

Abstract
Polyhydroxyalkanoate (PHA) synthases (PhaCs) catalyze the formation of biodegradable PHAs that are considered as an ideal alternative to nonbiodegradable synthetic plastics. However, study of PhaC has been challenging because the rate of PHA chain elongation is much faster than that of initiation. This difficulty along with lack of a structure has become the main hurdle to understand and engineer PhaCs for economical PHA production. Here we reported the synthesis of two carbadethia CoA analogs, sT-CH2-CoA 26a and sTet-CH2-CoA 26b as well as sT-aldehyde 29 as new PhaC inhibitors. Study of these analogs with PhaECAv revealed that 26a/b and 29 are competitive and mixed inhibitors, respectively. It was observed that CoA moiety and PHA chain extension can increase binding affinity, which is consistent with the docking study. Estimation from Kic of 26a/b predicts that a CoA analog attached with an octameric-HB chain may facilitate the formation of a kinetically well-behaved synthase.
Keywords: PHA synthases, inhibitors, synthesis design, chemoenzymatic approach, molecular docking
Introduction
Polyhydroxyalkanoates (PHAs) are polyoxoesters that serve as carbon and energy storage materials in cells under nutrient-limited conditions with excess carbon sources.[1] Up till now, 150 structurally different monomers have been found to be polymerized into PHAs by different bacterial strains.[2] PHAs are considered as environmentally friendly materials because they can be synthesized from renewable resources and are biodegradable.[3] They have been marketed as an ideal alternative to non-biodegradable petroleum-based plastics.[4] Recently, significant progress has been made in the application of PHAs as high-technology materials in medical fields.[5] However, their commercialization is limited mainly due to high costs incurred during their production.[6]
As shown in Scheme 1, PHA synthases (PhaCs) catalyze the polymerization of 3-R-hydroxyalkyl CoA thioester to form PHAs with concomitant release of CoA. They can be divided into four classes depending on their subunit composition and substrate specificity.[7] While class I and II consist of a single subunit (PhaC), class III and IV contain two subunits (PhaEC for class III and PhaRC for class IV). Although PhaE and PhaR subunits are required for enzyme activity, their exact role is still unclear. Class I and III synthases use only the short chain length (C3−C5) substrates (e.g. R-3-hydroxybutyrate CoA: HBCoA) while class II and IV prefer the medium chain length (C6 and greater) substrates. We are interested in class I and III synthases because they share same catalytic mechanism[1b] and their substrates are synthetically accessible. PHA synthases from Ralstonia eutropha (PhaCRe)[8] and Allochromatium vinosum (PhaECAv)[9] have been employed as the prototypic class I and III enzymes, respectively.
Scheme 1.
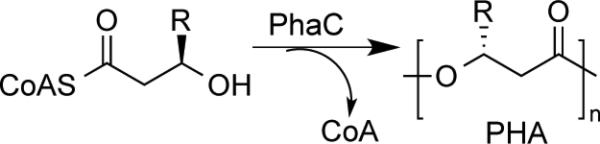
Formation of PHAs catalyzed by PhaCs.
It is known that PhaCs play crucial roles in substrate recognition as well as in controlling PHA chain length and polydispersity.[10] However, study of PhaC has been challenging because the rate of PHA chain elongation is much faster than that of initiation.[1b] Furthermore, despite much effort, the crystal structure of PHA synthases is still unavailable. All of these limit our ability to understand and rationally engineer PhaCs so that the PHAs can be produced in an economically competitive fashion. Therefore, we set our goal to determine the requirements of a probe that can not only facilitate the formation of kinetically well-behaved synthases, but also enhance PhaC crystallization.
Saturated trimer-CoA (sTCoA)[11] shown in Scheme 2 has been employed extensively in PhaC mechanistic study.[1b] It can act as an artificial primer to uniformly load the synthases, which results in the formation of proteins that have comparable rates of PHA chain initiation and elongation.[12] However, the attached saturated trimer (sT-) chain is unstable and can be cleaved off from the protein through hydrolysis catalyzed by the synthases. It has been proposed that the active site of PHA synthases consist of a substrate entrance channel and a product exit channel.[13] Full occupancy of these channels would suppress the hydrolysis and result in a kinetically well-behaved enzyme, which could also facilitate the formation of PhaC with high physical purity for crystallization purposes. In order to estimate the channel length, the binding property of sTCoA has to be characterized. However, this turned out to be difficult and expensive because significant amount of tritium-labelled sTCoA ([3H]-sTCoA)[11] is required. Therefore, to avoid the high cost and safety concerns associated with radioactive chemicals, we decided to prepare a nonhydrolyzable carbadethia sTCoA analog (sT-CH2-CoA) 26a as a PhaC inhibitor to evaluate sT-CoA binding property. The carbadethia analog of saturated tetramer-CoA (sTet-CH2-CoA) 26b was also synthesized to enable the estimation. Additionally, saturated trimer aldehyde (sT-aldehyde) 29 was prepared in order to investigate the importance of CoA in substrate binding as well as whether this moiety could be eliminated to simplify the synthesis in future.
Scheme 2.
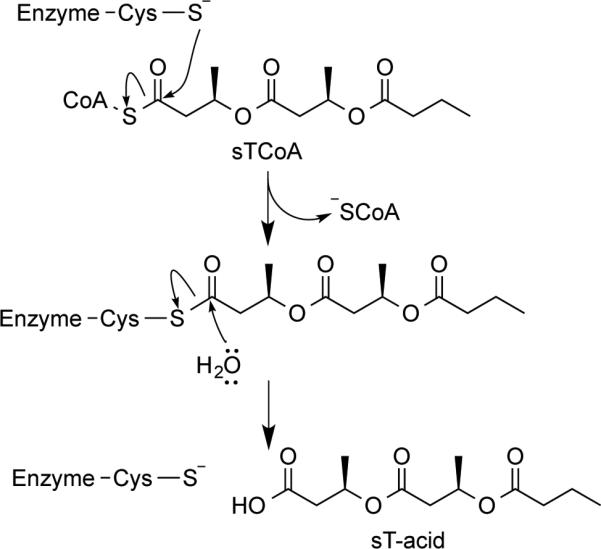
Acylation of PhaCs by sTCoA and PhaC-catalyzed hydrolysis.
Furthermore, among various strategies that can be envisaged to enhance protein crystallization is complexation with ligands,[14] which has been widely used in drug discovery to design new molecules.[15] It has also been reported that structures of ligand-binding proteins can be employed in computational protein engineering to generate mutants with artificial functions.[16] Therefore, the inhibitors described here will contribute to our efforts to generate a ligand library that could be used to enhance PhaC crystallization for its first structure.
Results and Discussion
Chemoenzymatic synthesis of carbadethia analog 26
Coenzyme A (CoA) esters are among the most important small molecules that are involved in a variety of biological processes including fatty acid biosynthesis, carbohydrate catabolism, and generation of secondary metabolites.[17] CoA is also a major regulator of energy metabolism that is closely related to cellular development, aging, and cancers.[18] Therefore, even seventy years after its discovery by Lipmann,[19] CoA is still actively pursued by scientists and synthesis of its analogs remains as a major tool to decipher the aforementioned biological pathways at the molecular level.[17d] Although elucidation of CoA biosynthesis has greatly facilitated introduction of the adenosine nucleotide into CoA analogs,[20] synthesis of pantothenate-based precursors to enzymatic conversions remains difficult and specific to the proteins of interest. Furthermore, among various CoA analogs, preparation of the carbadethia derivatives that have a methylene group in place of the sulfur atom has been proven the most challenging.[21]
Chemical synthesis of the key intermediate and enzymatic precursor, pantetheine derivative 17 is described in Scheme 3. The terminal alcohol 3 was prepared by a nucleophilic acyl substitution of amide 1[22] with the Grignard reagent 2 generated in situ from 3-chloropropan-1-ol.[23] Subsequent to acetylation, the carbonyl group in 4 was protected with ethylene glycol to give an intermediate 5. The terminal hydroxyl group in 6 was converted into an amino group in 8 through a Mitsunobu reaction[24] involving a phthalimide derivative 7 followed by hydrazine hydrolysis. Coupling between an amine 8 and acid 9 yielded an amide 10 in the presence of EDCI and HOBT. Hydrogenation of 10 catalyzed by Pd/C gave an alcohol 11 in a good yield.
Scheme 3.

Chemical synthesis of precursors to enzymatic reactions: a) 2 (1.1 equiv.), THF, 0 °C, 50 min, then aq. NH4Cl, 86%; b) Ac2O (2.0 equiv.), pyridine (3.0 equiv.), 4-(dimethylamino)pyridine (DMAP, 0.05 equiv.), CH2Cl2, 12 hrs, 97%; c) ethylene glycol (10 equiv.), CH(OEt)3 ( 4.0 equiv.), camphorsulfonic acid (CSA, 0.05 equiv.), 55 °C, 8 hrs, 65%; d) 2 M NaOH, 4 hrs, 70%; e) Ph3P (1.1 equiv.), phthalimide (1.1 equiv.), diisopropyl diazene-1,2-dicarboxylate (DIAD, 1.1 equiv.), THF, 0°C, 12 hrs, 90%; f) N2H4•H2O (3.0 equiv.), EtOH, reflux, 3 hrs, 90%; g) 9 (1.1 equiv.), Et3N (2.5 equiv.), N-ethyl- N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI, 1.5 equiv.), hydroxybenzotriazole (HOBT, 1.5 equiv.), CH2Cl2, 12 hrs, 60%; h) H2 (1 atm), 10% Pd-C (0.15 equiv.), 5 hrs, 74%; i) 12 (3.0 equiv.), 2,4,6-trichlorobenzoyl chloride (TCBC, 3.0 equiv.), Et3N (3.5 equiv.), DMAP (3.5 equiv.), CH2Cl2, 17 hrs, 99%; j) same as (h), 89%; k) n = 1: n-butyryl chloride (3.0 equiv.), DMAP (3.4 equiv.), 0 °C to r.t., 24 hrs, 43%; n = 2: 15 (1.6 equiv.), (COCl)2 (3.2 equiv.), N, N′-dimethylformamide (DMF, one drop), CH2Cl2, 2hrs, 83%; l) 1M HCl, MeCN, 2.5 hrs, 96% for n = 1, 83% for n = 2.
Much effort has been made in the formation of ester 16a. Initially, we tried to couple the alcohol 11 and saturated dimer-acid (sD-acid) 15[25] to directly generate ester 16a described in Scheme 4 (dotted line). Numerous coupling reagents, such as (COCl)2/DMF and PyBOP/DIPEA were screened for this step and resulted in low yields. However, when DCBC or TCBC (Yamaguchi reagent)[26] was used, the reaction was clean (solid line in Scheme 4). The product was isolated in 60% and 82% yields for DCBC and TCBC, respectively. However, compound characterization revealed that the product was ester 18, which is one HB unit (boxed in Scheme 4) shorter from the expected ester 16a. Moreover, significant amount of crotonic acid was recovered from the same reaction. Therefore, a mechanism shown in Scheme 5 is proposed. The Yamaguchi reagent TCBC was used to activate sD-acid 15 to give an anhydride 19. Addition of DMAP would displace the 2,4,6-trichlorobenzoyl (TCB) group in 19 to form an amide 20 bearing a positive charge. Subsequent intramolecular cyclization of 20 would generate a six-membered ring derivative 21 that could undergo nucleophilic attack by DMAP to form an intermediate 22. Elimination by the TCB anion would result in the formation of crotonic acid as well as an activated amide 23. Nucleophilic attack by the alcohol 11 would yield the observed product 18. Structure of 18 was confirmed by 1H and 13C-NMR. As far as we know, this is the first example of intramolecular cyclization followed by elimination discovered in the Yamaguchi esterification.
Scheme 4.

Unexpected formation of ester 18: a) (COCl)2, catalytic DMF; or (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), N,N′-diisopropylethylamine (DIPEA); b) (i) 2,4-dichlorobenzoyl chloride (DCBC) or 2,4,6-trichlorobenzoyl chloride (TCBC), Et3N, THF; (ii) DMAP, CH2Cl2.
Scheme 5.

Proposed mechanism for the formation of ester 18
Finally, as described in Scheme 3, ester 16 was generated from 11 in three steps that included Yamaguchi esterification between acid 12[27] and alcohol 11,hydrogenation of ester 13 to remove a benzyl (Bn) group, and another esterification between alcohol 14 and n-butyryl chloride or sD-acid 15. The enzymatic precursor 17 was obtained after acid hydrolysis in acetonitrile for 2.5 hrs. It has to be pointed out that workup for most pantetheine derivatives is tedious, which usually involves an ion exchange chromatography to neutralize the acid followed by lyophilization to remove water.[28] However, due to the presence of hydrophobic PHA chain in 17, its workup was simple and could be readily achieved by using ethyl acetate as the extraction solvent.
Enzymatic synthesis to convert the pantetheine derivative 17 to a carbadethia CoA analog 26 was described in Scheme 6 by employing three enzymes involved in CoA biosynthesis: a pantothenate kinase from Staphylococcus aureus (SaPanK),[29] a phosphopantetheine adenylyltransferase (EcCoaD),[30] and a dephospho-CoA kinase (EcCoaE)[31] from Escherichia coli. These enzymes can accept a wide spectrum of substrates and have been extensively used in the synthesis of CoA analogs.[20c, 21a, 32] In order to monitor the enzymatic transformation, the reactions were initially carried out stepwise in a small scale. While the enzymatic precursor 17 and phosphopantetheine derivative 24 were followed at 220 nm, the 3′-dephospho-CoA analog 25 and final carbadethia CoA analog 26 were monitored at 260 nm. It can be seen from the Figure 1 that, with sequential additions of SaPanK, EcCoaD, and EcCoaE, the resulting products were eluted faster in reverse-phase HPLC (RP-HPLC) chromatography due to increasing number of phosphate groups present in the structure. After the identities of 24 and 25 were confirmed, the carbadethia CoA analog 26 was prepared in a large scale using three enzymes simultaneously and purified by semi-preparative RP-HPLC. All compounds including 24, 25, and 26 were fully characterized by 1H-, 13C-, and 31P-NMR and HRMS.
Scheme 6.

Enzymatic conversion to form carbadethia CoA analog 26. Each compound in this scheme is designated as a and b when n equals to 1 and 2, respectively.
Figure 1.

HPLC profiles of enzymatic conversions: from 17a to 26a (A) and from 17b to 26b (B). The peaks in black, red, green, and purple represent compounds 17, 24, 25, and 26 shown in Scheme 5, respectively. The reaction progress was monitored by HPLC with an analytical column (Luna C18-2, 5 μm, 4.6 mm × 250 mm) that was eluted at 1 mL/min using a linear gradient from 10 to 90% methanol in 10.0 mM ammonium acetate (pH 4.00) over 60 min.
Synthesis of sT-aldehyde
In order to investigate the importance of CoA moiety in substrate binding, sT-aldehyde 29 was prepared as the PhaC inhibitor. Since aldehydes have been widely employed as the complexed ligands for structural study of enzymes that involve cysteine as the catalytic residue,[33] sT-aldehyde could also help our efforts in PhaC crystallization. Therefore, 29 was prepared according to the approach shown in Scheme 7. Starting with saturated trimer-acid (sT-acid) 27,[11] the carboxylic group was reduced by H3B:SMe2 to yield saturated trimer-alcohol (sT-alcohol) 28. Subsequent Swern oxidation[34] gave the desired sT-aldehyde 29 in a total 67% yield for two steps. The final compound was confirmed by 1H- and 13C-NMR and HRMS.
Scheme 7.

Chemical synthesis of sT-aldehyde 29.
Inhibition study with PhaECAv
While class I and III PHA synthases use the same substrates and are thought to share similar mechanism for polymerization, their kinetics are quite different from each other.[1b] The class I enzyme has a characteristic lag phase followed by a fast phase.[12] The cause of the lag phase is still unknown though protein dimerization has been suggested to relate with this phenomenon.[12] The class III enzyme exhibits biphasic kinetics with a fast phase followed by a slow phase.[35] In order to avoid complications resulting from the lag phase in Michaelis-Menten kinetics analysis, the inhibition study was only carried out with class III synthase PhaECAv.
Inhibition studies with PhaECAv were performed with sT-CH2-CoA 26a, sTet-CH2-CoA 26b, and sT-aldehyde 29. The enzyme was assayed by monitoring CoA release using 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB).[12] Since all analogs contain a carbonyl group, time-dependent experiments were performed in order to see whether a hemithioacetal could be formed between the cysteine and carbonyl group. It was found that pre-incubation of PhaECAv with the analogs did not inactivate the enzyme (data not shown). Therefore, continuous DTNB assays were carried out with the synthase by varying concentrations of inhibitors and substrate HBCoA. The rates of the reactions were determined by the slope of the initial fast phase. The obtained data were fitted to different inhibition modes (competitive, noncompetitive, uncompetitive and mixed) using SigmaPlot. The results are shown in Figure 2 and summarized in Table 1.
Figure 2.

Lineweaver-Burk plots of the PhaECAv activity in the presence of inhibitors sT-CH2-CoA 26a (A), sTet-CH2-CoA 26b (B), and sT-aldehyde 29 (C) at different concentrations. Assays were performed in duplicate.
Table 1.
Summary of inhibition constant (Ki) and binding energy (ΔGb) with PhaECAv
| Inhibitors | Kic (mM) | Kiu (mM) | ΔGb (kcal/mol) |
|---|---|---|---|
| 26a | 0.60 ± 0.03 | –6.50 | |
| 26b | 0.50 ± 0.03 | –6.70 | |
| 29 | 3.13 ± 0.47 | 15.0 ± 0.73 | –4.80 |
Lineweaver-Burk plots reveal that both sT-CH2-CoA 26a and sTet-CH2-CoA 26b are competitive inhibitors. The competitive inhibition constants (Kic) for 26a and 26b are 0.60 and 0.50 mM, respectively. These Kic values are larger than the Michaelis-Menten constant (KM) of the natural substrate HBCoA (0.13 mM), which indicates that the synthase has higher affinity with HBCoA than with product-like inhibitors 26a/b. Moreover, a slight decrease in Kic (0.10 mM) was observed when the PHA chain extends from trimer to tetramer. This may suggest that the product binding affinity increases during initial chain elongation. As described earlier, full occupancy of the substrate entrance and product exit channels is expected to eliminate hydrolysis, a side reaction catalyzed by the synthases. To calculate the channel length, it is hypothesized that an inhibitor that can fill the channel would have a Kic similar to KM of HBCoA. Therefore, assuming each additional HB unit would decrease Kic value by 0.10 mM, it is predicted that a carbadethia CoA analog with an octameric-HB chain would reach a Kic close to 0.13 mM. This prediction agrees well with the observed result that a primed PhaECAv has a trimer to decamer chain attached during PHA re-initiation.[13] Thus, a CoA analog attached with an octamer chain is proposed to be a good probe that can potentially facilitate the formation of kinetically well-behaved synthases. Preparation of such analog is under way.
The Lineweaver-Burk lines of sT-aldehyde 29 do not intersect at the 1/[HBCoA] axis or 1/v axis, which indicates that sT-aldehyde is a mixed inhibitor. Its Kic and Kiu (uncompetitive inhibition constant) constants are 3.13 and 15.0 mM, respectively. Thus, the inhibition mode of sT-aldehyde is quite different from carbadethia CoA analogs, which may be attributed to the absence of CoA moiety in sT-aldehyde. As summarized in Table 1, the Kic of sT-aldehyde is at least 5-fold higher than that of carbadethia CoA analogs, which shows that the CoA moiety is indeed important for and has much larger influence on substrate binding. Additionally, the Kiu of sT-aldehyde is 5-fold higher than its Kic, suggesting the existence of a second binding site. These observed phenomena are consistent with the docking study described below.
Molecular docking and implications
Molecular docking was performed in order to unveil the structural basis for the observed results from inhibition study. Since PhaC crystal structure is unavailable, homology models were built using CPHmodels3.0,[36] SWISS-MODEL[37] and I-TASSER.[38] Three crystal structures of proteins that have considerable sequence similarity to PhaECAv around active site cysteine (~40%) were used: human gastric lipase,[39] esterase from Sulfolobus solfataricus,[40] and dog gastric lipase (DGL).[41] As shown in Figure 3A, an overlay of the generated models reveals that these structures have similar backbone folds with minor differences near the active site. An I-TASSER predicted structure was selected for docking since it had the highest score for structural quality. As depicted in Figure 3B, the active site is deeply buried[35] and located in a pocket where the substrate entrance and product exit channels may exist.
Figure 3.

Homology modelling of PhaECAv. (A) Overlay of three separately prepared models. Protein backbones within 6 Å of the displayed active site residues (C149, D302, and H331) are shown as Cα atom traces. Blue: model produced by CPH models server; purple: model produced by the Swiss-Model server; green: top-ranked model produced by I-TASSER server; (B) Surface representation of the active site pocket. The model is produced by I-TASSER. The active site residues are shown as sticks.
Automated docking by AutoDock Vina[42] resulted in a predicted binding mode where sT-CH2-CoA occupies almost the same space as the grey-colored detergent and inhibitor molecules bound to DGL (PDB: 1K8Q, Figure 4A). However, their positions are different, which could be caused by the fact that two molecules were used to mimic the triglyceride substrate in DGL while only one large molecule sT-CH2-CoA was docked in PhaECAv. Additionally, the −SH on C149 is 7.70 Å away from the carbonyl group and their directions are pointing perpendicular to each other. This suggests that the synthase must go through additional conformational changes in order to bring the nucleophile (−SH) and electrophile (carbonyl) close enough to have the polymerization reactions. Nevertheless, the generated docking models can still be used to evaluate binding interactions between the substrate and enzyme. As depicted in Figure 4B, the CoA moiety in sT-CH2-CoA is responsible for five H-bonds (black dotted lines) and hydrophobic interactions with L76, V77, F260, and F263, which is absent from the docking model with sT-aldehyde (Figure 4C). This explains the difference in predicted binding energy (ΔGb) and observed Ki. As summarized in Table 1, the ΔGb of sT-CH2-CoA is −1.70 kcal/mol lower than that of sT-aldehyde, which is translated into a 5.2-fold increase in the observed Kic (0.6 vs. 3.13 mM). Therefore, it can be concluded that presence of CoA moiety will significantly increase binding affinity. Furthermore, two of five H-bonds are formed between S252 and phosphate groups present in CoA, which is consistent with a recent experimental observation by Ushimaru et al.[43] Interactions between the PHA chain and hydrophobic residues including L184, V189, M229, L232, L233, L304, and I332 also contribute to substrate binding. Extension of chains from trimer to tetramer can potentially enhance this type of hydrophobic interactions, which was supported by the observed slight decrease in ΔGb (from −6.50 to −6.70 kcal/mol) and Kic (from 0.60 to 0.50 mM).
Figure 4.

Docking study. (A) Overlay of docking model (green: catalytic residues; yellow: sT-CH2-CoA) and DGL (1KQ8, blue) complexed with an inhibitor and detergent (grey); (B) Binding mode of sT-CH2-CoA. H-bonds are represented by dashed lines; (C) Binding of sT-aldehyde. Two modes with the highest ΔGb are shown here. H-bond is represented by a dashed line.
Docking study of the sT-aldehyde resulted in multiple binding modes, among which the ones with the lowest ΔGb are shown in Figure 4C. At least two binding sites are available for sT-aldehyde. One is close to the catalytic triad and the other is at the mouth of the binding pocket, which may explain the inhibition mode of sT-aldehyde is mixed. In addition to the long-range H-bonds between the terminal carbonyl/ester group and Y74/I249 (3.3−3.5Å), binding affinity mainly comes from the hydrophobic interactions between PHA chain and residues such as M82, V230, L233, L249, F250, and I332.
Conclusion
In an effort to find out the requirements of a probe that can facilitate the formation of kinetically well-behaved synthases as well as enhance protein crystallization, two non-hydralyzable carbadethia CoA analogs, sT-CH2-CoA, and sTet-CH2-CoA were prepared as PhaC inhibitors through a chemoenzymatic approach. During the synthesis of ester 16a, it was discovered that the Yamaguchi esterification between the alcohol 11 and sD-acid 15 resulted in the loss of one HB unit from the expected product when DMAP was present (pathway b in Scheme 4). A mechanism involving intramolecular cyclization followed by elimination is proposed to explain the formation of ester 18 (Scheme 5). Finally, the desired ester 16a was prepared in three steps from 11 in a good yield. A third PhaC inhibitor, sT-aldehyde 29 was synthesized in order to study the importance of CoA moiety. Inhibition studies with PhaECAv reveal that carbadethia CoA analogs and sT-aldehyde are competitive and mixed inhibitors, respectively. Presence of a CoA moiety results in a tighter binding and has a much larger influence on binding affinity than PHA chain, which is consistent with the docking study. Therefore, CoA moiety must be included in a designed probe. Based on the Kic values of sT-CH2-CoA and sTet-CH2-CoA, it is predicted that a CoA analog with an octamer chain will facilitate the formation of a kinetically well-behaved synthase that should have comparable rates of PHA chain initiation and elongation. Moreover, the inhibitors presented here are being used for PhaC crystallization. The work is in progress and will be reported in due course.
Experimental Section
General information
All chemicals were purchased at the highest purity grade. All solvents were anhydrous. All reactions were performed under argon atmosphere unless otherwise specified. Thin layer chromatography (TLC) was performed using 60 mesh silica gel plates and visualization was performed using short wavelength UV light (254 nm) and basic KMnO4 staining. HPLC was performed with a Waters Breeze 2 system consisting of a 1525 pump and a 2998 photodiode array detector. Absorbance was recorded on an Agilent Cary 100 UV-Vis spectrophotometer or Molecular Devices SpectraMax Plus 384. NMR spectra were recorded on a Varian 400 MHz spectrometer. Chemical shifts of proton (1H NMR) and carbon (13C NMR) were reported in ppm relative to the residual solvent peaks except that methanol was employed as the external reference for 13C NMR when D2O was used. Chemical shifts of phosphorus (31P NMR) were reported in ppm relative to the external reference of 85% H3PO4. High resolution mass spectrometry (HRMS) was recorded on a Q-Star Elite spectrometer manufactured by Applied Biosystems.
Protein purification and enzyme assay
His-tagged pantothenate kinases including SaPanK, EcCoaD, and EcCoaE were purified according to the published methods.[29, 32b] Their specific activities (SA) were measured at 45, 27, and 20 µmol min−1mg−1 at 25 °C for SaCoaA, EcCoaD, and EcCoaE, respectively. PHA synthase PhaECAv was purified following the published procedures.[9b] The SA was measured at 338 µmol min−1mg−1 at 30 °C.
Chemoenzymatic synthesis of sT-CH2-CoA 26a and sTet-CH2-CoA 26b (R)-6-(benzyloxy)-1-hydroxyheptan-4-one 3
MeMgBr (3.0 M in Et2O, 19.6 mL, 58.4 mmol) was added drop-wise to a solution of 3-chloropropan-1-ol (5.50 g, 58.4 mmol) in THF (140 mL) cooled to −20 °C. The mixture was then warmed to room temperature and transferred to a new flask containing Mg turnings (0.50 g, 87.6 mmol). After added 70 μL 1, 2-dibromoethane, the solution was heated at reflux for 1.5 hrs. to generate the Grignard reagent 2. This reagent was transferred to a dropping funnel and added to a solution of amide 1 (12.6 g, 53.1 mmol) in THF (175 mL) with 20 min at 0 °C. The resulting mixture was stirred for another 30 min and quenched with saturated NH4Cl (aq.) (50.0 mL) at 0 °C. The mixture was then extracted with EtOAc (30.0 mL × 3). The organic extracts were combined and washed with brine (60.0 mL), dried with anhydrous MgSO4, and concentrated to dryness. The residue was purified by silica gel chromatography eluting with hexane/EtOAc (3/1) to yield 3 (10.8 g, 86.0%) as colorless oil; 1H NMR (400 MHz, CDCl3) δ: 7.31 (m, 5H), 4.50 (dd, 1H, J = 12.0 Hz), 4.06 (m, 1H), 3.61 (quart, 2H, J = 7.0 Hz), 2.78 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.58 (m, 2H), 2.48 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 1.83 (m, 2H), 1.81 (t, 1H, J = 7.0 Hz, OH), 1.24 (d, 3H, J = 8.0 Hz); 13C NMR (400 MHz, CDCl3) δ: 210.0, 138.5, 128.5, 127.8, 71.9, 71.0, 62.1, 50.1, 40.8, 26.4, 20.0.
(R)-2-(3-(2-(2-(benzyloxy)propyl)-1,3-dioxolan-2-yl)propyl)isoindoline-1,3-dione 7
To a mixture of compound 6 (3.00 g, 10.7 mmol), Ph3P (2.30 g, 11.8 mmol) and phthalimide (1.70 g, 11.8 mmol) in THF (100 mL) in ice bath was added a solution of DIAD (2.40 g, 11.8 mmol) in 5.00 mL THF. The reaction mixture was stirred for 12 hrs. and then the solvent was removed. The residue was purified by silica gel chromatography eluting with hexane/EtOAc (10/1) to give compound 7 (4.00 g, 90%) as a white solid; 1H NMR (400 MHz, CDCl3) δ: 7.83 (m, 2H), 7.70 (m, 2H), 7.34-7.24 (m, 5H), 4.50 (dd, 2H, J = 12.0 Hz), 3.90 (m, 4H), 3.66 (m, 3H), 2.03 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 1.74 (m, 4H), 1.69 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 1.23 (d, 3H, J = 4.0 Hz); 13C NMR (400 MHz, CDCl3) δ: 168.3, 138.9, 134.1, 133.8, 132.1, 128.3, 127.7, 127.3, 123.4, 123.1, 110.4, 71.7, 70.3, 64.9, 64.8, 44.1, 38.1, 34.9, 23.0, 21.0.
(R)-N-(3-(3-(2-((R)-2-(benzyloxy)propyl)-1,3-dioxolan-2-yl)propylamino)-3-oxopropyl)-2,2,5,5-tetra methyl-1,3-dioxane-4-carboxamide 10
To a solution of amine 8 (3.00 g, 10.7 mmol) in CH2Cl2 (120 mL) was added Et3N (3.70 mL, 26.9 mmol), acid 9 (3.30 g, 12.9 mmol), EDCI (3.00 g, 16.1 mmol), and HOBT (2.10 g, 16.1 mmol) at r.t. The mixture was stirred for 12 hrs. and then diluted with CH2Cl2 (100 mL). The organic layer was washed sequentially with saturated aqueous NaHCO3 (100 mL) and water (100 mL) and then dried with Na2SO4. The solvent was removed and the residue was purified by silica gel chromatograph eluting with hexane/EtOAc (1/1) to give 10 (3.30 g, 60%) as colorless oil; 1H NMR (400 MHz, CDCl3) δ: 7.33-7.27 (m, 5H), 7.03 (m, 1H), 5.79 (m, 1H), 4.49 (dd, 2H, J = 12.0 Hz), 4.07 (s, 1H), 3.92 (m, 4H), 3.69 (m, 2H), 3.50 (m, 2H), 3.27 (d, 1H, J = 12.0 Hz), 3.17 (quart, 2H, J = 8.0 Hz), 2.31 (t, 2H, J = 6.0 Hz), 2.01 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 1.68 (m, 3H), 1.57 (m, 2H), 1.46 (s, 3H), 1.41 (s, 3H), 1.24 (s, 3H, J = 4.0 Hz), 1.04 (s, 3H), 0.97 (s, 3H); 13C NMR (400 MHz, CDCl3) δ: 170.8, 170.0, 138.8, 128.3, 127.6, 127.5, 110.5, 99.0, 77.1, 71.8, 71.4, 70.3, 64.8, 64.6, 43.8, 39.5, 35.9, 34.9, 33.0, 29.5, 23.7, 22.2, 20.9, 18.9, 18.7.
(R)-((R)-1-(2-(3-(3-((R)-2,2,5,5-tetramethyl-1,3-dioxane-4-carboxamido)propanamido)propyl)-1,3-diox olan-2-yl)propan-2-yl) 3-(benzyloxy)butanoate 13
To a solution of compound 12 (0.70 g, 3.50 mmol) and Et3N (0.60 mL, 4.10 mmol) in THF was added TCBC (0.50 mL, 3.50 mmol) at r.t. The mixture was stirred overnight and the Et3N·HCl solid was removed by filtration. The filtrate was concentrated to dryness and the residue was re-dissolved in CH2Cl2 (15.0 mL). To the above solution was added DMAP (0.50 g, 4.10 mmol) and alcohol 11 (0.50 g, 1.20 mmol) in CH2Cl2 (5.00 mL). The mixture was stirred for additional 2 hrs and then concentrated to dryness. The residue was purified by silica gel chromatograph eluting with hexane/EtOAc (1/2) to give ester 13 (0.70 g, 99 %) as pale yellow oil; 1H NMR (400 MHz, CDCl3) δ: 7.32-7.27 (m, 5H), 7.04 (m, 1H,), 5.90 (m, 1H), 5.13 (m, 1H), 4.53 (dd, 1H, J = 12.0 Hz), 4.07 (s, 1H), 4.02 (m, 1H), 3.87 (m, 4H), 3.68 (d, 1H, J = 12.0 Hz), 3.56 (m, 2H), 3.28 (d, 1H, J = 12.0 Hz), 3.18 (m, 2H), 2.58 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.39 (m, 3H), 1.98 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 1.72 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 1.53 (m, 2H), 1.46 (s, 3H), 1.42(s, 3H), 1.26 (d, 3H, J = 4.0 Hz), 1.24 (d, 3H, J = 4.0 Hz), 1.04 (s, 3H), 0.97 (s, 3H); 13C NMR (400 MHz, CDCl3) δ: 170.9, 170.0, 138.5, 128.3, 127.7, 127.5, 109.9, 99.0, 77.1, 72.1, 71.4, 70.9, 67.4, 64.8, 42.6, 42.4, 39.5, 35.9, 34.9, 34.7, 33.0, 29.5, 23.7, 22.2, 21.3, 19.8, 18.9, 18.7.
(R)-((R)-1-(2-(3-(3-((R)-2,2,5,5-tetramethyl-1,3-dioxane-4-carboxamido)propanamido)propyl)-1,3-dioxo lan-2-yl)propan-2-yl) 3-(butyryloxy)butanoate 16a
To a mixture of alcohol 14 (0.40 g, 0.70 mmol) and DMAP (0.30 g, 2.40 mmol) in CH2Cl2 (20.0 mL) was added n-butyryl chloride (0.20 mL, 2.10 mmol) at 0 °C. The reaction mixture was stirred for 24 hrs at r.t. The solvent was removed and the residue was purified by silica gel chromatography eluting with hexane/ EtOAc (1/2) to give ester 16a (170.0 mg, 43%) as colorless oil; 1H NMR (400 MHz, CDCl3) δ: 7.05 (m, 1H), 6.04 (m, 1H), 5.27 (m, 1H), 5.10 (m, 1H), 4.07 (s, 1H), 3.91 (m, 4H), 3.68 (d, 1H, J = 12.0 Hz), 3.54 (m, 2H), 3.28 (m, 3H), 2.58 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.44 (m, 3H), 2.24 (t, 2H, J = 8.0 Hz), 1.98 (dd, 1H, J = 12.0 Hz, 8.0 Hz), 1.72 (dd, 1H, J = 12.0 Hz, 4.0 Hz), 1.64 (m, 6H), 1.46 (s, 3H), 1.42 (s, 3H), 1.29 (d, 3H, J = 8.0 Hz), 1.22 (d, 3H, J = 4.0 Hz), 1.04 (s, 3H), 0.97 (s, 3H), 0.93 (t, 3H, J = 6.0 Hz); 13C NMR (400 MHz, CDCl3) δ: 172.9, 171.0, 170.1, 169.7, 110.0, 99.1, 77.2, 71.5, 67.7, 67.2, 64.9, 64.8, 42.6, 41.4, 39.6, 36.4, 36.1, 35.0, 34.7, 33.0, 29.5, 23.8, 22.2, 21.3, 20.0, 19.0, 18.8, 18.5, 13.7.
(R)-((R)-4-oxo-4-((R)-1-(2-(3-(3-((R)-2,2,5,5-tetramethyl-1,3-dioxane-4 carboxamido)propanamido)propyl)-1,3-dioxolan-2-yl)propan-2-yloxy)butan-2-yl) 3-(butyryloxy)butanoate 16b
To a mixture of compound 15 (0.14 g, 0.80 mmol) and (COCl)2 (0.10 mL, 1.60 mmol) in CH2Cl2 (5.00 mL) was added one drop of DMF. The resulting mixture was stirred for 2 hrs. and concentrated in vacuum under argon. The residue was re-dissolved in CH2Cl2 (5.00 mL) and transferred to a solution consisting of 14 (0.30 g, 0.50 mmol) and pyridine (0.10 mL, 1.60 mmol) in CH2Cl2 (5.00 mL). After stirring for 3 hrs., the reaction mixture was evaporated to dryness under vacuum and the residue was purified by silica gel chromatography eluting with hexane/EtOAc (1/2) to give 16b (0.30 g, 83%) as colorless oil; 1H NMR (400 MHz, CDCl3) δ: 7.04 (m, 1H), 6.22 (m, 1H), 5.22 (m, 2H), 5.04 (m, 1H), 4.01 (s, 1H), 3.85 (m, 4H), 3.62 (d, 1H, J = 12.0 Hz), 3.48 (m, 2H), 3.20 (m, 3H), 2.55 (dd, 1H, J = 12.0 Hz, 8.0 Hz), 2.40 (m, 4H), 2.18 (t, 2H, J = 8.0 Hz), 1.93 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 1.72 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 1.58 (m, 6H), 1.40 (s, 3H), 1.36 (s, 3H), 1.22 (m, 6H), 1.16 (d, 3H, J = 4.0 Hz), 0.97 (s, 3H), 0.91 (s, 3H), 0.88 (t, 3H, J = 8.0 Hz); 13C NMR (400 MHz, CDCl3) δ: 172.8, 171.0, 170.0, 169.5, 169.4, 109.9, 99.1, 77.2, 71.5, 67.7, 67.0, 64.8, 42.5, 41.1, 41.0, 39.5, 36.3, 36.0, 34.9, 34.7, 33.0, 29.5, 23.9, 22.2, 21.3, 19.9, 18.9, 18.7, 18.4, 13.7.
(R)-((R)-7-(3-((R)-2,4-dihydroxy-3,3-dimethylbutanamido)propanamido)-4-oxoheptan-2-yl) 3-(butyry loxy)butanoate 17a
A mixture of compound 16a (0.14 g, 0.20 mmol) in CH3CN (7.00 mL) and 1N HCl (7.00 mL) was stirred for 2.5 hrs. at r.t. The reaction mixture was then extracted with EtOAc (15.0 mL × 2). The organic extracts were combined, washed sequentially with saturated aqueous NaHCO3 and brine, and dried with Na2SO4. The solvent was removed and the residue was purified by silica gel chromatography eluting with CH2Cl2/MeOH (20/1) to give 17a (0.12 g, 96%) as pale yellow oil; 1H NMR (400MHz, CDCl3) δ: 7.44 (m, 1H), 6.58 (m, 1H), 5.23 (m, 2H), 4.42 (br, 1H, OH), 3.98 (d, 1H, J = 4.0 Hz), 3.84 (br, 1H, OH), 3.54 (quart, 2H, J = 8.0 Hz), 3.46 (s, 2H), 3.21 (m, 2H), 2.75 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.45 (m, 7H), 2.23 (t, 2H, J = 8.0 Hz), 1.74 (quint, 2H, J = 7.0 Hz), 1.61 (sext, 2H, J = 8.0 Hz), 1.24 (m, 6H), 0.98 (s, 3H), 0.92 (t, 3H, J = 8.0 Hz), 0.90 (s, 3H); 13C NMR (400 MHz, CDCl3) δ: 207.6, 173.9, 173.2, 171.8, 169.8, 77.7, 71.0, 67.6, 67.2, 48.7, 41.3, 40.5, 39.5, 39.0, 36.5, 36.0, 23.3, 21.6, 20.6, 20.2, 20.0, 18.5, 13.8.
(R)-((R)-7-(3-((R)-2,4-dihydroxy-3,3-dimethylbutanamido)propanamido)-4-oxoheptan-2-yl) 3-((R)-3-(butyryloxy)butanoyloxy)butanoate 17b
The procedures and reaction scale were the same as the synthesis of compound 17a described above. Compound 17b (0.12 g, 83%) was obtained as colorless oil; 1H NMR (400 MHz, CDCl3) δ: 7.36 (m, 1H), 6.29 (m, 1H), 5.25 (m, 3H), 3.99 (s, 1H), 3.58 (m, 2H), 3.49 (s, 2H), 3.23 (m, 2H), 2.76 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.57 (m, 2H), 2.47 (m, 7H), 2.24 (t, 2H, J = 4.0 Hz), 1.77 (m, 2H), 1.63 (sext, 2H, J = 8.0 Hz), 1.27 (m, 9H), 1.03 (s, 3H), 0.93 (m, 6H); 13C NMR (400 MHz, CDCl3) δ: 207.4, 173.9, 173.0, 171.8, 169.6, 77.5, 70.9, 67.7, 67.6, 67.1, 48.6, 41.1, 40.5, 39.4, 38.9, 36.4, 23.2, 21.4, 20.5, 20.1, 20.0, 19.9, 19.8, 18.5, 13.7.
Enzymatic synthesis and HPLC purification
A 2-mL reaction mixture contained enzymatic precursor (20.0 mM), ATP (50.0 mM), MgCl2 (10.0 mM), SaCoaA (80.0 μg), EcCoaD (80.0 μg) and EcCoaE (80.0 μg) in 100 mM Tris-HCl (pH 7.60). The reaction was initiated by addition of the enzymes and incubated at 25 °C for 3 h. The reaction was stopped by heating the mixture in a 95 °C water bath for 5 min, and the precipitated protein was removed by centrifugation (14,000 rpm × 5 min). The supernatant was loaded onto a semi-preparative HPLC column (Luna C18-2, 5 μm, 10 mm × 250 mm) that was eluted at 3.00 mL/min using a linear gradient from 10 to 90% methanol in 10.0 mM ammonium acetate (pH 4.00) over 60 min. The fractions containing the product were pooled, concentrated in vacuo, and lyophilized to give a white powder.
Carbadethia CoA analog 26a
20.0 mg, 50% yield; HPLC: t = 29 min; 1H NMR (400MHz, D2O) δ: 8.57 (s, 1H), 8.29 (s, 1H), 6.20 (d, 1H, J = 4.0 Hz), 5.26 (m, 2H), 4.88 (s, 1H), 4.62 (s, 1H), 4.28 (s, 2H), 4.04 (s, 1H), 3.86 (m, 1H), 3.60 (m, 1H), 3.49 (m, 2H), 3.14 (t, 2H, J = 8.0 Hz), 2.89 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.79 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 2.67 (m, 2H), 2.58 (m, 2H), 2.48 (t, 2H, J = 6.0 Hz), 2.33 (t, 2H, J = 8.0 Hz), 1.72 (quint, 2H, J = 8.0 Hz), 1.60 (sext, 2H, J = 8.0 Hz), 1.28 (m, 6H), 0.91 (m, 6H), 0.80 (s, 3H); 13C NMR (101 MHz, D2O) δ: 212.9, 176.3, 174.8, 173.8, 172.4, 155.0, 152.1, 149.3, 140.3, 118.7, 86.8, 83.5, 74.3, 73.9, 72.0, 68.7, 68.5, 65.5, 47.9, 40.8, 40.3, 38.7, 36.2, 35.7, 22.7, 21.0, 19.2, 18.4, 18.2, 13.1; 31P NMR (161 MHz, D2O) δ: 0.45 (s, 1P), −10.73 (d, 1P, J = 19.3 Hz), −11.23 (d, 1P, J = 19.3 Hz); HRMS: calc. for C34H55N7O21P3− [M−H]−: 990.2664, found: 990.2701.
Carbadethia CoA analog 26b
26.0 mg, 60% yield; HPLC: t = 34 min; 1H NMR (400 MHz, D2O) δ: 8.56 (s, 1H), 8.28 (s, 1H), 6.19 (d, 1H, J = 8.0 Hz), 5.26 (m, 2H), 4.87 (s, 1H), 4.62 (s, 1H), 4.28 (s, 2H), 4.04 (s, 1H), 3.86 (m, 1H), 3.60 (m, 1H), 3.49 (m, 2H), 3.14 (t, 2H, J = 8.0 Hz), 2.89 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.80 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 2.63 (m, 6H), 2.48 (t, 2H, J = 6.0 Hz), 2.33 (t, 2H, J = 8.0 Hz), 1.71 (quint, 2H, J = 8.0 Hz), 1.60 (sext, 2H, J = 8.0 Hz), 1.26 (m, 9H), 0.92 (m, 6H), 0.79 (s, 3H); 13C NMR (400 MHz, D2O) δ: 212.7, 176.2, 174.8, 173.8, 172.3, 155.3, 152.4, 149.4, 140.2, 118.7, 86.7, 83.5, 74.3, 74.0, 69.0, 68.7, 68.4, 65.5, 47.9, 40.7, 40.2, 38.8, 36.3, 35.6, 22.7, 21.0, 19.2, 19.1, 18.4, 18.2, 13.1; 31P NMR (161 MHz, D2O) δ: 0.21 (s, 1P), −10.71 (d, 1P, J = 17.7 Hz), −11.19 (d, 1P, J = 17.7 Hz); HRMS: calc. for C38H61N7O23P3− [M−H]−: 1076.3032, found: 1076.3013.
For intermediates of enzymatic conversions, they were separated by RP-HPLC in the same manner as the carbadethia CoA analogs 26a/b described above.
Phosphopantetheine derivative 24a
HPLC: t = 39 min; 1H NMR (400MHz, D2O) δ: 5.30 (m, 2H), 4.07 (s, 1H), 3.82 (dd, 1H, J = 12.0 Hz, 8.0 Hz), 3.60 (dd, 1H, J = 12.0 Hz, 4.0 Hz), 3.54 (m, 2H), 3.19 (t, 2H, J = 8.0 Hz), 2.90 (m, 2H), 2.70 (m, 3H), 2.52 (t, 2H, J = 8.0 Hz), 2.37 (t, 2H, J = 8.0 Hz), 1.76 (m, 2H), 1.63 (q, 2H, J = 8.0 Hz), 1.30 (m, 6H), 1.00 (s, 3H), 0.92 (m, 6H); 13CNMR (101 MHz, D2O) δ: 213.1, 176.7, 176.3, 175.0, 173.9, 172.5, 74.7, 71.2, 70.1, 68.8, 68.5, 47.9, 40.8, 40.3, 38.8, 36.3, 35.7, 35.6, 22.7, 21.0, 19.4, 19.2, 18.8, 18.2, 13.1, 13.0; 31PNMR (161 MHz, D2O) δ: 0.86 (s, 1P); HRMS: calc. for C24H42N2O12P− [M−H]−: 581.2481, found: 581.2507.
Phosphopantetheine derivative 24b
HPLC: t = 42 min; 1H NMR (400MHz, D2O) δ: 5.30 (m, 2H), 4.07 (s, 1H), 3.80 (dd, 1H, J = 12.0 Hz, 8.0 Hz), 3.53 (m, 3H), 3.18 (t, 2H, J = 8.0 Hz), 2.90 (m, 2H), 2.68 (m, 6H), 2.51(m, 2H), 2.36 (t, 2H, J = 8.0 Hz), 1.75 (m, 2H), 1.63 (q, 2H, J = 8.0 Hz), 1.29 (m, 9H), 1.00 (s, 3H), 0.92 (m, 6H); 13CNMR (101 MHz, D2O) δ: 212.9, 176.3, 175.0, 173.9, 172.3, 74.7, 71.1, 69.1, 68.7, 68.4, 47.9, 4.7, 40.2, 38.7, 36.3, 35.6, 22.7, 21.0, 19.2, 19.0, 18.7, 18.2, 13.0; 31PNMR (161 MHz, D2O) δ: 1.07 (s, 1P); HRMS: calc. for C28H48N2O14P− [M−H]−: 667.2849, found: 667.2830.
3′-dephospho-CoA analog 25a
HPLC: t = 37 min; 1H NMR (400MHz, D2O) δ: 8.54 (s, 1H), 8.29 (s, 1H), 6.16 (d, 1H, J = 4.0 Hz), 5.25 (m, 2H), 4.74 (s, 1H), 4.55 (t, 2H, J = 4.0 Hz), 4.41 (s, 1H), 4.25 (s, 2H), 4.03 (s, 1H), 3.86 (m, 1H), 3.59 (m, 1H), 3.48 (m, 2H), 3.13 (t, 2H, J = 8.0 Hz), 2.88 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.78 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 2.65 (m, 2H), 2.56 (m, 2H), 2.47 (t, 2H, J = 6.0 Hz), 2.33 (t, 2H, J = 6.0 Hz), 1.70 (m, 2H), 1.60 (m, 2H), 1.26 (m, 6H), 0.93 (m, 6H), 0.92 (s, 3H); 13CNMR (101 MHz, D2O) δ: 212.8, 176.2, 174.8, 173.8, 172.4, 155.1, 152.0, 140.1, 87.2, 84.0, 74.4, 74.2, 72.0, 70.4, 68.6, 65.3, 47.8, 40.7, 40.1, 38.6, 38.4, 36.3, 36.1, 35.6, 35.5, 22.6, 20.9, 19.2, 19.0, 18.3, 18.1, 13.0; 31PNMR (161 MHz, D2O) δ: −9.81(d, 1P, J = 21.7 Hz), −10.23 (d, 1P, J = 21.7 Hz); HRMS: calc. for C34H54N7O18P2− [M−H]−: 910.3006, found: 910.2994.
3′-dephospho-CoA analog 25b
HPLC: t = 39 min; 1H NMR (400MHz, D2O) δ: 8.60 (s, 1H), 8.36 (s, 1H), 6.18 (d, 1H, J = 4.0 Hz), 5.27 (m, 2H), 4.77 (s, 1H), 4.56 (t, 2H, J = 4.0 Hz), 4.43 (s, 1H), 4.27 (s, 2H), 4.05 (s, 1H), 3.88 (m, 1H), 3.61 (m, 1H), 3.49 (m, 2H), 3.15 (t, 2H, J = 8.0 Hz), 2.90 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.80 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 2.62 (m, 6H), 2.58 (m, 2H), 2.47 (t, 2H, J = 6.0 Hz), 2.33 (t, 2H, J = 6.0 Hz), 1.72 (m, 2H), 1.61 (m, 2H), 1.30 (m, 9H), 0.93 (m, 6H), 0.82 (s, 3H); 13CNMR (101 MHz, D2O) δ: 212.8, 176.4, 176.3, 174.8, 173.9, 172.5, 172.8, 153.5, 149.7, 141.1, 87.6, 84.2, 74.7, 70.5, 69.0, 68.6, 68.4, 47.9, 40.9, 40.7, 40.2, 38.7, 36.2, 35.7, 35.6, 22.7, 21.0, 19.2, 19.1, 19.0, 18.4, 18.2, 13.0; 31PNMR (161 MHz, D2O) δ: −9.81(d, 1P, J = 19.3 Hz), −10.05 (d, 1P, J = 19.3 Hz); HRMS: calc. for C38H60N7O20P2−[M−H]−: 996.3374, found: 996.3412.
Chemical synthesis of sT-aldehyde 29
sT-alcohol 28
To a solution of sT-acid 27 (0.20 g, 0.80 mmol) in THF (2.0 mL) was added BH3:Me2S (2.0 M in THF, 0.80 mL, 1.6 mmol) at 0 °C. After stirring for 4 hrs at r.t., MeOH (2.0 mL) was added to the reaction mixture and followed by extraction with EtOAc (10.0 mL). The organic layer was washed with brine, dried with Na2SO4, and concentrated to dryness. The residue was purified by silica gel chromatography eluting with CH2Cl2/MeOH (100/1 to 20/1) to give 3 as colorless oil (0.20 g, 95%); 1H NMR (400 MHz, CDCl3) δ: 5.28 (m, 1H), 5.12 (m, 1H), 3.58 (m, 2H), 2.61 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.50 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 2.28 (br, 1H, OH), 2.23 (t, 2H, J = 8.0 Hz), 1.74 (m, 2H), 1.62 (sext, 2H, J = 8.0 Hz), 1.28 (d, 3H, J = 8.0 Hz), 1.25 (d, 3H, J = 8.0 Hz), 0.92 (t, 3H, J = 8.0 Hz); 13C NMR (400 MHz, CDCl3) δ: 173.0, 170.8, 68.7, 67.2, 58.8, 41.3, 39.1, 36.4, 20.5, 20.1, 18.5, 13.7.
sT-aldehyde 29
A solution of DMSO (0.10 mL, 1.60 mmol) in dry CH2Cl2 (1.00 mL) was cooled to −78 °C, to which oxalyl chloride (0.10 g, 0.80 mmol) was added slowly. After stirring for 1 h, sT-alcohol 28 (50.0 mg, 0.20 mmol) was added to the reaction mixture followed by addition of Et3N (0.30 mL, 2.00 mmol). After stirring for additional 1 h, the reaction mixture was diluted with EtOAc (5.00 mL). The organic layer was washed with water (1.00 mL × 2), dried with Na2SO4, and concentrated to dryness. The residue was purified by silica gel chromatography eluting with hexane/EtOAc (5/1 to 5/1) to give 29 as colorless oil (35.0 mg, 70%); 1H NMR (400 MHz, CDCl3) δ: 5.38 (m, 1H), 5.25 (m, 1H), 2.74 (dd, 1H, J = 16.0 Hz, 8.0 Hz), 2.61 (m, 2H), 2.47 (dd, 1H, J = 16.0 Hz, 4.0 Hz), 2.24 (t, 2H, J = 8.0 Hz), 1.63 (sext, 2H, J = 8.0 Hz), 1.31 (d, 2H, J = 8.0 Hz), 1.28 (d, 2H, J = 8.0 Hz), 0.94 (t, 3H, J = 8.0 Hz); 13C NMR (400 MHz, CDCl3) δ: 199.3, 173.0, 169.7, 67.1, 66.3, 49.6, 41.2, 36.4, 20.2, 20.1, 18.6, 13.8; HRMS: calc. for C12H21O5+ [M+H]+: 245.1384, found: 245.1396.
Inhibition study with PhaECAv
A continuous assay was carried out at 30 °C in a final volume of 160 μL consisting of 100 mM KPi (pH 7.80), 0.30 mM DTNB, 2 mg/mL BSA, 5.80 nM wt-PhaECAv, HBCoA (0.025 to 1.60 mM) and inhibitor at different concentrations. Formation of 3-thio-6-nitrobenzoate dianion was monitored by the absorbance at 412 nm and quantified using an extinct coefficient of 13.7 mM−1cm−1. The rates of reactions were determined by the slope of the initial fast phase. Each data point was done in duplicate. The data were analyzed by SigmaPlot and fitted to Michaelis-Menten equation for different inhibition modes.
Homology modelling of PhaECAv and docking study
For the construction of homology model and docking study, three online servers were used that included http://www.cbs.dtu.dk/services/CPHmodels, http://swissmodel.expasy.org, and http://zhanglab.ccmb.med.umich.edu/I-TASSER. The model with the highest C-score was selected if multiple models were generated. CPHmodels3.0 used human gastric lipase 1HLG as a template while Swiss model chose esterase 2RAU. The I-TASSER made a hybrid model based on lipases (1K8Q and 1HLG), esterase 2RAU, and hydrolase 3OM8. Docking was carried out with the I-TASSER model using AutoDock Vina. A search space of 32 × 28 × 28 Å, spanning the enzyme's active site was used. All other parameters were set to default values. The docking algorithm resulted in multiple binding modes, of which the one with the lowest binding energy (ΔGb) is shown in Figure 4. Figures were prepared using PyMOL software (www.pymol.org).
Supplementary Material
Acknowledgements
This project was supported by startup fund from Kansas State University and awards from Terry C. Johnson Center for Basic Cancer Research. W. Z. was partially supported by a postdoctoral fellowship under the Kansas INBRE, P20 GM103418. We thank Prof. C.B. Aakeröy for helpful discussion and reading the manuscript.
References
- 1.a Steinbuchel A, Hein S. Adv. Biochem. Eng./Biotechnol. 2001;71:81–123. doi: 10.1007/3-540-40021-4_3. [DOI] [PubMed] [Google Scholar]; b Stubbe J, Tian J. Nat. Prod. Rep. 2003;20:445–457. doi: 10.1039/b209687k. [DOI] [PubMed] [Google Scholar]
- 2.Steinbuchel A, Lutke-Eversloh T. Biochem. Eng. J. 2003;16:81–96. [Google Scholar]
- 3.Jendrossek D, Handrick R. Annu. Rev. Microbiol. 2002;56:403–432. doi: 10.1146/annurev.micro.56.012302.160838. [DOI] [PubMed] [Google Scholar]
- 4.Chen GQ. Plastics from bacteria: natural functions and applications. Springer; Heidelberg, New York: 2010. [Google Scholar]
- 5.Wu Q, Wang Y, Chen GQ. Artif. Cell. Blood Sub. 2009;37:1–12. doi: 10.1080/10731190802664429. [DOI] [PubMed] [Google Scholar]
- 6.Kunasundari B, Sudesh K. Express Polym. Lett. 2011;5:620–634. [Google Scholar]
- 7.Rehm BHA, Steinbuchel A. Int. J. Biol. Macromol. 1999;25:3–19. doi: 10.1016/s0141-8130(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 8.Gerngross TU, Snell KD, Peoples OP, Sinskey AJ, Csuhai E, Masamune S, Stubbe J. Biochemistry. 1994;33:9311–9320. doi: 10.1021/bi00197a035. [DOI] [PubMed] [Google Scholar]
- 9.a Liebergesell M, Steinbuchel A. Eur. J. Biochem. 1992;209:135–150. doi: 10.1111/j.1432-1033.1992.tb17270.x. [DOI] [PubMed] [Google Scholar]; b Muh U, Sinskey AJ, Kirby DP, Lane WS, Stubbe J. Biochemistry. 1999;38:826–837. doi: 10.1021/bi9818319. [DOI] [PubMed] [Google Scholar]
- 10.a Chuah JA, Tomizawa S, Yamada M, Tsuge T, Doi Y, Sudesh K, Numata K. Appl. Environ. Microbiol. 2013;79:3813–3821. doi: 10.1128/AEM.00564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nomura CT, Taguchi S. Appl. Microbiol. Biotechnol. 2007;73:969–979. doi: 10.1007/s00253-006-0566-4. [DOI] [PubMed] [Google Scholar]; c Sim SJ, Snell KD, Hogan SA, Stubbe J, Rha CK, Sinskey AJ. Nat. Biotechnol. 1997;15:63–67. doi: 10.1038/nbt0197-63. [DOI] [PubMed] [Google Scholar]; d Takase K, Matsumoto K, Taguchi S, Doi Y. Biomacromolecules. 2004;5:480–485. doi: 10.1021/bm034323+. [DOI] [PubMed] [Google Scholar]; e Tsuge T, Saito Y, Narike M, Muneta K, Normi YM, Kikkawa Y, Hiraishi T, Doi Y. Macromol. Biosci. 2004;4:963–970. doi: 10.1002/mabi.200400075. [DOI] [PubMed] [Google Scholar]; f Tsuge T, Watanabe S, Shimada D, Abe H, Doi Y, Taguchi S. FEMS Microbiol. Lett. 2007;277:217–222. doi: 10.1111/j.1574-6968.2007.00958.x. [DOI] [PubMed] [Google Scholar]
- 11.Jia Y, Yuan W, Wodzinska J, Park C, Sinskey AJ, Stubbe J. Biochemistry. 2001;40:1011–1019. doi: 10.1021/bi002219w. [DOI] [PubMed] [Google Scholar]
- 12.Wodzinska J, Snell KD, Rhomberg A, Sinskey AJ, Biemann K, Stubbe J. J.Am.Chem.Soc. 1996;118:6319–6320. [Google Scholar]
- 13.Tian JM, Sinskey AJ, Stubbe J. Biochemistry. 2005;44:8369–8377. doi: 10.1021/bi050331u. [DOI] [PubMed] [Google Scholar]
- 14.Hassell AM, An G, Bledsoe RK, Bynum JM, Carter HL, Deng SJJ, Gampe RT, Grisard TE, Madauss KP, Nolte RT, Rocque WJ, Wang LP, Weaver KL, Williams SP, Wisely GB, Xu R, Shewchuk LM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007;63:72–79. doi: 10.1107/S0907444906047020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danley DE. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006;62:569–575. doi: 10.1107/S0907444906012601. [DOI] [PubMed] [Google Scholar]
- 16.Tinberg CE, Khare SD, Dou JY, Doyle L, Nelson JW, Schena A, Jankowski W, Kalodimos CG, Johnsson K, Stoddard BL, Baker D. Nature. 2013;501:212–216. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a Dewick PM. Medicinal Natural Products : A Biosynthetic Approach. 3rd ed. Wiley; Hoboken: 2008. [Google Scholar]; b Engel C, Wierenga R. Curr. Opin. Struc. Biol. 1996;6:790–797. doi: 10.1016/s0959-440x(96)80009-1. [DOI] [PubMed] [Google Scholar]; c Knudsen J, Jensen MV, Hansen JK, Faergeman NJ, Neergaard TBF, Gaigg B. Mol. Cell. Biochem. 1999;192:95–103. doi: 10.1007/978-1-4615-4929-1_11. [DOI] [PubMed] [Google Scholar]; d Mishra PK, Drueckhammer DG. Chem. Rev. 2000;100:3283–3309. doi: 10.1021/cr990010m. [DOI] [PubMed] [Google Scholar]
- 18.a Triola G, Waldmann H, Hedberg C. ACS Chem. Biol. 2012;7:87–99. doi: 10.1021/cb200460u. [DOI] [PubMed] [Google Scholar]; b Brownell JE, Allis CD. Curr. Opin. Genet. Dev. 1996;6:176–184. doi: 10.1016/s0959-437x(96)80048-7. [DOI] [PubMed] [Google Scholar]
- 19.Lipmann F, Kaplan NO. J. Biol. Chem. 1946;162:743–744. [Google Scholar]
- 20.a La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Chem. Biol. 2004;11:195–201. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]; b Gehring AM, Lambalot RH, Vogel KW, Drueckhammer DG, Walsh CT. Chem. Biol. 1997;4:17–24. doi: 10.1016/s1074-5521(97)90233-7. [DOI] [PubMed] [Google Scholar]; c Dai M, Feng YG, Tonge PJ. J. Am. Chem. Soc. 2001;123:506–507. doi: 10.1021/ja003406k. [DOI] [PubMed] [Google Scholar]; d Strauss E, Begley TP. ChemBioChem. 2005;6:284–286. doi: 10.1002/cbic.200400340. [DOI] [PubMed] [Google Scholar]
- 21.a Tosin M, Spiteller D, Spencer JB. ChemBioChem. 2009;10:1714–1723. doi: 10.1002/cbic.200900093. [DOI] [PubMed] [Google Scholar]; b Tautz L, Retey J. Eur. J. Org. Chem. 2010:1728–1735. doi: 10.1002/ejoc.200901410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paterson I, Coster MJ, Chen DYK, Gibson KR, Wallace DJ. Org. Biomol. Chem. 2005;3:2410–2419. doi: 10.1039/b504148a. [DOI] [PubMed] [Google Scholar]
- 23.Cahiez G, Alexakis A, Normant JF. Tetrahedron Lett. 1978:3013–3014. [Google Scholar]
- 24.Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Chem. Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 25.Li P, Chakraborty S, Stubbe J. Biochemistry. 2009;48:9202–9211. doi: 10.1021/bi901329b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- 27.Seebach D, Brandli U, Schnurrenberger P, Przybylski M. Helv. Chim. Acta. 1988;71:155–167. [Google Scholar]
- 28.Clarke KM, Mercer AC, La Clair JJ, Burkart MD. J. Am.Chem.Soc. 2005;127:11234–11235. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]
- 29.Leonardi R, Chohnan S, Zhang YM, Virga KG, Lee RE, Rock CO, Jackowski S. J.Biol.Chem. 2005;280:3314–3322. doi: 10.1074/jbc.M411608200. [DOI] [PubMed] [Google Scholar]
- 30.Geerlof A, Lewendon A, Shaw WV. J. Biol.Chem. 1999;274:27105–27111. doi: 10.1074/jbc.274.38.27105. [DOI] [PubMed] [Google Scholar]
- 31.Mishra PK, Park PK, Drueckhammer DG. J. Bacteriol. 2001;183:2774–2778. doi: 10.1128/JB.183.9.2774-2778.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a Meier JL, Mercer AC, Rivera H, Burkart MD. J. Am. Chem. Soc. 2006;128:12174–12184. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]; b Strauss E, Begley TP. J. Biol. Chem. 2002;277:48205–48209. doi: 10.1074/jbc.M204560200. [DOI] [PubMed] [Google Scholar]; c van der Westhuyzen R, Strauss E. J. Am. Chem. Soc. 2010;132:12853–12855. doi: 10.1021/ja106204m. [DOI] [PubMed] [Google Scholar]; d Strauss E, de Villiers M, Rootman I. Chemcatchem. 2010;2:929–937. [Google Scholar]
- 33.a Pietsch M, Chua KCH, Abell AD. Curr. Top. Med. Chem. 2010;10:270–293. doi: 10.2174/156802610790725489. [DOI] [PubMed] [Google Scholar]; b Wang QM, Chen SH. Curr. Protein Pept. Sci. 2007;8:19–27. doi: 10.2174/138920307779941523. [DOI] [PubMed] [Google Scholar]
- 34.Mancuso AJ, Huang SL, Swern D. J. Org. Chem. 1978;43:2480–2482. [Google Scholar]
- 35.Jia Y, Kappock TJ, Frick T, Sinskey AJ, Stubbe J. Biochemistry. 2000;39:3927–3936. doi: 10.1021/bi9928086. [DOI] [PubMed] [Google Scholar]
- 36.Nielsen M, Lundegaard C, Lund O, Petersen TN. Nucleic Acids Res. 2010;38:W576–W581. doi: 10.1093/nar/gkq535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roussel A, Canaan S, Egloff MP, Riviere M, Dupuis L, Verger R, Cambillau C. J. Biol. Chem. 1999;274:16995–17002. doi: 10.1074/jbc.274.24.16995. [DOI] [PubMed] [Google Scholar]
- 40.She Q, Singh RK, Confalonieri F, Zivanovic Y, Allard G, Awayez MJ, Chan-Weiher CCY, Clausen IG, Curtis BA, De Moors A, Erauso G, Fletcher C, Gordon PMK, Heikamp-de Jong I, Jeffries AC, Kozera CJ, Medina N, Peng X, Thi-Ngoc HP, Redder P, Schenk ME, Theriault C, Tolstrup N, Charlebois RL, Doolittle WF, Duguet M, Gaasterland T, Garrett RA, Ragan MA, Sensen CW, Van der Oost J. Proc. Natl. Acad. Sci. U. S. A. 2001;98:7835–7840. doi: 10.1073/pnas.141222098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roussel A, Miled N, Berti-Dupuis L, Riviere M, Spinelli S, Berna P, Gruber V, Verger R, Cambillau C. J. Biol. Chem. 2002;277:2266–2274. doi: 10.1074/jbc.M109484200. [DOI] [PubMed] [Google Scholar]
- 42.a Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trott O, Olson AJ. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ushimaru K, Sangiambut S, Thomson N, Sivaniah E, Tsuge T. Appl. Microbiol. Biotechnol. 2013;97:1175–1182. doi: 10.1007/s00253-012-4089-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.