

Abstract
A novel microfluidic chromatography device coupled with tandem mass spectrometry (LC–MS/MS) was utilized for the multiplex analysis of 5 steroids (testosterone, dihydrotestosterone, progesterone, cortisol, cortisone) in human serum. The use of microfluidics allowed for reduction of the chromatographic flow rate to 3 μl/min with overall method run times comparable to standard flow LC–MS/MS methods reported in the literature, corresponding to a 150 fold decrease in solvent consumption. Furthermore, a simple sample preparation protocol was employed requiring injection of only 0.5 μl of sample, corresponding to a 100–400 fold increase in on-column sensitivity as compared to published standard flow assays. The measured LOQ for both testosterone and progesterone was 0.4 ng/mL, representing an improvement over reported literature values obtained by standard flow methods employing comparable sample preparation and large injection volumes. The LOQs for cortisol (1.9 ng/mL), cortisone (0.3 ng/mL), and dihydrotestosterone (1.4 ng/mL) were all within a biologically relevant range. A comparison of clinical serum samples was performed for the analysis of testosterone using this microfluidic LC–MS/MS assay and the Beckman Access II automated antibody-based measurement system. The immunoassay results were systematically higher due to matrix interference which was easily resolved with the increased chromatographic resolution obtained in the microflow LC–MS/MS assay.
Keywords: Steroid, MRM, Microflow, Serum, Immunoassay, LC–MS/MS
1. Introduction
Steroids can be measured directly from biofluids using multiple techniques including radioimmunoassay (RIA), enzyme-linked immunosorbent assay (ELISA), chemiluminescent immunoassay (CLIA), and more recently, liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS). Of these, CLIA has become the most widespread method due to advantages including automation, high-throughput, ease of use, and a relatively low level of required technical expertise. However, it suffers from serious shortcomings including susceptibility to sample matrix effects (lipemia, hemolysis) and lack of specificity due to antibody cross-reactivity with other endogenous steroids or lipids [1–4]. Confirming this lack of specificity, numerous authors have reported a systematic overestimation in values obtained from immunoassays as compared to LC–MS/MS [2,5–8]. Furthermore, some steroids are not suited to analysis by immunoassay for all demographics. For example, the measurement of testosterone in women and children suffers from reliability issues with the traditional immunoassay and displays significant cross-platform variability [4,9]. This is of such serious concern in the field of endocrinology that the Endocrine Society has recently issued a position statement on the pitfalls of measuring low levels of testosterone and the need for improved techniques [4].
LC–MS/MS is becoming an increasingly important analytical method for clinical assays [3,10]. The precision, sensitivity, and throughput of this approach enables accurate, multiplexed measurement of analytes in biofluids, and allows for long-term serial monitoring of patients [11]. This is particularly important for real time monitoring applications, for example, clinicians monitoring steroid levels in patients with endocrine disorders where assay results have a direct impact on the treatment plan [12,13]. The specific LC–MS/MS approach that has been applied to the analysis of steroids and hormones is called multiple reaction monitoring (MRM) [14]. In this approach, the parent ion of the analyte is isolated and fragmented within the mass spectrometer and the resultant daughter ions are monitored and quantified. Detection of the daughter ion(s) enables increased specificity and sensitivity to the analyte of interest and allows for manual validation against a stable isotope labeled internal standard. Many routine clinical laboratories have adopted this technology on a large scale including ARUP National Reference Laboratory, Mayo Clinic, and Quest Diagnostics. Using this technique, steroids are routinely measured in diverse biological matrices including serum, plasma, urine, and saliva. Depending on the matrix, required sensitivity, and instrumentation, sample preparation may be as simple as dilution or protein precipitation or more laborious clean-up steps such as liquid–liquid extraction (LLE) or solid-phase extraction (SPE) or even derivatization may be required [3].
Most clinical LC–MS/MS assays employ an instrument configuration using a standard electrospray ionization (ESI) source coupled to a triple quadrupole mass spectrometer [3], allowing for fast run times (from 5 to 15 min) and multiplexing. Typical flow rates are in the range of 600–1000 μL/min and injection volumes are upwards of 100–400 μL per sample [3,8,13,15]. This approach consumes a high level of both solvent and sample, increasing the cost of the assay and limiting the number of replicates that can be analyzed. The use of microfluidics for this application is appealing to reduce solvent and sample consumption. In this study, we have utilized novel microfluidic LC–MS/MS technology for the multiplex analysis of 5 steroids in human serum. The primary advantage of this approach is the reduction of solvent and sample consumption. Specifically, the use of microfluidics allows for a decrease of the chromatographic flow rate to 3 μL/min while method run times remain comparable to standard flow LC–MS/MS methods reported in the literature [3], corresponding to a 150 fold decrease in solvent consumption. Furthermore, a 100–300 fold increase in on-column sensitivity was achieved with injection of only 0.5 μL of extracted sample. Importantly, this increased sensitivity was realized using a simple and high throughput sample preparation protocol (i.e. LLE and/or SPE and/or derivatization was not required). Technical parameters including ion suppression, % recovery, limit of detection (LOD), limit of quantitation (LOQ), and interday/intraday variation were evaluated. A comparison of clinical serum samples was performed for the analysis of testosterone using both the novel microfluidic LC–MS/MS assay and the Beckman Access II automated antibody-based measurement system.
2. Methods
2.1. Materials
Cortisol, cortisone, progesterone, testosterone, dihydrotestosterone, 2,3,4-13C3 progesterone, 9,11,12,12-d4 cortisol, and 2,2,4,6,6,12,12,-d7 cortisone were purchased from Sigma (St. Louis, MO) and 16,16,17-d3 testosterone and 16,16,17-d3 dihydrotestosterone were purchased from Cerilliant (Round Rock, TX). Steroid-free serum was purchased from MP Biomedicals (Salon, OH). Formic acid (98%), HPLC-grade water and methanol were purchased from Sigma.
2.2. Preparation of standard solutions and quality controls
Each standard steroid (1 mg) was dissolved in 1 mL methanol (stock solution) and stored at −20 °C. A master mix was prepared by spiking steroid-free serum with each steroid at 10 μg/mL followed by precipitation of 100 μL spiked serum with 370 μL methanol containing 10 ng/mL final concentration of each labeled internal standard (IS; d3 testosterone, d3 dihydrotestosterone, 13C3 progesterone, d4 cortisol, d7 cortisone). Subsequent serial dilutions were performed using steroid-free serum precipitated with IS spiked methanol. The 10 ng/mL concentration was used as an in-house QC. Concentrations reported hereafter refer to serum steroid concentrations unless otherwise stated.
2.3. Sample preparation
Patient serum was obtained from men being screened for enrollment into a study of the effects of testosterone supplementation and resistance exercise on physical function and body composition. The protocol was approved by the Colorado Multiple Institutional Review Board and all volunteers provided informed consent to participate.
Patient serum (100 μL) was pipetted into 500 μL 96-well plates (VWR, Radnor, PA). Protein was precipitated from serum samples with 3.7 volumes ice-cold methanol spiked with internal standards as described above. Plates were incubated at −80 °C for 30 min, centrifuged at 3270 × g for 10 min, and supernatant was collected. The collected supernatant was centrifuged a second time and the resulting supernatant was transferred into 350 μL 96-well plates (Waters, Milford, MA).
2.4. MS methods
The LC MS/MS system consisted of a Waters nanoACQUITY UPLC coupled to at Waters TQ-S mass spectrometer fitted with a Trizaic source. The instrument was operated in positive electrospray ionization mode using MassLynx V4.1 SCN810 (Waters). Chromatography was performed on a 150 μM × 50 mm Trizaic nanotile packed with BEH C18 1.7 μm. Injections were 0.5 μL using partial loop mode. The initial solvent composition was 90% A (0.1% formic acid in water) and 10% B (0.1% formic acid in methanol). The mobile phase gradient was: 10–55% B from 0.25 to 1 min, 55–95% B from 1 to 7.5 min, 95–10% B from 7.5 to 8 min, and a hold at 10% B from 8 to 12 min. The flow rate was 3.06 μL/min and the column was heated to 45 °C. The cone voltage and collision energy were optimized for each compound using the on board Intellistart system in the TQ-S; the parameters are listed in Table 1. The capillary voltage was 3.2 kV, source temperature was 100 °C, the source offset was 50 V, and the collision gas was argon. Dwell times for all compounds was 0.011 s. The MRM transitions for each compound are listed in Table 1. Patient samples were randomized across two 96-well plates and injected in duplicate from individual wells.
Table 1.
MRM transitions and instrument settings for each compound.
| Compound | Time window (min) | Transition | Cone (V) | Collision energy (V) |
|---|---|---|---|---|
| MRM functions and settings for detecting steroids | ||||
| Testosterone | 4.1–5.3 | 289.24 > 97.03 | 50 | 20 |
| Dihydrotestosterone | 4.5–5.5 | 291 > 255 | 46 | 14 |
| d3 testosterone | 4.1–5.3 | 292.2 > 97.03 | 50 | 20 |
| d3 dihydrotestosterone | 4.5–5.5 | 294.1 > 258.2 | 46 | 14 |
| Progesterone | 4.9–6 | 315 > 109 | 20 | 26 |
| 13C3 progesterone | 4.9–6 | 318.2 > 112.2 | 20 | 26 |
| Cortisone | 3.5–4.6 | 361 > 163.05 | 25 | 30 |
| Cortisol | 3.5–5 | 363 > 327.14 | 25 | 16 |
| d4 cortisol | 3.5–5 | 367.2 > 331 | 25 | 22 |
| d7 cortisone | 3.5–4.6 | 368.2 > 169 | 25 | 22 |
2.5. Quality control
To ensure long-term signal stability and data quality, QC samples containing testosterone and cortisone were monitored at the beginning of each run and every 12 h within runs. The following metrics were monitored: concentration (RSD < 20%) and retention time (±1%). If a failure in QC samples occurred, the analysis was stopped until adequate QC was achieved.
2.6. Evaluation of assay performance
Ion suppression was assessed by comparing standards in methanol vs. standards spiked into precipitated steroid-free serum (n = 5). Serial dilutions were performed in IS methanol or IS methanol precipitated steroid-free serum, respectively for final concentrations of 1–250 ng/mL. Percent Recovery was compared between (1) spike then precipitate: standards spiked into steroid-free serum then precipitated with IS methanol and diluted in IS methanol precipitated steroid-free serum or (2) precipitate then spike: IS methanol precipitated steroid-free serum spiked with standards and diluted with IS methanol precipitated steroid-free serum for final concentrations of 2, 10, 20, and 500 ng/mL (n = 5). Percent recovery was calculated as: mean spike then precipitate/mean precipitate then spike * 100. Extraction reproducibility (% CV) was evaluated on the spike then precipitate samples at the same concentrations (n = 5). Interday and intraday variation (% CV) were evaluated over the course of 8 consecutive days (n = 4 per day) at 2, 10, and 20 ng/mL using the spike then precipitate method. Limit of detection (LOD) and limit of quantitation (LOQ) were calculated for each compound with concentrations ranging from 0.05 to 500 ng/mL (n = 5) as follows: LOD = standard deviation (blank) * 3/slope of regression line and LOQ = standard deviation (blank) * 10/slope of regression line. All experiments were performed in 96-well plates.
2.7. Beckman access II automated antibody assay
The Access testosterone assay is a competitive binding immunoenzymatic assay and was performed using the manufactures protocol. Briefly, patient serum was added to a reaction vessel along with Sample Treatment Solution, mouse monoclonal anti-testosterone antibody, testosterone alkaline phosphatase conjugate, and paramagnetic beads coated with goat anti-mouse polyclonal antibody. Testosterone in the serum is released from the carrier proteins by the Sample Treatment Solution and competes with the testosterone alkaline phosphatase conjugate for binding sites on the specific anti-testosterone monoclonal antibody. The resulting antigen–antibody complexes were then bound to the solid phase by the capture antibody. After incubation, separation was performed in a magnetic field and included a washing step to remove materials not bound to the beads. A chemiluminescent substrate, Lumi-Phos* 530, was added to the reaction vessel and light generated by the reaction was measured with a luminometer. Testosterone concentrations were determined by fitting the luminesce signal to a pre-determined multi-point calibration curve.
2.8. Data analysis
Peak areas for each compound were normalized to the peak area for the corresponding IS in each sample. Quantification of patient samples and QCs were done using linear regression against a standard curve in MassLynx (500, 250, 100 ng/ml with cortisol and cortisone; 20, 10, 2, 1, 0.4 ng/ml with cortisol, cortisone, testosterone, dihydrotestosterone, progesterone; labeled internal standards at 10 ng/mL each). Bland–Altman analysis between testosterone levels from the LC–MS/MS and Beckman Access II antibody assay was performed in GraphPad Prism.
3. Results and discussion
3.1. Technical parameters
Chromatographic separation of the five compounds is shown in Fig. 1 (average peak width 6 s). Matrix effects and ion suppression were evaluated by comparing neat standards in methanol vs. neat standards spiked into protein precipitated steroid-free serum. Ion suppression ranged from 3% to 67% across compounds and concentrations (Fig. 2), with levels generally below 40%. Ion suppression is thus accounted for through our use of an internal standard for each compound [16]. Calibration curves for all compounds have an r2 = 0.99 or greater (see cortisone calibration curve, Supplemental Material Fig. 1). Percent recovery after extraction ranged from 20% to 90%, with most compounds and concentrations close to 60%; extraction reproducibility ranged from 13% to 35% CV with most samples having a % CV less than 25% (Table 2). Within-run/day (intraday) and between-run/day (interday) % CVs were measured at 2, 10, and 20 ng/mL (Table 3). Intraday variation ranged from 0.5% to 18% CV; 2 ng/mL had higher % CV in general than the 10 and 20 ng/mL (n = 4). Interday variation was measured over 8 consecutive days with 4 replicates per concentration per day, and ranged from 5% to 23% CV with lower variation at 10 and 20 ng/mL. LOD and LOQ were calculated to be 3× or 10×, respectively, the standard deviation of the matrix blank/slope of the regression line (Table 4).
Fig. 1.

Overlay of MRM transitions. Chromatograms showing chromatographic separation of testosterone, dihydrotestosterone, progesterone, cortisone, and cortisol spiked to 20 ng/mL each in steroid-free serum precipitated with IS methanol.
Fig. 2.

Ion suppression. Neat standards were compared to standards spiked in precipitated steroid-free serum (n = 5).
Table 2.
% Recovery and extraction reproducibility. Comparison of spike then precipitate vs. precipitate then spike. % CVs are for precipitate then spike (n = 5).
| 2 ng/ml | 10 ng/ml | 20 ng/ml | 500 ng/ml | |
|---|---|---|---|---|
| Testosterone | 66.7 | 61.1 | 58.0 | N/A |
| % CV | 25.2 | 25.7 | 24.6 | N/A |
| Dihydrotestosterone | 59.7 | 57.5 | 56.8 | N/A |
| % CV | 13.6 | 25.4 | 17.0 | N/A |
| Progesterone | 66.0 | 63.6 | 60.8 | N/A |
| % CV | 19.4 | 20.5 | 22.0 | N/A |
| Cortisone | 50.6 | 60.7 | 54.5 | 96.4 |
| % CV | 17.9 | 22.2 | 16.3 | 25.9 |
| Cortisol | 19.7 | 58.0 | 53.3 | 90.3 |
| % CV | 32.8 | 32.6 | 35.3 | 26.5 |
% CV for spike then precipitate: extraction reproducibility.
Table 3.
Interday and intraday variation (% CV) were evaluated over the course of 8 consecutive days (n = 4 per day) at 2, 10, and 20 ng/ml using the spike then precipitate method.
| No. | Analyte | Concentration (ng/ml) | Intraday (% CV) | Interday (% CV) |
|---|---|---|---|---|
| 1 | Testosterone | 2 | 18.04 | 21.86 |
| 10 | 2.06 | 11.18 | ||
| 20 | 2.2 | 8.3 | ||
| 2 | Dihydrotestosterone | 2 | 9.38 | 23.32 |
| 10 | 3.86 | 22.58 | ||
| 20 | 1.52 | 8.33 | ||
| 3 | Progesterone | 2 | 3.9 | 11.29 |
| 10 | 1.04 | 7.83 | ||
| 20 | 0.97 | 7.37 | ||
| 4 | Cortisone | 2 | 5.7 | 11.18 |
| 10 | 4.29 | 8.45 | ||
| 20 | 3.12 | 5.84 | ||
| 5 | Cortisol | 2 | 11.22 | 15.44 |
| 10 | 5.9 | 20.94 | ||
| 20 | 0.49 | 6.19 |
Table 4.
Limits of detection and quantitation.a
| Analyte | Microflow method LOD (ng/ml) | Microflow method LOQ (ng/ml) | Published LOQ (ng/ml) |
|---|---|---|---|
| Published | |||
| Testosterone | 0.12 | 0.41 | 0.6 [12] |
| Dihydrotestosterone | 0.42 | 1.40 | 0.85 [12] |
| Progesterone | 0.03 | 0.40 | 2 [12] |
| Cortisone | 0.09 | 0.29 | 0.5 [16] |
| Cortisol | 0.57 | 1.90 | 0.27 [12] |
All values from samples prepared via protein precipitation.
3.2. Sensitivity
The LOQ for testosterone, progesterone, and cortisone achieved by our microflow LC–MS/MS assay were similar to or lower than values reported in the literature (Table 4). Dihydrotestosterone and cortisol displayed slightly higher LOQs as compared to values reported from protein precipitation or LLE (Table 4), although still well within the levels of clinical relevance [3,17]. Importantly, these sensitivity levels were achieved with only 0.5 μL of extracted sample injected on column as compared to assays performed in standard flow regimes that typically require injection of 50–200 μL of extracted sample. Thus, by moving to a microflow regime we have observed an effectively increase in on-column sensitivity of 100–400 fold. Furthermore, the reported sensitivity for each compound was obtained using a simple, high-throughput, and cost effective protein precipitation step for steroid extraction. The reported LOQs (Table 4) are within the range of biologically relevant levels for a majority of clinical demographics while consuming significantly less sample than traditional flow assays [3]. While improved sensitivity has been reported it is at the expense of additional time and cost involved in targeted extraction usually involving SPE, LLE, derivatization, or some combination of these [7,18–20]. The use of targeted compound specific extraction protocols coupled with the use of microfluidics could additionally improve the reported LOQs for applications requiring lower sensitivity (e.g. testosterone levels in young females) [15].
3.3. Comparison of LC–MS/MS and immunoassay for the analysis of testosterone
Serum from 24 male patients was analyzed via LC–MS/MS and the Beckman Access II automated immunoassay system. This data is presented in a Bland Altman plot which shows the relationship between the mean serum testosterone measured via LC–MS/MS and the Beckman immunoassay on the x-axis and the percent difference of the LC–MS/MS value from immunoassay on the y-axis (Fig. 3a). There was a significant Pearson’s correlation between the two assays (r = 0.78, p < 0.0001), however, the absolute concentrations measured by LC–MS/MS were systematically lower than the immunoassay by approximately half Fig. 3b. In fact, for 7 of the samples, no detectable levels of testosterone were observed by LC–MS/MS however levels between 1 and 3 ng/mL were measured for these samples by immunoassay.
Fig. 3.

Comparison of testosterone measurement by LC–MS/MS and Beckman access II. (a) Bland Altman plot of percent differences in serum testosterone levels (immunoassay minus LC–MS/MS) against the average of the two methods. The dashed lines represent the upper and lower 95% confidence limits. (b) Pearson’s correlation analysis and linear regression between the two assays.
Non-specific binding is a known issue for immunoassays of steroids and is likely the cause of the higher values in these results. Epitestosterone is an epimer of testosterone and thus the two compounds have the exact same mass and structure and differ only in stereochemistry [1,8,15]. According to Beckman literature, the testosterone immunoassay has not been tested for cross-reactivity with epitestosterone. In the LC–MS/MS assay, there is a clear interference present in the testosterone assay (Fig. 4). The MRM method utilized in this assay is very specific to the mass of the analyte and the mass of a daughter ion specific to the analyte. Consequently, if an interference peak is observed, it must have the same intact mass and structural similarity to generate the same daughter ion. This, for example, would be indicative of a structural isoform or an epimer such as epitestosterone. With sufficient chromatographic resolution, such isoforms can be often be separated and distinguished as shown in Fig. 4. It is likely that the peak at 4.58 min is epitestosterone (or an alternative structural isomer of testosterone) as previously shown by Rauh and French [1,8,15] and the peak at 4.4 min is an unknown compound that Rauh could not resolve due to their peak width of 24 s vs. the 6 s peak width obtained in our microflow assay. Hence, by exploiting the specificity of the LC–MS/MS assay and the superior resolution of our chromatographic system we can separate this signal from our true testosterone measurement. This specificity is not possible with immunoassay techniques which will inflate measured levels due to non-specific detection of structural and/or stereochemical isomers.
Fig. 4.
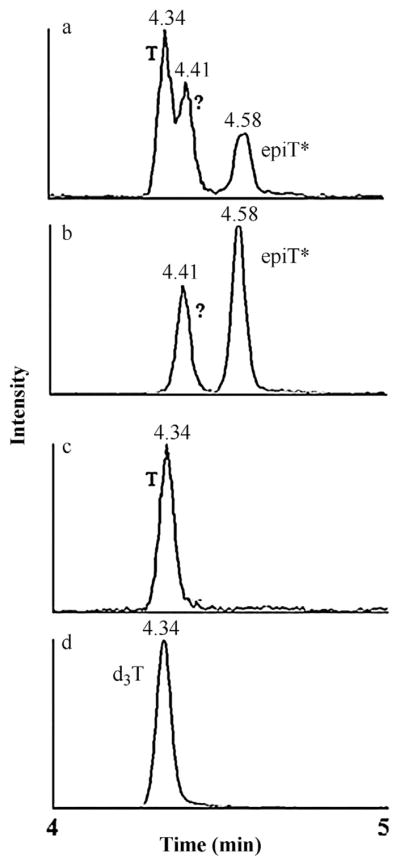
Peaks observed in the LC–MS/MS testosterone assay when monitoring for transition 289 > 97. (a and b) Chromatographic separation of testosterone (4.34 min), an unknown compound (4.41 min) and EpiT* (4.58 min) in patient serum, (c) standard testosterone (4.34 min), (d) internal standard d3T (4.34 min) d3T: labeled internal standard; EpiT: epitestosterone; T: testosterone; *EpiT putatively identified according to Rauh et al., 2006, 2009 and French 2013.
4. Conclusion
We have demonstrated the use of a novel microfluidic liquid chromatography device coupled with mass spectrometry detection for the absolute quantitation of steroids in human serum. The assay was fully qualified and compared with a gold standard clinical immunoassay for the analysis of testosterone. The superb specificity of the LC–MS/MS assay allowed for the distinction of testosterone and its structural isomers yielding more accurate results than the immunoassay which suffers from non-specific interactions and systematic bias [2]. By moving to a microflow regime we were able to reduce solvent consumption by 150 fold and sample consumption by 100–400 fold as compared to standard flow methods. Importantly, decreasing the sample injection volume did not negatively impact the LOD or LOQ as compared to published methods, corresponding to a 100–400 fold increase in on-column sensitivity. Furthermore, the reported LOQs were obtained using a simple protein precipitation step whereas many of the published methods require more cumbersome and costly liquid–liquid or solid-phase extractions [3,15,18,19,21,22]. In conclusion, this novel microflow LC–MS/MS assay provides significant advantages such as low sample and solvent consumption, minimal sample preparation, and improved chromatographic resolution, while still maintaining comparable LOD/LOQs to standard flow LC–MS/MS assays.
Supplementary Material
Abbreviations
- RIA
radioimmunoassay
- ELISA
enzyme-linked immunosorbent assay
- CLIA
chemiluminescent immunoassay
- LC–MS/MS
liquid chromatography with tandem mass spectrometry
- LLE
liquid–liquid extraction
- SPE
solid phase extraction
- ESI
electrospray ionization
- LOD
limit of detection
- LOQ
limit of quantitation
- MRM
multiple reaction monitoring
- T
testosterone
- epiT
epitestosterone
- RSD
relative standard deviation
- QC
quality control
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jchromb.2013.06.031.
References
- 1.Rauh M. Mol Cell Endocrinol. 2009;301:272. doi: 10.1016/j.mce.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Wang C, Catlin DH, Demers LM, Starcevic B, Swerdloff RS. J Clin Endocrinol Metab. 2004;89:534. doi: 10.1210/jc.2003-031287. [DOI] [PubMed] [Google Scholar]
- 3.Kushnir MM, Rockwood AL, Roberts WL, Yue B, Bergquist J, Meikle AW. Clin Biochem. 2011;44:77. doi: 10.1016/j.clinbiochem.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. J Clin Endocrinol Metab. 2007;92:405. doi: 10.1210/jc.2006-1864. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong M, Liu AH, Harbeck R, Reisdorph R, Rabinovitch N, Reisdorph N. J Chromatogr B: Anal Technol Biomed Life Sci. 2009;877:3169. doi: 10.1016/j.jchromb.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taieb J, Mathian B, Millot F, Patricot MC, Mathieu E, Queyrel N, Lacroix I, Somma-Delpero C, Boudou P. Clin Chem. 2003;49:1381. doi: 10.1373/49.8.1381. [DOI] [PubMed] [Google Scholar]
- 7.Cawood ML, Field HP, Ford CG, Gillingwater S, Kicman A, Cowan D, Barth JH. Clin Chem. 2005;51:1472. doi: 10.1373/clinchem.2004.044503. [DOI] [PubMed] [Google Scholar]
- 8.Rauh M, Groschl M, Rascher W, Dorr HG. Steroids. 2006;71:450. doi: 10.1016/j.steroids.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 9.Rothman MS, Carlson NE, Xu M, Wang C, Swerdloff R, Lee P, Goh VH, Ridgway EC, Wierman ME. Steroids. 2011;76:177. doi: 10.1016/j.steroids.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grebe SK, Singh RJ. Clin Biochem Rev. 2011;32:5. [PMC free article] [PubMed] [Google Scholar]
- 11.Stanczyk FZ, Clarke NJ. J Steroid Biochem Mol Biol. 2010;121:491. doi: 10.1016/j.jsbmb.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Legro RS, Schlaff WD, Diamond MP, Coutifaris C, Casson PR, Brzyski RG, Christman GM, Trussell JC, Krawetz SA, Snyder PJ, Ohl D, Carson SA, Steinkampf MP, Carr BR, McGovern PG, Cataldo NA, Gosman GG, Nestler JE, Myers ER, Santoro N, Eisenberg E, Zhang M, Zhang H. J Clin Endocrinol Metab. 2010;95:5305. [Google Scholar]
- 13.Ray JA, Kushnir MM, Rockwood AL, Meikle AW. Clin Chim Acta. 2011;412:1221. doi: 10.1016/j.cca.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 14.Wooding KM, Auchus RJ. Mol Cell Endocrinol. 2013 doi: 10.1016/j.mce.2012.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.French D. Clin Chim Acta. 2013;415:109. doi: 10.1016/j.cca.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Annesley TM. Clin Chem. 2003;49:1041. doi: 10.1373/49.7.1041. [DOI] [PubMed] [Google Scholar]
- 17.Ionita IA, Fast DM, Akhlaghi F. J Chromatogr B: Anal Technol Biomed Life Sci. 2009;877:765. doi: 10.1016/j.jchromb.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Rossi C, Calton L, Hammond G, Brown HA, Wallace AM, Sacchetta P, Morris M. Clin Chim Acta. 2010;411:222. doi: 10.1016/j.cca.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Keski-Rahkonen P, Huhtinen K, Poutanen M, Auriola S. J Steroid Biochem Mol Biol. 2011;127:396. doi: 10.1016/j.jsbmb.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Shi RZ, van Rossum HH, Bowen RA. Clin Biochem. 2012;45:1706. doi: 10.1016/j.clinbiochem.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Janzen N, Sander S, Terhardt M, Peter M, Sander J. J Chromatogr B: Anal Technol Biomed Life Sci. 2008;861:117. doi: 10.1016/j.jchromb.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Koal T, Schmiederer D, Pham-Tuan H, Rohring C, Rauh M. J Steroid Biochem Mol Biol. 2012;129:129. doi: 10.1016/j.jsbmb.2011.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.