

Abstract
Heat-labile enterotoxin (LT) is an important virulence factor expressed by enterotoxigenic Escherichia coli. The route of LT secretion through the outer membrane and the cellular and extracellular localization of secreted LT were examined. Using a fluorescently labeled receptor, LT was found to be specifically secreted onto the surface of wild type enterotoxigenic Escherichia coli. The main terminal branch of the general secretory pathway (GSP) was necessary and sufficient to localize LT to the bacterial surface in a K-12 strain. LT is a heteromeric toxin, and we determined that its cell surface localization was mediated by the its B subunit independent of an intact GM1 ganglioside binding site and that LT binds lipopolysaccharide and GM1 concurrently. The majority of LT secreted into the culture supernatant by the GSP in E. coli associated with vesicles. Only a mutation in hns, not overexpression of the GSP or LT, caused an increase in vesicle yield, supporting a specific vesicle formation machinery regulated by the nucleoid-associated protein HNS. We propose a model in which LT is secreted by the GSP across the outer membrane, secreted LT binds lipopolysaccharide via a GM1-independent binding region on its B subunit, and LT on the surface of released outer membrane vesicles interacts with host cell receptors, leading to intoxication. These data explain a novel mechanism of vesicle-mediated receptor-dependent delivery of a bacterial toxin into a host cell.
Enterotoxigenic Escherichia coli (ETEC)1 is an important pathogen responsible for traveler’s diarrhea and >700,000 childhood deaths annually because of diarrhea in third world countries (1–4). ETEC produces two toxins implicated in the etiology of diarrhea, heat stabile toxin and heat-labile enterotoxin (LT) (1, 5). LT, which is encoded on a relatively uncharacterized 60-kb virulence plasmid (1), is one of a group of AB5 toxins (6, 7) that also includes Shiga toxin, pertussis toxin, and cholera toxin (CT). These heteromeric toxins consist of a catalytic A subunit (LTA) and a pentamer of receptor-binding B subunits (LTB) (8, 9). In addition to structural homology, LT shares 80% sequence homology with the Vibrio cholerae toxin CT (10, 11). The ring-shaped B pentamer of both LT and CT mediates binding to the host epithelial receptor GM1 (12–14). LT is more promiscuous than CT in that it can also bind other receptors containing a terminal galactose (13). After binding, the receptor/toxin complex is internalized, and LTA is trafficked to the cytosol (13, 15, 16). Upon entry into the cytosol, the catalytic subunit constitutively activates adenylate cyclase, resulting in water and electrolyte efflux from the host cells (17).
One major difference between the CT and LT lies in the fact that although CT is secreted from the cell, LT reportedly remains periplasmic (5, 18–20). Some studies have also found LT to be associated with membranes extracellularly (3, 21, 22). Despite the equivalent activity that CT and LT exhibit in bioassays, disease caused by ETEC is much less severe than that caused by V. cholerae (2). This finding suggests that the difference between V. cholerae and ETEC virulence may depend on the efficiency of toxin secretion and the delivery mechanism (1).
The main terminal branch of the general secretory pathway (GSP) is a well conserved set of proteins encoded by 13–15 genes clustered in an operon that allow secretion through the outer membrane (23–25). CT secretion in V. cholerae progresses through the three steps of the type II secretion pathway: translocation across the inner membrane, folding in the periplasm, and secretion through the outer membrane (23–31). Extracellular CT secretion occurs through the GSP (32). An examination of many Gram-negative bacteria including those from genera Pseudomonas, Klebsiella, Erwinia, and Vibrio has shown that homologs of this pathway (Xcp, Pul, Out, and Eps, respectively) are responsible for the secretion of a large host of soluble extracellular proteases and toxins across the outer membrane (24).
When introduced on an exogenous plasmid, LT is secreted solubly from V. cholerae in the same manner as CT (18, 29, 30), whereas K-12 E. coli transformed with a CT-expressing plasmid does not secrete CT (30, 33). These results suggest that E. coli does not possess or express the secretion apparatus present in V. cholerae. However, these experiments have not been conducted in ETEC where a secretion system may be active. Enterotoxigenic and K-12 E. coli contain a gsp gene cluster homologous to the eps genes encoding the secretion machinery for CT, but these genes are not expressed by K-12 strains under laboratory conditions (34, 35).2 Lack of expression of these genes may be attributed to the transcription factor HNS, which has been shown to negatively regulate the GSP (36) as well as other virulent factors including LT (37). Transformation of hns-deficient E. coli K-12 with a plasmid encoding the full gsp operon along with one of its substrates, chitinase, is sufficient to cause the secretion of chitinase (36). In ETEC, the deletion of leoA causes a buildup of LT in the periplasm, a decrease of LT in the supernatant, and a decrease of toxicity in vivo (38). The molecular role that the leoA gene product plays in LT secretion remains uncharacterized.
Although not secreted from ETEC in a soluble form, LT has been observed to be associated with LPS in a particulate fraction of the culture supernatant (3, 39). These observations were explained by the reports that LT was associated with ETEC vesicles (21, 22, 40, 41). Most Gram-negative bacteria shed portions of their outer membrane into the cell culture supernatant (41–49). These spherical outer membrane fragments termed “vesicles” are composed of LPS, lipids, proteins, and toxins. Protease- and toxin-containing vesicles from E. coli, Shigella flexneri, Pseudomonas aeruginosa, Borrelia burgdorferi, Actinobacillus actinomycetemcomitans, and Helicobacter pylori interact with bacteria and mammalian cells via an adherence and/or fusion mechanism (39, 49–54).3 This activity suggests that membrane vesicles may be natural vehicles for intercellular transport of virulence factors to host cells during a bacterial infection.
Previously, we have shown that active LT is enriched in and present on the surface of the ETEC vesicle (21). Upon further examination of the data presented by Kolling and Matthews (46), we propose that in addition to its internal localization, Shiga toxin is similarly localized to the surface of vesicles derived from enterohemmorragic E. coli. In addition, the toxins VacA and leukotoxin have been found to be displayed on the surface of vesicles derived from H. pylori and A. actinomycetemcomitans, respectively (53, 54). In this work, we present data demonstrating that LT can be secreted extracellularly by the GSP and that LT remains localized to the surface of the cell bound to LPS through LTB independent of the GM1 binding pocket. The interaction of LPS with LT neither enhances nor attenuates toxicity. We propose that LT is tethered by LPS to the ETEC vesicle and encourages intimate contact between the vesicle and the host cells, leading to intoxication.
EXPERIMENTAL PROCEDURES
Strains and Media
Bacterial strains and plasmids used in this investigation are listed in Table I. Strains were grown in LB (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.0) and maintained on LB agar. Transformations were performed using a modified CaCl2 protocol (55). Antibiotics were added as required at the following concentrations: 100 μg/ml ampicillin, 50 μg/ml kanamycin, and 35 μg/ml chloramphenicol. Isopropyl-1-thio-β-D-galactopyranoside (IPTG) (1.0 mM) was used to induce the expression of LTB constructs. Y1 adrenal cells (ATCC number CCL-79) were maintained in F12K media supplemented with 2.5% fetal calf serum and 12% horse serum as per ATCC instruction. Unless specified, reagents were purchased from Fisher.
Table I.
Strains and plasmids used in this study
| Name/alias | Genotype | Relevant characteristics | CFS toxicitya | Ref. |
|---|---|---|---|---|
| Strains | ||||
| E9034A | Wild-type ETEC (LT+) | + | 73 | |
| E9034P | ETEC cured of virulence plasmid (LT−) | − | 73 | |
| H10407 | Wild-type ETEC (ATCC 35401) (LT+) | + | 38 | |
| ΔleoA | H10407ΔleoA | H10407 mutant defective in LT secretion (LT+) | − | 38 |
| MC4100 | MC4100 | K-12 E. coli | − | 36 |
| GSP | MC4100/pCHAP4278 | K-12 expressing GSP | − | This work |
| GSP/LT | MC4100/pCHAP4278/pPLT | K-12 expressing GSP and LT | + | This work |
| Δhns | MC4100 hns1001 :: kan | K-12 with KanR cassette insertion into hns, KanR | − | 36 |
| Δhns/LT | MC4100 hns1001 :: kan/pPLT | K-12 with KanR cassette insertion into hns, expressing LT, KanR, AmpR | − | This work |
| Δhns/GSP | MC4100 hns1001 :: kan/pCHAP4278 | K-12 with KanR cassette insertion into hns, expressing wt GSP, KanR, CmR | − | This work |
| Δhns/GSP/LT | MC4100 hns1001 :: kan/pCHAP4278/pPLT | K-12 with KanR cassette insertion into hns, expressing wt GSP and wt LT, KanR, CmR, AmpR | + | This work |
| Δhns/ΔGSP | MC4100 hns1001 :: kan/pCHAP4280 | K-12 with KanR cassette insertion into hns, expressing mutant GSP, KanR, CmR | − | This work |
| Δhns/ΔGSP/LT | MC4100 hns1001 :: kan/pCHAP4280/pPLT | K-12 with KanR cassette insertion into hns, expressing mutant GSP and LT, KanR, CmR, AmpR | − | This work |
| Δhns/GSP/LTB | MC4100 hns1001 :: kan/pCHAP4278/pMMB68 | K-12 with KanR cassette insertion into hns, expressing wt GSP and wt LTB, KanR, CmR, AmpR | ND | This work |
| Δhns/GSP/LTBG33D | MC4100 hns1001 :: kan/pCHAP4278/pTRH64 | K-12 with KanR cassette insertion into hns, expressing wt GSP and GM1 binding mutant LTBG33D, KanR, CmR, AmpR | ND | This work |
| Plasmids | ||||
| pWD600 | eltAB and ~4 kb of uncharacterized downstream DNA, TetR | 74 | ||
| pUC19 | Cloning vector, AmpR | |||
| LT | pPLT | eltAB under control of native promoter, cloned from pWD600 into pUC19, AmpR | This work | |
| GSP | pCHAP4278 | gspAB and gspC-R under control of native promoter, CmR | 36 | |
| ΔGSP | pCHAP4280 | gspAB and gspC-RΔDE under control of native promoter, CmR | 36 | |
| LTB | pMMB68 | eltB under control of IPTG-inducible promoter | 59 | |
| LTBG33D | pTRH64 | eltBG33D under control of IPTG-inducible promoter | 59 | |
Toxicity of the cell-free supernatant (CFS) was determined using Y1 cells. +, toxicity score 2–4; −, toxicity score 0–1; ND, not determined. n ≥ 3.
Construction of pPLT
A 2.0-kb fragment containing eltAB including its promoter (37) was generated by PCR from the template plasmid pWD600 using primers PstL (5′-AACGACGAGCGTGAC-3′) and 3′LT-Kpn (5′-TTTTTTGGTCCTAGTTTTCCATACTGATTGCCGC-3′). The fragment was cut with PstI and KpnI and cloned into similarly digested pUC19 (New England Biolabs), resulting in pPLT.
BODIPY-GM1 Labeling
Cells were grown to mid-log phase (A600 ~0.3) and serially plated for colony forming units (CFU). 30 ml of cells were centrifuged at 6000 × g for 10 min, washed once in ice-cold HEPES (50 mM, pH 6.8), and resuspended in 1.0 ml of HEPES. Cells (100 μl) were mixed with 100 μl of 300 nM BODIPY-GM1 (Molecular Probes) in methanol or 100 μl of methanol, and then mixtures were incubated on ice for 30 min. Cells were pelleted and washed three times in ice-cold HEPES. Cells were resuspended in 200 μl of HEPES, and 100 μl was applied to duplicate 96-well microtiter plates. Fluorescence was measured on a FLUOstar Galaxy fluorometer (BMG Labtechnologies) and normalized to cell number (RFU/CFU).
Bacterial Immunoprecipitation Assay
Strains Δhns/GSP, Δhns/GSP/LTB, Δhns/GSP/LTBG33D (1.5 ml, log phase) were incubated overnight at 4 °C with and without rabbit anti-CT antibody (Sigma) (485 μg) that cross-reacted with LT. The mixture was incubated for 120 min at 4 °C with and without protein A-Sepharose (Sigma) (100 μl, 50% slurry in HEPES buffer), and A600 was measured initially (0 min) and after 240 min using a Spec-20 (Spectronic Instruments). The amount of cells that settled was determined by dividing the A600 at 240 min by the A600 at 0 min. The amount of cells that were specifically immunoprecipitated by the antibody was defined as the ratio of cells that settled with beads and antibody to cells that settled without beads and/or antibody. There was no difference in the amount of cells that settled in the presence and absence of beads or antibody for the control strain (Δhns/GSP).
Membrane Stability Assays
For deoxycholate (DOC) sensitivity, cultures were streaked on 0.5% DOC/LB plates and grown overnight (56). To assay RNase leakage, cultures were streaked on 1.5% RNA/LB plates and grown overnight. Plates were then covered in cold 10% trichloroacetic acid to detect RNA (57).
LT-Bacterial Surface Binding Assay
E9034P was grown to mid-log phase, pelleted, washed twice in cold HEPES, and resuspended in 800 μl of 0.1 M Tris, pH 7.5. Cells (400 μl, 2 × 107 CFU/ml) were incubated at 37 °C for 15 min with either 0.1 mg/ml Pronase (Roche Molecular Biochemicals) in Tris or 0.1 mg/ml Pronase plus 1× EDTA-free Complete protease inhibitor (Roche Molecular Biochemicals) in a total volume of 500 μl. Cells were pelleted, washed twice in HEPES, and resuspended in 400 μl of HEPES. Cells (50 μl) were incubated with 0.5 μg of LT (List Biologicals) for 30 min on ice, pelleted, washed three times in ice-cold HEPES, and incubated in 40 μl of 1× SDS-PAGE sample buffer at 25 °C for 5 min. 20 μl was applied to 15% SDS-PAGE and then immunoblotted for LT. A standard immunoblotting protocol was performed (55) using anti-CT antibody, anti-rabbit-horseradish peroxidase (Sigma), and the ECL detection reagents (Amersham Biosciences).
To assay inhibitors of exogenous LT binding to cells, E9034P (1.0 ml) was grown to mid-log phase, pelleted, washed twice in cold HEPES, and resuspended in 0.5 ml of HEPES. LT (0.5 μg, 60 pmol) was incubated at 25 °C for 30 min with HEPES or a 10-fold molar excess of galactose, lactose, glucose, mannose, phosphatidylethanolamine, phosphatidylglycerol, E. coli LPS (O55 and Ra), E. coli Lipid A, or GM1 (Sigma). Washed E9034P cells (50 μl) were then incubated with the LT mixtures for 30 min on ice. Cells were pelleted, washed, resuspended, and analyzed by immunoblotting for LT as described above. NIH Image was used for quantitative densitometry of the immunoblots, and the values were normalized to the amount of LT bound in incubations with buffer.
Toxicity Assay
Y1 cells (105/well) were plated in 96-well polystyrene plates (Corning) and allowed to adhere for 2–4 h. The initial growth medium was removed from the Y1 cells and replaced with samples diluted with fresh F12K medium. Cell-free supernatants were prepared from a late log-phase culture (A600 = 0.8) by pelleting cells at 10,000 × g for 10 min and sterilizing the supernatant using a 0.45-μm sterile filter (Amicon). Cell-free supernatants (100 μl) were diluted to 200 μl in F12K and incubated with Y1 cells. Morphology was scored blind 18 h later. Scores: 1 = <25% rounding; 2 = 26–50% rounding; 3 = 51–75% rounding; and 4 = >76% rounding (58). All of the toxicity assays were performed in duplicate.
To assay the effect of soluble LPS on LT toxicity, LT (6.0 nmol, 600 pmol, and 60 pmol) was incubated at 25 °C for 30 min with HEPES, or a 10-, 100-, or 1000-fold molar excess of O55 LPS or GM1. Samples were diluted to 500 μl with Y1 media, and 200 μl was placed on Y1 cells. For the fractionation assay, cell-free supernatants from Δhns/GSP/LT and Δhns/GSP or soluble LT (1 μg/ml) were filtered through a 100-kDa Microcon filter (Amicon) to obtain supernatants and filtrates. Samples were diluted to 500 μl with Y1 media, and 200 μl was placed on Y1 cells. Scores were averaged, and the concentrations of LT in the total and filtrate fractions were extrapolated from a soluble LT standard curve.
Determination of Vesicle Yield
LB (500 ml) was inoculated with 1:100 dilutions of overnight cultures and grown at 37 °C to A600 = 0.5. A portion of the cells was plated for CFU determination. Cells were pelleted at 10,000 × g for 10 min. Supernatants were subjected to high speed centrifugation at 40,000 × g for 60 min. Vesicle pellets were resuspended in HEPES and sterile-filtered using a 0.45-μm ultra-free filter (Amicon). Protein concentrations were determined using Coomassie Plus (Pierce). The yield per CFU values were normalized to the yield of the Δhns strain.
RESULTS
Cell Surface Detection of LT
LT is present on the surface of vesicles produced by growing ETEC (21). Because vesicles are expected to form by pinching off of the outer membrane, we reasoned that components bound to the vesicle surface are probably also bound to the cell surface. To determine whether LT is bound to the surface of growing ETEC, we developed a detection assay using BODIPY-GM1, a fluorescently labeled membrane-impermeable receptor for LT. BODIPY-GM1 was incubated with early to mid-log phase ETEC, the cells were washed, and cell-associated fluorescence was measured. BODIPY-GM1 labeling of E9034A and E9034P, a virulence plasmid-cured derivative of E9034A that lacks LT, was compared (Fig. 1A). In contrast to E9034A, very little fluorescence was associated with E9034P. We also tested an isogenic pair of strains, H10407, a wild type ETEC, and ΔleoA, a mutant that accumulates LT in the periplasm and prevents LT secretion into the supernatant (38). Any LT in the ΔleoA supernatant would be a result of cell lysis and would be available for “nonspecific” cell association. BODIPY-GM1 associated with ΔleoA cells 7 times less than with H10407 cells (Fig. 1A). These results showed that GM1-accessible LT is bound to the surface of wild type ETEC and that the exterior localization of LT did not result from cell lysis.
Fig. 1. Detection of cell surface-associated LT.

A, wild type ETEC and mutant cells grown to log phase were incubated with BODIPY-GM1. Fluorescence was determined, and values were normalized to cell number (RFU/CFU). Data shown are from one reproducible representative experiment. n ≥ 3. B, as indicated, HNS and Δhns K-12-expressing GSP, ΔGSP, LT, LTB, and/or LTBG33D from plasmids were incubated with BODIPY-GM1, and RFU/CFU was determined as in A. Error bars = mean ± S.E.; n ≥ 3. C, immunoprecipitation of cells because of surface-associated LTB. Values shown are fold over nonspecific settling seen in the background strain (Δhns/GSP). Error bars = ± S.E.; n = 7.
LT Secretion via the GSP
Because LT and CT secretion depended on the native gsp gene cluster in V. cholerae (18, 32), we wanted to investigate whether the GSP secretion machinery is responsible for LT secretion across the outer membrane and, consequently, the association of LT to the cell surface in E. coli. Wild type ETEC strains are not ideal for the dissection of this secretory pathway, because ETEC may possess multiple gsp loci in its chromosome and in its multiple uncharacterized virulence plasmids and the gsp and LT operons are repressed by HNS (36, 37). MC4100, a K-12 strain, has successfully been used to study GSP-dependent secretion of another substrate, chitinase (36). Although K-12 E. coli contain gsp genes, their level of expression appeared to be insufficient for the secretion of chitinase. The presence of a low copy number gsp-carrying plasmid (pCHAP4278) coupled with derepression of gsp genes in an hns-deficient background strain (designated here as Δhns) was found to be necessary for chitinase secretion. Therefore, we used MC4100 and Δhns as “clean” background strains in which to study LT secretion via the GSP. We transformed Δhns with a plasmid encoding either a functional GSP (pCHAP4278) to create Δhns/GSP or the gsp gene cluster with a deletion in gspD and gspE (pCHAP4280) to create Δhns/ΔGSP (Table I). Strains Δhns, Δhns/GSP, and Δhns/ΔGSP were transformed with pPLT, a plasmid encoding LT under the control of its native promoter to create Δhns/LT, Δhns/GSP/LT, and Δhns/ΔGSP/LT, respectively. LT production was equivalent for all strains carrying pPLT as measured by Y1 cell toxicity of total cell lysates (data not shown). The addition of the plasmid-expressed GSP moderately slowed the growth rate of Δhns, and this was compounded by the addition of plasmid-expressed LT (data not shown). To confirm that the slowed growth rate was not a result of cell lysis, we examined the membrane integrity of each of our strain constructs using two established assays, DOC resistance, and periplasmic RNase leakage (56, 57). None of the strains exhibited periplasmic leakage; however, ΔleoA was completely unable to grow on DOC, and several of the GSP- and LT-expressing strains were moderately sensitive to DOC (Table II). Therefore, GSP and LT expression increase detergent sensitivity of the outer membrane but do not compromise the outer membrane barrier.
Table II.
Membrane stability of strains
| Strain | Growth on DOC platea | RNaseb |
|---|---|---|
| H10407 | ++ | − |
| ΔleoA | − | − |
| MC4100 | ++ | − |
| Δhns | ++ | − |
| Δhns/LT | + | − |
| GSP | ++ | − |
| GSP/LT | + | − |
| Δhns/GSP | + | − |
| Δhns/GSP/LT | + | − |
| Δhns/ΔGSP | + | − |
| Δhns/ΔGSP/LT | + | − |
++, thick growth; +, growth; −, no growth.
−, no detectable RNAse activity in area surrounding bacterial growth.
The presence of LT on the surface of the gsp and LT isogenic strains was investigated using the BODIPY-GM1 binding assay. Strains GSP, Δhns, Δhns/GSP, and Δhns/ΔGSP did not bind BODIPY-GM1 (data not shown). Externally bound LT was detected on Δhns/GSP/LT, which contains both GSP and LT plasmids (Fig. 1B). Strain GSP/LT, which has a functional HNS and contains both GSP and LT plasmids bound 5-fold less BODIPY-GM1 than Δhns/GSP/LT, probably because of repression of both GSP and LT expression. Expression of chromosomal GSP (Δhns/LT) or of the mutant GSP (Δhns/ΔGSP/LT) resulted in a 346- and 30-fold respective reduction in the amount of LT secreted to the surface (Fig. 1B). Therefore, BODIPY-GM1 labeling of the LT-expressing strains depended on functional GSP expression, strengthening the argument that cell lysis does not cause LT association with the outer membrane. Together, these results show that LT is actively secreted through the GSP and remains associated with the surface of bacteria after secretion.
LTB Mediates Binding to Bacterial Surface
As LT is composed of A and B subunits, it was necessary to determine which subunit was responsible for cell surface localization. Expression of only the LTB pentamer in the Δhns/GSP strain (Δhns/GSP/LTB) resulted in cell surface localization of LTB as detected by BODIPY-GM1 (Fig. 1B), indicating that the A subunit was unnecessary for LT binding to the outer membrane. As expected, BODIPY-GM1 did not bind Δhns/GSP/LTBG33D cells that express LTB with a point mutation that is defective in GM1 binding (Fig. 1B) (59). To examine whether the GM1 binding pocket of LTB was involved in cell surface binding, we developed an immunoprecipitation assay that did not rely on BODIPY-GM1 binding. The quantity of Δhns/GSP/LTB and Δhns/GSP/LTBG33D cells that were specifically immunoprecipitated was similar and 3–4-fold more than nonspecific precipitation of the background strain (Δhns/GSP), demonstrating that LTBG33D was also bound to the bacterial cell surface (Fig. 1C). The detection of LTBG33D bound to the surface of cells indicates that the intact GM1 binding pocket of LTB is unnecessary to mediate that interaction.
LT association with the surface of the K-12 strains expressing GSP suggested that this interaction is not mediated by a factor specific to the surface of ETEC and that LT probably bound to protein or LPS present in the outer leaflet of the outer membrane. To investigate the nature of this interaction, we examined whether exogenously applied LT could bind to the surface of a bacterium and whether binding would be inhibited by protease treatment. Untreated and protease-treated E9034P cells were incubated with LT, washed, and analyzed by SDS-PAGE and immunoblotting for the presence of LT (Fig. 2A). Samples were examined by SDS-PAGE and Coomassie Blue staining before and after protease treatment to ensure that proteolysis occurred (data not shown). LT bound to the surface of untreated bacteria and those treated with active or inactivated protease (Fig. 2A). In fact, protease treatment slightly increased the amount of LT bound to the surface of the cells, indicating that not only are proteins unlikely to be involved in the binding of LT to the outer membrane, they may mask binding sites for LT.
Fig. 2. Binding of exogenous LT to the surface of LT-deficient ETEC.

A, immunoblot of LT bound to untreated or pretreated E9034P. Incubation with LT and cell pretreatments with Pronase or inhibitor-inactivated Pronase are indicated. B, densitometric analysis of immunoblots of E9034P incubated with buffer (no LT), LT and buffer (B), or LT preincubated with the following compounds: galactose (Gal); phosphatidylglycerol (PG); phosphatidylethanolamine (PE); O55 LPS (O55); Ra LPS (Ra); Lipid A (A); and monosialoganglioside (GM1). Error bars = mean ± S.E.; n ≥ 3. *, p < 0.005 by Student’s t test.
LT Binds LPS
We next examined the role of LPS in LT association with bacteria by testing the ability of various sugars and lipids to block the cell-LT association. LT was preincubated with 10-fold molar excess of the sugars glucose, galactose, lactose, and mannose, and the lipids phosphatidylglycerol, phosphatidylethanolamine, LPS from an O55 and a Ra mutant E. coli, Lipid A, and GM1 ganglioside. E9034P cells were then incubated with the LT/sugar or LT/lipid mixture. A 10-fold or greater excess of the simple sugars including galactose and lactose previously shown to bind soluble LT (60, 61) or the lipids (phosphatidylethanolamine, phosphatidylglycerol, and Lipid A) was unable to block binding (Fig. 2B, and data not shown). GM1, which previously has been determined to bind and inactivate LT (21, 62–64), did not block the ability of LT to bind to the cells. Only LPS was able to block the binding of LT to the surface of E9034P. Whereas the lipid core of LPS (Lipid A) was not able to inhibit LT binding, LPS that lacks O-antigen (Ra) did block binding, indicating that LT binding requires at least the LPS core sugars but not the O-antigen. These data along with the protease insensitivity of the interaction indicate that LPS, independent of O-antigen, mediates the association of LT with the surface of cells, supporting the results described above that LT is capable of binding both LPS and GM1 simultaneously.
We tested whether LPS could block the toxicity of LT in the same manner as GM1 or whether that association could enhance LT toxicity. LT toxicity is commonly assayed using Y1 adrenal mouse cells (58), and we determined that LPS and GM1 alone (up to 6.0 nmol) were nontoxic to Y1 cells (data not shown). Y1 cells were treated with 6 nmol, 600 pmol, and 60 pmol of LT to elicit varying degrees of toxic response (Fig. 3, black, gray, and hatched bars). When LT was preincubated with a 10-fold excess of GM1, the toxicity of LT was blocked (Fig. 3, last group). Preincubation of LT with up to a 1000-fold molar excess of LPS, however, had no effect on the toxicity of LT (Fig. 3, LPS groups). Thus, although soluble LPS can block the binding of LT to the surface of bacteria (Fig. 2B), LPS does not inhibit or enhance the interaction of LT with its host cell surface receptor GM1.
Fig. 3. Binding to LPS does not affect LT toxicity.

LT preincubated with buffer or an excess of LPS or GM1 in the amounts indicated was applied to Y1 cells and scored for toxicity. Black bars, 6.0 nmol LT; gray bars, 600 pmol LT; hatched bars, 60 pmol LT. Error bars = mean ± S.E.; n = 3.
LT Secreted by the GSP Is Vesicle-associated
Previous work has demonstrated that LT secreted by ETEC is associated with vesicles (21, 22, 40, 41). To investigate whether this association of LT with vesicles was simply because of a lack of GSP expression, we wanted to determine whether LT secreted by the GSP is soluble or associated with vesicles. We first examined the toxicity of unfractionated cell-free supernatants from the strains constructed in this work using the Y1 adrenal cell assay. The toxicity of the MC4100 supernatants clearly depended on the expression of LT and the presence of a functional plasmid-expressed GSP (Table I). To differentiate whether this toxicity was soluble or associated with vesicles, we fractionated the cell-free supernatant of strain Δhns/GSP/LT. Y1 cell toxicity was used to assay soluble LT and cell-free supernatants before and after filtration (Fig. 4A). After filtration of a pure soluble LT sample, all of the activity was detected in the 100-kDa filtrate, whereas purified vesicles did not enter the filtrate (data not shown). Before filtration, the average LT activity in the total cell-free supernatant of the LT- and GSP-expressing strain (Δhns/GSP/LT) was equivalent to 139 pg/ml as extrapolated from the soluble LT toxicity standard curve (Fig. 4A). After filtration, the average LT toxicity score dropped to a value equivalent to 6 pg/ml (Fig. 4A). Thus, 95% of secreted LT activity is associated with material that is >100 kDa and behaves like vesicles. Morphological changes were specific for LT as cell-free supernatant from Δhns/GSP had no effect on Y1 cells. These data demonstrate that active LT secreted by the GSP is associated with vesicles.
Fig. 4. GSP-secreted LT is associated with vesicles and vesicle production is HNS-regulated.

A, toxicity of total and size-fractionated (100-kDa filtrate) cell-free supernatants from cultures of Δhns/GSP/LT (n = 16) (black bars) and Δhns/GSP (n = 8) (gray bars). Error bars = mean ± S.E. Inset, standard curve of LT toxicity on Y1 cells. Error bars = mean ± S.E.; n ≥ 6. B, vesicle yields per CFU of indicated strains compared with Δhns. Error bars = mean ± S.E.; n = 6.
Vesicle Production Is Regulated by HNS
Because vesicle production may be linked to protein secretory pathways, we also examined whether the strains containing various secretion constructs had altered outer membrane vesicle production. When total vesicles produced per CFU were compared, we determined that the deletion of hns caused a 3-fold increase in vesiculation (Fig. 4B). The addition of plasmid-born GSP, mutant GSP, and/or LT had no effect on the total vesicle production compared with the Δhns parent strain. Therefore, the vesicle production level in E. coli is not dramatically affected by a specific backup in the LT secretion pathway but is affected by pleiotropic effects of the hns mutation.
DISCUSSION
This work describes the first comprehensive study of the bacterial secretion mechanism of heat-labile enterotoxin in E. coli. The mode by which LT is released by ETEC to interact with host cells has long been a paradox. Previously, it was observed that LT remained in the periplasmic space and therefore was unavailable to intoxicate cells. However, LT had also been detected extracellularly and associated with LPS and, more specifically, with vesicles (3, 21, 22, 39). Given the role of the GSP in the secretion of CT and the fact that the gsp operon has been detected in ETEC and other pathogenic E. coli (34, 65),2 we considered that the GSP might be responsible for the secretion of LT to the cell surface.
In previous work, we discovered that active LT is present on the surface of ETEC vesicles and that ETEC vesicles could bind immobilized receptor (21). In this study, we developed the BODIPY-GM1 assay to detect externally bound LT, and we found that LT was associated specifically with the surface of ETEC cells, which are the source of the outer membrane vesicles. LT could become associated with the cell surface through two possible mechanisms: nonspecific association resulting from cell lysis or a specific transport step across the outer membrane. Three lines of evidence demonstrated that cell lysis was not the means by which LT became bound to the cell surface: the lack of binding of BODIPY-GM1 to ΔleoA cells, GSP-dependent BODIPY-GM1 labeling of cells, and the lack of periplasmic leakage from our strains. Thus, surface-localized LT specifically depended on the co-expression of LT and the functional plasmid-encoded GSP. These data indicate that the GSP is necessary and sufficient for the secretion of LT, LT remains associated with the surface of the cell after secretion, and LT is capable of simultaneously associating with the outer membrane and the GM1 receptor.
Because LT localized to the outside of cells, either protein or lipid components of the outer membrane were mediating toxin binding. The outer leaflet of the outer membrane is composed of LPS, protein, and possibly minor amounts of phospholipid. Bacterial proteins have been shown to bind to lipids (66); therefore, we tested common membrane phospholipids and found that they did not inhibit LT binding. The association of LT with the bacterial surface was resistant to protease and inhibitable with soluble LPS, indicating that LT binds to LPS in the outer membrane. We reasoned that because LT binds GM1 by a terminal galactose and agarose beads through this same sugar (61), LT might be binding LPS on the surface of E. coli through a galactose in the O-antigen. Therefore, we were surprised to discover that LT bound to the surface of E9034P, which has a polymannose O-antigen (67), K-12 (expressing the GSP), which does not express O-antigen, and Ra LPS. In addition, none of the simple sugars such as mannose, glucose, galactose, or lactose inhibited LT binding. Therefore, LT may recognize sugar complexes in LPS in the O-antigen (if it is present) or in the core.
The fact that BODIPY-GM1 bound cells in an LTB-dependent manner demonstrates that LTB can simultaneously bind LPS and GM1 receptor. LTBG33D was found to be associated with cells to the same extent as wild type LTB; therefore, the intact GM1 binding site is not required for the LTB-LPS interaction. The presence of independent GM1 and LPS binding sites on LTB is supported by our findings that GM1, not LPS, was able to block the toxicity of LT, and conversely, LPS but not GM1 inhibited LT binding to bacteria. Based on these data, one LTB subunit potentially could bind both LPS and GM1 simultaneously via distinct sites. However, LPS and GM1 are sterically large molecules; thus, we propose that concurrent LT binding to LPS and GM1 occurs by LPS engaging one or more LTB subunits of the pentamer and GM1 engaging one or more of the remaining LTB subunits.
We have proposed that vesicles are one means by which active LT can be delivered to eukaryotic cells, a conclusion that is supported by previous observations (3, 22, 39). However, we considered that vesicles may not be the only form of transport and that in in vitro culture conditions, a soluble LT secretion pathway (e.g. the GSP) was either not expressed or not functional. To address whether LT would be secreted solubly if the GSP pathway was available, we assayed the culture supernatant of E. coli expressing LT and the GSP in the derepressing hns background strain. LT from the supernatant of Δhns/GSP/LT was retained on a 100-kDa filter, indicating that LT secreted by the GSP remains associated with vesicles rather than being liberated solubly into the medium. Thus, although CT is secreted in a soluble form by the GSP, LT secreted by the GSP is exclusively associated with outer membrane vesicles. The difference between the extracellular localization of these two similar toxins may be attributed to the fact that CT is less promiscuous in that it can only bind monosialo-GM1 (12) or that the LPS core structures of E. coli and V. cholerae differ (68, 69).
Very little is currently known regarding the regulation of vesicle production by pathogenic bacteria. We investigated whether HNS, LT, or GSP expression affected vesicle production, because it was possible that an increase in vesicle cargo could induce vesicle formation. Our data indicate that only HNS expression affected the amount of vesicles produced. The hns-dependent increase in vesicle yield could be either a primary or secondary effect. HNS could regulate a specific machinery that mediates the formation of vesicles. Thus, in an hns mutant, a negative regulator of vesicle production could have been lost, resulting in an increase in vesicle production. Alternatively, the deletion of hns may result in derepression of periplasmic and membrane proteins, and the increase of proteins may result in a bulk flow effect on vesiculation, i.e. more protein results in more vesicles. The latter model is unlikely because introduction of the GSP and LT expression plasmids did not affect vesiculation levels. Thus, we favor a model in which an hns-regulated specific machinery exists for the formation of outer membrane vesicles.
Based on this work, we proposed the following model for LT secretion in K-12, and we further postulate that the same mechanism occurs in ETEC during physiological conditions that down-regulate HNS expression. Prior to this work, the early stages of LT secretion had been elucidated. LTA and LTB are secreted as monomers through the inner membrane via the Sec pathway (Fig. 5, step 1) (31). Once in the periplasm, the subunits assemble to form the AB5 toxin (Fig. 5, step 2) (70). Folded, assembled, and active periplasmic LT is secreted via the GSP through GspD, a gated outer membrane pore (Fig. 5, step 3) (71). On the exterior of the cell, LT binds to LPS via a novel binding region on its B subunit and remains associated with the surface of the cell (Fig. 5, step 4). The outer membrane of the bacterium bud vesicles that contain luminal LT and display surface LT (Fig. 5, step 5). LT on the surface of vesicles can bind GM1 on the host epithelium and LPS on the vesicle simultaneously, tethering the vesicle to the host (Fig. 5, step 6). The interaction of LT with GM1 may lead to further interaction between the host cell and the vesicle, similar to the association between LPS and CD14 that allows subsequent association between LPS and TLR4 (72). The LT-induced bridge causes the host cell to internalize the vesicle and intoxicate the cells (21).3 Further studies are necessary to determine how LT interacts with the GSP, how LT binds LPS, and how vesicle production is regulated.
Fig. 5. Model of LT secretion across the outer membrane of ETEC and interaction between vesicles carrying LT and host cells.
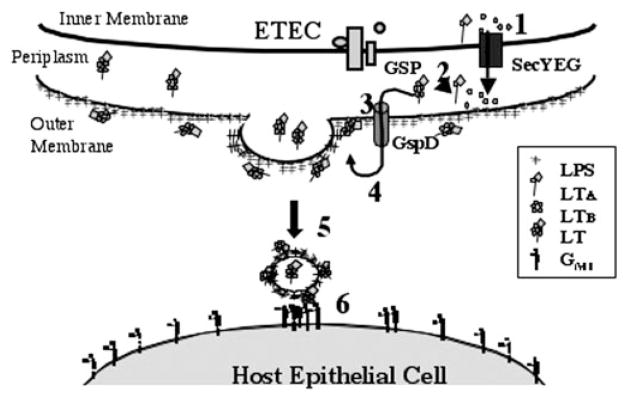
After secretion by the GSP, LT binds to LPS on the cell surface via LTB. Consequently, vesicles released from the cell have LT on their surface that can act as a tether between GM1 on the host cell and LPS on the vesicle. Internalization of vesicles and their associated LT cause toxicity in the host cell. Steps are outlined under “Discussion.”
Acknowledgments
We thank M. Rosser, J. Plank, and S. Bauman for critical review of this paper and suggestions; W. Dallas for the plasmid pWD600; J. Fleckenstein for the strain ΔleoA; O. Francetic for the plasmids pCHAP4278 and pCHAP4280 and strain Δhns; J. Giron for the strains E9034A and -P; and T. Hirst for the plasmids pMMB68 and pTRH64.
Footnotes
This work was supported by a Burroughs Wellcome career award (to M. J. K.) and an institutional research grant of the American Cancer Society.
The abbreviations used are: ETEC, enterotoxigenic E. coli; GM1, Galβ1,3GalNAcβ1–4(NeuAcα2,3)4Galβ1,4Glc-ceramide; LT, heat-labile enterotoxin; CT, cholera toxin; LTA and LTB, LT subunits A and B; GSP, general secretory pathway; CFU, colony forming units; RFU, relative fluorescence units; LPS, lipopolysaccharide; DOC, deoxycholate; IPTG, isopropyl-1-thio-β-D-galactopyranoside.
A. L. Horstman and M. J. Kuehn, unpublished data
K. Mason, N. Kesty, and M. J. Kuehn, submitted for publication.
References
- 1.Spangler BD. Microbiol Rev. 1992;56:622–647. doi: 10.1128/mr.56.4.622-647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunkel SL, Robertson DC. Infect Immun. 1979;25:586–596. doi: 10.1128/iai.25.2.586-596.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wensink J, Gankema H, Jansen WH, Guinee PA, Witholt B. Biochim Biophys Acta. 1978;514:128–136. doi: 10.1016/0005-2736(78)90082-2. [DOI] [PubMed] [Google Scholar]
- 4.Institute of Medicine. Disease of Importance in Developing Countries. II. National Academy Press; Washington, D. C: 1986. pp. 159–169. [Google Scholar]
- 5.Gyles CL. Can J Microbiol. 1992;38:734–746. doi: 10.1139/m92-120. [DOI] [PubMed] [Google Scholar]
- 6.So M, Dallas WS, Falkow S. Infect Immun. 1978;21:405–411. doi: 10.1128/iai.21.2.405-411.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dallas WS, Gill DM, Falkow S. J Bacteriol. 1979;139:850–858. doi: 10.1128/jb.139.3.850-858.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merritt EA, Hol WG. Curr Opin Struct Biol. 1995;5:165–171. doi: 10.1016/0959-440x(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 9.Sixma TK, Kalk KH, van Zanten BA, Dauter Z, Kingma J, Witholt B, Hol WG. J Mol Biol. 1993;230:890–918. doi: 10.1006/jmbi.1993.1209. [DOI] [PubMed] [Google Scholar]
- 10.Mekalanos JJ, Swartz DJ, Pearson GD, Harford N, Groyne F, de Wilde M. Nature. 1983;306:551–557. doi: 10.1038/306551a0. [DOI] [PubMed] [Google Scholar]
- 11.Dallas WS, Falkow S. Nature. 1980;288:499–501. doi: 10.1038/288499a0. [DOI] [PubMed] [Google Scholar]
- 12.Fukuta S, Magnani JL, Twiddy EM, Holmes RK, Ginsburg V. Infect Immun. 1988;56:1748–1753. doi: 10.1128/iai.56.7.1748-1753.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fishman PH, Pacuszka T, Orlandi PA. Adv Lipid Res. 1993;25:165–187. [PubMed] [Google Scholar]
- 14.Holmgren J, Fredman P, Lindblad M, Svennerholm AM, Svennerholm L. Infect Immun. 1982;38:424–433. doi: 10.1128/iai.38.2.424-433.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lencer WI, de Almeida JB, Moe S, Stow JL, Ausiello DA, Madara JL. J Clin Invest. 1993;92:2941–2951. doi: 10.1172/JCI116917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lencer WI, Hirst TR, Holmes RK. Biochim Biophys Acta. 1999;1450:177–190. doi: 10.1016/s0167-4889(99)00070-1. [DOI] [PubMed] [Google Scholar]
- 17.Verlinde CL, Merritt EA, Van den Akker F, Kim H, Feil I, Delboni LF, Mande SC, Sarfaty S, Petra PH, Hol WG. Protein Sci. 1994;3:1670–1686. doi: 10.1002/pro.5560031006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirst TR, Sanchez J, Kaper JB, Hardy SJ, Holmgren J. Proc Natl Acad Sci U S A. 1984;81:7752–7756. doi: 10.1073/pnas.81.24.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirst TR, Holmgren J. Proc Natl Acad Sci U S A. 1987;84:7418–7422. doi: 10.1073/pnas.84.21.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirst TR, Holmgren J. J Bacteriol. 1987;169:1037–1045. doi: 10.1128/jb.169.3.1037-1045.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horstman AL, Kuehn MJ. J Biol Chem. 2000;275:12489–12496. doi: 10.1074/jbc.275.17.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wai SN, Takade A, Amako K. Microbiol Immunol. 1995;39:451–456. doi: 10.1111/j.1348-0421.1995.tb02228.x. [DOI] [PubMed] [Google Scholar]
- 23.Russel M. J Mol Biol. 1998;279:485–499. doi: 10.1006/jmbi.1998.1791. [DOI] [PubMed] [Google Scholar]
- 24.Pugsley AP. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lory S. Curr Opin Microbiol. 1998;1:27–35. doi: 10.1016/s1369-5274(98)80139-2. [DOI] [PubMed] [Google Scholar]
- 26.Economou A. Trends Microbiol. 1999;7:315–320. doi: 10.1016/s0966-842x(99)01555-3. [DOI] [PubMed] [Google Scholar]
- 27.Danese PN, Silhavy TJ. Annu Rev Genet. 1998;32:59–94. doi: 10.1146/annurev.genet.32.1.59. [DOI] [PubMed] [Google Scholar]
- 28.De Gier JW, Valent QA, Von Heijne G, Luirink J. FEBS Lett. 1997;408:1–4. doi: 10.1016/s0014-5793(97)00402-x. [DOI] [PubMed] [Google Scholar]
- 29.Leece R, Hirst TR. J Gen Microbiol. 1992;138:719–724. doi: 10.1099/00221287-138-4-719. [DOI] [PubMed] [Google Scholar]
- 30.Connell TD, Metzger DJ, Wang M, Jobling MG, Holmes RK. Infect Immun. 1995;63:4091–4098. doi: 10.1128/iai.63.10.4091-4098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palva ET, Hirst TR, Hardy SJ, Holmgren J, Randall L. J Bacteriol. 1981;146:325–330. doi: 10.1128/jb.146.1.325-330.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandkvist M, Michel LO, Hough LP, Morales VM, Bagdasarian M, Koomey M, DiRita VJ. J Bacteriol. 1997;179:6994–7003. doi: 10.1128/jb.179.22.6994-7003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pearson GD, Mekalanos JJ. Proc Natl Acad Sci U S A. 1982;79:2976–2980. doi: 10.1073/pnas.79.9.2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stojiljkovic I, Schonherr R, Kusters JG. J Bacteriol. 1995;177:1892–1895. doi: 10.1128/jb.177.7.1892-1895.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Francetic O, Pugsley AP. J Bacteriol. 1996;178:3544–3549. doi: 10.1128/jb.178.12.3544-3549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Francetic O, Belin D, Badaut C, Pugsley AP. EMBO J. 2000;19:6697–6703. doi: 10.1093/emboj/19.24.6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trachman JD, Maas WK. J Bacteriol. 1998;180:3715–3718. doi: 10.1128/jb.180.14.3715-3718.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleckenstein JM, Lindler LE, Elsinghorst EA, Dale JB. Infect Immun. 2000;68:2766–2774. doi: 10.1128/iai.68.5.2766-2774.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gankema H, Wensink J, Guinee PA, Jansen WH, Witholt B. Infect Immun. 1980;29:704–713. doi: 10.1128/iai.29.2.704-713.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofstra H, Witholt B. J Biol Chem. 1984;259:15182–15187. [PubMed] [Google Scholar]
- 41.Hoekstra D, van der Laan JW, de Leij L, Witholt B. Biochim Biophys Acta. 1976;455:889–899. doi: 10.1016/0005-2736(76)90058-4. [DOI] [PubMed] [Google Scholar]
- 42.Devoe IW, Gilchrist JE. J Exp Med. 1973;138:1156–1167. doi: 10.1084/jem.138.5.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dorward DW, Garon CF, Judd RC. J Bacteriol. 1989;171:2499–2505. doi: 10.1128/jb.171.5.2499-2505.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grenier D, Mayrand D. Infect Immun. 1987;55:111–117. doi: 10.1128/iai.55.1.111-117.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadurugamuwa JL, Beveridge TJ. J Bacteriol. 1995;177:3998–4008. doi: 10.1128/jb.177.14.3998-4008.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kolling GL, Matthews KR. Appl Environ Microbiol. 1999;65:1843–1848. doi: 10.1128/aem.65.5.1843-1848.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, Clarke AJ, Beveridge TJ. J Bacteriol. 1998;180:5478–5483. doi: 10.1128/jb.180.20.5478-5483.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pettit RK, Judd RC. Mol Microbiol. 1992;6:729–734. doi: 10.1111/j.1365-2958.1992.tb01522.x. [DOI] [PubMed] [Google Scholar]
- 49.Saunders NB, Shoemaker DR, Brandt BL, Moran EE, Larsen T, Zollinger WD. Infect Immun. 1999;67:113–119. doi: 10.1128/iai.67.1.113-119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shoberg RJ, Thomas DD. Infect Immun. 1993;61:3892–3900. doi: 10.1128/iai.61.9.3892-3900.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kadurugamuwa JL, Beveridge TJ. Antimicrob Agents Chemother. 1998;42:1476–1483. doi: 10.1128/aac.42.6.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadurugamuwa JL, Beveridge TJ. Microbiology. 1999;145:2051–2060. doi: 10.1099/13500872-145-8-2051. [DOI] [PubMed] [Google Scholar]
- 53.Fiocca R, Necchi V, Sommi P, Ricci V, Telford J, Cover TL, Solcia E. J Pathol. 1999;188:220–226. doi: 10.1002/(SICI)1096-9896(199906)188:2<220::AID-PATH307>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 54.Kato S, Kowashi Y, Demuth DR. Microb Pathog. 2002;32:1–13. doi: 10.1006/mpat.2001.0474. [DOI] [PubMed] [Google Scholar]
- 55.Sambrook J, Fritsch EF, Maniatis T. In: Molecular Cloning: A Laboratory Manual. 2. Ford N, editor. 1–3. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. pp. 1.82–1.84.pp. 18.64–18.74. [Google Scholar]
- 56.Sun TP, Webster RE. J Bacteriol. 1986;165:107–115. doi: 10.1128/jb.165.1.107-115.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bernadac A, Gavioli M, Lazzaroni JC, Raina S, Lloubes R. J Bacteriol. 1998;180:4872–4878. doi: 10.1128/jb.180.18.4872-4878.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donta ST, Moon HW, Whipp SC. Science. 1974;183:334–336. doi: 10.1126/science.183.4122.334. [DOI] [PubMed] [Google Scholar]
- 59.Nashar TO, Webb HM, Eaglestone S, Williams NA, Hirst TR. Proc Natl Acad Sci U S A. 1996;93:226–230. doi: 10.1073/pnas.93.1.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sixma TK, Pronk SE, Kalk KH, van Zanten BA, Berghuis AM, Hol WG. Nature. 1992;355:561–564. doi: 10.1038/355561a0. [DOI] [PubMed] [Google Scholar]
- 61.Clements JD, Finkelstein RA. Infect Immun. 1979;24:760–769. doi: 10.1128/iai.24.3.760-769.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ricci LC, de-Siqueira PS, Castro AF. Braz J Med Biol Res. 1992;25:805–807. [PubMed] [Google Scholar]
- 63.Merritt EA, Sixma TK, Kalk KH, van Zanten BA, Hol WG. Mol Microbiol. 1994;13:745–753. doi: 10.1111/j.1365-2958.1994.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 64.Merritt EA, Sarfaty S, van den Akker F, L’Hoir C, Martial JA, Hol WG. Protein Sci. 1994;3:166–175. doi: 10.1002/pro.5560030202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt H, Henkel B, Karch H. FEMS Microbiol Lett. 1997;148:265–272. doi: 10.1111/j.1574-6968.1997.tb10299.x. [DOI] [PubMed] [Google Scholar]
- 66.Khursigara C, Abul-Milh M, Lau B, Giron JA, Lingwood CA, Foster DE. Infect Immun. 2001;69:6573–6579. doi: 10.1128/IAI.69.11.6573-6579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cox AD, Brisson JR, Varma V, Perry MB. Carbohydr Res. 1996;290:43–58. doi: 10.1016/0008-6215(96)00135-8. [DOI] [PubMed] [Google Scholar]
- 68.Raetz CR. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 69.Yu J, Webb H, Hirst TR. Mol Microbiol. 1992;6:1949–1958. doi: 10.1111/j.1365-2958.1992.tb01368.x. [DOI] [PubMed] [Google Scholar]
- 70.Hardie KR, Lory S, Pugsley AP. EMBO J. 1996;15:978–988. [PMC free article] [PubMed] [Google Scholar]
- 71.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. J Biol Chem. 1999;274:10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 72.Giron JA, Gomez-Duarte OG, Jarvis KG, Kaper JB. Gene (Amst) 1997;192:39–43. doi: 10.1016/s0378-1119(97)00039-5. [DOI] [PubMed] [Google Scholar]
- 73.Dallas WS. Infect Immun. 1983;40:647–652. doi: 10.1128/iai.40.2.647-652.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]