulation. BV2 cells were pre-treated with 20 μM VU0360172 for 1 h prior to LPS stimulation (50 ng/ml), and samples were collected for analysis of M1 (iNOS) and M2 (arginase 1 and Ym1) markers after 24 h. Compared to control-treated cells there was a significant increase in iNOS (M1) expression following LPS stimulation (P < 0.001; Fig. 5a,b), which was significantly reduced by VU0360172 treatment (P < 0.001). In addition, BV2 microglia treated with VU0360172 alone had reduced iNOS expression when compared to control-treated cells (P < 0.05). Notably, pre-treatment with VU0360172 prior to LPS stimulation resulted in a significant increase in expression of M2 markers, arginase 1 (P < 0.001; Fig. 5a,c), and Ym1 (P < 0.001; Fig. 5a,d), in BV2 microglia. These data indicate that the mGluR5 PAM, VU0360172, can repolarize microglia towards an M2 activation state following LPS stimulation.
Fig. 5.

a–d VU0360172 treatment alters BV2 microglial polarization following LPS stimulation. a Western blots for M1 polarization (iNOS) and M2 (arginase 1 and Ym1) markers in LPS-stimulated BV2 microglia with/without VU0360172 treatment. b Quantification of iNOS protein expression. LPS significantly increased iNOS expression (*** P < 0.001) when compared to control-treated samples, whereas VU0360172 treatment significantly reduced iNOS protein (+++ P < 0.001). VU0360172 alone treatment significantly reduced iNOS protein (* P < 0.05) when compared to control-treated samples. c Quantification of arginase 1 protein expression. VU0360172 treatment prior to LPS resulted in a significant increase in arginase 1 protein (*** P < 0.001) compared to LPS treatment alone. d Quantification of Ym1 protein expression. VU0360172 treatment prior to LPS resulted in a significant increase in Ym1 protein (*** P < 0.001) compared to LPS treatment alone. Analysis by 2-way ANOVA, followed by post-hoc adjustments using a Bonferonni multiple comparisons test. Mean ± S.E.M. (n = 4/treatment)
VU0360172 Attenuates NOX2 Expression in Reactive Microglia at 28 Days Post-Injury and Promotes an M2 Activation Phenotype
Previously, we showed that knockout of NOX2 abolished the anti-inflammatory effects of mGluR5 stimulation by CHPG in activated microglia [16], and that CHPG treatment following CCI resulted in significantly reduced microglial activation and NOX2 expression at 4 months post-injury [13]. Here, we evaluated NOX2 expression in microglia in vehicle- and VU0360172-treated CCI groups by double immunofluorescence staining for gp91phox (NOX2) and Iba1 (microglia). gp91phox was highly expressed in Iba1-positive microglia in the injured cortex of vehicle-treated CCI group at 28 days post-injury, whereas its expression was markedly reduced in the VU0360172-treated CCI group (Fig. 6a). Analysis of gp91phox/Iba1 co-localization demonstrated that VU0360172 treatment significantly reduced gp91phox expression (P < 0.05) at 28 days post-injury (Fig. 6c). We evaluated the expression of CD68, a marker of highly activated phagocytic microglia [38], in each group. CD68 expression was highly up-regulated in the vehicle-treated CCI group (Fig. 6b), and its expression was significantly reduced in the VU0360172-treated CCI group (P < 0.05) (Fig. 6b,d). We also analyzed iNOS and arginase 1 expression in the injured cortex at 28 days post-injury as phenotypic markers of M1- and M2-polarized microglia [37], respectively. We performed double immunofluorescence staining for either iNOS or arginase 1 in combination with CD11b to confirm the expression of phenotypic markers in microglia. iNOS was highly expressed in CD11b-positive microglia in the injured cortex of vehicle-treated CCI group at 28 days post-injury, whereas its expression was reduced markedly in the VU0360172-treated CCI group (Fig. 7a). Analysis of iNOS/CD11b co-localization demonstrated that VU0360172 treatment significantly reduced iNOS expression (P < 0.05) (Fig. 7c). In contrast, arginase 1 expression was markedly increased in the VU0360172-treated CCI group (Fig. 7b), and analysis of arginase 1/CD11b co-localization demonstrated that VU0360172 treatment significantly increased arginase 1 expression (P < 0.05) when compared with the vehicle-treated CCI group (Fig. 7d). In summary, our in vitro and in vivo studies indicate that VU0360172 has significant anti-inflammatory properties, and administration of VU0360172 starting at 3 h post-injury reduces the expression of NOX2, CD68 and iNOS in highly activated microglia at 28 days post-injury, and repolarizes microglia towards an M2 phenotype with high arginase 1 expression.
Fig. 6.

a–d VU0360172 treatment reduces the expression of NOX2 and CD68 in microglia at 28 days post-injury. a, c Double immunofluorescence staining for the NOX2 subunit, gp91phox (green), and microglia (Iba-1; green) in the injured cortex of vehicle- and VU0360172-treated CCI samples at 28 days post-injury. Analysis of the Mander’s overlap coefficient for gp91phox+/Iba1+ demonstrated that VU0360172 treatment significantly reduced the expression of NOX2 in microglia (* P < 0.05) when compared to the vehicle-treated CCI group. b, d Immunofluorescence staining for the CD68 (magenta) in the injured cortex of vehicle- and VU0360172-treated CCI samples at 28 days-post injury. VU0360172 treatment significantly reduced the expression of CD68 (* P < 0.05) when compared to the vehicle-treated CCI group. Unpaired t-test. Mean ± S.E.M. (total of 35–45 sections from n = 5/group)
Fig. 7.

a–d VU0360172 treatment reduces iNOS expression and up-regulates arginase 1 expression in microglia at 28 days post-injury. a, c Double immunofluorescence staining for iNOS (red) and microglia (CD11b; magenta) in the injured cortex of vehicle- and VU0360172-treated CCI samples at 28 days post-injury. Analysis of the Mander’s overlap coefficient for iNOS+ /CD11b+ demonstrated that VU0360172 treatment significantly reduced iNOS expression in microglia (* P < 0.05) when compared to the vehicle-treated CCI group. b, d Double immunofluorescence staining for arginase 1 (green) and microglia (CD11b; magenta) in the injured cortex of vehicle- and VU0360172-treated CCI samples at 28 days post-injury. Analysis of the Mander’s overlap coefficient for arginase 1+ /CD11b+ demonstrated that VU0360172 treatment significantly increased the expression of arginase 1 in microglia (* P < 0.05) when compared to the vehicle-treated CCI group. Unpaired t-test. Mean ± S.E.M. (total of 35–45 sections from n = 5/group)
Discussion
Allosteric modulators of mGluR5 represent promising drug candidates for the treatment of various psychiatric disorders and neurodegenerative diseases [2, 3], as well as CNS injury [4]. In the present study, systemic administration of the mGluR5 PAM VU0360172, starting at 3 h after CCI, significantly reduced the lesion volume, attenuated delayed hippocampal neurodegeneration, and improved motor function recovery. These effects were reversed by prior administration of the mGluR5 antagonist MTEP, indicating that the neuroprotection actions of VU0360172 were mediated through mGluR5 receptors.
The present study and previously published reports from our laboratory, and others, demonstrate that selective activation of mGluR5 has powerful neuroprotective effects in experimental models of CNS injury [12–17]. Following moderate-to-severe spinal cord contusion injury in rats we demonstrated that intrathecal CHPG administration improved motor recovery, reduced lesion volume, and spared white matter loss [14]. Recently we showed that central administration of CHPG at 30 min post-injury resulted in significantly improved sensorimotor and cognitive recovery and reduced lesion volumes after CCI [16]. Furthermore, we also demonstrated that when CHPG was administered to CCI mice at 1 month post-injury it arrested the expansion of the lesion over time, reduced white matter loss and hippocampal neurodegeneration, and improved functional recovery at 4 months [13]. Both acute and delayed CHPG treatment reduced chronic microglial activation and the expression of NOX2 in microglia after CCI [13, 16]. In addition, when we co-administered the NOX2 inhibitor, apocynin, in combination with CHPG there were no additive neuroprotective effects, suggesting that both drugs may act on the same molecular pathway [16].
Unfortunately, all known selective mGluR5 orthosteric agonists, including CHPG, have certain therapeutic limitations; these include lack of selectivity due to the high level of conservation in the ligand binding region, poor penetration through the blood-brain barrier, and rapid desensitization of the mGluR5 receptor following interaction with a direct agonist [22]. These challenges led to renewed efforts in recent years to develop new potent, selective and brain-penetrant mGluR5 PAMs [32]. These compounds increase mGluR5 activity by binding to less conserved regions of the receptor outside of the ligand binding region and stabilize the receptor’s active configuration; this results in higher selectivity and reduces the likelihood for desensitization [22]. mGluR5 PAMs allosterically potentiate receptor function; in physiological conditions they do not activate the target receptor by themselves but rather shift the concentration response for orthosteric agonists to the left [39].
VU0360172 is a novel, orally bioavailable mGluR5 PAM with an excellent pharmacokinetic profile [32]. VU0360172 has increased potency and functional selectivity when compared with CHPG, and it readily penetrates the CNS with peak brain levels achieved within 30 min following systemic (i.p.) administration (Cmax = 7 μM for 10 mg/kg dose) [40]. These properties markedly enhance the translational potential of this class of drugs as neuroprotective agents, and we therefore set out to determine if systemic administration of VU0360172 could improve functional and histological outcomes after TBI. We chose to administer 50 mg/kg VU0360172 at 3 h and 24 h post-injury in order to maintain mGluR5 receptor stimulation because prior pharmacokinetic analysis demonstrated an approximate 10-fold decrease in VU0360172 levels in the brain at 6 h post-administration [40]. These studies demonstrated that systemic delivery of VU0360172 starting at 3 h post-injury significantly improved sensorimotor recovery after CCI. Furthermore, stereological assessments demonstrated that treatment significantly reduced lesion volumes and hippocampal neuronal loss compared to vehicle-treated controls. Both functional and histological outcomes were blocked by co-administration of MTEP, suggesting that the therapeutic actions of VU0360172 were mediated by selective stimulation of mGluR5 receptors. The lack of effect of VU0360172 on cognitive recovery (data not shown) may reflect only partial hippocampal neuroprotection; future studies will focus on optimization of dose and duration of administration, as well as the use of additional more sensitive behavioral tests, to assess the effect of VU0360172 treatment on cognitive function recovery after CCI.
Consistent with our prior studies using the mGluR5 orthosteric agonist [13, 16], VU0360172 treatment modulated chronic microglial activation after CCI. Specifically, treatment significantly reduced the expression of CD68 and NOX2 in highly activated microglia within the cortical lesion at 28 days post-injury. NOX2 has been implicated as a common and essential mechanism of microglial-mediated neurotoxicity in neurodegenerative diseases [41], and we have demonstrated chronic expression of NOX2 in microglia up to 1 year following TBI [21]. Prior in vitro studies demonstrated that selective activation of mGluR5 in microglia inhibited NOX2 expression and activity, as well as reduced microglial-mediated neurotoxicity in a co-culture system [20]. In addition, studies using siRNAs for NOX2 and microglial cultures from NOX2-deficient mice showed that NOX2 was downstream of mGluR5 in microglia and a target for its neuroprotective actions [16, 20]. Our in vitro studies using BV2 microglia and primary microglia demonstrated that VU0360172 significantly reduced NO and TNFα release from LPS- and IFNγ-stimulated microglia at lower concentrations than previously shown for CHPG [19, 20] (20 μM for VU0360172 vs 2000 μM for CHPG). The lowest concentrations at which VU0360172 was able to significantly inhibit microglial activation were not cytotoxic; however, at concentrations of 50 μM and higher it negatively impacted BV2 microglial cell viability. These in vitro studies indicate that VU0360172 suppresses pro-inflammatory signaling pathways in microglia, and support prior studies that demonstrated anti-inflammatory actions of first generation mGluR5 PAMs (DFP, CDPPB) in LPS, IFNγ [31], and fibrinogen [42] models of microglial activation.
Microglia, like peripheral macrophages, have multiple activation phenotypes [43, 44], and depending on the stimuli in their local microenvironment they can be polarized to have distinct molecular phenotypes and effector functions [37]. For example, LPS or IFNγ, promote a ‘classical’ phenotype (M1), which produces high levels of pro-inflammatory cytokines and oxidative metabolites that are essential for host defense and phagocytic activity [38]. However, excessive M1-polarization can lead to exacerbation of injury and progressive tissue destruction (i.e., the M1 neurotoxic phenotype). Conversely, microglial activation in response to anti-inflammatory cytokines such as IL-4 or IL-10, promote ‘alternative’ (M2a) or ‘acquired deactivated’ (M2c) phenotypes, respectively [45, 46]. M2 microglia express specific markers such as arginase 1, mannose receptor (CD206), chitinase 3-like 3 (Ym1) and TGFβ [45], among others. M2 microglia promote repair processes, such as angiogenesis and extracellular matrix remodeling, while also suppressing destructive immune responses [37]. Using an LPS model of microglial activation we demonstrated that VU0360172 treatment attenuated M1 activation (reduced iNOS expression) and increases M2 activation (increased arginase 1 and Ym1 expression), which indicates that mGluR5 PAM signaling modulates microglia polarization in vitro. Furthermore, systemic administration of VU0360172 after CCI resulted in a significant decrease in the expression of iNOS and NOX2 in M1-polarized microglia in the injured cortex, and a concomitant increase in the expression of arginase 1 in M2-polarized microglia, thereby shifting the balance between M1/M2 microglial activation states towards an M2 phenotype at 28 days post-injury. Given that M2 polarized microglia are anti-inflammatory and display properties that may mediate tissue remodeling and repair [37], it is plausible that the increase expression of the M2 pro-repair microglial activation phenotype in the CCI cortex of VU0360172 treated mice may be, in part, responsible for improved motor recovery and reduced cortical tissue loss observed in this group.
In conclusion, our study underscores the neuroprotective effects of selective stimulation of mGluR5 receptors following experimental TBI, and suggests that such effects, in part, may reflect their ability to modulate the neurotoxic effects of posttraumatic neuroinflammation. The fact that an mGluR5 PAM is protective following systemic administration, and that this drug class is being actively studied for other neurological conditions, makes them promising compounds for the treatment of clinical TBI.
Electronic supplementary material
(PDF 213 kb)
Acknowledgments
We thank Marie Hanscom for expert technical assistance. This work was supported by a grant from the NIH/NINDS to AIF (5R01NS037313), and by a 2013 UMB Pilot & Exploratory Interdisciplinary Research (IDR) Award to B.S. and F.X.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nickols HH, Conn PJ. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis. 2014;61:55–71. doi: 10.1016/j.nbd.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicoletti F, Bockaert J, Collingridge GL, et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60:1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrnes KR, Loane DJ, Faden AI. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics. 2009;6:94–107. doi: 10.1016/j.nurt.2008.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen JW, Knoblach SM, Faden AI. Activation of group I metabotropic glutamate receptors reduces neuronal apoptosis but increases necrotic cell death in vitro. Cell Death Differ. 2000;7:470–476. doi: 10.1038/sj.cdd.4400678. [DOI] [PubMed] [Google Scholar]
- 6.Allen JW, Eldadah BA, Faden AI. Beta-amyloid-induced apoptosis of cerebellar granule cells and cortical neurons: exacerbation by selective inhibition of group I metabotropic glutamate receptors. Neuropharmacology. 1999;38:1243–1252. doi: 10.1016/S0028-3908(99)00044-1. [DOI] [PubMed] [Google Scholar]
- 7.Fei Z, Zhang X, Bai HM, Jiang XF, Wang XL. Metabotropic glutamate receptor antagonists and agonists: potential neuroprotectors in diffuse brain injury. J Clin Neurosci. 2006;13:1023–1027. doi: 10.1016/j.jocn.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 8.Szydlowska K, Kaminska B, Baude A, Parsons CG, Danysz W. Neuroprotective activity of selective mGlu1 and mGlu5 antagonists in vitro and in vivo. Eur J Pharmacol. 2007;554:18–29. doi: 10.1016/j.ejphar.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 9.Movsesyan VA, Stoica BA, Faden AI. MGLuR5 activation reduces beta-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J Neurochem. 2004;89:1528–1536. doi: 10.1111/j.1471-4159.2004.02451.x. [DOI] [PubMed] [Google Scholar]
- 10.Vincent AM, TenBroeke M, Maiese K. Metabotropic glutamate receptors prevent programmed cell death through the modulation of neuronal endonuclease activity and intracellular pH. Exp Neurol. 1999;155:79–94. doi: 10.1006/exnr.1998.6966. [DOI] [PubMed] [Google Scholar]
- 11.Zhu P, DeCoster MA, Bazan NG. Interplay among platelet-activating factor, oxidative stress, and group I metabotropic glutamate receptors modulates neuronal survival. J Neurosci Res. 2004;77:525–531. doi: 10.1002/jnr.20175. [DOI] [PubMed] [Google Scholar]
- 12.Bao WL, Williams AJ, Faden AI, Tortella FC. Selective mGluR5 receptor antagonist or agonist provides neuroprotection in a rat model of focal cerebral ischemia. Brain Res. 2001;922:173–179. doi: 10.1016/S0006-8993(01)03062-1. [DOI] [PubMed] [Google Scholar]
- 13.Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation. 2012;9:43. doi: 10.1186/1742-2094-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrnes KR, Stoica B, Riccio A, Pajoohesh-Ganji A, Loane DJ, Faden AI. Activation of metabotropic glutamate receptor 5 improves recovery after spinal cord injury in rodents. Ann Neurol. 2009;66:63–74. doi: 10.1002/ana.21673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen T, Zhang L, Qu Y, Huo K, Jiang X, Fei Z. The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int J Mol Med. 2012;29:630–636. doi: 10.3892/ijmm.2011.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI. Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. J Neurotrauma. 2013;30:403–412. doi: 10.1089/neu.2012.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang JW, Wang HD, Cong ZX, Zhang XS, Zhou XM, Zhang DD. Activation of metabotropic glutamate receptor 5 reduces the secondary brain injury after traumatic brain injury in rats. Biochem Biophys Res Commun. 2013;430:1016–1021. doi: 10.1016/j.bbrc.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 18.Loane DJ, Stoica BA, Faden AI. Metabotropic glutamate receptor-mediated signaling in neuroglia. Wiley Interdiscip Rev Membr Transp Signal. 2012;1:136–150. doi: 10.1002/wmts.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrnes KR, Stoica B, Loane DJ, Riccio A, Davis MI, Faden AI. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia. 2009;57:550–560. doi: 10.1002/glia.20783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loane DJ, Stoica BA, Pajoohesh-Ganji A, Byrnes KR, Faden AI. Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. J Biol Chem. 2009;284:15629–39. doi: 10.1074/jbc.M806139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Homayoun H, Moghaddam B. Group 5 metabotropic glutamate receptors: role in modulating cortical activity and relevance to cognition. Eur J Pharmacol. 2010;639:33–39. doi: 10.1016/j.ejphar.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 23.Kinney GG, Burno M, Campbell UC, et al. Metabotropic glutamate subtype 5 receptors modulate locomotor activity and sensorimotor gating in rodents. J Pharmacol Exp Ther. 2003;306:116–123. doi: 10.1124/jpet.103.048702. [DOI] [PubMed] [Google Scholar]
- 24.Lecourtier L, Homayoun H, Tamagnan G, Moghaddam B. Positive allosteric modulation of metabotropic glutarnate 5 (rnGlu5) receptors reverses N-methyl-d-aspartate antagonist-induced alteration of neuronal firing in prefrontal cortex. Biol Psychiatry. 2007;62:739–746. doi: 10.1016/j.biopsych.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B. Functional interaction between NMDA and mGlu5 receptors: effects on working memory, instrumental learning, motor behaviors, and dopamine release. Neuropsychopharmacology. 2004;29:1259–1269. doi: 10.1038/sj.npp.1300417. [DOI] [PubMed] [Google Scholar]
- 26.Kinney GG, O'Brien JA, Lemaire W, et al. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 27.Gravius A, Dekundy A, Nagel J, More L, Pietraszek M, Danysz W. Investigation on tolerance development to subchronic blockade of mGluR5 in models of learning, anxiety, and levodopa-induced dyskinesia in rats. J Neural Transm. 2008;115:1609–1619. doi: 10.1007/s00702-008-0098-4. [DOI] [PubMed] [Google Scholar]
- 28.Gass JT, Olive MF. Positive allosteric modulation of mGluR5 receptors facilitates extinction of a cocaine contextual memory. Biol Psychiatry. 2009;65:717–20. doi: 10.1016/j.biopsych.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olive MF. Cognitive effects of Group I metabotropic glutamate receptor ligands in the context of drug addiction. Eur J Pharmacol. 2010;639:47–58. doi: 10.1016/j.ejphar.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayala JE, Chen YL, Banko JL, et al. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology. 2009;34:2057–2071. doi: 10.1038/npp.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue F, Stoica BA, Hanscom M, Kabadi SV, Faden AI. Positive allosteric modulators (PAMs) of metabotropic glutamate receptor 5 (mGluR5) attenuate microglial activation. CNS Neurol Disord Drug Targets. 2013;13:558–566. doi: 10.2174/18715273113126660199. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez AL, Grier MD, Jones CK, et al. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fox GB, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Z, Loane DJ, Murray MG, 2nd, Stoica BA, Faden AI. Comparing the predictive value of multiple cognitive, affective, and motor tasks after rodent traumatic brain injury. J Neurotrauma. 2012;29:2475–2489. doi: 10.1089/neu.2012.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–1411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loane DJ, Pocivavsek A, Moussa CE, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nature Med. 2009;15:377–379. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 39.de Paulis T, Hemstapat K, Chen Y, et al. Substituent effects of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides on positive allosteric modulation of the metabotropic glutamate-5 receptor in rat cortical astrocytes. J Med Chem. 2006;49:3332–3344. doi: 10.1021/jm051252j. [DOI] [PubMed] [Google Scholar]
- 40.Noetzel MJ, Rook JM, Vinson PN, et al. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol. 2012;81:120–133. doi: 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piers TM, Heales SJ, Pocock JM. Positive allosteric modulation of metabotropic glutamate receptor 5 down-regulates fibrinogen-activated microglia providing neuronal protection. Neurosci Lett. 2011;505:140–145. doi: 10.1016/j.neulet.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 44.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 45.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 213 kb)