

Abstract
Background
Progressive Hemifacial Atrophy (PHA) is an acquired, typically unilateral, facial distortion with unknown etiology. The true incidence of this disorder has not been reported, but it is often regarded as a subtype of localized scleroderma. Historically, a debate existed whether PHA is a form of linear scleroderma, called morphea en coup de sabre (ECDS), or whether these conditions are inherently different processes or appear on a spectrum (; Adv Exp Med Biol 455:101–4, 1999; J Eur Acad Dermatol Venereol 19:403–4, 2005). Currently, it is generally accepted that both diseases exist on a spectrum of localized scleroderma and often coexist.
The pathogenesis of PHA has not been delineated, but trauma, autoimmunity, infection, and autonomic dysregulation have all been suggested. The majority of patients have initial manifestations in the first two decades of life; however, late presentations in 6th and 7th decades are also described [J Am Acad Dermatol 56:257–63, 2007; J Postgrad Med 51:135–6, 2005; Neurology 61:674–6, 2003]. The typical course of PHA is slow progression over 2-20 years and eventually reaching quiescence.
Systemic associations of PHA are protean, but neurological manifestations of seizures and headaches are common [J Am Acad Dermatol 56:257–63, 2007; Neurology 48:1013–8, 1997; Semin Arthritis Rheum 43:335–47, 2013]. As in many rare diseases, standard guidelines for imaging, treatment, and follow-up are not defined.
Methods
This review is based on a literature search using PubMed including original articles, reviews, cases and clinical guidelines. The search terms were “idiopathic hemifacial atrophy”, “Parry-Romberg syndrome”, “Romberg’s syndrome”, “progressive hemifacial atrophy”, “progressive facial hemiatrophy”, “juvenile localized scleroderma”, “linear scleroderma”, and “morphea en coup de sabre”. The goal of this review is to summarize clinical findings, theories of pathogenesis, diagnosis, clinical course, and proposed treatments of progressive hemifacial atrophy using a detailed review of literature.
Inclusion- and exclusion criteria
Review articles were used to identify primary papers of interest while retrospective cohort studies, case series, case reports, and treatment analyses in the English language literature or available translations of international literature were included.
Keywords: Progressive hemifacial atrophy, Facial atrophy, Facial hemiatrophy, Parry-Romberg syndrome, Romberg’s syndrome, Morphea, Scleroderma
History and nomenclature
First described by Parry in 1825, and Romberg in 1846, this constellation of craniofacial findings was labeled as progressive hemifacial atrophy by Eulenberg in 1871. Other names used to describe this disorder include Parry-Romberg syndrome (PRS), idiopathic hemifacial atrophy, progressive facial hemiatrophy (PFH), and Romberg’s syndrome. Interestingly, evidence of the disease dates back to ancient Egypt with mummies exhibiting craniofacial dysmorphism consistent with PHA [1-11].
Epidemiology
Studies to determine true incidence and epidemiologic characteristics of PHA have been elusive due to the rarity of the disease, a lack of standardized criteria for diagnosis [12], and the overlapping features of PHA and ECDS [13].
PHA is more often seen in the female sex, and similarly, morphea is typically described as having a female predilection [14]. In one recent series of 32 patients, 66% were female [15]. This is a similar ratio seen in other recent studies, including one with 54 patients, and another with 22 patients [1,13]. Older studies have shown a slightly higher female prevalence. In the aforementioned review of 772 cases, the female to male ratio of 3:1 [16]. This was replicated in a recent global survey of PHA (3:1); however, there may have been some responder bias leading to the higher female prevalence [5].
Clinical course and associations/complications
PHA usually presents in the first 20 years of life, although, late-onset disease has also been described [4,15,17,18]. A Mayo Clinic study of 54 patients, grouping ECDS and PHA, showed an average age of onset of 13.6 years with a median of 10.5 years and a range of 0.3 to 75 years. These findings have been corroborated by other large reports [1,5,13,17,19].
After initial presentation, the disorder is usually slowly progressive but self-limited [12]. The disease typically “burns out” in 2-10 years before becoming stationary [16,20,21]. While a majority of patients experience halting of the facial atrophy, in the aforementioned global internet survey, 26% of patients reported disease acceleration. 68% of these cases were women and experienced worsening of facial hemiatrophy during pregnancy (9 women) or after childbirth (8 women). Of the progressive disease cases, stress (26%) and surgery (8%) were identified as possible triggers for acceleration [5]. However, the level of disease activity of each responder could not be assessed by the author. The slowly progressive nature of the disease, and at which point patients are on the spectrum of disease activity, must be taken into account to distinguish disease acceleration versus normal evolution.
Clinical description
PHA refers to hemifacial atrophy of the skin and craniofacial tissue inferior to the forehead, typically involving dermatomes of one or more branches of the fifth cranial nerve [12,22]. The atrophy affects subcutaneous tissue, fat, muscle, and osteocartilaginous structures creating a sunken hemiface appearance [12]. (Figure 1a and b) Epidermal cutaneous involvement is minimal, but the tongue, gingiva, teeth, and palate may also be involved [13,16,18,23].
Figure 1.
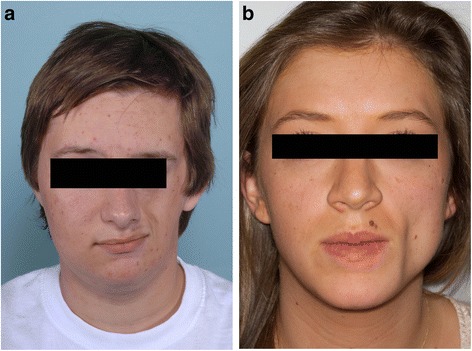
Progressive Hemifacial Atrophy. a: Hemifacial atrophy affecting subcutaneous tissue, fat, muscle, and osteocartilaginous structures. b: Left-sided sunken appearance secondary to subcutaneous and osseous atrophy.
ECDS manifests as a lesion of linear depression generally located on the frontoparietal scalp or paramedian forehead [24]. It is usually a linear unilateral depression, which may extend below the forehead, involving the nose, medial cheek, and sometimes the upper lip, although rare bilateral cases have been described. Involved skin is hyperpigmented, shiny, firm, and displays alopecia [13,25]. The atrophic shiny plaque may be associated with the scalp or madarosis. Hence, a resemblance of this furrow to a “stroke from a sword”. (Figure 2a and b) While the two may coexist in the same patient, (Figure 3a and b) clinical features are typically used to distinguish PHA from ECDS [12,21,26].
Figure 2.
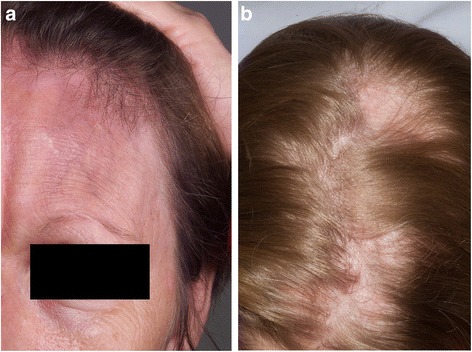
Morphea En Coup de Sabre. a: Atrophic shiny plaque with scalp and eyebrow alopecia. b: Scalp linear alopecia in a patient with ECDS.
Figure 3.
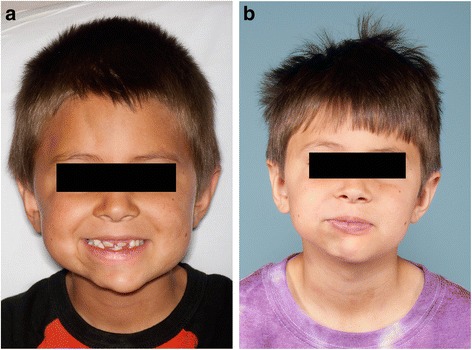
Morphea ECDS and PHA. a: Patient demonstrating concurrent right ipsilateral morphea ECDS and PHA. b: Same patient with concurrent morphea ECDS and PHA 2 years later.
PHA is associated with multiple extracutaneous findings, of which neurologic complications are the most common. Seizures and headaches are the most common neurologic symptoms in patients with PHA. In patients with epileptic activity, simple or complex partial seizures originating in the ipsilateral cerebral cortex are most often seen, and can be refractory to treatment. In patients with other forms of localized and systemic scleroderma, seizures, headaches, and other central nervous system findings may sometimes be seen, but peripheral and autonomic neuropathies tend to predominate [1,7,15,17,27-30].
Cranial neuropathies involving cranial nerves III, V, VI, and VII, have also been described in patients with PHA [19,31-34]. Secondary trigeminal neuralgia has been reported due to impingement of the nerve by destruction of bony structures, as well as vascular inflammation and damage resulting in facial pain that can be chronic and poorly responsive to treatment [35,36]. Additionally, speech may be affected in PHA patients resulting in dysarthria or aphasia [37,38]. Cognitive impairment and an increase in behavioral disorders have also been noted [15,39]. Depending on the degree of atrophy, changes to intracranial tissue and vessels may also result in hemiparesis, dysesthesias, and paresthesias [34,37,40-42].
Vascular damage in the form of infarction, hemorrhage, and white matter hyper-density can be seen in patients with PHA and is most likely due to vascular inflammation [1,41,43]. Dysplastic vessel formation has also been demonstrated in damaged areas of the cerebellum in PHA patients and may also contribute to intracranial findings [44,45]. Vascular damage, dysplastic vessels, and changes in vessel diameter may lead to cerebellar atrophy and production of neurologic symptoms. Some of this may be reversible with treatment [39,46]. Rare intracranial vascular malformations may be seen in patients with PHA and usually consist of dilation of intracranial blood vessels [40,47-49].
Ocular problems may be seen in patients with PHA; these can result in mild visual impairment to blindness [50]. Because of atrophy involving deeper tissue, enophthalmos has been frequently described and often results in loss of function of the orbitalis muscle. Other tissues altered by the atrophic process include the eye itself, the eyelid, and other extraocular muscles. The atrophy may be so severe that restrictive strabismus may result [51-54]. Additionally, patients with PHA have sometimes been noted to have retinal vasculitis, thus further supporting a possible vascular pathogenesis. Less common findings of neuroretinitis, uveitis, papillitis, glaucoma, cataracts, pigmentary changes of the retina and optical fundus, and iris heterochromia have also been demonstrated in some patients with PHA [51,55-61].
Teeth abnormalities and involvement of the mandible and masticatory muscles is frequently seen in patients with PHA. Shorter crowns and roots and crowding of teeth have been seen, as well. Secondary to maxillary and mandibular hypoplasia, chewing, smiling, and speech may be affected, although hemiatrophy and disturbances of other oral structures such as the tongue, lips, salivary glands, and gingiva can also contribute to the development of these symptoms. In those with dental involvement, infection is frequent. Pain secondary to masticatory muscle spasm, temporomandibular joint pain, and locking of the jaw has also been reported [52,62-69].
Skeletal hypoplasia of involved areas of the skull is common, although the clinical significance of this is unknown [52,70]. Atrophy of the extremities, both ipsilaterally and contralaterally, as well as the trunk, has been described in rare cases [12,18,71-73]. Although the reason for this peripheral atrophy is unknown, one such affected patient had normal magnetic resonance angiography in the atrophic limb with only reduction in volume of muscle and fat, making an ischemic or vascular process unlikely in that case [12].
Relationship of morphea ECDS and PHA
Multiple studies have tried distinguishing PHA and ECDS based on clinical and histopathologic criteria [2,13,19]. Thomas Lehman mentioned two key clinically differentiating factors: paramedian atrophy of PHA without significant induration of the overlying skin and associated atrophy extending down the side of the face with tongue and mandibular involvement. ECDS, in his opinion, is associated with cutaneous induration in the area of the scalp and does not extend below the forehead [74,75]. His disclaimer, that most cases lack clear distinction, is echoed by multiple authors and is consistent with the thought that these entities lie on the same spectrum of disease [2,3].
Some authors have proposed that these two diseases may be differentiated clinically and histologically. In one series of 13 cases of ECDS and 9 cases of PHA, cases were retrospectively evaluated using photos and histopathologic slides [13]. The authors found that the main clinical difference between the two entities was cutaneous sclerosis (present in 8/13 of ECDS and 0 PHA), hyperpigmentation, and alopecia in ECDS, versus total hemifacial involvement and ocular changes in PHA. Histopathologically, however, there was significant overlap as connective tissue fibrosis was seen in all cases with ECDS and in 2/9 cases of PHA. Adnexal atrophy was seen in 11/13 of ECDS and 3/9 of PHA while mononuclear cell infiltrates was present in all ECDS biopsies and 6/9 PHA. Another case series that followed 71 ECDS and PHA patients over a 20 year timespan, found that the clinical morphological pattern of the disease presentation changed progressively such that clear distinctions between the two entities often disappeared over time [3] (Table 1).
Table 1.
Comparison of morphea ECDS and PHA
| Morphea en Coup de Sabre | Progressive Hemifacial Atrophy | |
|---|---|---|
| Average Age (years) | 10 | 13.6 |
| Gender (F:M) | 2:1 – 3:1 | 3:1 |
| Clinical Features | -Cutaneous induration/sclerosis | -Paramedian atrophy |
| -Scalp to forehead^ | -No overlying skin induration | |
| -Hyperpigmentation | -Atrophy may extend down entire face | |
| -Alopecia (scalp/eyebrow) | ||
| Histopathologic Features~ | -Dermal Sclerosis | -Dermal sclerosis |
| -Adnexal atrophy | -Fat atrophy | |
| -Mononuclear cell infiltrates* | -Decrease in adnexal structures | |
| -Mononuclear cell infiltrates* | ||
| Extracutaneous Associations | -Atrophy subcutaneous tissue, fat, muscle, and osteocartilaginous structures | |
| -Atrophy and deformity of the tongue, teeth, and gingiva | ||
| -Cranial neuropathies | ||
| -Vision loss | ||
| -Seizure disorder |
Female (F).
Male (M).
*Histopathologic features depend on disease activity.
^May extend below the forehead and involve the nose, medial cheek, and upper lip.
~Connective tissue fibrosis, adnexal/fat atrophy, and mononuclear cell infiltrates all seen ECDS > PHA although considerable overlap may be seen.
Duymaz et al. proposed clinical criteria when evaluating a patient with facial atrophy. PHA was suggested when the patient presented with unilateral facial atrophy and lacked previous induration, inflammation, or cutaneous atrophy and sclerosis. Scleroderma ECDS was favored if unilateral band-like changes of sclerosis with hyperpigmentation in the frontoparietal area with associated induration was noted. Involvement of the area above the eyebrow without extension inferiorly, with cutaneous sclerosis and marked deformity, was noted to better describe ECDS [12]. However, histopathologic findings were not examined.
Often, clinical findings of both are present in the same individual [1,17,43,74,76]. In fact, 28 to 42% of patients have been reported to have coexisting ECDS and PHA [1,17,76]. In one review of 235 patients with localized scleroderma at Mayo Clinic from 1923 to 1954, facial hemiatrophy was associated with 41.3% of the 29 localized scleroderma cases involving the frontal, frontoparietal, or facial regions [77]. A large literature review of 772 cases mentioned ECDS as a potential initial presentation in some cases. It is possible that some of these patients may have had coexisting disease or only ECDS [16]. In another evaluation of 58 cases of ECDS, 20 developed signs of PHA [2]. Additionally, progression or transition of ECDS into PHA in the same physical location has also been illustrated in a handful of cases [2,3].
In another large series of ECDS and PHA patients, the authors concluded that their findings support these two entities as being on a spectrum of disease. 71.4% of the 28 patients with PHA in their study had facial sclerosis, with 53.6% having en coup de sabre lesions. Additionally, they found that of the 41 patients with ECDS, 36.6% also had PHA. Histologically, the biopsy specimens of PHA patients who did not have cutaneous sclerosis showed findings consistent with morphea [1]. Several prior studies had similar results [17,78].
In summary, while there may be some features that clinically distinguish ECDS from PHA, clinical and histopathologic findings may often overlap in the same patient, thus supporting the belief that ECDS and PHA fall on the same spectrum of disease.
Diagnosis
The diagnosis of PHA is a largely clinical one, supported by other findings such as histopathology and imaging. No universal diagnostic criteria are accepted. As discussed above, the characteristic clinical features of PHA which allow for its diagnosis is the presence of unilateral idiopathic facial atrophy, typically involving the lower face, without significant epidermal change. Deeper involvement of bone, teeth, tongue, and gingiva may also be present (Table 1). Patients are typically classified as having ECDS if they demonstrate linear scleroderma of the frontoparietal scalp with involvement of medial or paramedian forehead with possible extension of the sclerosis onto the scalp, nasal sidewall, and maxilla [1]. The cutaneous sclerosis, with possible hyperpigmentation and alopecia of ECDS, has been suggested as a potential differentiating factor between PHA and ECDS in those who desire to distinguish between them [13]. However, the boundaries between these entities are blurred as seen in patients with cutaneous sclerosis developing more significant atrophy than typically seen in ECDS, and conversely, those with atrophy without significant epidermal change, going on to develop cutaneous sclerosis [3].
Histopathology of affected areas in PHA, with or without clinical findings of a sclerotic process, shows homogenized dermal sclerosis, fat atrophy, decrease in adnexal structures, and perivascular plasma cells and lymphocytes [1,13,79] (Table 1). On the other hand, biopsy specimens in ECDS have been noted to show hyperpigmentation, alopecia, and cutaneous sclerosis. Atrophy of epidermis, dermis, adnexal structures and subcutaneous tissue has been seen in both PHA (with and without dermal sclerosis) and ECDS. More specific changes may be seen in biopsy specimens of patients with PHA including: sparse to exuberant lymphocytic infiltrates in the dermis, and degenerative changes of vascular endothelium have also been noted in some studies [1,2,13,17,78,80,81]. Hence, biopsies often may not be helpful in the diagnosis of these entities. Since skin biopsy is infrequently done in cases of PHA, photographical records should be maintained and followed throughout the duration of follow-up. As PHA slowly progresses until the disease plateaus, photographical evaluation is helpful in following the disease course. Additionally, if rapid changes in skin induration or overall clinical presentation are noted, a biopsy may be considered. Electroencephalogram (EEG) changes may be seen in patients who have a known seizure disorder and also in those who do not. EEG findings may or may not be associated with intracranial structural or imaging changes [7,30].
Laboratory work up of patients with PHA, as well as other types of localized scleroderma, tends to be largely unrevealing. A positive anti-nuclear antibody is the most common laboratory abnormality, with approximately 25-52% of patients having an elevated titer [17,22,23,78]. Serology for rheumatoid factor, anti-Scl-70, C-reactive protein (CRP), anti-dsDNA antibody, extractable nuclear antigen screening, genotyping for HLA-B27, and anti-cardiolipin antibodies are rarely abnormal and of limited value in PHA patients. Rheumatoid factor has been shown to be elevated in localized scleroderma with extracutaneous involvement and those with arthritis. However, it is currently unknown whether any PHA patients regularly demonstrate an elevated rheumatoid factor [22,23]. Peripheral eosinophilia and an elevated erythrocyte sedimentation rate have also been reported in association with localized scleroderma, but not with PHA [22,23].
Ultrasound can be used to detect presence of sclerosis, monitor disease activity, and treatment progress by measuring dermal thickness and echogenicity of the affected areas. Color Doppler ultrasound has the added benefit of measuring dermal blood flow, an increase of which indicates active disease. In patients with PHA, assessment of the ipsilateral parotid gland and salivary glands may also reveal increased blood flow and a hypoechoic appearance, indicating inflammation of the glands and active disease [82-84].
Due to the frequency of neurologic complications in PHA, baseline imaging may be performed, particularly in those patients that have neurologic symptoms. Typical findings on MRI show hyperintense white matter lesions on T2-weighted sequences. These are most prominent ipsilaterally but are often seen bilateral despite lack of skin involvement on the contralateral side. (Figure 4a and b) These findings often do not progress despite progressive skin and skeletal involvement. Cerebral atrophy, encephalomalacia, and cystic degeneration have been seen on intracranial imaging of patients with PHA [37]. Computed tomography (CT) scanning may also be used, but is less useful for evaluation of brain parenchyma than MRI. Typically, calcifications and changes in density of white matter can be seen on CT [15,30,85].
Figure 4.
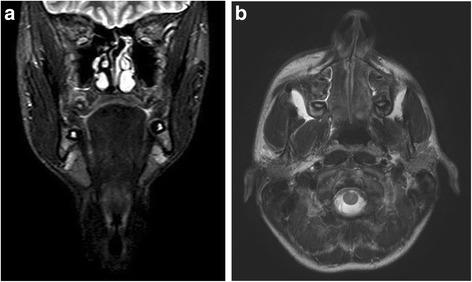
MRI of Progressive Hemifacial Atrophy. a: Coronal view from MRI demonstrating left hemifacial atrophy of soft tissue and osseous structures. b: Axial view from MRI demonstrating left hemifacial atrophy of soft tissue and osseous structures.
Whether or not abnormalities are demonstrated on MRI, not all patients will have corresponding clinical neurologic findings. Additionally, epileptic activity may occur prior or in the absence of any abnormal findings on MRI [30]. Skin findings are also not predictive of CNS involvement [15,19,85]. Because a direct correlation with CNS symptoms, skin findings, and MRI is not always demonstrated, clinical evaluation on an individual case-by-case basis is necessary to determine the need for imaging, whether at baseline, or in follow-up [30,37,85].
Clinically silent white matter findings do occur [86]. Determining the significance of these may be challenging. Single-photo emission CT (SPECT) may be a unique way to test the regional blood flow in the presumingly involved area, as regional blood flow hypoperfusion in the parietooccipital area of the affected hemisphere has been described in PHA patients with temporal lobe epilepsy [87]. Conversely, relative increases in regional cerebral blood flow (CBF) of affected cortical hemisphere on SPECT may be associated with to preserved cortical function and normal psychomotor development [86]. Even if extensive white matter abnormalities are demonstrated on MRI, preserved cortical function may be demonstrated by SPECT, proton MR spectroscopy (1H-MR spectroscopy), and diffuse tensor imaging (DTI) [15]. These findings were confirmed in an analysis of 19 PHA and 7 ECDS patients with MRI, angio-MRI, and SPECT for CBF [19]. In this study, the authors found that CNS involvement in patients with PHA may be seen irrespective of when cutaneous indurations were first noted clinically, and a diagnosis of PHA was made. Furthermore, the SPECT may be abnormal in PHA and ECDS whose MRI findings were unrevealing [19]. While observation of CNS symptoms and initial evaluation with MRI when indicated are often favored, the addition of a CBF analysis such as SPECT may give additional detail on the overall CNS involvement and prognosis in this patient population.
If there is a suspicion of a seizure disorder, electroencephalography may be done. In those patients with EEG abnormalities, findings are usually ipsilateral to the clinical involvement and most commonly show localized activity [1,30,88].
As discussed above, ocular manifestations of PHA can be potentially threatening to vision and are common. Ophthalmology should therefore be consulted for evaluation of vision, restrictive strabismus, ocular muscle strength, glaucoma, and inflammatory pathologies associated with the eye and PHA [50,51,61]. Ocular imaging has demonstrated retinal edema and optical disc swelling in some patients, which likely also contributes to visual impairment and loss [89].
Dental involvement in PHA is common, making early evaluation critical in reducing deformity and related complications to the teeth, mandible, maxilla, and other oral structures. Serial panoramic radiographs and photographs can be useful in monitoring disease progress until there is stabilization of disease. This can also assist in planning for surgery and orthodontic appliances [71,90].
Etiopathogenesis
A leading theory is that PHA is an autoimmune disorder. This is supported by the presence of other autoimmune conditions such as systemic lupus erythematosus (SLE), generalized myopathy, and rheumatoid arthritis (RA) in some patients with PHA [91-97]. Additionally, 17% and 10% of survey respondents with PHA to a survey described a medical history of vitiligo and thyroid dysfunction, respectively [5]. Other autoimmune conditions seen in these responders were inflammatory bowel disease (5%), RA (4%), ankylosing spondylitis (2%), and SLE (2%) [5]. The possible autoimmune etiology is also supported serologically due to the frequent presence of anti-nuclear antibodies, although serologies are of limited use in diagnosis, as discussed above.
Another leading theory in the pathogenesis of morphea form disorders such as PHA is that of vascular dysfunction. Vascular changes may invoke collagen production and extracellular matrix proliferation seen in morphea and systemic sclerosis [91]. It has also been proposed that ECDS and PHA may be related to a neuro-vasculitis [2,46,47,74]. A study of 41 PHA patients with light microscopy performed on 19 tissue specimens and ultrastructural analysis on 6 patients led to the theory that lymphocytic neuro-vasculitis caused by chronic cell-mediated vascular injury and incomplete endothelial regeneration along branches of the trigeminal nerve is central to the pathogenesis of PHA [81]. While not proven, this may contribute to the pathogenesis of this disorder.
There have also been reports of PHA following injury leading to a possible trauma-induced hypothesis [98,99]. In fact, many of the studies focusing on PHA and ECDS have noted trauma in a significant cohort of their study population. The large survey found 27% of responders had a childhood head injury; however, only 12% of responders had injuries that seemed relevant to the authors [5]. Similarly, Sommer et al. found 33% had a preceding injury including sclerotic lesions after a hematoma and secondary to an insect bite [17]. Caution must be used when interpreting this data, however, as these are relatively common occurrences, and there may be a fair amount of responder bias.
Infectious etiologies have also been proposed in the pathogenesis of PHA. It has been suggested that a Bell’s palsy or herpes zoster in the trigeminal distribution may be associated with PHA; however, studies have not been able to corroborate this theory [100]. The coexistence of Borrelia burgdorferi antibodies and Lyme infection have been described in European patients with morphea; however, recent studies, in the United Kingdom, United States, and Turkey have not supported this association [101-105]. Of the 12 German cases of PHA reviewed by Sommer et al, none had antibodies to Borrelia [17]. A recent study of 21 PHA patients and 6 with ECDS, along with 21 matched controls in Mexico did not show an association between Anti-Borrelia IgG antibodies and PHA, although Lyme disease is uncommon in this geographic location [106]. Other studies from various locations in North America also failed to show a direct association between Borrelia and morphea [107-111].
Lastly, sympathetic hyperactivity and dysregulation has been proposed as a potential pathogenic mechanism in the development of PHA [18,112,113]. A captivating animal experiment by Resende et al. followed cats, dogs, and rabbits for one year after ablation of the superior cervical ganglion at the age of 30 days old. The authors found the altered animals to have evidence of localized alopecia, corneal ulceration, keratitis, strabismus, enophthalmos, ocular atrophy, hemifacial atrophy and slight bone atrophy on the side of the sympathectomy, all potential findings seen in PHA patients [114]. PHA was also induced following a similar experiment in a rat model in 1960 [115]. These associations, however, have not been demonstrated in human patients.
With the current body of evidence, an acquired etiology to PHA is accepted, but familial cases have also rarely been described [113,116,117]. The aforementioned global survey of individuals with PHA found six patients (3%) had family members with facial asymmetry, with only one of these six responders with a first degree relative demonstrating involvement [5]. Autosomal dominant inheritance with incomplete penetrance has been suggested, but no support for this hypothesis has been provided to this point [118,119]. Hence, genetic counseling is recommended in individual cases of PHA. The lack of an etiology consensus and explanation of pathogenesis of PHA also make a defined pattern of inheritance unlikely. In summary, a distinct etiology of PHA is still elusive; however, a combination of autoimmune, vascular, and autonomic dysfunction is likely.
Differential diagnosis
The differential diagnosis of PHA includes other forms of juvenile localized scleroderma, Rasmussen encephalitis, Barraquer-Simons syndrome, congenital hemiatrophy, and primary hemifacial hypertrophy.
Rasmussen encephalitis (RE) presents with unilateral cerebral hemisphere destruction with intractable focal seizures and progressive neurologic deficits. Pathology from the involved cerebral tissue shows chronic inflammation [120]. The similar ages of onset, unilateral characteristics, and epilepsy occurrence may account for the multiple reports of associations of RE and PHA and the difficulty of differentiating these processes [41,120-124]. In fact, some authors suggest an overlap in the disorders, especially in the cases of PHA that are complicated by seizures [41,120]. Patients with RE alone usually present with hemiparesis and intractable seizures [123]. The clinical hemiatrophy has only been described in PHA patients with coexisting RE or ECDS [41,120,122,123,125]. Additionally, brain biopsies of RE show distinct findings of neuronal loss, reactive blood vessels, and a pale background with neutrophils and chronic inflammatory cells [123]. Brain biopsies are not typically done in PHA. The clinical, and if needed, histopathological differences separate these two syndromes; however, coexistence of RE and PHA is possible.
Baraquer-Simons syndrome is an acquired partial progressive cephalothoracic lipodystrophy that presents with a gradual onset of symmetrical bilateral subcutaneous fat loss from the face, neck, upper extremities, thorax, and abdomen but sparing the lower extremities. Central nervous system findings of deafness, epilepsy, and intellectual disability have also been described [126]. The bilateral nature of this disease and systemic involvement of the kidneys may differentiate these processes. However, associations of Baraquer-Simons lipodystrophy and scleroderma are also described [127].
Hemifacial hypertrophy, formerly termed primary hemifacial hypertrophy, is a rare asymmetric enlargement of half of the head without enlargement of other body parts [128,129]. While there is unilateral face enlargement instead of atrophy as seen in PHA, this disorder may be considered in the clinical differential diagnosis of an asymmetric unilateral facial deformity.
Other clinical mimickers of PHA are fat necrosis, whether from infection such as bulbar poliomyelitis [130], trauma, or connective tissue disease, and congenital deformities such as “wry neck” [131]. Congenital hemiatrophy has been used interchangeably with PHA; however, it is likely a different process [16,132].
Treatment
Treatment for PHA can be challenging. The primary aim is to stop the active disease process. Methotrexate (MTX) is the standard therapy for active disease. MTX dosing is not standardized and ranges from 0.3-1 milligrams/kilogram/week (mg/kg/wk) with a maximum dose of 25 mg weekly in either an oral or injectable form. The MTX is often combined with oral prednisone over the first three months due to the fact that the methotrexate has a delayed effect on inflammation and fibrosis. Most regimens call for two months of prednisone at 1 mg/kg/day with a taper to be done during the third month. Pulsed high dose IV methylprednisone has also been explored using 1000 mg for three days monthly for 6 months. The goal with this approach is to gain the anti-inflammatory effects of corticosteroids without the large side-effect profile. A long course of therapy is typically required as relapse is frequently seen with shorter courses of therapy. The specific length of therapy required to reduce relapse is unknown, and likely varies from patient to patient. Current evidence supports a 12-24 month course of methotrexate being most effective in inducing prolonged remission [1,23,83,132-136].
Isolated case reports of other immunosuppressive agents such as mycophenolate mofetil, cyclosporine, and cyclophosphamide have shown variable success in patients who have failed treatment with MTX [135,137,138]. The use of antimalarial agents has also been described with some efficacy in a limited number of cases [1]. Ultraviolet-A (UVA) and psoralen combined with ultraviolet A (PUVA) therapy have been shown to be effective in treatment of localized scleroderma [139,140]. PUVA has been reported to arrest disease activity in isolated reports of PHA [103,141].
Surgical treatment for PHA often requires a multi-specialty approach with repeated procedures, depending on degree of involvement. The therapeutic goal of surgery for PHA patients is to minimize psychosocial effects, and to correct the appearance and function of involved facial structures [90,142-145]. Timing of surgical intervention in patients with PHA has been debated. Most experts recommend that procedural therapy be delayed until disease progression has halted or plateaued in order to avoid multiple surgeries as defects progress and because of the need for a stable skeletal foundation [146-152]. However, others argue for earlier intervention despite active disease due to the psychosocial difficulties facing patients with PHA, promotion of normal development of facial structures, and because most patients with PHA require multiple surgeries [144,145]. Ego-dystonic feelings, loss of confidence due to appearance, and bullying are the primary reasons behind pursuing surgical therapy [145]. In one study patients who had earlier surgical intervention had higher satisfaction scores [144], however the utility and timing of surgical procedures must be evaluated on an individual basis.
If surgical therapy is indeed delayed until disease burns out, proper mandibular development and parallelism of facial planes can be guided with orthodontic rehabilitation and orthodontic appliances [142,143]. For mild to moderate atrophy, fat grafting, as well as lipoinjection and other soft tissue fillers, can be employed [142,144,149,153,154]. For more severe atrophy, a combined approach of skeletal and soft tissue augmentation is often recommended. Bone paste cranioplasty, autologous fat infections, dermal fat grafts and adipofascial flaps can also be used to correct large volume atrophy [144,145,155]. Additionally, the use of bone grafts and biocompatible porous polyethylene implants can be used to correct skeletal deformity [145,154,155]. Eyebrow lifting, Z-plasty, lip repair, nasal reconstruction, eyebrow repair, face-lift, lip augmentation, hair transplant, and other adjuvant procedures can also be used to create a better cosmetic outcome [144,145,155].
Conclusion
Progressive hemifacial atrophy, or Parry-Romberg syndrome, is a slowly progressive and self-limited dysplasia causing unilateral craniofacial atrophy. The close association with morphea en coup de sabre has been investigated, but no consensus on the pathogenesis of PHA is available. We reviewed literature addressing PHA and summarized the relevant findings.
Limitations
Since the review focuses on English literature, several large studies have been left out including 100 cases from Mussinelli, Magri, and Origlio [156,157], and large German reviews by Möbius in 1895, [158] Marburg [159], and Cassirer in 1912 [160] were excluded. Multiple international case series were reviewed by Roddi et al., in 1994 [21], Archambault and Fromm in 1932, [18] Wartenberg in 1945 [75], and Blair O. Rogers in 1964 [16]. These publications were reviewed in detail.
Consent
Written informed consent was obtained for the publication of this report and any accompanying images.
Abbreviations
- (PHA)
Progressive Hemifacial Atrophy
- (ECDS)
morphea en coup de sabre
- (PFH)
Progressive facial hemiatrophy
- (PRS)
Parry-Romberg syndrome
- (MRI)
Magnetic resonance imaging
- (RA)
Rheumatoid arthritis
- (SLE)
Systemic lupus erythematosus
- (ANA)
Autoantibodies against nuclear
- (RF)
Rheumatoid factor
- (RE)
Rasmussen encephalitis
- (SPECT)
Single-photo emission computed tomography
- (1H-MR spectroscopy)
Proton MR spectroscopy
- (DTI)
Diffuse tensor imaging
- (CBF)
Cerebral blood flow
- (CNS)
Central nervous system
- (MTX)
Methotrexate
- (mg)
Milligrams
- (kg)
Kilogram
- (wk)
Week
- (UVA)
Ultraviolet-A
- (PUVA)
Psoralen combined with ultraviolet A
- (MEDPOR) (trade name)
Biocompatible, porous polyethylene implants
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ST participated in the sequence alignment. ST, NP, MT conceived of the study, participated in its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Stanislav N Tolkachjov, Email: Tolkachjov.stanislav@mayo.edu.
Nirav G Patel, Email: patel.nirav@mayo.edu.
Megha M Tollefson, Email: Tollefson.Megha@mayo.edu.
References
- 1.Tollefson MM, Witman PM. En coup de sabre morphea and Parry-Romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol. 2007;56(2):257–63. doi: 10.1016/j.jaad.2006.10.959. [DOI] [PubMed] [Google Scholar]
- 2.Blaszczyk M, Jablonska S. Linear scleroderma en Coup de Sabre. Relationship with progressive facial hemiatrophy (PFH) Adv Exp Med Biol. 1999;455:101–4. [PubMed] [Google Scholar]
- 3.Jablonska S, Blaszczyk M. Long-lasting follow-up favours a close relationship between progressive facial hemiatrophy and scleroderma en coup de sabre. J Eur Acad Dermatol Venereol. 2005;19(4):403–4. doi: 10.1111/j.1468-3083.2005.00979.x. [DOI] [PubMed] [Google Scholar]
- 4.Mendonca J, Viana SL, Freitas F, Lima G. Late-onset progressive facial hemiatrophy (Parry-Romberg syndrome) J Postgrad Med. 2005;51(2):135–6. [PubMed] [Google Scholar]
- 5.Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology. 2003;61(5):674–6. doi: 10.1212/wnl.61.5.674. [DOI] [PubMed] [Google Scholar]
- 6.Dupont S, Catala M, Hasboun D, Semah F, Baulac M. Progressive facial hemiatrophy and epilepsy: a common underlying dysgenetic mechanism. Neurology. 1997;48(4):1013–8. doi: 10.1212/wnl.48.4.1013. [DOI] [PubMed] [Google Scholar]
- 7.Amaral TN, Peres FA, Lapa AT, Marques-Neto JF, Appenzeller S. Neurologic involvement in scleroderma: a systematic review. Semin Arthritis Rheum. 2013;43(3):335–47. doi: 10.1016/j.semarthrit.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Parry CH. Collections from the unpublished medical writings of the late Caleb Hillier Parry. London: Underwoods; 1825. 478.
- 9.Romberg MH, Henoch, E.H. Krankheiten des nervensystems (IV: Trophoneurosen). Klinische ergebnisse. Berlin: Albert Förstner; 1846. 75–81.
- 10.Eulenburg A. Hemiatrophia facialis progressiva. Lehrbuch der functionellen nervenkrankheiten auf physiologischer basis. Berlin: Verlag von August Hirschwald; 1871. 712–26.
- 11.Appenzeller O, Stevens JM, Kruszynski R, Walker S. Neurology in ancient faces. J Neurol Neurosurg Psychiatry. 2001;70(4):524–9. doi: 10.1136/jnnp.70.4.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duymaz A, Karabekmez FE, Keskin M, Tosun Z. Parry-Romberg syndrome: facial atrophy and its relationship with other regions of the body. Ann Plast Surg. 2009;63(4):457–61. doi: 10.1097/SAP.0b013e31818bed6d. [DOI] [PubMed] [Google Scholar]
- 13.Orozco-Covarrubias L, Guzman-Meza A, Ridaura-Sanz C, Carrasco Daza D, Sosa-de-Martinez C, Ruiz-Maldonado R. Scleroderma ‘en coup de sabre’ and progressive facial hemiatrophy. Is it possible to differentiate them? J Eur Acad Dermatol Venereol. 2002;16(4):361–6. doi: 10.1046/j.1468-3083.2002.00442.x. [DOI] [PubMed] [Google Scholar]
- 14.Peterson LS, Nelson AM, Su WP, Mason T, O’Fallon WM, Gabriel SE. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960-1993. J Rheumatol. 1997;24(1):73–80. [PubMed] [Google Scholar]
- 15.Chiu YE, Vora S, Kwon EK, Maheshwari M. A significant proportion of children with morphea en coup de sabre and Parry-Romberg syndrome have neuroimaging findings. Pediatr Dermatol. 2012;29(6):738–48. doi: 10.1111/pde.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers BO. Progressive facial hemiatrophy: Romberg’s disease; a review of 772 cases. Third International Congress Plastic Surgery, Excerpta Medica, Amsterdam. 1964. p. 681. [Google Scholar]
- 17.Sommer A, Gambichler T, Bacharach-Buhles M, von Rothenburg T, Altmeyer P, Kreuter A. Clinical and serological characteristics of progressive facial hemiatrophy: a case series of 12 patients. J Am Acad Dermatol. 2006;54(2):227–33. doi: 10.1016/j.jaad.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 18.Archambault LS, Fromm NK. Progressive facial hemiatrophy. Arch Neurol Psychiatry. 1932;27:529. [Google Scholar]
- 19.Blaszczyk M, Krolicki L, Krasu M, Glinska O, Jablonska S. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol. 2003;30(9):1997–2004. [PubMed] [Google Scholar]
- 20.Rogers BO. Progressive hemifacial atrophy: Romberg’s disease. In: Converse JM, editor. Reconstructive plastic surgery, vol 4. 2. Philadelphia: Saunders; 1977. p. 2337. [Google Scholar]
- 21.Roddi RR E, Gilbert PM, van der Meulen JCH. Progressive hemifacial atrophy: historical review and actual features related to a new classification of facial hypoplasia and atrophy. Eur J Plast Surg. 1994;17(4):178–83. [Google Scholar]
- 22.Zulian F. Scleroderma in children. Pediatr Clin N Am. 2005;52(2):521–45. doi: 10.1016/j.pcl.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Zulian F, Athreya BH, Laxer R, Nelson AM, de Oliveira SK F, Punaro MG, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology. 2006;45(5):614–20. doi: 10.1093/rheumatology/kei251. [DOI] [PubMed] [Google Scholar]
- 24.Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma) Mayo Clin Proc. 1995;70(11):1068–76. doi: 10.4065/70.11.1068. [DOI] [PubMed] [Google Scholar]
- 25.Torok E, Ablonczy E. Morphoea in children. Clin Exp Dermatol. 1986;11(6):607–12. doi: 10.1111/j.1365-2230.1986.tb00517.x. [DOI] [PubMed] [Google Scholar]
- 26.Singh G, Bajpai HS. Progressive facial hemiatrophy. Dermatologica. 1969;138(4):288–91. doi: 10.1159/000253994. [DOI] [PubMed] [Google Scholar]
- 27.El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67(4):769–84. doi: 10.1016/j.jaad.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 28.Sartori S, Martini G, Calderone M, Patrizi A, Gobbi G, Zulian F. Severe epilepsy preceding by four months the onset of scleroderma en coup de sabre. Clin Exp Rheumatol. 2009;27(3 Suppl 54):64–7. [PubMed] [Google Scholar]
- 29.Muchnick RS, Aston SJ, Rees TD. Ocular manifestations and treatment of hemifacial atrophy. Am J Ophthalmol. 1979;88(5):889–97. doi: 10.1016/0002-9394(79)90567-1. [DOI] [PubMed] [Google Scholar]
- 30.Yano T, Sawaishi Y, Toyono M, Takaku I, Takada G. Progressive facial hemiatrophy after epileptic seizures. Pediatr Neurol. 2000;23(2):164–6. doi: 10.1016/s0887-8994(00)00168-5. [DOI] [PubMed] [Google Scholar]
- 31.Sathornsumetee S, Schanberg L, Rabinovich E, Lewis D, Jr, Weisleder P. Parry-Romberg syndrome with fatal brain stem involvement. J Pediatr. 2005;146(3):429–31. doi: 10.1016/j.jpeds.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 32.Gambichler T, Kreuter A, Hoffmann K, Bechara FG, Altmeyer P, Jansen T. Bilateral linear scleroderma “en coup de sabre” associated with facial atrophy and neurological complications. BMC Dermatol. 2001;1:9. doi: 10.1186/1471-5945-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viana M, Glastonbury CM, Sprenger T, Goadsby PJ. Trigeminal neuropathic pain in a patient with progressive facial hemiatrophy (parry-romberg syndrome) Arch Neurol. 2011;68(7):938–43. doi: 10.1001/archneurol.2011.126. [DOI] [PubMed] [Google Scholar]
- 34.Holland KE, Steffes B, Nocton JJ, Schwabe MJ, Jacobson RD, Drolet BA. Linear scleroderma en coup de sabre with associated neurologic abnormalities. Pediatrics. 2006;117(1):e132–6. doi: 10.1542/peds.2005-0470. [DOI] [PubMed] [Google Scholar]
- 35.Borodic GE, Caruso P, Acquadro M, Chick S. Parry-Romberg syndrome vasculopathy and its treatment with botulinum toxin. Ophthal Plast Reconstr Surg. 2014;30(1):e22–5. doi: 10.1097/IOP.0b013e31828de9c0. [DOI] [PubMed] [Google Scholar]
- 36.Dalla Costa G, Colombo B, Dalla Libera D, Martinelli V, Comi G. Parry Romberg syndrome associated with chronic facial pain. J Clin Neurosci. 2013;20(9):1320–2. doi: 10.1016/j.jocn.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 37.Cory RC, Clayman DA, Faillace WJ, McKee SW, Gama CH. Clinical and radiologic findings in progressive facial hemiatrophy (Parry-Romberg syndrome) AJNR Am J Neuroradiol. 1997;18(4):751–7. [PMC free article] [PubMed] [Google Scholar]
- 38.Stone J, Franks AJ, Guthrie JA, Johnson MH. Scleroderma “en coup de sabre”: pathological evidence of intracerebral inflammation. J Neurol Neurosurg Psychiatry. 2001;70(3):382–5. doi: 10.1136/jnnp.70.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grosso S, Fioravanti A, Biasi G, Conversano E, Marcolongo R, Morgese G, et al. Linear scleroderma associated with progressive brain atrophy. Brain Dev. 2003;25(1):57–61. doi: 10.1016/s0387-7604(02)00147-x. [DOI] [PubMed] [Google Scholar]
- 40.Strenge H, Cordes P, Sticherling M, Brossmann J. Hemifacial atrophy: a neurocutaneous disorder with coup de sabre deformity, telangiectatic naevus, aneurysmatic malformation of the internal carotid artery and crossed hemiatrophy. J Neurol. 1996;243(9):658–60. doi: 10.1007/BF00878663. [DOI] [PubMed] [Google Scholar]
- 41.Paprocka J, Jamroz E, Adamek D, Marszal E, Mandera M. Difficulties in differentiation of Parry-Romberg syndrome, unilateral facial sclerodermia, and Rasmussen syndrome. Childs Nerv Syst. 2006;22(4):409–15. doi: 10.1007/s00381-005-1262-x. [DOI] [PubMed] [Google Scholar]
- 42.Malandrini A, Dotti MT, Federico A. Selective ipsilateral neuromuscular involvement in a case of facial and somatic hemiatrophy. Muscle Nerve. 1997;20(7):890–2. doi: 10.1002/(sici)1097-4598(199707)20:7<890::aid-mus16>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 43.Menni S, Marzano AV, Passoni E. Neurologic abnormalities in two patients with facial hemiatrophy and sclerosis coexisting with morphea. Pediatr Dermatol. 1997;14(2):113–6. doi: 10.1111/j.1525-1470.1997.tb00216.x. [DOI] [PubMed] [Google Scholar]
- 44.Chung MH, Sum J, Morrell MJ, Horoupian DS. Intracerebral involvement in scleroderma en coup de sabre: report of a case with neuropathologic findings. Ann Neurol. 1995;37(5):679–81. doi: 10.1002/ana.410370519. [DOI] [PubMed] [Google Scholar]
- 45.Chbicheb M, Gelot A, Rivier F, Roubertie A, Humbertclaude V, Coubes P, et al. [Parry-Romberg’s syndrome and epilepsy] Rev Neurol. 2005;161(1):92–7. doi: 10.1016/s0035-3787(05)84980-x. [DOI] [PubMed] [Google Scholar]
- 46.Miedziak AI, Stefanyszyn M, Flanagan J, Eagle RC., Jr Parry-Romberg syndrome associated with intracranial vascular malformations. Arch Ophthalmol. 1998;116(9):1235–7. doi: 10.1001/archopht.116.9.1235. [DOI] [PubMed] [Google Scholar]
- 47.Bosman T, Van Bei Jnum J, Van Walderveen MA, Brouwer PA. Giant intracranial aneurysm in a ten-year-old boy with parry romberg syndrome. A case report and literature review. Interv Neuroradiol. 2009;15(2):165–73. doi: 10.1177/159101990901500205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aoki T, Tashiro Y, Fujita K, Kajiwara M. Parry-Romberg syndrome with a giant internal carotid artery aneurysm. Surg Neurol. 2006;65(2):170–3. doi: 10.1016/j.surneu.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 49.Catala M. Progressive intracranial aneurysmal disease in a child with progressive hemifacial atrophy (Parry-Romberg disease): case report. Neurosurgery. 1998;42(5):1195–6. doi: 10.1097/00006123-199805000-00161. [DOI] [PubMed] [Google Scholar]
- 50.Miller MT, Sloane H, Goldberg MF, Grisolano J, Frenkel M, Mafee MF. Progressive hemifacial atrophy (Parry-Romberg disease) J Pediatr Ophthalmol Strabismus. 1987;24(1):27–36. doi: 10.3928/0191-3913-19870101-07. [DOI] [PubMed] [Google Scholar]
- 51.Dawczynski J, Thorwarth M, Koenigsdoerffer E, Schultze-Mosgau S. Interdisciplinary treatment and ophthalmological findings in Parry-Romberg syndrome. J Craniofac Surg. 2006;17(6):1175–6. doi: 10.1097/01.scs.0000236440.20592.be. [DOI] [PubMed] [Google Scholar]
- 52.Balan P, Gogineni SB, Shetty SR, D’Souza D. Three-dimensional imaging of progressive facial hemiatrophy (Parry-Romberg syndrome) with unusual conjunctival findings. Imaging Sci Dent. 2011;41(4):183–7. doi: 10.5624/isd.2011.41.4.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khan AO. Restrictive strabismus in Parry-Romberg syndrome. J Pediatr Ophthalmol Strabismus. 2007;44(1):51–2. doi: 10.3928/01913913-20070101-09. [DOI] [PubMed] [Google Scholar]
- 54.Nasser O, Greiner K, Amer R. Unilateral optic atrophy preceding Coats disease in a girl with Parry-Romberg syndrome. Eur J Ophthalmol. 2010;20(1):221–3. doi: 10.1177/112067211002000132. [DOI] [PubMed] [Google Scholar]
- 55.Bellusci C, Liguori R, Pazzaglia A, Badiali L, Schiavi C, Campos EC. Bilateral Parry-Romberg syndrome associated with retinal vasculitis. Eur J Ophthalmol. 2003;13(9–10):803–6. doi: 10.1177/1120672103013009-1014. [DOI] [PubMed] [Google Scholar]
- 56.Ong K, Billson FA, Pathirana DS, Clifton-Bligh P. A case of progressive hemifacial atrophy with uveitis and retinal vasculitis. Aust N Z J Ophthalmol. 1991;19(4):295–8. doi: 10.1111/j.1442-9071.1991.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 57.La Hey E, Baarsma GS. Fuchs’ heterochromic cyclitis and retinal vascular abnormalities in progressive hemifacial atrophy. Eye. 1993;7(Pt 3):426–8. doi: 10.1038/eye.1993.84. [DOI] [PubMed] [Google Scholar]
- 58.Yildirim O, Dinc E, Oz O. Parry-Romberg syndrome associated with anterior uveitis and retinal vasculitis. Can J Ophthalmol. 2010;45(3):289–90. doi: 10.3129/i09-241. [DOI] [PubMed] [Google Scholar]
- 59.Cohen JS. Congenital nonprogressive facial hemiatrophy with ipsilateral eye abnormalities and juvenile glaucoma. Ann Ophthalmol. 1979;11(3):413–6. [PubMed] [Google Scholar]
- 60.Ibarra Perez C. [Parry-Romberg syndrome with glaucoma and pregnancy] Gac Med Mex. 2001;137(3):289. [PubMed] [Google Scholar]
- 61.Miller MT, Spencer MA. Progressive hemifacial atrophy. A natural history study. Trans Am Ophthalmol Soc. 1995;93:203–15. [PMC free article] [PubMed] [Google Scholar]
- 62.Szcepanska I, Wilczynska M, Kakolewska-Maczynska J. [Facial asymmetry, changes in the masticatory organ and evaluation of thickness of soft tissues by the ultrasonic method in cases of secondary facial hemiatrophy following scleroderma] Czasopismo stomatologiczne. 1974;27(4):375–84. [PubMed] [Google Scholar]
- 63.Talacko AA, Reade PC. Hemifacial atrophy and temporomandibular joint pain-dysfunction syndrome. Int J Oral Maxillofac Surg. 1988;17(4):224–6. doi: 10.1016/s0901-5027(88)80044-4. [DOI] [PubMed] [Google Scholar]
- 64.Kaufman MD. Masticatory spasm in facial hemiatrophy. Ann Neurol. 1980;7(6):585–7. doi: 10.1002/ana.410070614. [DOI] [PubMed] [Google Scholar]
- 65.O’Flynn S, Kinirons M. Parry-Romberg syndrome: a report of the dental findings in a child followed up for 9 years. Int J Paediatr Dent. 2006;16(4):297–301. doi: 10.1111/j.1365-263X.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- 66.Fayad S, Steffensen B. Root resorptions in a patient with hemifacial atrophy. J Endod. 1994;20(6):299–303. doi: 10.1016/s0099-2399(06)80821-6. [DOI] [PubMed] [Google Scholar]
- 67.Tsuda N, Yamamoto K, Fukusako T, Morimatsu M. [A case of unilateral lingual atrophy and ipsilateral muscular atrophy supplied by trigeminal nerve–in relation to progressive facial hemiatrophy] Rinsho Shinkeigaku. 1991;31(9):1007–9. [PubMed] [Google Scholar]
- 68.Schwartz RA, Tedesco AS, Stern LZ, Kaminska AM, Haraldsen JM, Grekin DA. Myopathy associated with sclerodermal facial hemiatrophy. Arch Neurol. 1981;38(9):592–4. doi: 10.1001/archneur.1981.00510090086013. [DOI] [PubMed] [Google Scholar]
- 69.Rees TD, Tabbal N, Aston SJ. Tongue-flap reconstruction of the lip vermilion in hemifacial atrophy. Plast Reconstr Surg. 1983;72(5):643–7. doi: 10.1097/00006534-198311000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Moore MH, Wong KS, Proudman TW, David DJ. Progressive hemifacial atrophy (Romberg’s disease): skeletal involvement and treatment. Br J Plast Surg. 1993;46(1):39–44. doi: 10.1016/0007-1226(93)90063-h. [DOI] [PubMed] [Google Scholar]
- 71.Lakhani PK, David TJ. Progressive hemifacial atrophy with scleroderma and ipsilateral limb wasting (Parry-Romberg syndrome) J R Soc Med. 1984;77(2):138–9. doi: 10.1177/014107688407700215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chapman MS, Peraza JE, Spencer SK. Parry-Romberg syndrome with contralateral and ipsilateral extremity involvement. J Cutan Med Surg. 1999;3(5):260–2. doi: 10.1177/120347549900300509. [DOI] [PubMed] [Google Scholar]
- 73.Rees TD. Facial atrophy. Clin Plast Surg. 1976;3(4):637–46. [PubMed] [Google Scholar]
- 74.Lehman TJ. The Parry Romberg syndrome of progressive facial hemiatrophy and linear scleroderma en coup de sabre. Mistaken diagnosis or overlapping conditions? J Rheumatol. 1992;19(6):844–5. [PubMed] [Google Scholar]
- 75.Wartenberg R. Progressive facial hemiatrophy. Arch Neurol Psychiatr. 1945;54:75–96. [Google Scholar]
- 76.Jun JH, Kim HY, Jung HJ, Lee WJ, Lee SJ, Kim do W, et al. Parry-romberg syndrome with en coup de sabre. Ann Dermatol. 2011;23(3):342–7. doi: 10.5021/ad.2011.23.3.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Christianson HB, Dorsey CS, O'Leary PA, Kierland RR. Localized scleroderma: a clinical study of 235 cases. Arch Dermatol. 1956;74:629. doi: 10.1001/archderm.1956.01550120049012. [DOI] [PubMed] [Google Scholar]
- 78.Falanga V, Medsger TA, Jr, Reichlin M, Rodnan GP. Linear scleroderma. Clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. 1986;104(6):849–57. doi: 10.7326/0003-4819-104-6-849. [DOI] [PubMed] [Google Scholar]
- 79.Taylor HM, Robinson R, Cox T. Progressive facial hemiatrophy: MRI appearances. Dev Med Child Neurol. 1997;39(7):484–6. doi: 10.1111/j.1469-8749.1997.tb07469.x. [DOI] [PubMed] [Google Scholar]
- 80.Pinheiro TP, Silva CC, Silveira CS, Botelho PC, Pinheiro M, Pinheiro JJ. Progressive Hemifacial Atrophy–case report. Med Oral Patol Oral Cir Bucal. 2006;11(2):E112–4. [PubMed] [Google Scholar]
- 81.Pensler JM, Murphy GF, Mulliken JB. Clinical and ultrastructural studies of Romberg’s hemifacial atrophy. Plast Reconstr Surg. 1990;85(5):669–74. [PubMed] [Google Scholar]
- 82.Wortsman X, Wortsman J, Sazunic I, Carreno L. Activity assessment in morphea using color Doppler ultrasound. J Am Acad Dermatol. 2011;65(5):942–8. doi: 10.1016/j.jaad.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 83.Zulian F, Cuffaro G, Sperotto F. Scleroderma in children: an update. Curr Opin Rheumatol. 2013;25(5):643–50. doi: 10.1097/BOR.0b013e3283641f61. [DOI] [PubMed] [Google Scholar]
- 84.Weibel L, Howell KJ, Visentin MT, Rudiger A, Denton CP, Zulian F, et al. Laser Doppler flowmetry for assessing localized scleroderma in children. Arthritis Rheum. 2007;56(10):3489–95. doi: 10.1002/art.22920. [DOI] [PubMed] [Google Scholar]
- 85.Doolittle DA, Lehman VT, Schwartz KM, Wong-Kisiel LC, Lehman JS, Tollefson MM. CNS imaging findings associated with Parry-Romberg syndrome and en coup de sabre: correlation to dermatologic and neurologic abnormalities. Neuroradiology. 2014. doi:10.1007/s00234-014-1448-6 [DOI] [PubMed]
- 86.Okumura A, Ikuta T, Tsuji T, Kato T, Fukatsu H, Naganawa S, et al. Parry-Romberg syndrome with a clinically silent white matter lesion. AJNR Am J Neuroradiol. 2006;27(8):1729–31. [PMC free article] [PubMed] [Google Scholar]
- 87.DeFelipe J, Segura T, Arellano JI, Merchan A, DeFelipe-Oroquieta J, Martin P, et al. Neuropathological findings in a patient with epilepsy and the Parry-Romberg syndrome. Epilepsia. 2001;42(9):1198–203. doi: 10.1046/j.1528-1157.2001.45800.x. [DOI] [PubMed] [Google Scholar]
- 88.Kister I, Inglese M, Laxer RM, Herbert J. Neurologic manifestations of localized scleroderma: a case report and literature review. Neurology. 2008;71(19):1538–45. doi: 10.1212/01.wnl.0000334474.88923.e3. [DOI] [PubMed] [Google Scholar]
- 89.Raina UK, Seth A, Gupta R, Goel N, Gupta A, Ghosh B. Parry-Romberg syndrome studied by spectral-domain optical coherence tomography. Ophthalmic Surg Lasers Imaging Retina. 2014;45 Online:E1–4. doi: 10.3928/23258160-20140115-01. [DOI] [PubMed] [Google Scholar]
- 90.You KH, Baik HS. Orthopedic and orthodontic treatment of Parry-Romberg syndrome. J Craniofac Surg. 2011;22(3):970–3. doi: 10.1097/SCS.0b013e31820fe339. [DOI] [PubMed] [Google Scholar]
- 91.Sartori-Valinotti JC, Tollefson MM, Reed AM. Updates on morphea: role of vascular injury and advances in treatment. Autoimmune Diseases. 2013;2013:467808. doi: 10.1155/2013/467808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roddi R, Riggio E, Gilbert PM, Vaandrager JM, van der Meulen JC. Progressive hemifacial atrophy in a patient with lupus erythematosus. Plast Reconstr Surg. 1994;93(5):1067–72. doi: 10.1097/00006534-199404001-00027. [DOI] [PubMed] [Google Scholar]
- 93.Pietkiewicz P, Gornowicz-Porowska J, Bowszyc-Dmochowska M, Dmochowski M. Pemphigoid gestationis in a female with progressive facial hemiatrophy: microchimerism as a speculative shared background is disputable. Pol J Pathol. 2012;63(1):71–4. [PubMed] [Google Scholar]
- 94.Poniecki A, Bernacka K, Hryszko S. [Romberg-type hemiatrophy in a case of severe rheumatoid arthritis] Wiad Lek. 1971;24(13):1313–5. [PubMed] [Google Scholar]
- 95.Gonul M, Dogan B, Izci Y, Varol G. Parry-Romberg syndrome in association with anti-dsDNA antibodies: a case report. J Eur Acad Dermatol Venereol. 2005;19(6):740–2. doi: 10.1111/j.1468-3083.2005.01290.x. [DOI] [PubMed] [Google Scholar]
- 96.Garcia-de la Torre I, Castello-Sendra J, Esgleyes-Ribot T, Martinez-Bonilla G, Guerrerosantos J, Fritzler MJ. Autoantibodies in Parry-Romberg syndrome: a serologic study of 14 patients. J Rheumatol. 1995;22(1):73–7. [PubMed] [Google Scholar]
- 97.Kleiner-Baumgarten A, Sukenik S, Horowitz J. Linear scleroderma, hemiatrophy and systemic lupus erythematosus. J Rheumatol. 1989;16(8):1141–3. [PubMed] [Google Scholar]
- 98.Asher SW, Berg BO. Progressive hemifacial atrophy: report of three cases, including one observed over 43 years, and computed tomographic findings. Arch Neurol. 1982;39(1):44–6. doi: 10.1001/archneur.1982.00510130046011. [DOI] [PubMed] [Google Scholar]
- 99.Fry JA, Alvarellos A, Fink CW, Blaw ME, Roach ES. Intracranial findings in progressive facial hemiatrophy. J Rheumatol. 1992;19(6):956–8. [PubMed] [Google Scholar]
- 100.Wolf SM. MA Verity Neurological complications of progressive facial hemiatrophy. J Neurol Neurosurg Psychiatry. 1974;37(9):997–1004. [Google Scholar]
- 101.Abele DC, Bedingfield RB, Chandler FW, Given KS. Progressive facial hemiatrophy (Parry-Romberg syndrome) and borreliosis. J Am Acad Dermatol. 1990;22(3):531–3. doi: 10.1016/s0190-9622(08)80402-1. [DOI] [PubMed] [Google Scholar]
- 102.Stern HS, Elliott LF, Beegle PH., Jr Progressive hemifacial atrophy associated with Lyme disease. Plast Reconstr Surg. 1992;90(3):479–83. doi: 10.1097/00006534-199209000-00020. [DOI] [PubMed] [Google Scholar]
- 103.Baskan EB, Kacar SD, Turan A, Saricaoglu H, Tunali S, Adim SB. Parry-Romberg syndrome associated with borreliosis: could photochemotherapy halt the progression of the disease? Photodermatol Photoimmunol Photomed. 2006;22(5):259–61. doi: 10.1111/j.1600-0781.2006.00238.x. [DOI] [PubMed] [Google Scholar]
- 104.Sahin MT, Baris S, Karaman A. Parry-Romberg syndrome: a possible association with borreliosis. J Eur Acad Dermatol Venereol. 2004;18(2):204–7. doi: 10.1111/j.1468-3083.2004.00862.x. [DOI] [PubMed] [Google Scholar]
- 105.Goodlad JR, Davidson MM, Gordon P, Billington R, Ho-Yen DO. Morphoea and Borrelia burgdorferi: results from the Scottish Highlands in the context of the world literature. Mol Pathol. 2002;55(6):374–8. doi: 10.1136/mp.55.6.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gutierrez-Gomez C, Godinez-Hana AL, Garcia-Hernandez M, Suarez-Roa Mde L, Toussaint-Caire S, Vega-Memije E, et al. Lack of IgG antibody seropositivity to Borrelia burgdorferi in patients with Parry-Romberg syndrome and linear morphea en coup de sabre in Mexico. Int J Dermatol. 2014;53(8):947–51. doi: 10.1111/ijd.12105. [DOI] [PubMed] [Google Scholar]
- 107.Daoud MS, Su WP, Leiferman KM, Perniciaro C. Bullous morphea: clinical, pathologic, and immunopathologic evaluation of thirteen cases. J Am Acad Dermatol. 1994;30(6):937–43. doi: 10.1016/s0190-9622(94)70113-x. [DOI] [PubMed] [Google Scholar]
- 108.Colome-Grimmer MI, Payne DA, Tyring SK, Sanchez RL. Borrelia burgdorferi DNA and Borrelia hermsii DNA are not associated with morphea or lichen sclerosus et atrophicus in the southwestern United States. Arch Dermatol. 1997;133(9):1174. [PubMed] [Google Scholar]
- 109.Fan W, Leonardi CL, Penneys NS. Absence of Borrelia burgdorferi in patients with localized scleroderma (morphea) J Am Acad Dermatol. 1995;33(4):682–4. doi: 10.1016/0190-9622(95)91311-4. [DOI] [PubMed] [Google Scholar]
- 110.Dillon WI, Saed GM, Fivenson DP. Borrelia burgdorferi DNA is undetectable by polymerase chain reaction in skin lesions of morphea, scleroderma, or lichen sclerosus et atrophicus of patients from North America. J Am Acad Dermatol. 1995;33(4):617–20. doi: 10.1016/0190-9622(95)91281-9. [DOI] [PubMed] [Google Scholar]
- 111.De Vito JR, Merogi AJ, Vo T, Boh EE, Fung HK, Freeman SM, et al. Role of Borrelia burgdorferi in the pathogenesis of morphea/scleroderma and lichen sclerosus et atrophicus: a PCR study of thirty-five cases. J Cutan Pathol. 1996;23(4):350–8. doi: 10.1111/j.1600-0560.1996.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 112.Lonchampt P, Emile J, Pelier-Cady MC, Cadou B, Barthelaix A. Central sympathetic dysregulation and immunological abnormalities in a case of progressive facial hemiatrophy (Parry-Romberg disease) Clin Auton Res. 1995;5(4):199–204. doi: 10.1007/BF01824007. [DOI] [PubMed] [Google Scholar]
- 113.Anderson PJ, Molony D, Haan E, David DJ. Familial Parry-Romberg disease. Int J Pediatr Otorhinolaryngol. 2005;69(5):705–8. doi: 10.1016/j.ijporl.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 114.Resende LA, Dal Pai V, Alves A. [Experimental study of progressive facial hemiatrophy: effects of cervical sympathectomy in animals] Rev Neurol. 1991;147(8-9):609–11. [PubMed] [Google Scholar]
- 115.Moss ML, Crikelair GF. Progressive facial hemiatrophy following cervical sympathectomy in the rat. Arch Oral Biol. 1960;1:254–8. doi: 10.1016/0003-9969(60)90052-2. [DOI] [PubMed] [Google Scholar]
- 116.Ignatowicz R, Michalowicz R, Kmiec T, Jozwiak S. [Familial form of Parry-Romberg syndrome] Pol Tyg Lek. 1985;40(2):47–9. [PubMed] [Google Scholar]
- 117.Leao M, da Silva ML. Progressive hemifacial atrophy with agenesis of the head of the caudate nucleus. J Med Genet. 1994;31(12):969–71. doi: 10.1136/jmg.31.12.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Johnson RH, Spaulding JM. Disorders of the autonomic nervous system. Chapter 7. Some disorders of regional circulation. Contemp Neurol Ser. 1974;11:114–28. [PubMed] [Google Scholar]
- 119.Lewkonia RM, Lowry RB. Progressive hemifacial atrophy (Parry-Romberg syndrome) report with review of genetics and nosology. Am J Med Genet. 1983;14(2):385–90. doi: 10.1002/ajmg.1320140220. [DOI] [PubMed] [Google Scholar]
- 120.Carreno M, Donaire A, Barcelo MI, Rumia J, Falip M, Agudo R, et al. Parry Romberg syndrome and linear scleroderma in coup de sabre mimicking Rasmussen encephalitis. Neurology. 2007;68(16):1308–10. doi: 10.1212/01.wnl.0000259523.09001.7a. [DOI] [PubMed] [Google Scholar]
- 121.Echenne B, Sebire G. Parry Romberg syndrome and linear scleroderma en coup de sabre mimicking Rasmussen encephalitis. Neurology. 2007;69(24):2274. doi: 10.1212/01.wnl.0000295701.56822.6f. [DOI] [PubMed] [Google Scholar]
- 122.Longo D, Paonessa A, Specchio N, Delfino LN, Claps D, Fusco L, et al. Parry-Romberg syndrome and Rasmussen encephalitis: possible association. Clinical and neuroimaging features. J Neuroimaging. 2011;21(2):188–93. doi: 10.1111/j.1552-6569.2009.00398.x. [DOI] [PubMed] [Google Scholar]
- 123.Shah JR, Juhasz C, Kupsky WJ, Asano E, Sood S, Fain D, et al. Rasmussen encephalitis associated with Parry-Romberg syndrome. Neurology. 2003;61(3):395–7. doi: 10.1212/wnl.61.3.395. [DOI] [PubMed] [Google Scholar]
- 124.Straube A, Padovan CS, Seelos K. [Parry-Romberg syndrome and Rasmussen syndrome: only an incidental similarity?] Nervenarzt. 2001;72(8):641–6. doi: 10.1007/s001150170066. [DOI] [PubMed] [Google Scholar]
- 125.Menascu S, Padeh S, Hoffman C, Ben-Zeev B. Parry-Romberg syndrome presenting as status migrainosus. Pediatr Neurol. 2009;40(4):321–3. doi: 10.1016/j.pediatrneurol.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 126.Simsek-Kiper PO, Roach E, Utine GE, Boduroglu K. Barraquer-Simons syndrome: a rare clinical entity. Am J Med Genet A. 2014;164(7):1756–60. doi: 10.1002/ajmg.a.36491. [DOI] [PubMed] [Google Scholar]
- 127.Payapvipapong K, Niumpradit N, Nakakes A, Buranawuti K. A rare case of acquired partial lipodystrophy (Barraquer-Simons syndrome) with localized scleroderma. Int J Dermatol. 2014;53(1):82–4. doi: 10.1111/j.1365-4632.2011.05435.x. [DOI] [PubMed] [Google Scholar]
- 128.Bergman JA. Primary hemifacial hypertrophy. Review and report of a case. Arch Otolaryngol. 1973;97(6):490–4. doi: 10.1001/archotol.1973.00780010504015. [DOI] [PubMed] [Google Scholar]
- 129.Gorlin RJ, Meskin LH. Congenital hemihypertrophy. Review of the literature and report of a case with special emphasis on oral manifestations. J Pediatr. 1962;61:870–9. doi: 10.1016/s0022-3476(62)80198-x. [DOI] [PubMed] [Google Scholar]
- 130.Antoniades K, Giannouli T, Vahtsevanos K. Hemifacial atrophy secondary to poliomyelitis. Int J Oral Maxillofac Surg. 1997;26(3):215–6. doi: 10.1016/s0901-5027(97)80823-5. [DOI] [PubMed] [Google Scholar]
- 131.Crikelair GF, Moss ML, Khuri A. Facial hemiatrophy. Plast Reconstr Surg Transplant Bull. 1962;29:5–13. doi: 10.1097/00006534-196201000-00002. [DOI] [PubMed] [Google Scholar]
- 132.Kreuter A, Gambichler T, Breuckmann F, Rotterdam S, Freitag M, Stuecker M, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141(7):847–52. doi: 10.1001/archderm.141.7.847. [DOI] [PubMed] [Google Scholar]
- 133.Zulian F, Vallongo C, Patrizi A, Belloni-Fortina A, Cutrone M, Alessio M, et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea) J Am Acad Dermatol. 2012;67(6):1151–6. doi: 10.1016/j.jaad.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 134.Goldberg-Stern H, deGrauw T, Passo M, Ball WS., Jr Parry-Romberg syndrome: follow-up imaging during suppressive therapy. Neuroradiology. 1997;39(12):873–6. doi: 10.1007/s002340050525. [DOI] [PubMed] [Google Scholar]
- 135.Korkmaz C, Adapinar B, Uysal S. Beneficial effect of immunosuppressive drugs on Parry-Romberg syndrome: a case report and review of the literature. South Med J. 2005;98(9):940–2. doi: 10.1097/01.smj.0000177355.43001.ff. [DOI] [PubMed] [Google Scholar]
- 136.Hashkes PJ, Becker ML, Cabral DA, Laxer RM, Paller AS, Rabinovich CE, et al. Methotrexate: new uses for an old drug. J Pediatr. 2014;164(2):231–6. doi: 10.1016/j.jpeds.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 137.Martini G, Ramanan AV, Falcini F, Girschick H, Goldsmith DP, Zulian F. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil. Rheumatology. 2009;48(11):1410–3. doi: 10.1093/rheumatology/kep244. [DOI] [PubMed] [Google Scholar]
- 138.Bergler-Czop B, Lis-Swiety A, Brzezinska-Wcislo L. Scleroderma linearis: hemiatrophia faciei progressiva (Parry-Romberg syndrom) without any changes in CNS and linear scleroderma “en coup de sabre” with CNS tumor. BMC Neurol. 2009;9:39. doi: 10.1186/1471-2377-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kreuter A, Hyun J, Stucker M, Sommer A, Altmeyer P, Gambichler T. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma. J Am Acad Dermatol. 2006;54(3):440–7. doi: 10.1016/j.jaad.2005.11.1063. [DOI] [PubMed] [Google Scholar]
- 140.Kerscher M, Volkenandt M, Gruss C, Reuther T, von Kobyletzki G, Freitag M, et al. Low-dose UVA phototherapy for treatment of localized scleroderma. J Am Acad Dermatol. 1998;38(1):21–6. doi: 10.1016/s0190-9622(98)70533-x. [DOI] [PubMed] [Google Scholar]
- 141.Gambichler T, Kreuter A, Rotterdam S, Altmeyer P, Hoffmann K. Linear scleroderma ‘en coup de sabre’ treated with topical calcipotriol and cream psoralen plus ultraviolet A. J Eur Acad Dermatol Venereol. 2003;17(5):601–2. doi: 10.1046/j.1468-3083.2003.00626.x. [DOI] [PubMed] [Google Scholar]
- 142.de Vasconcelos CM, do Nascimento GJ, Andrade E, Andrade M, Sobral AP. Association of aesthetic and orthodontic treatment in Parry-Romberg syndrome. J Craniofac Surg. 2010;21(2):436–9. doi: 10.1097/SCS.0b013e3181cfe917. [DOI] [PubMed] [Google Scholar]
- 143.Grippaudo C, Deli R, Grippaudo FR, Di Cuia T, Paradisi M. Management of craniofacial development in the Parry-Romberg syndrome: report of two patients. Cleft Palate Craniofac J. 2004;41(1):95–104. doi: 10.1597/02-066. [DOI] [PubMed] [Google Scholar]
- 144.Slack GC, Tabit CJ, Allam KA, Kawamoto HK, Bradley JP. Parry-Romberg reconstruction: optimal timing for hard and soft tissue procedures. J Craniofac Surg. 2012;23(7 Suppl 1):1969–73. doi: 10.1097/SCS.0b013e318258bd11. [DOI] [PubMed] [Google Scholar]
- 145.Palmero ML, Uziel Y, Laxer RM, Forrest CR, Pope E. En coup de sabre scleroderma and Parry-Romberg syndrome in adolescents: surgical options and patient-related outcomes. J Rheumatol. 2010;37(10):2174–9. doi: 10.3899/jrheum.100062. [DOI] [PubMed] [Google Scholar]
- 146.Jurkiewicz MJ, Nahai F. The use of free revascularized grafts in the amelioration of hemifacial atrophy. Plast Reconstr Surg. 1985;76(1):44–55. doi: 10.1097/00006534-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 147.Cervelli V, Gentile P. Use of platelet gel in Romberg syndrome. Plast Reconstr Surg. 2009;123(1):22e–3. doi: 10.1097/PRS.0b013e318194d242. [DOI] [PubMed] [Google Scholar]
- 148.Cervelli V, Gentile P. Use of cell fat mixed with platelet gel in progressive hemifacial atrophy. Aesthet Plast Surg. 2009;33(1):22–7. doi: 10.1007/s00266-008-9223-x. [DOI] [PubMed] [Google Scholar]
- 149.Grimaldi M, Gentile P, Labardi L, Silvi E, Trimarco A, Cervelli V. Lipostructure technique in Romberg syndrome. J Craniofac Surg. 2008;19(4):1089–91. doi: 10.1097/SCS.0b013e318176354a. [DOI] [PubMed] [Google Scholar]
- 150.Siebert JW, Longaker MT. Secondary craniofacial management following skeletal correction in facial asymmetry. Application of microsurgical techniques. Clin Plast Surg. 1997;24(3):447–58. [PubMed] [Google Scholar]
- 151.Siebert JW, Anson G, Longaker MT. Microsurgical correction of facial asymmetry in 60 consecutive cases. Plast Reconstr Surg. 1996;97(2):354–63. doi: 10.1097/00006534-199602000-00013. [DOI] [PubMed] [Google Scholar]
- 152.Roddi R, Riggio E, Gilbert PM, Hovius SE, Vaandrager JM, van der Meulen JC. Clinical evaluation of techniques used in the surgical treatment of progressive hemifacial atrophy. J Craniomaxillofac Surg. 1994;22(1):23–32. doi: 10.1016/s1010-5182(05)80292-6. [DOI] [PubMed] [Google Scholar]
- 153.Slack GC, Tabit CJ, Allam KA, Kawamoto HK, Bradley JP. Parry-romberg reconstruction: beneficial results despite poorer fat take. Ann Plast Surg. 2014;73(3):307–10. doi: 10.1097/SAP.0b013e31827aeb0d. [DOI] [PubMed] [Google Scholar]
- 154.Hu J, Yin L, Tang X, Gui L, Zhang Z. Combined skeletal and soft tissue reconstruction for severe Parry-Romberg syndrome. J Craniofac Surg. 2011;22(3):937–41. doi: 10.1097/SCS.0b013e31820fe27d. [DOI] [PubMed] [Google Scholar]
- 155.Yu-Feng L, Lai G, Zhi-Yong Z. Combined treatments of facial contour deformities resulting from Parry-Romberg syndrome. J Reconstr Microsurg. 2008;24(5):333–42. doi: 10.1055/s-2008-1080536. [DOI] [PubMed] [Google Scholar]
- 156.Mussinelli F, Magri F, Origlio A. L’emiatrofia facciale progressiva (I) Rivista Oto-Neuro-Oftal. 1964;39:3. [Google Scholar]
- 157.Mussinelli F, Magri F, Origlio A. L’emiatrofia facciale progressiva (II) Rivista Oto-Neuro-Oftal. 1965;40:105. [Google Scholar]
- 158.MÖbius. Der umschriebene Gesichtsschwund. In: Nothnagel CWH, editor. Specielle Pathologie und Therapie. Vienna: A. HÖlder; 1895.
- 159.Marburg O. Die Hemiatrophia facialis progressiva. In: Nothnagel CWH, editor. Specielle Pathologie und Therapie. Vienna: A. HÖlder; 1912. [Google Scholar]
- 160.Cassirer R. Die vasomotorisch-trophischen Neurosen. 2. Berlin: S. Karger; 1912. [Google Scholar]