

Abstract
Among the high bone mass disorders, the osteopetroses reflect osteoclast failure that prevents skeletal resorption and turnover leading to reduced bone growth and modeling and characteristic histopathological and radiographic findings. We report an 11-year-old boy with a new syndrome that radiographically mimics osteopetrosis but features rapid skeletal turnover. He presented at age 21 months with a parasellar, osteoclast-rich giant cell granuloma. Radiographs showed a dense skull, generalized osteosclerosis, and cortical thickening, medullary cavity narrowing, and diminished modeling of tubular bones. His serum alkaline phosphatase was > 5,000 IU/L (normal < 850). After partial resection, the granuloma re-grew but then regressed and stabilized during three years of uncomplicated pamidronate treatment. His hyperphosphatasemia transiently diminished but all bone turnover markers, especially those of apposition, remained elevated. Two years after pamidronate therapy stopped, BMD z-scores reached + 9.1 and + 5.8 in the lumbar spine and hip, respectively, and iliac crest histopathology confirmed rapid bone remodeling. Serum multiplex biomarker profiling was striking for low sclerostin. Mutation analysis was negative for activation of LRP4, LRP5, or TGFβ1 and for defective SOST, OPG, RANKL, RANK, SQSTM1, or sFRP1. Microarray showed no notable copy number variation. Studies of his non-consanguineous parents were unremarkable. The etiology and pathogenesis of this unique syndrome are unknown.
Keywords: biomarker profiling, giant cell granuloma, osteosclerosis, sclerostin
II) Introduction
Mendelian diseases that cause high bone mass reveal genes that importantly regulate skeletal homeostasis.(1,2) For several, enhanced osteoblast (OB) activity leads to strong bones. Examples include the endosteal hyperostoses that gradually add good quality bone to the subperiosteal and especially endosteal surfaces of cortices.(1,2) Among them, sclerosteosis and van Buchem disease reflect autosomal recessive (AR) transmission of loss-of-function mutations that compromise the structure and expression, respectively, of the gene that encodes the OB inhibitor sclerostin (SOST).(2) Consequently, bone formation mediated by low-density lipoprotein receptor-related protein 5 (LRP5) is enhanced.(2,3) Similarly, autosomal dominant (AD) “Worth type” endosteal hyperostosis(1) increases skeletal mass and strength, but from heterozygous gain-of-function mutation of LRP5.(3–5) In these endosteal hyperostoses, tubular bone “modeling” (external shaping) is essentially normal.(6) Conversely, wide and weak bones characterize the extremes of skeletal remodeling, including, when excessive, in juvenile Paget’s disease (JPD), Camurati-Engelmann disease (progressive diaphyseal dysplasia), or McCune-Albright syndrome,(6) and when halted from osteoclast (OC) failure in osteopetrosis (OPT).(7) The OPT skeleton is brittle despite hyperostosis (cortical bone thickening) and osteosclerosis (increased trabecular bone) stemming from the accumulation of calcified cartilage synthesized during endochondral bone formation within wide “undertubulated” metaphyses (Erlenmeyer flask deformity),(6) failure of osteons to interconnect, “hardening” of hydroxyapatite crystals, and, perhaps, poor healing of microfractures.(7)
We report a new syndrome that features a parasellar giant cell granuloma (GCG) and rapid skeletal turnover associated with radiographic findings indicating OPT.
III) Materials and Methods
A) Patient
This 11-year-old boy English boy weighed 3.6 kg when delivered at term to a 32-year-old G3, P2-3 woman after an uncomplicated pregnancy. Early milestones were on time. His parents were not consanguineous. The father was 6′ 3″ tall, and healthy; mother was 5′ 7″ tall, and had psoriasis and iritis. A brother and sister were well.
At age 2⅓ years, he became clumsy and an ophthalmologist noted vertical nystagmus. Magnetic resonance imaging (MRI) showed a well-defined, 5 × 5 × 6 cm, partially cystic, parasellar mass encroaching on his optic foramina.
At age 28 months, transpalatal subtotal resection of the mass revealed a GCG with abundant OCs (see Histopathological Findings). Before surgery, his serum alkaline phosphatase (ALP) was 5,598 IU/L (Nl, 177 – 1,036), 25-hydroxyvitamin D 16 ng/ml (Nl, 15 – 60), and IGF-1 was normal. One day and one month later, ALP was 1,883 and 5,640 IU/L, respectively. Serum TSH and free T4 were normal. Three months postoperatively, MRI showed that solid tissue had replaced the cystic component of the tumor, and that the mass had regrown to 6 × 5 × 4 cm.
At age 2¾ years, the patient struck his head and developed vomiting and left side paralysis. Imaging revealed a cerebellar infarct. Clotting studies were normal. Aspirin, 150 mg po every other day, was taken until age 7 years with physical rehabilitation for six months. Strength and function largely recovered. Serum ALP remained elevated at 5,266 IU/L, although calcium was 2.45 mmol/L (Nl, 2.15 – 2.74), 1,25-dihydroxyvitamin D 83 pmol/L (Nl, 43 – 144), and parathyroid hormone (PTH) 8.7 pmol/L (Nl, 1.1 – 6.9). Serum angiotensin converting enzyme was 85 U/L (Nl, 18 – 66).
At age 3 years, a rheumatologist found no antinuclear, antineutrophil cytoplasmic, or antiphospholipid antibodies and a negative rheumatoid factor. High dose adrenocorticotrophin stimulation testing showed adequate adrenal responsiveness. A chest radiograph, skeletal survey, and bone scintigraphy demonstrated diffuse osteosclerosis and mild modeling errors of his tubular bones (see Radiological Findings). The thick cortices and narrow medullary spaces of long bones suggested an endosteal hyperostosis, but mutation analysis for van Buchem disease and LRP5 activation was negative (courtesy, Dr. Wim van Hul, Antwerp, Belgium), and we then assessed the radiographic changes as consistent with OPT. Single photon emission computed tomography (SPECT) revealed intense activity in the mid-line skull base (see Radiological Findings). Because the GCG had regrown, pamidronate (PMD) was given intravenously (1 mg/kg/dose) for three consecutive days every three months from ages 3 to 6 years.
At age 5 years, SPECT revealed significant reduction in radionuclide accumulation within the patient’s head, and by age 6 years the GCG had regressed to 4.9 × 2.4 × 4.9 cm (see Radiological Findings). As the mass was then stable and the PMD infusions caused pruritus, treatment was decreased and stopped.
Bone turnover markers (BTMs) had not been measured before PMD administration. After 2 ½ years of PMD treatment, serum ALP activity decreased from 5,818 to 2,223 IU/L, but then rebounded. After 3 ¾ years of treatment, all BTMs were markedly elevated, especially those reflecting apposition (courtesy of Professor William Fraser, Royal Liverpool University Hospital, U.K.): serum procollagen type 1 amino-terminal propeptide (P1NP) was 2,654 mcg/L (Nl, 20 – 76), osteocalcin 285 mcg/L (Nl, 7 – 32), and carboxy-terminal collagen crosslinks (CTX) 3.5 mcg/L (Nl, 0.1 – 0.5). First-void morning urine (ARUP Consult®) showed a pyridinoline/creatinine (CRT) ratio of 644 μmole/mol (Nl, 117 – 325) and a deoxypyridinoline (DPD)/CRT ratio of 160 μmol/mol (Nl, 20 – 75). Thus, the pyridinoline/DPD ratio was elevated at 4.0 (Nl, 0.19 – 0.25) compared to age-matched controls.
At age 7 years, lethargy, pallor, recurrent viral illnesses, and headaches led to another adrenocorticotropin with normal results. However, his afternoon cortisol level was low (< 100 nmol/L) and the symptoms responded to 5 mg hydrocortisone orally each day at 4 pm. Subsequent testing demonstrated an impaired cortisol response, and the dose was increased to full replacement.
At age 8 years, following written consent, the patient was admitted to the Research Center at Shriners Hospital for Children, St. Louis, MO, USA where he received his ad libitum calcium intake of 950 mg/day estimated from a 7-day food record (1998 US RDA 800 mg). Diffuse evening leg pains had lessened with hydrocortisone therapy. Vision was impaired with decreased extra-ocular movement on the left. Sharp headaches occurred throughout the day, usually during writing, but he made good grades. Audiology had been reassuring. He reported normal smell and taste and no heat or cold intolerance, polydipsia, or polyuria. Shedding of deciduous teeth had begun normally. Two permanent mandibular incisors had erupted. His weight was 29 kg (76th centile), height 135 cm (87th centile), arm span 134 cm, sitting height 71 cm, and head circumference 53 cm (70th centile). Blood pressure was 98/63 mm Hg, pulse 96/min, respirations 24/min, and temperature 36.4° C (oral). There was pallor, mild hypertelorism, and his left eye could not adduct. His mandible was small. Overgrown maxillary gingiva was noted. Mild knock-knee deformity was apparent (Figure 1). Vision was markedly and moderately impaired in his left and right eye, respectively. Cranial nerves II and left III were compromised. The sclerae were white and fundi unremarkable. Strength and muscle tone were normal.
Figure 1. Patient at Age 8 Years.

There is pronounced micrognathia, mild hypertelorism, and mild lateral femoral and anterior tibial bowing.
Fasting serum was used for a biochemical profile (Dade Behring Dimension Xpand instrument; Siemens Health Care Diagnostics, Inc., Los Angeles, CA, USA) and to assay bone-specific alkaline phosphatase (BAP) by ELISA (Quidel Comp., San Diego, CA, USA) and osteocalcin (OCN: Kit #LKN1; Siemens Health Care Diagnostics). Results were compared to published reference ranges and Research Center values from 34 healthy children.(8) Free DPD (Immulite 1000 Pyrilinks – D Kit; Siemens Medical Solutions Diagnostics Ltd., Lianberis, Gwynedd, UK) was measured in a 24-hr urine collection. To explore for an OPT, we looked for serum elevations of the brain isoenzyme of creatine kinase (CK-BB: Kit #K20; Sebia, Norcross, GA, USA),(9) tartrate-resistant acid phosphatase, TRACP-5b (Kit #8033, Quidel; Los Angeles, CA, USA), lactate dehydrogenase (LDH), and aspartate aminotransferase (AST).(8) Serum calcium, ionized calcium, inorganic phosphate (Pi), CRT, and PTH were normal, but 25-hydroxyvitamin D was < 8 ng/ml (Nl, 19 – 58) (Supplementary Appendix, Table 1). Routine urinalysis and complete blood count were normal. Prothrombin time was increased at 12.5 seconds (Nl, 9.7 – 11.4), partial thromboplastin time 31 seconds (Nl, 24 – 34), international normalized ratio 1.2, and platelet function testing 88 seconds (Nl, 70 – 170). Two 24-hour urine collections revealed calcium/CRT ratios of 233 and 298 mg/g (183 and 328 mg/L; 4.7 and 5.4 mg/kg body weight, respectively), and CRT clearances of 135 and 120 ml/min/1.73m2. Serum TRACP5b was elevated at 40 U/L (Nl, 6 – 27) consistent with accelerated bone turnover or OC accumulation in OPT.(7–9) However, total CK was 76 U/L (Nl, 31 – 152), CK-BB 9.8% (Nl, 0 – 11), LDH 211 U/L (Nl, 141 – 237), and AST 19 U/L (Nl, 0 – 36)(8,9) (see Discussion). All BTMs were substantially elevated, especially those of bone apposition: serum ALP was 3,486 U/L (Nl, 218- 499 ), BAP 2,542 (Nl, 26 – 259), and urine DPD/CRT 177 μmol/mmol (Nl, 14 – 41) (Supplementary Appendix, Table 1). Serum multiplex biomarker profiling (SMBP)(10) of the patient and his parents was performed at Amgen, Inc., Thousand Oaks, CA, USA (Supplementary Appendix) using one “batch” assay with results contrasted to 36 gender-matched healthy pediatric and adult controls (12 males and 24 females). He also underwent DXA (Hologic QDR-4500A; Waltham, MA) and transiliac crest biopsy using a 5 mm internal diameter Bordier trephine after two 3-day “labels” of demeclocycline for non-decalcified histology (see Results).
Upon discharge from the Research Center, the patient was repleted with vitamin D and felt well with no significant bone-related symptoms. Two maxillary molars were extracted uneventfully. MRI demonstrated a stable GCG. Height velocity was normal, and stature appropriate for parental height. Serum IGF-1 was 20 nmol/L (Nl, 14 – 74) and gonadotropin profile expectedly prepubertal, but free T4 became low at 8.3 pmol/L (Nl, 9 – 19) with no compensatory increase in TSH ---- suggesting progressive loss of pituitary function. Then, he suffered new headaches and generalized limb pains. From ages 9 – 11 years, his serum ALP increased steadily from 6200 to 6400 to 9049 IU/L (Nl, 202 – 1151).
B) Gene Studies
Genomic DNA was purified from peripheral blood leukocytes (Gentra Puregene DNA extraction kit: Invitrogen; Carlsbad, CA, USA). PCR and sequencing in both directions (Applied Biosystems 3130, Foster City, CA) examined all coding exons and adjacent mRNA splice sites of candidate genes using published and unpublished methods and primers(11–23) (available upon request). DNA sequences were evaluated using AlignX software (Vector NTI, Invitrogen; Carlsbad, CA, USA) and by inspecting the electropherograms.
Because our patient had no biochemical or histopathological evidence of OPT (see Results), mutations were not sought for those genes.(24) Instead, we sequenced the exons and splice sites of SOST encoding sclerostin (sclerosteosis),(11) TNFRSF11B encoding OPG (JPD),(14) TNFRSF11A encoding RANK (disorders of constitutive RANK activation),(19) SQSTM1 encoding sequestosome 1 (Paget’s bone disease),(20) TGFβ1 encoding transforming growth factor beta 1 (Camurati-Engelmann disease),(16–18) exons 2 – 4 of LRP5(21–23) (Worth-type “high bone mass” endosteal hyperostosis), and exons 25 – 26 of LRP4 (bone overgrowth syndrome).(5) We also tested TNFSF11 encoding RANKL(13) and sFRP1(12) encoding secreted frizzled-related protein 1 for an activating mutation.
Microarray copy number analysis was performed with the Affymetrix SNP 6.0 chip (Laboratory for Clinical Genomics, Washington University School of Medicine, St. Louis, MO, USA) and Partek Genomics Suite (Partek, St. Louis, MO, USA).
IV) Results
A) Radiological Findings
1) Radiographs
Skeletal radiographs spanned ages 2¾ – 8 years and the PMD therapy and were consistently indicative of an OPT.(6,7)
a) Before PMD Treatment
At age 2¾ years, OPT was suggested by a sclerotic skull (including the base and orbits, with thickened diploic space), generalized osteosclerosis with uniform involvement of entire vertebrae, and thick cortices, narrow medullary cavities, and mild modeling errors in tubular bones (Figure 2). The ribs and clavicles were dense with slight widening and small medullary cavities. The pelvis and scapulae were uniformly sclerotic. There was slight lateral femoral bowing. No “rugger-jersey spine” or “bone-in-bone” changes that might suggest AD or AR OPT, respectively, were noted.(6,7)
Figure 2. Radiographic Findings Before PMD Therapy.

At age 3 years, the skeleton shows features consistent with OPT. These include diffuse osteosclerosis and hyperostosis, a skull base that is very sclerotic (a), laterally bowed femora (b), mild modeling abnormalities including an Erlenmeyer flask deformity of the proximal humerus (c), diffusely sclerotic spine (d), and femoral medullary cavities narrowing and thick cortices (e).
b) After PMD Treatment
At age 8 years, two years after PMD therapy stopped, the diffuse osteosclerosis was greater (Supplementary Appendix). Long bone cortices were thick with narrow medullary cavities and most showed slightly more undertubulation with mild Erlenmeyer flask appearances of the proximal and distal femurs, proximal and distal tibiae, fibulae, and humeri. Anterior tibial bowing indicated skeletal weakness, but there was no spondylolysis or spondylolithesis common in OPT.(7,25)
2) DXA
At age 3¾ years, after beginning PMD, the first DXA study revealed a lumbar spine BMD z-score of + 3.2. At age 5½ years, the spine BMD z-score was + 6.9, and the total body (including head) z-score was + 5.2 (DXA: GE-Lunar Prodigy,® Madison, WI, USA). At age 8 years, DXA (Hologic QDR-4500A) L1 - L4 spine z-score was + 9.1, total hip z-score was + 5.8 calculated from Kelly et al,(26) but reference ranges for z-scores were not available for left forearm (total) 0.451 g/cm2, whole body (excluding head) 1.025 g/cm2, or head 2.067 g/cm2.
3) Bone Scintigraphy
At age 3 years, before PMD treatment, increased tracer uptake involved the midline skull base in the region of the sphenoid bone, apparently extending into the suprasellar aspect of the GCG above the floor of the anterior cranial fossa.
At age 5 years, the skull appeared thick with increased radioisotope uptake in the base, face (greater on the left), maxilla, mandible, and neck.
4) Magnetic Resonance Imaging
At age 3 years, before PMD treatment, the intracranial mass was isotense on T1 imaging, of intermediate intensity on T2 imaging, and markedly enhanced after contrast. At age 5 years, the skull was thick, but the brain, ventricular size, and optic nerves were unremarkable. There was no Chiari malformation or intracranial calcification. High signal intensity in the sphenoid sinus matched the GCG (Supplementary Appendix). MRI findings spanning ages 8 – 11 years remained essentially unchanged with the GCG stable in size.
B) Histopathological Findings
1) Giant Cell Granuloma
The partially resected parasellar mass was a GCG. It contained ovoid and spindle cells with a zonal variation in cellularity and many scattered OC-like cells in a collagenous stroma with small foci of reactive bone and osteoid at the periphery (Figure 3A,B). The margins were expansile rather than invasive. The overlying sinonasal mucosa showed chronic inflammation. The OC-like cells expressed TRACP and cathepsin K (CTSK) (Figure 3C,D). The hypocellular areas appeared more edematous and collagenous. Scattered normal mitoses were seen without necrosis.
Figure 3. Giant Cell Grauloma.

At age 2-⅓ years, incomplete resection of the parasellar mass revealed findings consistent with a GCRG.
a. Low magnification (x 40) showing zones of pale collagenous tissue (asterisks) and darker, more cellular, tissue with hemorrhage (arrowheads) and many OCs (arrows).
b. High magnification (x 100) shows many multinucleate OCs (arrows) in a collagenous stroma with osteoid (arrowheads).
c,d. Immunohistochemical labeling shows strong expression of TRACP (C) and cathepsin K (D) in multinucleate OCs [magnification ~ x 200].
2) Iliac Crest
The right iliac crest was biopsied to secure the most representative skeletal histopathology seen radiographically. The surgeon reported very hard bone. Features of rapid bone remodeling included “cortical trabecularization” (i.e., no distinction between cortical and trabecular bone), with essentially the entire specimen being high-density trabecular bone (Figure 4A). The trabeculae had excess osteoid covering approximately 80% of their surface, and in many places this osteoid had increased thickness, was irregular, and extended into the center of trabeculae (Figure 4B). Goldner trichrome staining demonstrated abundant, plump, active-appearing OBs covering most of the osteoid surfaces (Figure 4C). OCs were also easily readily, and most appeared to be resorbing bone although a few were round and separate from bone surfaces consistent with prior exposure to PMD.(27) Fluorescence studies demonstrated good integration of demeclocycline labels, with well-demarcated and broadly-spaced bands adjacent to linear osteoid, as well as more sinuous labels at deeply placed osteoid (Figure 4D). Toluidine blue staining (not illustrated) showed no cartilage. The medullary space was not fibrotic, although hematopoietic elements were scarce. Cumulatively, these findings excluded OPT,(7) and instead indicated rapid bone turnover. High-power light microscopy did not suggest any inclusion bodies in the OCs.
Figure 4. Iliac Crest.

At age 8 years, biopsy was performed after “tetracycline labeling”.
a. Low-power image of entire specimen, stained with Goldner trichrome, shows no clear demarcation between cortex and trabeculae (scale bar, 2 mm).
b. Higher power image of the central portion of the specimen, stained with von Kossa, shows that unmineralized bone matrix (osteoid, red) covers most of the bone surface, and is also present around osteocyte lacunae deep in the trabeculae (scale bar, 500 μm).
c. Goldner trichrome stain shows numerous OCs, as well as osteoblasts (arrowheads), indicating high bone turnover (scale bar, 200 μm) (arrows).
d. Fluorescence microscopy reveals abundant, widely-spaced tetracycline labels (arrowheads) along most osteoid-covered bone surfaces, new bone formation more deeply, and irregular tetracycline incorporation (arrows) (scale bar, 200 μm).
C) Genetic Studies
No mutations were found in the genes that encode SOST, LRP5 (exons 2 – 4), LRP4 (exons 25 – 26), OPG, RANKL, RANK, SQSTM1, TGFβ1, and sFRP1. Microarray showed no notable copy number variation.
D) Serum Multiplex Biomarker Profiling
A distinctly low level of SOST was noted (Figure 5). Also, OPN and cathepsin K (CTSK) levels were clearly elevated. Conspicuous elevation of all BTMs (apposition greater than resorption) was revealed despite normal levels of PTH, TGFβ1, PGE2, OPG, RANKL, and DKK1 (Supplementary Appendix).
Figure 5. Serum Multiplex Biomarker Profiling.
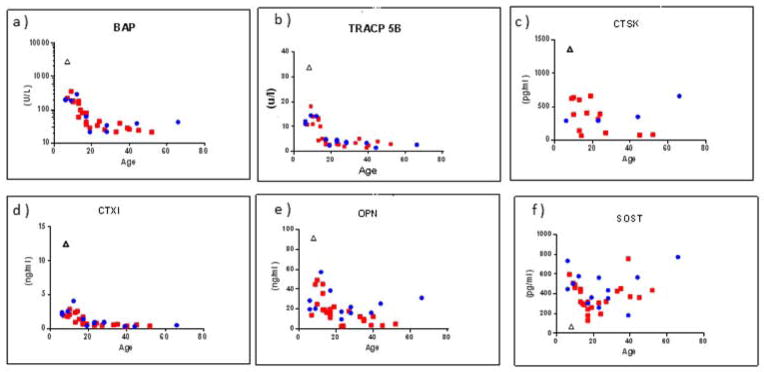
Select profiles (Supplementary Appendix) represent elevation of all BTMs, especially those of apposition, including BAP (a), TRACP-5b (b), CTX1 (c), CTSK (d), and OPN (e), whereas SOST is distinctly low (f).
E) Parent Studies
Skeletal radiographs were unremarkable. DXA (Hologic QDR 4500-A) revealed normal spine and hip BMD z-scores. Fasting sera had normal levels of ALP, BAP, and OCN. SMBP revealed no abnormalities reminiscent of their son’s findings (Supplementary Appendix).
V) Discussion
Elevated bone mass has a wide-ranging differential diagnosis (Supplementary Appendix: Table 5)(28) that is usually approachable from the radiographic features, including the anatomic distribution of the dense bone, whether hyperostosis and/or osteosclerosis are present, and if bone modeling and/or integrity are altered.(6,7,28–30) For the mendelian disorders, a diagnosis is now usually achievable by mutation analysis,(2,31,32) but new high bone mass diseases (some heritable) remain unreported and unexplained (MPW: personal observation). Our patient’s radiographic findings excluded many genetic disorders; e.g., pycnodysostosis (open fontanel, distal phalangeal absorption), dysosteosclerosis (metaphyseal osteosclerosis, platyspondyly), sclerosteosis (bone syndactyly), van Buchem disease (distribution and appearance of hyperostosis and osteosclerosis), Camurati-Engelmann disease (progressive diaphyseal dysplasia), etc.(6,7,28,33) The endosteal hyperostoses were dismissed because the skeleton models well and is strong, BTMs are normal or only modestly increased,(2,28) and mutation analyses proved negative.
Instead, our patient’s radiographs before PMD treatment indicated an OPT. He had a thick skull, significant generalized osteosclerosis, and wide cortices, narrow medullary spaces, and diminished modeling of major long bones.(6,7) Also, his appendicular skeleton seemed weak with tibial bowing.(7) Radiographs showed that his high BMD z-scores were not an artifact from expanded bones assessed by this areal (gm/cm2) measurement. He had received a relatively high dose of PMD, 12 mg/kg/yr, that may have exacerbated his radiographic features of OPT.(25,27) Erlenmeyer flask deformity can occur from Gaucher’s disease, craniometaphyseal dysplasia, and other disorders,(33,34) but these entities seemed excluded. A rugger jersey spine and dense skull base occur in adults with AD OPT (Albers-Schönberg disease), but not in young children. “Endobones”, a finding of severe OPT, was understandably absent in our patient because of his high bone turnover. Cranial nerve palsies from impaired widening of their foramina, and myelophthisis from small medullary spaces,(7) are extraskeletal complications of severe OPT, but our patient’s neurological deficits were attributable to his stroke and GCG, and he manifested no bleeding, anemia, infections, or hematological disturbances. We found no report of GCG in OPT. Now, most OPTs are understood genetically,(24,32,35) featuring disruption of production or secretion of protons (H+) by OCs into the resorption space,(7) but especially rare AR “OC-poor” OPTs reflect impaired osteoclastogenesis from deactivation of TNFSF11(13) or TNFRSF11A(19) encoding RANKL and RANK, respectively. Excluding OPT in our patient was important because we would use a bisphosphonate for the OC-like cells in his GCG. Although, antiresorptives are now so potent that prolonged excesses during growth can cause OPT,(25,27) our patient’s BTMs were always elevated and OPT was excluded when iliac crest histopathology showed rapid remodeling without retained calcified cartilage encased within trabeculae.(7)
Rapid bone remodeling causes skeletal weakness.(28) Remodeling is fastest in juvenile Paget’s disease (JPD) that features a “cotton-wool” pattern in the calvarium,(33,36) wide undertubulated osteopenic bones with irregular cortical thickening, a disorganized trabecular pattern that sometimes becomes radiographically dense, bone deformities, and fractures.(36) Most JPD reflects AR loss-of-function mutations within TNFRSF11B which encodes OPG (“JPD1”),(36) though rarely there is a heterozygous insertional duplication within exon 1 of TNFRSF11A encoding RANK (“JPD2”),(37) or the etiology is unknown.(36,37) Our patient had no mutations of OPG or RANK, and his radiographic findings did not match JPD.(14,36) Hence, we also excluded mutations in SQSTM1 associated with Paget’s bone disease (PBD)(20) and TNFSF11 (encoding RANKL) and sFRP1 (encoding frizzled related protein-1 which binds RANKL and inhibits OC formation).(12) Microarray copy number analysis too was unrevealing. Whole exome sequencing is planned,(35) but our studies of his non-consanguineous parents did not support a genetic etiology.
Central GCG is a benign interosseous lesion typically of the maxilla and mandible found before age 20 years.(38) GCGs can be locally aggressive and recur following resection.(39) In 2013, a t(1;17;18) as well as other random numerical chromosomal changes were reported.(40) Lesions histologically identical to central GCG occur in cherubism, Noonan syndrome, and neurofibromatosis, type 1.(41) Our patient’s mass was typical of a GCG, including a zonal architecture with a predominance of mononuclear cells and many multinucleate OCs that expressed TRACP and CTSK.
OCs accumulate in GCG (sometimes referred to as GC reparative granuloma),(42) brown tumors from hyperparathyroidism, and GC tumor of bone (GCT). In the craniofacial region, these three lesions share clinical, radiological, and histopathologic features.(42) Of interest, GCGs and GCTs complicate PBD with its one or more foci of rapidly remodeling bone.(43,44) Furthermore, we reported “extraskeletal osteoclastomas” in an African-American woman with PBD and neurofibromatosis, type 1,(45) and in a Korean man with familial PBD.(46) It is possible, therefore, that our patient’s rapid bone turnover predisposed his GCG. GCT of bone seems to offer a paradigm of stromal-hematopoietic cellular interactions,(47) where RANK and RANKL are both expressed.(48) Alternatively, his GCG could be elaborating some systemic factor(s) causing his skeletal disease. However, we found no report that GCGs release systemic factors. Sequential SMBP would have been interesting spanning resection of his GCG. An abrupt reduction of his serum ALP followed the surgery, and later SMBP revealed elevated BTMs, particularly those of bone apposition. High serum TRACP5b and CTSK levels were consistent with his accelerated skeletal turnover and were also present in his GCG. SMBP showed a low ratio between the type I collagen formation marker, CICP, and the collagen degradation maker, CTX1, of just 38 (mean ± SD; 148 ± 52 for healthy individuals across all ages), due to elevation of CTX1. This suggested our patient produced normal amounts of type I collagen, but degraded it rapidly. Nevertheless, he had normal serum levels of RANKL and OPG and unremarkable DKK-1, TGFβ1, PGE2, and FGF-23 levels. The very low SOST level despite a structurally intact SOST gene may indicate decreased production or enhanced degradation of SOST and explain his rapid bone apposition. Alternatively, his rapid skeletal turnover with impaired osteocyte formation (the major source of SOST)(49) might explain this finding.
Regression and then stability of our patient’s GCG during PMD therapy was intriguing. Regression has occurred for GCT cells exposed in vitro to bisphosphonate,(50) and for GCGs treated with intra-lesional steroid, calcitonin, or interferon alpha.(39,41) Regression and then stability of his GCG with PMD treatment occurred despite only modest transient decrements in BTMs, and no radiologic evidence that his skeleton had improved. We used PMD cautiously hoping to control his GCG, but anticipated that his skeleton would respond as has benefitted JPD.(39) However, two years after we stopped his PMD therapy, BTMs were again markedly elevated, iliac crest histopathology confirmed the presence of rapid bone remodeling, and the highest BMD values were recorded, although our patient’s GCG remained stable. Glucocorticoid replacement for pituitary-centered adrenal insufficiency probably did not influence his skeletal disease, although “extraskeletal osteoclastomas” respond to pharmacologic doses of dexamethasone.(35,36) Denosumab, an anti-RANKL monoclonal antibody, was not available when we treated our patient with PMD. In 2013, it was approved by the FDA for treatment of GCT,(51–53) and has been given “off-label” to children with various skeletal disorders including osteogenesis imperfecta,(54) fibrous dysplasia,(55) JPD,(56) and post-transplantation hypercalcemia in OPT,(57) and to adults with GCG.(58) There would be concern for abrupt hypocalcemia, because significant hypocalcemia has complicated treatment of JPD with denosumab(56) or zolendronate,(59) and a rapid increase in his already elevated bone mass. We do not know if our patient’s rapid remodeling skeleton and GCG represented an “OC-autonomous” disease treatable by marrow cell transplantation, so anti-resorptive therapy clearly seemed safest.
In summary, although the radiographic features of our patient’s high bone mass disease indicated an OPT, iliac crest histology documented rapid skeletal remodelling concordant with elevated BTMs. However, no mutations associated with accelerated skeletal turnover were identified. His GCG with many OC-like cells and his bone disease indicated systemic enhancement of osteoclastogenesis. SMBP showed a striking low but unexplained serum SOST level that suggested deficiency of this inhibitor of WNT signalling explained his high bone mass. Whether his GCG could be enhancing bone remodelling systemically, or was a consequence, is uncertain. The etiology and pathogenesis of this new and intriguing syndrome remain unknown.
Supplementary Material
Acknowledgments
Supported by Shriners Hospitals for Children, The Clark and Mildred Cox Inherited Metabolic Bone Disease Research Fund, The Hypophosphatasia Research Fund, The Frederick S. Upton Foundation, The Barnes-Jewish Hospital Foundation, and Amgen, Inc. Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number DK067145.
Dr. Ziafang Zhang, Ms. Margaret Huskey, and Ms. Shenghui Duan helped with mutation analyses. Ms. Denise Dwyer and Ms. Kay Stigall facilitated the SMBP. Dr. Steven Teitelbaum reviewed the skeletal histopathology. Dr. Nick Athanasou performed the immunohistochemical studies. Ms. Sharon McKenzie and Ms. Vivienne McKenzie helped prepare the manuscript.
Footnotes
Presented in part at the 33rd Annual Meeting of the American Society for Bone and Mineral Research, September 16 – 20, 2011, San Diego, CA, USA [J Bone Miner Res 26 (Suppl):156, 2011] and the 50th Annual Meeting of the European Society of Pediatric Endocrinology, September 25 – 28, 2011, Glasgow, UK [Horm Res Paediatr 76 (Suppl 2):64 – 65, 2011].
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures: The authors have nothing to disclose.
Contributor Information
Katherine L. Madson, Email: kmadson@shrinenet.org.
Steven Mumm, Email: smumm@dom.wustl.edu.
William H. McAlister, Email: mcalisterw@mir.wustl.edu.
Deborah V. Novack, Email: dnovack@path.wustl.edu.
Jo C. Blair, Email: Jo.Blair@alderhey.nhs.uk.
Timothy R. Helliwell, Email: trh@liverpool.ac.uk.
Marina Stolina, Email: mstolina@amgen.com.
Laurence J. Abernethy, Email: Laurence.Abernethy@alderhey.nhs.uk.
Nicholas J. Shaw, Email: NICK.SHAW@bch.nhs.uk.
VII) References
- 1.Whyte MP. Searching for gene defects that cause high bone mass. Am J Hum Genet. 1997;60:1309–1311. doi: 10.1086/515486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perdu B, Van Hul W. Sclerosing bone disorders. In: Thakker RV, Whyte MP, Eisman JA, Igarashi T, editors. Genetics of Bone Biology and Skeletal Disease. 2013. pp. 361–374. [Google Scholar]
- 3.Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone mass trait. Am J Hum Genet. 2002;70:11–9. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y, Warman ML, de Vernejoul MC, Bollerslev J, Van Hul W. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with anincreased bone density. Am J Hum Genet. 2003;72:763–771. doi: 10.1086/368277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leupin O, Piters E, Halleux G, Hu S, Kramer I, Morvan F, Bouwmesster T, Schirle M, Bueno-Lozano M, Fuentes FJR, Itin PH, boudin E, deFreitas F, Jennes K, Brannetti B, Charara N, Ebersbach H, Geisse S, Lu CX, Bauer A, Van Hul W, Kneissel M. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286:19489–19500. doi: 10.1074/jbc.M110.190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozlowski K, Beighton P. An aid to Radiodiagnosis. 2. Springer-Verlag London Limited; 1995. Gamut index of skeletal dysplasias. [Google Scholar]
- 7.Whyte MP. Osteopetrosis. In: Royce PM, Steinmann B, editors. Connective Tissue And Its Heritable Disorders: Molecular, Genetic, And Medical Aspects. 2. Wiley-Liss; New York, New York, USA: 2002. pp. 789–807. [Google Scholar]
- 8.Whyte MP, Kempa LG, McAlister WH, Zhang F, Mumm S, Wenkert D. Elevated serum lactate dehydrogenase isoenzymes and aspartate transaminase distinguish Albers-Schönberg osteopetrosis (chloride channel 7 deficiency) among the sclerosing bone disorders. J Bone Miner Res. 2010;25:2515–2526. doi: 10.1002/jbmr.130. [DOI] [PubMed] [Google Scholar]
- 9.Whyte MP, Chines A, Silva DP, Landt Y, Ladenson JH. Creatine kinase brain isoenzyme (BB-CK) presence in serum distinguishes osteopetroses among the sclerosing bone disorders. J Bone Miner Res. 1996;11:1438–1443. doi: 10.1002/jbmr.5650111010. [DOI] [PubMed] [Google Scholar]
- 10.Whyte MP, Wenkert D, Dwyer DC, Lacey DL, Stolina M. Systemic biomarker profiling of metabolic and dysplastic skeletal diseases using multiplex serum protein analyses (Abstract) J Bone Miner Res. 2010;25 (Suppl 1):S137. [Google Scholar]
- 11.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Häusler KD, Horwood NJ, Chuman Y, Fisher JL, Ellis J, Martin TJ, Rubin JS, Gillespie MT. Secreted frizzled-related protein-1 inhibits RANKL-dependent osteoclast formation. J Bone Miner Res. 2004;19(11):1873–81. doi: 10.1359/JBMR.040807. [DOI] [PubMed] [Google Scholar]
- 13.Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, Grabowski P, Del Fattore A, Messina C, Errigo G, Coxon FP, Scott DI, Teti A, Rogers MJ, Vezzoni P, Villa A, Helfrich MH. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nature Genet. 2007;39:960–962. doi: 10.1038/ng2076. [DOI] [PubMed] [Google Scholar]
- 14.Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, Mumm S. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med. 2002;347:175–84. doi: 10.1056/NEJMoa013096. [DOI] [PubMed] [Google Scholar]
- 15.Whyte MP, Wenkert D, McAlister WH, Novack DV, Nenninger AR, Zhang X, Huskey M, Mumm S. Dysosteosclerosis presents as an “osteoclast-poor” form of osteopetrosis: comprehensive investigation of a 3-year-old girl and literature review. J Bone Miner Res. 2010;25:2527–39. doi: 10.1002/jbmr.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whyte MP, Totty WG, Novack DV, Zhang X, Wenkert D, Mumm S. Camurati-Engelmann disease: unique variant featuring a novel mutation in TGFβ1 encoding transforming growth factor beta 1 and a missense change in TNFSF11 encoding RANK ligand. J Bone Miner Res. 2011;26:920–933. doi: 10.1002/jbmr.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssens K, Gershoni-Baruch R, Guañabens N, et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26:273–275. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- 18.Kinoshita A, Saito T, Tomita H, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000;26:19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- 19.Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, Moratto D, Mazzolari E, Clayton-Smith J, Orchard P, Coxon FP, Helfrich MH, Crockett JC, Mellis D, Vellodi A, Tezcan I, Notarangelo LD, Rogers MJ, Vezzoni P, Villa A, Frattini A. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008;83:64–76. doi: 10.1016/j.ajhg.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, Van Eerdewegh P, Recker RR, Johnson ML. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–9. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. New Eng J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 23.Whyte MP, Reinus WH, Mumm S. High-bone-mass disease and LRP5 (letter) N Engl J Med. 2004;350(20):2096–9. doi: 10.1056/NEJM200405133502017. [DOI] [PubMed] [Google Scholar]
- 24.Sobacchi C, Schulz A, Coxon FR, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Natures Reviews/Endocrinology. 2013;9:522–538. doi: 10.1038/nrendo.2013.137. [DOI] [PubMed] [Google Scholar]
- 25.Whyte MP, McAlister WH, Novack DV, Clements KL, Schoenecker PL, Wenkert D. Bisphosphonate-induced osteopetrosis: novel bone modeling defects, metaphyseal osteopenia, and osteosclerosis fractures after drug exposure ceases. J Bone Miner Res. 2008;23:1698–1707. doi: 10.1359/jbmr.080511. [DOI] [PubMed] [Google Scholar]
- 26.Kelly TL, Specker BL, Binkley T, et al. Pediatric BMD reference database for U.S. white children. Bone. 2005;36:s30. [Google Scholar]; Sawyer AJ, Bachrach LK, Fung EB, editors. Densitometry in Growing Patients: Guidelines for Clinical Practice. Humana Press; New Jersey: p. 203e204. [Google Scholar]
- 27.Whyte MP, Wenkert D, Clements KL, McAlister WH, Mumm S. Bisphosphonate-induced osteopetrosis. N Eng J Med. 2003;349:455–461. doi: 10.1056/NEJMoa023110. [DOI] [PubMed] [Google Scholar]
- 28.Whyte MP. Sclerosing bone disorders. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8. Chapter 93. The American Society for Bone and Mineral Research, Wiley-Blackwell; Ames, IA DC: 2013. p. 770. [Google Scholar]
- 29.Ihde LL, Forrester DM, Gottsegen CF, Masih S, Patel DB, Vachon LA, White EA, Matcuk GR. Sclerosing bone dysplasias: review and differentiation from other causes of osteosclerosis. RadioGraphics. 2011;31:1865–1882. doi: 10.1148/rg.317115093. [DOI] [PubMed] [Google Scholar]
- 30.Gregson CL, Hardcastle SA, Cooper C, Tobias JH. Friend or foe: high bone mineral density on routine bone density scanning, a review of causes and management. Rheumatology. 2013;52:968–985. doi: 10.1093/rheumatology/ket007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aggarwal S. Skeletal dysplasias with increased bone density: evolution of molecular pathogenesis in the last century. Gene. 2013;528:41–45. doi: 10.1016/j.gene.2013.04.069. [DOI] [PubMed] [Google Scholar]
- 32.Thakker RV, Whyte MP, Eisman J, Igarashi T, editors. Genetics of Bone Biology and Skeletal Disease. Elsevier (Academic Press); San Diego, CA: 2013. [Google Scholar]
- 33.Reeder MM. Comprehensive Lists of Roetgen Differential Diagnosis. Springer-Verlag Inc; New York: 1993. Reeder and Felson’s Gamuts in Bone, Joint and Spine Radiology. [Google Scholar]
- 34.Faden MA, Krakow D, Ezgu F, Rimoin DL, Lachman RS. The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am J Med Genet. 2009;149A:1334–1345. doi: 10.1002/ajmg.a.32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sule G, Campeau PM, Zhang VW, Nagamani SCS, Dawson BC, Grover M, Bacino CA, Sutton VR, Brunetti-Pierri N, Lu JT, Lemire E, Gibbs RA, Cohn DH, Cui H, Wong JL, Lee BH. Next-generation sequencing for disorders of low and high bone mineral density. Osteoporos Int. 2013;24:2253–2259. doi: 10.1007/s00198-013-2290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whyte MP. Mendelian Disorders of RANKL/OPG/RANK Signaling. In: Thakker RV, Whyte MP, Eisman J, Igarashi T, editors. Genetics of Bone Biology and Skeletal Disease. Elsevier (Academic Press); San Diego, CA: 2013. pp. 309–324. [Google Scholar]
- 37.Whyte MP, Tau C, McAlister WH, Zhang X, Novack DV, Santini-Araujo E, Preliasco V, Mumm S. Juvenile Paget’s disease caused by mutation of TNFRSF11A encoding RANK. (in manuscript) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaffe HI. Giant cell reparative granuloma, traumatic bone cyst, and fibrous (fibro-osseous) dysplasia of the jaw bones. Oral Surg. 1953;6:159–175. doi: 10.1016/0030-4220(53)90151-0. [DOI] [PubMed] [Google Scholar]
- 39.Nogueria RLM, Faria MHG, Osterne RLV, Cavalcante RB, Ribeiro RA, Rabenhorst SHB. Glucocorticoid and calcintonin receptor expression in central giant cell lesions: implications for therapy. Int J Oral Maxillofac Surg. 2012;41:994–1000. doi: 10.1016/j.ijom.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 40.Maron E, Kachko L, Brennan PA, Bodner L. Gytogenetics of Central Giant Cell Granuloma of the Mandible. J Oral Maxillofac Surg. 2013;71:1541–1544. doi: 10.1016/j.joms.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 41.deLange J, van den Akker HP, van den Berg H. Central giant cell granuloma of the jaw: a review of the literature with emphasis on therapy options. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104:603–615. doi: 10.1016/j.tripleo.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Borges BBP, Fornazieri MA, Correia de Araujo Bezerra AP, Martins LAL, de Rezende Pinna F, Voegels RL. Giant cell lesions in the craniofacial region: a diagnostic and therapeutic challenge. International Forum of Allergy & Rhinology. 2012;2:501–506. doi: 10.1002/alr.21050. [DOI] [PubMed] [Google Scholar]
- 43.Gianfrancesco F, Rendina D, Merlotti D, Esposito T, Amyere M, Formicola D, Muscariello R, De Filippo G, Strazzullo P, Nuti R, Vikkula M, Gennari G. Giant cell tumor occurring in familial Paget’s disease of bone: report of clinical characteristics and linkage analysis of a large pedigree. J Bone Miner Res. 2013;28:341–350. doi: 10.1002/jbmr.1750. [DOI] [PubMed] [Google Scholar]
- 44.Upchurch KS, Simon LS, Schiller AL, Rosenthal DI, Campion EW, Krane SM. Giant cell reparative granuloma of Paget’s disease of bone: a unique clinical entity. Annals of Internal Medicine. 1983;98:35–40. doi: 10.7326/0003-4819-98-1-35. [DOI] [PubMed] [Google Scholar]
- 45.Ziambaras K, Totty WA, Teitelbaum SL, Dierkes M, Whyte MP. Extraskeletal osteoclastomas responsive to dexamethasone treatment in Paget bone disease. Journal of Clinical Endocrinology and Metabolism. 1997;82:3826–3834. doi: 10.1210/jcem.82.11.4353. [DOI] [PubMed] [Google Scholar]
- 46.Kim GS, Kim SH, Cho JK, Park JY, Shin MJ, Shong YK, Lee K–U, Han H, Kim T–G, Teitelbaum SL, Reinus W, Whyte MP. Paget bone disease involving young adults in a three-generation Korean kindred. Medicine (Baltimore) 1997;76:157–169. doi: 10.1097/00005792-199705000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Robinson D, Einhorn TA. Giant cell tumor of bone: a unique paradigm of stromal-hematopoietic cellular interactions. J Cell Biochem. 1994;55:300–3. doi: 10.1002/jcb.240550305. [DOI] [PubMed] [Google Scholar]
- 48.Roux S, Amazit L, Meduri G, Guiochon-Mantel A, Milgrom E, Mariette X. RANK (receptor activator of nuclear factor kappa B) and RANK ligand are expressed in giant cell tumors of bone. Am J Clin Pathol. 2002;117:210–6. doi: 10.1309/BPET-F2PE-P2BD-J3P3. [DOI] [PubMed] [Google Scholar]
- 49.van Bezooijen RL, Roelen BAJ, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Löwik CWGM. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng YY, Huang L, Lee KM, Xu JK, Zheng MH, Jumta SM. Bisphosphonates induce apoptosis of stromal tumor cells in giant cell tumor of bone. Calcif Tissue Int. 2004;75:71–77. doi: 10.1007/s00223-004-0120-2. [DOI] [PubMed] [Google Scholar]
- 51.Chawla S, Henshaw R, Seeger L, Choy E, Blay JY, Ferrari S, Kroep J, Grimer R, Reichardt P, Rutkowski P, Schuetze S, Skubitz K, Staddon A, Thomas D, Qian Y, Jacobs I. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: interim analysis of an open-label, parallel-group, phase 2 study. doi: 10.1016/S1470-2045(13)70277-8. www.thelancet.com/oncology. Published online July 16, 2013 http://dx.doi.org/10.1016/S1470-2045(13)70277-8. [DOI] [PubMed]
- 52.Karras NA, Polgreen LE, Ogilvie C, Manivel JC, Skubitz KM, Lipsitz E. Denosumab treatment of metastatic giant-cell tumor of bone in a 10-year-old girl. Journal of Clinical Oncology 2013. 2013;31:e200–e202. doi: 10.1200/JCO.2012.46.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu SF, Adams B, Yu XC, Xu M. Denosumab and giant cell tumour of bone – a review and future management considerations. Current Oncology. 2013;20:442–447. doi: 10.3747/co.20.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Semler O, Netzer C, Hoyer-Kuhn H, Becker J, Eysel P, Schoenau E. First use of the RANKL antibody denosumab in osteogenesis imperfecta Type VI. J Musculoskelet Neuronal Interact. 2012;12:183–188. [PubMed] [Google Scholar]
- 55.Boyce AM, Chong WH, Yao J, Gafni RI, Kelly MH, Chamberlain CE, Bassim C, Cherman N, Ellsworth M, Kasa-Vubu JZ, Farley FA, Molinolo AA, Bhattacharyya N, Collins MT. Denosumab treatment for fibrous dysplasia. J Bone Minerl Res. 2012;27:1462–1470. doi: 10.1002/jbmr.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grasemann C, Schundeln M, Wieland R, Bergmann C, Wieczorek D, Zabel B, Schweiger B, Hauffa BP. Effects of denosumab on bone biochemistry and calcium metabolism in a girl with Juvenile Paget’s disease. J Clin Endocrinol Metab. 2013;98:3121–3126. doi: 10.1210/jc.2013-1143. [DOI] [PubMed] [Google Scholar]
- 57.Shroff R, Beringer O, Rao K, Hofbauer LC, Schulz A. Denosumab for post-transplantation hypercalcemia in osteopetrosis (letter to editor) N Engl J Med. 2012;367;18:1766–1767. doi: 10.1056/NEJMc1206193. [DOI] [PubMed] [Google Scholar]
- 58.Malmquist M, Schow SR. Treatment of central giant cell granuloma with denosumab therapy in two patients. Journal of Oral and Maxillofacial Surgery. 2013;71(9) Supplement 1:e75. doi: 10.1016/j.joms.2014.06.456. [DOI] [PubMed] [Google Scholar]
- 59.Polyzos SA, Anastasilakis AD, Litsas I, Efstathiadou Z, Kita M, Arsos G, Moralidis E, Papatheodorou A, Terpos E. Profound hypocalcemia following effective response to zoledronic acid treatment in a patient with juvenile Paget’s disease. J Bone Miner Metab. 2010;28:706–712. doi: 10.1007/s00774-010-0198-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.