

Abstract
S. marcescens FS14 was isolated from an Atractylodes macrocephala Koidz plant that was infected by Fusarium oxysporum and showed symptoms of root rot. With the completion of the genome sequence of FS14, the first comprehensive comparative-genomic analysis of the Serratia genus was performed. Pan-genome and COG analyses showed that the majority of the conserved core genes are involved in basic cellular functions, while genomic factors such as prophages contribute considerably to genome diversity. Additionally, a Type I restriction-modification system, a Type III secretion system and tellurium resistance genes are found in only some Serratia species. Comparative analysis further identified that S. marcescens FS14 possesses multiple mechanisms for antagonism against other microorganisms, including the production of prodigiosin, bacteriocins, and multi-antibiotic resistant determinants as well as chitinases. The presence of two evolutionarily distinct Type VI secretion systems (T6SSs) in FS14 may provide further competitive advantages for FS14 against other microbes. To our knowledge, this is the first report of comparative analysis on T6SSs in the genus, which identifies four types of T6SSs in Serratia spp.. Competition bioassays of FS14 against the vital plant pathogenic bacterium Ralstonia solanacearum and fungi Fusarium oxysporum and Sclerotinia sclerotiorum were performed to support our genomic analyses, in which FS14 demonstrated high antagonistic activities against both bacterial and fungal phytopathogens.
Introduction
The bacterial strain FS14 was isolated from an Atractylodes macrocephala Koidz plant infected by the pathogen Fusarium oxysporum, which is a causative agent of the root-rot disease [1]. The isolated red-pigmented bacteria were found to secret thermostable DNase and protease in our previous study [1]. With the combined use of morphological, biochemical and genetic characterization via 16S rDNA sequencing, the isolate was classified into the genus Serratia and designated as strain FS14. As the strain was found along with phytopathogens in diseased plants, we hypothesized that it has antagonistic potential against the pathogens.
Serratia is a genus of Gram-negative, facultatively anaerobic, rod-shaped bacteria of the Enterobacteriaceae family. They are ubiquitous organisms with diverse habitats including water, soil, plants, small mammals, and humans. The typical species of the genus is Serratia marcescens, well known as an opportunistic human pathogen involved in hospital acquired infections [2]. Serratia species with similar sites of isolation to strain FS14 include the endophytic S. proteamaculans found to promote plant growth via the production of specific compounds such as lipo-chitin oligosaccharides [3] and S. plymuthica which is considered a plant growth promoting rhizobacteria [4] and an antagonistic bacteria against phytopathogens since they produce a wide range of antimicrobial compounds [5].
Antagonistic bacteria exploit diverse strategies to outperform their competitors. The most characterized antimicrobial compound in Serratia is the red pigmented prodiginine, of which five types have been identified so far (prodigiosin, undecylprodigiosin, cycloprodigiosin, cyclononylprodigiosin, and butyl-meta-cyclo-heptylprodiginine) [6]. Prodigiosin is commonly produced by environmental isolates of S. marcescens, but not the clinical isolates [7]. In addition to its antibacterial, antifungal and antiprotozoal properties, prodiginine was recently reported to exhibit immunosuppressive and anticancer traits [8].
Another central strategy employed is manifested via the protein secretion systems, through which Gram-negative bacteria secrete effectors to their exterior [9]. In particular, the Type VI secretion system (T6SS), the most recently described of the six, has highly versatile functions, which include eukaryotic and bacterial cell targeting, gene regulation, conjugation and cellular adhesion [10]. Recent studies demonstrated that T6SSs in S. marcescens and other bacteria, e.g. Vibrio cholerae, Burkholderia thailandensis [11, 12], can target other bacterial competitors resulting in either growth inhibition or death.
Compared with Escherichia and Salmonella, which are also members of the Enterobacteriaceae family, there are relatively fewer—only 13—genomes of Serratia sequenced and reported to NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/): S. marcescens WW4 isolated from a paper machine [13], S. marcescens Db11 pathogen of drosophila, S. marcescens FGI 94 associated with leaf-cutter ant fungus garden [14], S. proteamaculans 568 with a detailed genome analysis on chitinase production [3], S. liquefaciens ATCC27592, S. fonticola RB-25 isolated from a waste landfill [15], three plant associated S. plymuthica strains AS13 [16], AS12, AS9, S13 [17] and 4Rx13 that exhibit plant-growth-promoting activities and Serratia symbiotica Cinara cedri uid82363 which coexists with B. aphidicola in aphid.
In this study, we present the first comprehensive comparative-genomic analysis of the genus Serratia, which helps to identify the antimicrobial compounds they secrete and other antagonistic genomic elements they harbor, e.g. Type VI secretion system, antibiotic-resistant genes and chitinases. In addition, we explore the antagonistic potential of strain FS14 against common fungal and bacterial phytopathogens.
Materials and Methods
Isolation and Total DNA extraction of Serratia marcescens FS14
Serratia marcescens FS14 was isolated in May 2009 from an Atractylodes macrocephala Koidz plant infected by Fusarium oxysporum in Jiangsu Province, China. A single isolate was grown in LB media and incubated at 28°C until they reached the exponential growth phase. The bacteria were then harvested by centrifugation and their genomic DNA was extracted according to the JGI bacterial DNA isolation CTAB protocol (http://my.jgi.doe.gov/general/protocols/JGI-Bacterial-DNA-isolation-CTAB-Protocol-2012.pdf).
Antibiotic-sensitivity tests
Antibiotic-sensitivity tests were performed by spreading bacterial suspensions on culture plates [18] and applying discs impregnated with different antibiotics (Hangzhoutianhe, China) including gentamycin (10 μg), penicillin G (10 IU), tetracycline (30 μg), rifampicin (5 μg), novobiocin (30 μg), neomycin (30 μg), spectinomycin (100 μg), ampicillin (10 μg), amoxicillin (10 μg), acetylspiramycin (30 μg), chloramphenicol (30 μg), erythromycin (15 μg), kanamycin (30 μg) and polymyxin B sulfate (300 μg). For all the above tests, each plate was incubated at 28°C for 24 hours under aerobic condition and the zones of inhibition were interpreted according to the manufacturer's instructions. All assays were performed in triplicate.
Pyrosequencing and complete genome assembly of S. marcescens FS14
To confirm the purity of S. marcescens FS14 genomic DNA, the 16S rDNA-specific region was amplified and 20 individual positive clones were sequenced by Genetic Analyzer 3130 (Invitrogen, Grand Island, US). BLASTn analysis [19] revealed that the S. marcescens FS14 16S rDNA sequence has a high similarity to that of species from the genus of Serratia available on GenBank. The quality of genomic DNA was evaluated by 0.8% agarose gel electrophoresis and Nanodrop2000 (Thermo Scientific, Waltham, US), and the quantity was evaluated by the Quant-iT Picogreen dsDNA kit (Invitrogen).
A whole genome shotgun library was generated with 500ng of S. marcescens FS14 genomic DNA. The shotgun sequencing procedure was performed using a Roche GS Junior Rapid Library Preparation Kit, per the manufacturer’s instructions (Roche, Basel, Switzerland). In addition, an 8kb span paired end library was generated with 15μg of S. marcescens FS14 genomic DNA. The paired end sequencing procedure was performed using a Roche GS Junior Paired End Library Preparation Kit, per the manufacturer’s instructions (Roche). One shotgun run and two paired end runs were performed on individual libraries prepared with the same genomic DNA sample. After sequencing, the raw data was assembled by Newbler 2.7 (Roche) with default parameters using paired end reads as an orientation guide. Primer pairs were designed along sequences flanking the gap regions and PCR gap-filling was followed by Sanger sequencing. The complete genome of S. marcescens FS14 was submitted to NCBI, GenBank accession number is CP005927, and it is publicly available for download at http://www.ncbi.nlm.nih.gov/nuccore/CP005927.
Genome annotation
Genome annotation and analysis of S. marcescens FS14 was performed using NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP) [20]. BLASTp [19] was applied to align the amino acid sequences against the COG database [21], VFDB database [22], and ARDB database [23]. Amino acid sequences with an alignment length over 90% of its own length and over 40% match identity (sequences annotated against COG database were filtered using over 20% match identity) were chosen and the descriptions of the best hits (with the highest alignment length percentage and match identity) were assigned as annotations of the predicted genes. All predicted amino acid sequences of S. marcescens FS14 were submitted to the KEGG database [24] for automatic pathway annotation (http://www.genome.jp/kaas-bin/kaas_main). All annotated pathways were manually downloaded and curated by in-house Perl scripts. PHAST (PHAge Search Tool)[25], available at http://phast.wishartlab.com/ was used for prophage identification. Genomic islands were predicted through IslandViewer [26] using the IslandPath-DIMOB method [27].
Phylogenetic and phylogenomic analysis
For phylogenomic analysis, 11 complete genomic sequences of Serratia spp. (S. marcescens WW4 [CP003959], S. marcescens Db11 [HG326223], S. marcescens FGI 94 [CP003942], S. proteamaculans 568 [CP000826], S. plymuthica AS13 [CP002775], S. plymuthica AS9 [NC_015567], S. plymuthica AS12 [NC_015566], S. plymuthica S13 [CP006566], S. plymuthica 4Rx13 [CP006250], S. liquefaciens ATCC27592 [CP006252], and S. fonticola RB-25 [CP007044]) were downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/). The genomic sequence of Serratia symbiotica Cinara cedri uid82363 has a genome size of only 1.8 Mb due to its endosymbiotic nature [28]; therefore, it was not included in the analysis. The orthologous genes were identified using BLAT [29] to match and align S. marcescens FS14 (CP005927) CDSs to all annotated genes from other Serratia spp. genomes. BLAT hits that were presented as single copies in Serratia spp. and cover over 90% of alignment length of FS14 genes were considered core genes. All of the core genes were aligned by MUSCLE [30] and randomly concatenated together. Finally, the phylogenomic tree was generated using these concatenated aligned genes with the GTR+G+I substitution model by MrBayes [31]. The chain length was set to 10,000,000 (1 sample/1000 generations). The first 2,000 samples were discarded as burn in after scrutinizing the trace files of two independent runs with Tracer v1.4 (http://tree.bio.ed.ac.uk/software/tracer/).
Since S. plymuthica AS9, AS12 and AS13 share a very high similarity, AS13 was chosen as the representative for all the following comparative analyses.
Pan genome analysis was performed using reciprocal BLAT between the ten Serratia spp. genomes, and the number of orthologs shared between them was calculated by in-house Perl scripts. Genomes of S. marcescens were aligned by Mauve [32] with Progressive Mauve [33] based on default parameters for investigation of potential genomic rearrangements.
The molecular phylogenetic tree was constructed using the maximum likelihood (ML) method (MEGA5) [34] based on the Poisson correction model [35] with concatenated amino acid sequences of two proteins, GyrB and RpoD, which are conserved in bacteria. They were retrieved from 16 different strains of Serratia and an E. coli K-12 publicly accessible on GenBank. A bootstrap consensus tree inferred from 100 replicates was chosen. The multiple alignments of protein sequences were built by CLUSTALW [36]. The same procedures and models were applied to a dataset consisting of 8 concatenated proteins—PigA(RedW), PigC(RedH), PigF(RedI), PigG(RedO), PigH(RedN), PigI(RedM), PigK(RedY) and PigM(RedV), all of which are homologues among different prodiginine-producing bacteria. Phylogenetic analysis of the Type III secretion system (T3SS) was performed based on a previous study [37]. Representatives of bacteria with T3SS were chosen to perform the phylogenetic analysis of T3SSs in Serratia. The multiple alignments of the T3SS ATPase sequences were built by CLUSTALW [36], and then a ML tree was constructed based on the Poisson correction model [35] with a bootstrap value of 100 by MEGA5 [34]. The T6SSs of Serratia were manually identified and the synteny study of the T6SS clusters was performed based on SyntTax [38] using default parameters and followed by manual confirmation.
Bacterial and fungal antagonistic bioassays
All biological samples involved in the bioassays designed to investigate the antibacterial and antifungal activities of S. marcescens FS14 were collected in Jiangsu province, China. The bacterial isolate, Ralstonia solanacearum NJ (RSNJ), was isolated from a tomato plant. The phytopathogenic fungus Fusarium oxysporum was isolated from the same plant that S. marcescens FS14 was isolated from, and Sclerotinia sclerotiorum was isolated from an infected Brassica napus ZY5. For fungal antagonistic assays, each fungus was grown for four days at 28°C and a 0.5cm-diameter agar with mycelia was cut and placed at a spot 2 cm away from the edge of the PDA plate. The overnight culture of FS14 was streaked across the middle of the plate [39]. Controls were set up without FS14. Fungal growth was observed for one week. All assays were performed in triplicate.
To prepare for the antibacterial competition assay, bacterial cultures were grown overnight in improved nutrient broth (1% glucose, 0.5% tryptone, 0.05% yeast extract and 0.3% beef extract) and adjusted to an OD600 of 0.5. For the biofilm culture, FS14 and RSNJ were mixed at a ratio of 1:1. 25μl of this mixture was spotted onto agar plates and incubated at 28°C. After 8-hour incubation, biofilm samples were recovered from the spot for quantification using sterile metal loops and then re-suspended in 1 ml improved nutrient broth. The recovery of viable cells is reported as the total number of recovered cells per biofilm co-culture spot. The control mixture was set up without FS14 and contained a 1:1 ratio of sterile nutrient broth to RSNJ. For the planktonic culture, FS14 and R. solanacearum NJ cells were adjusted to an OD600 of 0.1 and equal volumes (15 ml) of FS14 and RSNJ were mixed at a ratio of 1:1 in a 150 ml flask and cultured in a shaking incubator (200rpm at 28°C). One milliliter of culture was collected for quantification at 2, 4, 6, 8, 10, 12 and 24 hours after incubation. Then the quantities of FS14 and RSNJ were measured by serial dilutions and presented as Log CFU (colony-forming unit)/ml. Serial diluted samples were plated out on improved nutrient agar (w/v 1.5%) plates and CFU was counted after an incubation of 48 hours at 28°C. FS14 and RSNJ can be distinguished by color and antibiotic selection. Samples containing only FS14 or RSNJ were set up as controls. Cell number of each bacterium spotted onto the biofilm coculture plate at t = 0 hour is ~1 x 106, and concentration of each bacterium in the planktonic coculture at t = 0 hour is ~1.2 x 106 CFU/ml. Three independent experiments were performed for each assay.
Results and Discussion
Complete genome sequencing
The Serratia sp. FS14 genome is composed of a single circular chromosome of ~5.25 Mbp with a genomic GC content of 59.46% (S1 Fig) and a total of 4761 predicted coding sequences (CDSs). 91 tRNA genes and 6 rRNA operons were found in the FS14 genome. Genome statistics of the ten Serratia strains with complete genomes are shown in Table 1. FS14 has a similar genome size with S. marcescens WW4 and Db11. S. marcescens FGI 94 has a much smaller genome (~4.86 Mbp) with 4361 CDSs. CDSs that are absent in FGI94 are mainly transcriptional regulators, ATP-dependent transporters, chitinases and fimbrial biosynthetic proteins, all of which are not essential for bacterial survival [14, 40, 41, 42] and may not be necessary in its symbiotic lifestyle with the fungus; for instance, chitinases may antagonize its symbiotic fungus partner. This is partly in accordance to previous hypotheses that symbiotic bacteria genomes usually experience a reductive process and retained only the most essential functions [43, 44]. The genomic GC content of the analyzed Serratia strains ranges from 50.9% to 59.6%. The four S. marcescens strains have the highest GC content (around 59%) and S. fonticola RB-25 has the lowest GC content of 50.9%. No plasmid was identified in FS14, while S. marcescens WW4, S. proteamaculans 568, S. plymuthica 4Rx13 and S. liquefaciens ATCC27592 contained one plasmid each. No similarity can be observed between the plasmids in different strains of Serratia, which contributes to the variation of genomic content.
Table 1. Genome Statistics of S. marcescens FS14 and other Serratia spp..
| S. marcescens | S. proteamaculans | S. plymuthica | S.liquefaciens | S. fonticola | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FS14 | WW4 | Db11 | FGI 94 | 568 | AS13 | S13 | Rx13 | ATCC 27592 | RB-25 | |
| Genome size (Mb) | 5.24 | 5.24 | 5.11 | 4.86 | 5.5 | 5.44 | 5.47 | 5.4 | 5.28 | 5.49 |
| GC content % | 59.5 | 59.6 | 59.5 | 58.9 | 55 | 56 | 56.2 | 56.2 | 55.5 | 50.9 |
| No. of chromosome | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| No. of plasmid | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 |
| Predicted CDSs | 4761 | 4809 | 4709 | 4361 | 4942 | 4951 | 4991 | 4695 | 4894 | 4825 |
| No. of tRNAs | 91 | 79 | 88 | 83 | 85 | 87 | 85 | 81 | 79 | 77 |
| No. of rRNA operons | 6 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 6 | 7 |
| Percentage of ARDB annotated CDS % | 0.46 | 0.42 | 0.38 | 0.6 | 0.49 | 0.42 | 0.45 | 0.45 | 0.45 | 0.33 |
| Site of isolation | plant | paper machine | drosophila | Fungus garden | plant | plant | plant | soil | milk | landfill |
Taxonomic classification and comparative analysis of the genome
The bacterial isolate FS14 was classified into the genus Serratia according to its morphology, physiology, biochemistry, and 16S rDNA sequence [45]. In this study, taxonomic classification of strain FS14 was refined by two in silico analyses. In the phylogenetic tree constructed based on the two proteins conserved among bacteria—GyrB and RpoD [46, 47], Serratia sp. FS14 was found to form a close cluster with S. marcescens VGH107 (a clinical isolate), S. marcescens WW4 and S. marcescens Db11 with a high bootstrap value (100) distinct from clusters formed by other Serratia spp. (Fig 1A). Interestingly, S. marcescens FGI94 did not form a close cluster with the other strains of the same species.
Fig 1. Taxonomic classification of Serratia marcescens FS14.

(A) Maximum Likelihood Tree using Poisson correction model [35] and with 100 bootstrap replicates was constructed based on 2 conserved proteins among all bacteria—GyrB and RpoD—by MEGA 5 [34]. (B) The phylogenomic tree was constructed by MrBayes [17] using the random concatenation of 731 aligned core genes as the dataset and GTR + G+ I as the substitution model. The chain length was set to 10,000,000 (1 sample/1000 generations) whilst the burn-in was set as 2000. Posterior probabilities are denoted at nodes.
To better elucidate the taxonomic classification of FS14, a phylogenomic tree was constructed based on the 731 core genes (~19% of the FS14 genome) identified from the genome of FS14 and the other nine complete Serratia genomes publicly available. Serratia sp. FS14 was shown to cluster closely with S. marcescens WW4 with a high posterior probability (100) (Fig 1B), thus strongly advocating the notion that strain FS14 belongs to the species S. marcescens. Though FS14 was isolated from plants, it was unexpectedly found to be of a closer phylogenomic origin to the paper machine-isolated S. marcescens WW4 and the drosophila pathogen S. marcescens Db11 rather than the plant-associated S. plymuthica and S. proteamaculans [16, 48].
Mauve [32] analysis showed that the synteny between S. marcescens is not very conserved (S2 Fig). Chromosomal reorganizations are commonly observed along the whole stretch of the four genomes. In particular, a large region of ~3600–3700 kb in FS14 cannot be found in FGI 94, and further analysis identified these genes mainly as genomic factors associated with antagonistic traits of bacteria, such as the prodigiosin biosynthesis cluster, bacteriocins, iron uptake systems, chitinases and protein secretion systems, which, as previously mentioned, might not be necessary or even beneficial to its symbiotic lifestyle with fungus.
The pan-genome analysis demonstrated that the ten Serratia strains shared 1964 (~41% of the predicted CDSs in FS14) genes in their genomes and most of them were assigned with general cellular functions (S1 Table). As shown in the Venn diagram of the five representative Serratia genomes (Fig 2), they share 2171 CDSs, corresponding to 44% to 46% of all CDSs in these genomes. The unique CDS percentages are 13%, 15%, 18% and 21% for S. proteamaculans 568, S. liquefaciens ATCC27592, S. plymuthica AS13 and S. marcescens FS14, respectively. S. fonticola RB-25 has the highest number (~43%) of unique genes including a tellurium resistance gene cluster (Z042_12505–12590). A type I restriction-modification (RM) system, unique among the Serratia spp., was identified on a putative genomic island GI01 of FS14 (S1 Fig). This putative Type I RM system may protect specific DNA restriction sites from endonucleases and play a role in the regulation of gene expression, as previously reported for a similar Type I RM system (sharing ~72% to 93% identities) in Yersinia pseudotuberulosis [49]. Interestingly, S. marcescens FGI 94 was revealed to encode a larger percentage (5.41% of its total CDSs) of genes potentially contributing to virulence than the insect pathogen S. marcescens Db11 (4.93%) [50]. In addition, FGI 94 encodes a unique Type III secretion system (T3SS) gene cluster that has a much higher GC content (66.12%) than the rest of its genome (58.9%), implicating this cluster may have been acquired through horizontal gene transfer. Phylogenetic analysis supports that it may originate from bacteria of other genera (S3 Fig); however, caution must be taken as multiple gene loss events in other Serratia spp. are improbable, though not impossible. In general, most of the unique genes in each strain are hypothetical proteins or are associated with prophage regions. 33 putative prophage regions are identified across the ten Serratia genomes, and FS14 encodes a unique ~53kb prophage region of Enterobacteria phage BP-4795 on its genomic island GI02 (S1 Fig, S2 Table).
Fig 2. Venn diagram of five representative Serratia spp..

Five representative genomes, S. marcescens FS14 (CP005927), S. proteamaculans 568 (CP000826), S. plymuthica AS13 (CP002775), S. liquefaciens ATCC27592 (CP006252), and S. fonticola RB-25 (CP007044) were selected to illustrate the Venn diagram. The Venn diagram was not drawn in proportion; it sole purpose is to illustrate the common CDSs shared between the five strains. The overlapping regions represent CDSs shared with respective strains. The number outside the overlapping regions indicates the number of CDSs in each genome without homologs in other Serratia genomes.
Orthologs are genes in different species that have evolved from a common ancestral gene via speciation and often retain the same function in the course of evolution. Comparing orthologs is important to identify events of gene gain or loss. Clusters of orthologous groups (COGs) [51] provide genome-scale analysis of protein function prediction. Major COG-annotated class distribution is conserved between Serratia strains (S3 Table, S4 Fig). As expected, in all analyzed Serratia strains, the majority of COG-annotated genes are involved in basic cellular functions such as amino acid transport and metabolism (COG class [E]), transcription ([K]) and carbohydrate transport and metabolism ([G]); up to ~ 12% of the annotated genes have unknown ([S]) or predicted functions ([R])in the COG database.
Antagonistic effect on phytopathogens of S. marcescens FS14
Bioassays were designed to investigate the antagonistic activities of FS14 against plant bacterial and fungal pathogens. Fusarium oxysporum, Sclerotinia sclerotiorum and Ralstonia solanacearum were used as the indicator microorganisms in antagonistic assays following the methods in previous reports [52, 53].
In the bacterial-fungal confrontation assays, FS14 was co-inoculated with F. oxysporum and S. sclerotiorum, both of which are common plant pathogens. As shown in Fig 3, compared to the control without FS14, the mycelia of both fungi were clearly inhibited by the FS14 colony streaked across the agar plates as early as the second day of inoculation. The fungal mycelia also turned a darker color at the later stages of bacteria-fungi interaction. Both fungi did not grow past the FS14 colony, though the non-contact inhibition seemed to be more evident in F. oxysporum than Sclerotinia sclerotiorum. These results indicate that FS14 can significantly suppress the growth of phytopathogenic fungi through non-contact inhibition, an ability which may be attributed to extracellular secretion of antifungal substances such as chitinases.
Fig 3. Antagonistic activities of Serratia marcescens FS14 against fungi.

(A) Visualization of the FS14- Sclerotinia sclerotiorum (Sscl) and FS14-Fusarium oxysporum (Foxy) confrontation assays, 2–7 days after inoculation of Sscl (1st and 2nd rows) and Foxy (3rd and 4th rows) in the presence of S. marcescens FS14 (1st and 3rd rows) or in its absence (2nd or 4th rows); (B) Growth of F. oxysporum and S. sclerotiorum with and without FS14 were closely monitored. Challenging fungi were grown on PDA plates as described in Materials and Methods, the areas of growth of hyphae (in cm2) was measured. Numbers show an average of 3 plates, and error bars show standard errors of the means.
In the bacteria-bacteria competition assay, S. marcescens FS14 was co-cultured with a target bacterium R. solanacearum, a devastating phytopathogenic bacterium which causes wilt in a wide range of host plants [54]. In the biofilm culture, when R. solanacearum NJ (RSNJ) was co-cultured with FS14 at an initial cell ratio of 1:1 for an 8-hour incubation, the final ratio of recovered cells from the RSNJ and FS14 co-culture was ~1:104 (RSNJ: FS14). In comparison, a 106 times increase in the number of viable RSNJ cells was recovered when they were cultured with medium only after the same period of incubation (Fig 4A). A similar result was observed when FS14 and RSNJ were co-cultured at an initial ratio of 1:1 in the planktonic culture (Fig 4B). These results imply that S. marcescens FS14 also possesses an antagonistic potential against the bacterial phytopathogen.
Fig 4. Antagonistic activity of Serratia marcescens FS14 against Ralstonia solanacearum NJ.

(A) Recovery of viable R. solanacearum (RSNJ) cells after 8 hours of co-culture with (initial ratio of 1:1) and without FS14 at 28°C (see Materials and Methods for details); (B) The planktonic culture with quantities of S. marcescens FS14 (FS14) and R. solanacearum NJ (RSNJ) recorded at 0, 2, 4, 6, 8, 10, 12 and 24 hours after incubation. Numbers show an average of three replications, and error bars show standard errors of the means. a: the quantities of FS14 in the samples collected from the planktonic culture containing 1:1 of FS14 and RSNJ. b: the quantities of RSNJ in the samples collected from the planktonic culture containing 1:1 of FS14 and RSNJ.
In the comparative genome analysis of FS14, genomic elements which may be involved in its antagonistic activities against fungi or bacteria are identified. These include antibiotic-resistant genes, Type VI secretion systems, prodigiosin and other antimicrobial compounds, and chitinases.
Antibiotic-resistant traits
Many bacteria have evolved detoxification determinants or antibiotic resistance to counter antibiotics in competition against other microbes. Genomic analysis of S. marcescens FS14 showed that 22 (0.46%) out of 4761 predicted CDSs can be identified in the ARDB database (Table 2), suggesting it is a multi-drug resistant strain. The drosophila pathogen S. marcescens Db11 exhibits the highest number of antibiotic resistant genes (~0.6%, Table 1). Comparative study of FS14 and 9 other Serratia showed that all of them have potential resistance against four kinds of antibiotics: bacitracin, cephalosporin, fosmidomycin and penicillin. Interestingly, in the antibiotic sensitivity tests, FS14 exhibited resistance to vancomycin (Table 3), but the genomic analysis found no potential vancomycin-resistant genes in the FS14 genome, which may suggest that FS14 has either developed novel operons to resist vancomycin, or the antibiotic-resistant gene database is not comprehensive, or the presence of a multi-functional drug efflux pump is at work in the bacteria. Despite minor discrepancies, the experimental results are consistent with the genomic analysis and FS14 showed resistance against multiple antibiotics, which might contribute to its antagonistic effect against other microorganisms.
Table 2. Identification of potential antibiotic-resistant genes in Serratia spp.
| No. of CDSs annotated | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FS14 a | WW4 | Db11 a | FGI94 a | 568 b | AS13 c | S13 c | Rx13 c | ATCC 27592 d | RB-25 e | |
| acriflavin | 1 | 3 | 3 | 1 | 4 | 3 | 3 | 4 | 1 | 0 |
| acriflavine | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| amikacin | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| aminoglycoside | 2 | 1 | 0 | 2 | 2 | 1 | 2 | 1 | 1 | 1 |
| bacitracin | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| cephalosproin | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| chloramphenicol | 0 | 0 | 0 | 1 | 1 | 2 | 1 | 1 | 3 | 1 |
| deoxycholate | 1 | 2 | 1 | 2 | 3 | 2 | 2 | 3 | 2 | 1 |
| dibekacin | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| enoxacin | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 |
| fluoroquinolone | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| fosfomycin | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| fosmidomycin | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| kasugamycin | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| macrolide | 3 | 0 | 4 | 2 | 2 | 3 | 2 | 1 | 2 | 1 |
| na_antimicrobials | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| penicillin | 2 | 2 | 2 | 4 | 2 | 2 | 2 | 2 | 3 | 2 |
| streptogramin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| streptomycin | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 |
| teicoplanin | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| tetracycline | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| trimethoprim | 1 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| vancomycin | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 |
| Total | 22 | 20 | 18 | 26 | 24 | 24 | 21 | 21 | 22 | 16 |
a: S. marcescens;
b: S. proteamaculans;
c: S. plymuthica;
d: S.liquefaciens;
e: S. fonticola.
Table 3. The antibiotic sensitivity of S. marcescens FS14.
| Antibiotic | Sensitivity |
|---|---|
| azithromycin | I |
| acetyl spiramycin | R |
| amikacin | I |
| amoxicillin | R |
| ampicillin | R |
| cefradine | R |
| chlorampenicol | S |
| erythromycin | I |
| gentamicin | S |
| kanamycin | S |
| levofloxacin | S |
| lincomycin | R |
| neomycin | I |
| penicillin | R |
| polymyxin | I |
| rifampicin | R |
| spectinomycin | S |
| streptomycin | R |
| tetracycline | R |
| vancomycin | R |
R: Resistant; S: Sensitive; I: Intermediate.
The Type VI secretion system
Type VI secretion system (T6SS) is widely distributed amongst the Gram-negative bacteria. It is used to deliver antagonistic effectors to either adjacent bacteria or target eukaryotic cells [10]. T6SSs are encoded by variable gene clusters consisting of 13 core essential components (TssA-M), which form the apparatus that resembles an inverted bacteriophage puncturing device [55]. Two distinct T6SS clusters of 24-kb and 28-kb were identified in the FS14 genome, both found on a putative genomic island (GI03, S1 Fig, Fig 5). Mobile DNA elements (transposases, IS elements) are found between the two T6SS clusters, which indicates that the two T6SSs are likely to have been acquired by horizontal gene transfer or internal chromosomal rearrangements. No T6SSs were identified in S. liquefaciens ATCC 27592 and S. plymuthica strains, but the other Serratia evaluated also contain two T6SSs, with the exception of S. marcescens Db11, which has only one T6SS (S4 Table).
Fig 5. Genetic organizations of T6SS in Serratia and other bacteria.

(A), (B), (C) and (D) show the genetic organizations of the 11 T6SS clusters found in Serratia, which have different organizations and separate into four families (T6SS-a, T6SS-b, T6SS-c and T6SS-d). T6SS-a was aligned across S. proteamaculans 568, S. fonticola RB-25, S. marcescens Db11, FGI 94, FS14 and WW4 genomes at the corresponding loci. T6SS-b was found in S. marcescens FS14 and WW4. T6SS-c was only found in S. fonticola RB-25. T6SS-d was found in S. proteamaculans 568 and S. marcescens FGI 94. Each T6SS cluster contains conserved T6SS core component genes. Solid blue/green/purple/orange color boxes indicate the conserved T6SS core genes; gray boxes represent variable conserved genes and white boxes are unique genes. Details of the genetic organization of T6SS clusters in Serratia are listed in S4 Table.
Synteny analysis between the 11 T6SSs of the Serratia spp. showed that the T6SSs in Serratia separate into four families termed as T6SS-a, b, c and d (Fig 5). Only T6SS-a of the four evolutionarily distinct Serratia T6SSs is conserved among them. T6SS-a in strain Db10 (Db11) was previously shown to target bacterial competitors with antagonistic effectors, but is not required for virulence in eukaryotes [52]. T6SS-b in FS14 and WW4 shows synteny and homology with the T6SS of animal or soil-associated bacteria, such as Burkholderia thailandensis E264, which has five T6SSs including both anti-eukaryotic and antibacterial T6SSs [12]. T6SS-c in the S. fonticola RB25 genome is related to T6SSs of Yersinia pestis Antiqua, Photorhabdus asymbiotica ATCC43949 etc. in terms of composition, synteny and homology (Fig 5, data not shown). Serratia T6SS-d was found in S. marcescens FGI94 and S. proteamaculans 568.
Besides the gene variation in the core components, the accessory elements of T6SS clusters are highly variable; for example, T6SS-a of Serratia contains multiple accessory proteins, such as PpkA, PppA and Fha. Since the bacteria carrying T6SS gene clusters are found in diverse environments and the function of T6SS is highly versatile [10, 56, 57], these accessory proteins might be involved in regulation or might confer additional functions to the system [57]. Homologs of PpkA, PppA and Fha in T6SS-a was formerly characterized to play important roles in activation of T6SS at transcriptional or post-translational levels [58, 59]; however, the three regulatory components are absent from the other three T6SS families identified in Serratia (Fig 5, S4 Table). Moreover, some of the CDSs found within the T6SS cluster were reported as secretory effector proteins or self-resistance (‘immunity’) proteins, e.g. the Ssp proteins which are novel toxins recently identified in S. marcescens Db10 [60]. So it is speculated that other genes assigned with unknown functions in T6SS clusters are likely novel effectors or immunity proteins, whose roles remain to be experimentally verified.
It has been estimated that about one third of bacteria with a T6SS have multiple systems [55]. Multiple T6SSs in one organism, e.g. B. thailandensis, P. aeruginosa, may confer the species the ability to target multiple organisms [12, 61, 62, 63], allowing the secreting species to overcome rival bacteria and successfully colonize polymicrobial niches. The presence of T6SSs in FS14 may contribute to its antagonistic effect against microbial competitors to survive in polymicrobial habitats, but this hypothesis still requires further experimental elucidation.
Prodigiosin and other antimicrobial compounds
The ability of microorganisms to produce diverse antimicrobial compounds including antibiotics, bacteriocins and other secondary metabolites plays an important role in their antagonistic activities against other microbes [64, 65]. Prodiginines are a family of red-pigmented tripyrrole antibiotics produced by bacteria such as pigmented Serratia, Actinobacteria and some marine bacteria. A putative prodiginine gene cluster containing 14 CDSs was found in S. marcescens FS14, WW4 and S. plymuthica AS13, but the cluster was absent in the other Serratia genomes. Phylogenetic analysis with concatenated amino acid sequences of 8 proteins commonly found in the prodiginine-producing bacteria formed two main clades (Clade I and Clade II) (Fig 6). Clade I includes Serratia spp. and marine bacteria which produce prodigiosin or cycloprodigiosin, while Clade II includes Actinobacteria strains producing other kinds of prodiginines. Orthologs of FS14 were grouped under the clade of other prodigiosin-producing bacteria, and the FS14 pig cluster showed high amino acid sequence identities (~75%, S5 Table) with those in Serratia sp. ATCC 39006, which has been used as a model for the study of prodigiosin biosynthesis [66].
Fig 6. Phylogenetic tree of prodigiosin biosynthesis genes from all prodiginine-producing bacteria.

The Maximum Likelihood Tree with 100 bootstrap replicates was constructed based on Poisson correction model [35] with concatenated amino acid sequences of 8 proteins, PigA(RedW), PigC(RedH), PigF(RedI), PigG(RedO), PigH(RedN), PigI(RedM), PigK(RedY) and PigM(RedV) that are involved in prodiginine-biosynthesis and commonly found among 11 prodiginine-producing bacteria. Adjacent colored squares represent different kinds of prodiginines produced by the bacteria: prodigiosin (orange), undecylprodigiosin (red), cycloprodigiosin (purple), cyclononylprodigiosin (green), and butyl-meta-cyclo-heptylprodiginine (blue).
As shown in Fig 7, the synteny among the pig clusters of the S. marcescens FS14, WW4, S. plymuthica AS13 and ATCC3066 is conserved, except for a pigO present downstream of the cluster in ATCC39006. The pig cluster of S. marcescens FS14 is flanked by cueR and copA, genes involved in metal (copper)-dependent regulation and efflux (S5 Table, Fig 7). This observation is congruent with earlier studies [67] that the pig cluster was inserted between cueR and copA in the common ancestor of pigmented S. marcescens. In S. plymuthica AS13, the cueR and copA genes were directly adjacent to each other, but localized 20Kb downstream of the pig cluster. Moreover, a tRNA-Ser element was found directly adjacent to pigN in S. plymuthica AS13, hinting at the occurrence of a potential genomic rearrangement.
Fig 7. Synteny of the prodigiosin biosynthesis pig clusters among pigmented Serratia spp..

Genes are symbolized by arrows. The white arrows denote condensing enzymes; black arrows represent genes that encode proteins required for the biosynthesis of the monopyrroles; genes encoding proteins required for the biosynthesis of 4-methoxy-2,2-bipyrrole-5-carboxyaldehyde are in thin diagonally striped arrows; checkered arrows are used to highlight the additional pigO gene only found in ATCC39006; vertical striped arrows denote other flanking genes.
Many antibiotics produced by soil or plant associated bacteria have been reported to suppress diseases caused by bacterial pathogens [68]. The production of the red-pigmented prodigiosin in S. marcescens FS14 might contribute to its antagonistic effect against other bacteria. In addition, studies have shown that prodigiosin-producing isolates of S. marcescens are rarely found as human pathogens [7]; however, since S. marcescens FS14 shows a close relationship to a clinical strain and possesses multiple antibiotic resistant genes and T6SSs, further tests on its potential toxicity for humans including possible genetic modifications of the pathogenicity-related genomic elements should be carried out before further study and application.
The competition for limiting natural resources through producing bacteriocins to inhibit the growth of related organisms is also an antagonistic potential of bacteria [69, 70]. Within the ten Serratia genomes, 11 clusters were found to be involved in the biosynthesis of bacteriocin, colicin, entericidin, microcin and pyocin and one of the 11clusters is conserved across the genus (Table 4). FS14 has five bacteriocin gene clusters and one microcin H47 cluster (L085_22830–22835) of them is only found in the FS14 genome. Some of the bacteriocin operons also encode immunity proteins (e.g. L085_18570) as the cognate bacteriocins which may protect the producing bacterium from the inhibition by its own bacteriocin [69].
Table 4. Genes involved in potential bacterial competitions in Serratia.
| Pan-genome position | Description | Representing Loci | S. marcescens | S. proteamaculans | S. plymuthica | S. liquefaciens | S. fonticola | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FS14 | WW4 | Db11 | FGI 94 | 568 | AS13 | S13 | Rx13 | ATCC 27592 | RB-25 | |||
| Bacteriocin | ||||||||||||
| 451–452 | Entericidin | L085_02315–02320 | + | + | + | + | + | + | + | - | + | - |
| 2317–2319 | Microcin C | L085_11940–11950 | + | + | + | + | + | + | + | + | + | + |
| 3308–3309 | Microcin H47 | L085_17075–17080 | + | + | - | - | - | - | - | + | - | - |
| 3599–3600 | S-type pyocin | L085_18570–18585 | + | + | + | - | + | - | + | + | - | + |
| 4429–4430 | Microcin H47 | L085_22830–22835 | + | - | - | - | - | - | - | - | - | - |
| 6487–6491 | S-type pyocin | SOD_c04120–04170 | - | + | - | - | + | - | - | + | - | - |
| 7063–7064 | Microcin H47 | SOD_c38320–38330 | - | - | - | - | - | - | - | + | - | - |
| 12127–12133 | bacteriocin | SMDB11_1570–1575 | - | - | + | - | - | - | - | - | - | - |
| 12365–12366 | S-type pyocin | SMDB11_4214–4215 | - | - | + | - | - | - | - | - | - | - |
| Chitinase | ||||||||||||
| 996 | Chitinase A | L085_05105 | + | + | + | - | + | + | + | + | + | + |
| 2080 | Chitinase B | L085_10730 | + | + | + | - | + | + | + | + | + | - |
| 2922 | Chitinase A | L085_15120 | + | + | + | - | + | + | + | + | + | + |
| 4364 | Chitinase C | L085_22490 | + | + | + | + | + | + | + | + | + | + |
| 5302 | Chitinase C | SerAS13_2254 | - | - | - | - | - | + | - | + | - | - |
Chitinases
Other resources subjected to antagonistic activities include lytic enzymes such as chitinases. S. marcescens is known as one of the most efficient producers of chitinases, which degrade chitin, the main component of fungal cell walls and exoskeletons of insects [71, 72]. The chitinolytic activities of bacteria are mainly attributed to 3 essential enzymes: the processive chitinases chitinase A (ChiA), chitinase B (ChiB) essential for full degradation of chitin, and endo-acting chitinase C (ChiC) [73]. Five putative chitinases are found across Serratia genomes (Table 4) including two ChiC, and one of the ChiC is conserved across Serratia. An additional ChiC is found in the S. plymuthica AS13, S13 and S. fonticola RB-25 genomes. Two of the chitinases show high identities with the well-characterized ChiA in S. marcescens which is responsible for chitin hydrolysis and was demonstrated to control several important fungal phytopathogens [74, 75]. ChiB is not present in S. fonticola RB-25, suggesting that the bacteria may not degrade chitin efficiently or that a novel ChiB functioning gene may exist. Both ChiA and ChiB are absent in S. marcescens FGI 94, implicating that it may have lost the ability to degrade chitin when it adopted a commensal life with fungus. FS14 possesses 4 genes involved in the secretion of chitinases and they may play a critical role in its suppression of fungi necessary for its competitive lifestyle in diseased plants.
FS14 possesses a set of genes involved in the secretion of chitinases and they are expected to play an important role in the inhibition of mycelial extension, which is in agreement with the observations from our co-culture assays of FS14 and fungi. In fact, the culture filtrate of some S. plymuthica and S. marcescens has been used as a biocontrol agent against fungi because of their secretion of chitinases [76, 77].
Conclusions
In a nutshell, the complete genome of S. marcescens FS14 is 5.25Mb with a genomic GC content of 59.46%. The comparison of ten Serratia genomes has identified the universal core genes of Serratia and unique genes to each strain. S. marcescens FS14 possesses a unique Type I RM system, whereas FGI 94 has acquired a type III secretion system and S. fonticola RB-25 was found to have homologues of tellurium resistance genes. The presence of prodigiosin, bacteriocins, multi-drug resistant proteins and chitinases in Serratia suggests its antagonistic potential. The identification of two T6SS clusters offers further evidence that FS14 competes with other microbes, thus enhancing the survival of FS14 in various habitats. Compared to other Serratia, FS14 possesses both a prodigiosin cluster and two T6SSs. The antagonistic effect assays demonstrated that S. marcescens FS14 can suppress the growth of the vital plant pathogenic bacterium Ralstonia solanacearum and fungi Fusarium oxysporum and Sclerotinia sclerotiorum in vitro.
Supporting Information
From outer to inner layer: (1) nucleotide positions in kilobases (kb) (black); (2) COG database-annotated CDSs (light red); (3) ACLAME database-annotated potential horizontal transferring genes (orange: from plasmids, blue: from prophage and black: from virus); (4) ARDB-annotated potential drug resistant genes (green); (5) prodigiosin biosynthetic gene cluster (red), genes related to biosynthesis of chitinases (black), siderophores (blue); (6) tRNA region (purple), rRNA (red); (7) Predicted genomic islands (yellow); (8) GC density.
(PDF)
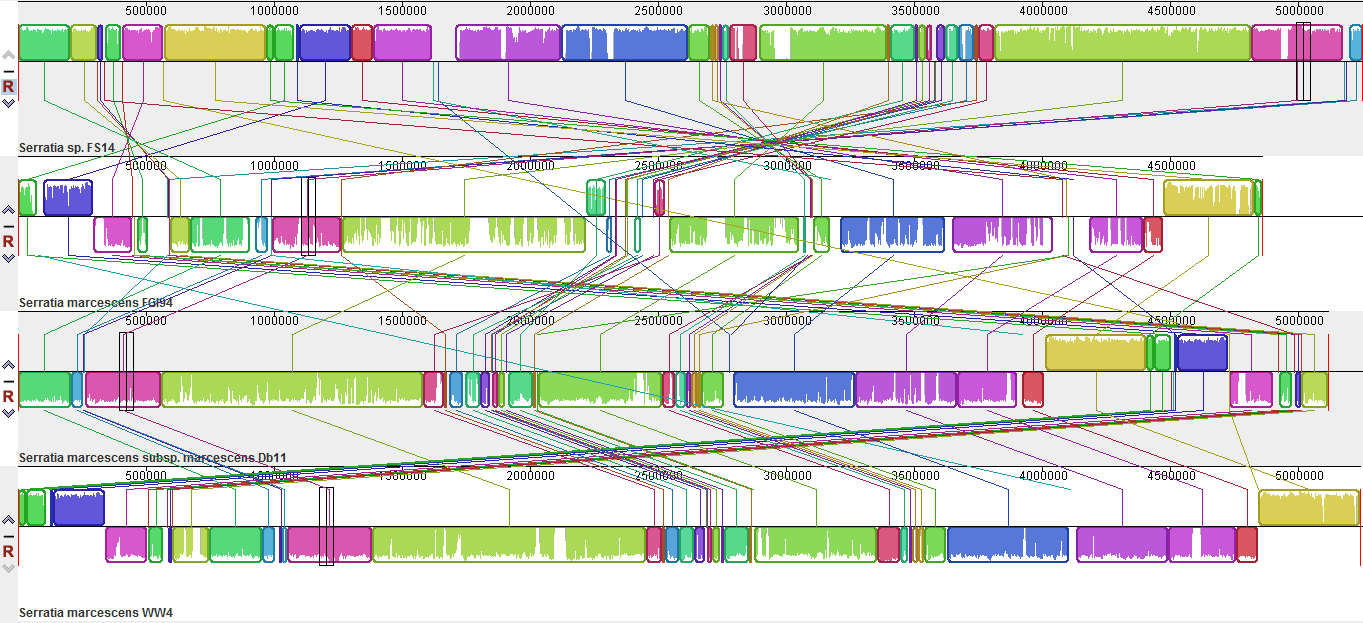
Progressive Mauve [33] alignment of S. marcescens FS14, FGI 94, WW4 and Db11 genome sequences with default parameters. Each same color block represents a locally collinear block (LCB) (i.e. homologous region shared by genomes without any rearrangements). Rearrangement of genomic regions was observed in the four genomes, in term of collinearity and their localization on the negative or positive strand (indicated by their genomic position below or above the black horizontal center line in the Mauve alignment, respectively).
(PNG)
{kind=link}
Maximum Likelihood Tree based on amino acid sequences of the conserved T3SS ATPase associated with S. marcescens FGI 94 constructed by 44-representive orthologs from each species using MEGA5. 7 different families of T3SS were identified. T3SS in S. marcescens FGI 94 was found in the Hrp1 family, which mainly composed of plant pathogens. The ATPase of the flagellum of E. coli was used as an outgroup. Bootstrap values are shown as percentages of 100 replicates, numbers at nodes represent bootstrap values, and only bootstrap values of >50 are shown.
(TIF)
COG-annotated genes of S. marcescens FS14 were compared to 9 other Serratia genomes: S. marcescens WW4 (CP003959), S. marcescens Db11 (HG326223), S. marcescens FGI 94 (CP003942), S. proteamaculans 568 (CP000826), S. plymuthica 4Rx13 (CP006250), S. plymuthica S13 (CP006566), S. plymuthica AS13 (CP002775), S. liquefaciens ATCC27592 (CP006252), and S. fonticola RB-25 (CP007044).
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
The work was supported by Bioinformatics Center of Nanjing Agricultural University. The author(s) wish to thank Dr. Raymond Hui for his help with the genome sequencing.
Data Availability
Genome data are deposited into NCBI GenBank (accession number: CP005927) and may be downloaded by following: http://www.ncbi.nlm.nih.gov/nuccore/CP005927.
Funding Statement
The work was supported by Bioinformatics Center of Nanjing Agricultural University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Zheng CC, Wang CL, Fu L, Wang WW, Xu DQ. Isolation and characterization of the Serratia sp. FS14 secreting thermostable DNase and protease.2011; 38: 228–236. [Google Scholar]
- 2. Hejazi A, Falkiner FR. Serratia marcescens. J Med Microbiol. 1997; 46: 903–912. [DOI] [PubMed] [Google Scholar]
- 3. Purushotham P, Arun PV, Prakash JS, Podile AR. Chitin binding proteins act synergistically with chitinases in Serratia proteamaculans 568. PLoS One. 2012; 7: e36714 10.1371/journal.pone.0036714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weise T, Thurmer A, Brady S, Kai M, Daniel R, Gottschalk G, et al. VOC emission of various Serratia species and isolates and genome analysis of Serratia plymuthica 4Rx13. FEMS Microbiol Lett. 2014; 352: 45–53. 10.1111/1574-6968.12359 [DOI] [PubMed] [Google Scholar]
- 5. De Vleesschauwer D, Höfte M. Using Serratia plymuthica to control fungal pathogens of plants. CAB Reviews. 2003; 2(46):189–257. [Google Scholar]
- 6. Williamson NR, Fineran PC, Leeper FJ, Salmond GP. The biosynthesis and regulation of bacterial prodiginines. Nat Rev Microbiol. 2006; 4: 887–899. [DOI] [PubMed] [Google Scholar]
- 7. Mahlen SD. Serratia infections: from military experiments to current practice. Clin Microbiol Rev. 2011; 24: 755–791. 10.1128/CMR.00017-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williamson NR, Fineran PC, Gristwood T, Chawrai SR, Leeper FJ, Salmond GP. Anticancer and immunosuppressive properties of bacterial prodiginines. Future Microbiol. 2007; 2: 605–618. [DOI] [PubMed] [Google Scholar]
- 9. Gerlach RG, Hensel M. Protein secretion systems and adhesins: the molecular armory of Gram-negative pathogens. Int J Med Microbiol. 2007; 297: 401–415. [DOI] [PubMed] [Google Scholar]
- 10. Coulthurst SJ. The Type VI secretion system—a widespread and versatile cell targeting system. Res Microbiol. 2013; 164: 640–654. 10.1016/j.resmic.2013.03.017 [DOI] [PubMed] [Google Scholar]
- 11. MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc Natl Acad Sci U S A. 2010; 107: 19520–19524. 10.1073/pnas.1012931107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwarz S, West TE, Boyer F, Chiang WC, Carl MA, Hood RD, et al. Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 2010; 6: e1001068 10.1371/journal.ppat.1001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung WC, Chen LL, Lo WS, Kuo PA, Tu J, Kuo CH. Complete Genome Sequence of Serratia marcescens WW4. Genome Announc. 2013; 1: e12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aylward FO, Tremmel DM, Starrett GJ, Bruce DC, Chain P, Chen A, et al. Complete Genome of Serratia sp. Strain FGI 94, a Strain Associated with Leaf-Cutter Ant Fungus Gardens. Genome Announc. 2013; 1: e23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ee R, Lim YL, Tee KK, Yin WF, Chan KG. Quorum sensing activity of Serratia fonticola strain RB-25 isolated from an ex-landfill site. Sensors (Basel). 2014; 14: 5136–5146. 10.3390/s140305136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neupane S, Finlay RD, Kyrpides NC, Goodwin L, Alstrom S, Lucas S, et al. Complete genome sequence of the plant-associated Serratia plymuthica strain AS13. Stand Genomic Sci. 2012; 7: 22–30. 10.4056/sigs.2966299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muller H, Furnkranz M, Grube M, Berg G. Genome Sequence of Serratia plymuthica Strain S13, an Endophyte with Germination- and Plant-Growth-Promoting Activity from the Flower of Styrian Oil Pumpkin. Genome Announc. 2013; 1: e00594–13. 10.1128/genomeA.00594-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buczolits S, Denner EB, Vybiral D, Wieser M, Kampfer P, Busse HJ. Classification of three airborne bacteria and proposal of Hymenobacter aerophilus sp. nov. Int J Syst Evol Microbiol. 2002; 52: 445–456. [DOI] [PubMed] [Google Scholar]
- 19. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity G, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS. 2008; 12: 137–141. 10.1089/omi.2008.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001; 29: 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012; 40: D641–D645. 10.1093/nar/gkr989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu B, Pop M. ARDB—Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009; 37: D443–D447. 10.1093/nar/gkn656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakaya A, Katayama T, Itoh M, Hiranuka K, Kawashima S, Moriya Y, et al. KEGG OC: a large-scale automatic construction of taxonomy-based ortholog clusters. Nucleic Acids Res. 2013; 41: D353–D357. 10.1093/nar/gks1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011; 39: W347–W352. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langille MG, Brinkman FS. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009; 25: 664–665. 10.1093/bioinformatics/btp030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsiao W, Wan I, Jones SJ, Brinkman FS. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics. 2003; 19: 418–420. [DOI] [PubMed] [Google Scholar]
- 28. Lamelas A, Gosalbes MJ, Manzano-Marin A, Pereto J, Moya A, Latorre A. Serratia symbiotica from the aphid Cinara cedri: a missing link from facultative to obligate insect endosymbiont. PLoS Genet. 2011; 7: e1002357 10.1371/journal.pgen.1002357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002; 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003; 19: 1572–1574. [DOI] [PubMed] [Google Scholar]
- 32. Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004; 14: 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010; 5: e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011; 28: 2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nei M, Kumar S. Molecular evolution and phylogenetics. 1st ed New York: Oxford University Press; 2000. [Google Scholar]
- 36. Fukami-Kobayashi K, Saito N. How to make good use of CLUSTALW. Tanpakushitsu Kakusan Koso. 2002; 47: 1237–1239. [PubMed] [Google Scholar]
- 37. Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006; 4: 811–825. [DOI] [PubMed] [Google Scholar]
- 38. Oberto J. SyntTax: a web server linking synteny to prokaryotic taxonomy. BMC Bioinformatics. 2013; 14: 4 10.1186/1471-2105-14-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mela F, Fritsche K, de Boer W, van Veen JA, de Graaff LH, van den Berg M, et al. Dual transcriptional profiling of a bacterial/fungal confrontation: Collimonas fungivorans versus Aspergillus niger. ISME J. 2011; 5: 1494–1504. 10.1038/ismej.2011.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012; 10: 13–26. [DOI] [PubMed] [Google Scholar]
- 41. Hansen AK, Moran NA. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc Natl Acad Sci U S A. 2011; 108: 2849–2854. 10.1073/pnas.1013465108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998; 396: 133–140. [DOI] [PubMed] [Google Scholar]
- 43. van Ham RC, Kamerbeek J, Palacios C, Rausell C, Abascal F, Bastolla U, et al. Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci U S A. 2003; 100: 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moran NA, Wernegreen JJ. Lifestyle evolution in symbiotic bacteria: insights from genomics. Trends Ecol Evol. 2000; 15: 321–326. [DOI] [PubMed] [Google Scholar]
- 45. Liu S, Ran T, Shen X, Xu L, Wang W, Xu D. Expression, crystallization and preliminary crystallographic data analysis of PigF, an O-methyltransferase from the prodigiosin-synthetic pathway in Serratia. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012; 68: 898–901. 10.1107/S1744309112024001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamamoto S, Kasai H, Arnold DL, Jackson RW, Vivian A, Harayama S. Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology. 2000; 146 (Pt 10): 2385–2394. [DOI] [PubMed] [Google Scholar]
- 47. Luque-Almagro VM, Acera F, Igeno MI, Wibberg D, Roldan MD, Saez LP, et al. Draft whole genome sequence of the cyanide-degrading bacterium Pseudomonas pseudoalcaligenes CECT5344. Environ Microbiol. 2013; 15: 253–270. 10.1111/j.1462-2920.2012.02875.x [DOI] [PubMed] [Google Scholar]
- 48. Grimont PA, Grimont F. The genus Serratia. Prokarytoes. 2006; 6: 219–244. [Google Scholar]
- 49. Pouillot F, Fayolle C, Carniel E. A putative DNA adenine methyltransferase is involved in Yersinia pseudotuberculosis pathogenicity. Microbiology. 2007; 153: 2426–2434. [DOI] [PubMed] [Google Scholar]
- 50. Flyg C, Kenne K, Boman HG. Insect pathogenic properties of Serratia marcescens: phage-resistant mutants with a decreased resistance to Cecropia immunity and a decreased virulence to Drosophila. J Gen Microbiol. 1980; 120: 173–181. [DOI] [PubMed] [Google Scholar]
- 51. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003; 4: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J Bacteriol. 2011; 193: 6057–6069. 10.1128/JB.05671-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mela F, Fritsche K, de Boer W, van Veen JA, de Graaff LH, van den Berg M, et al. Dual transcriptional profiling of a bacterial/fungal confrontation: Collimonas fungivorans versus Aspergillus niger. ISME J. 2011; 5: 1494–1504. 10.1038/ismej.2011.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Denny T. Plant pathogenic Ralstonia species In: Gnanamanickam SS, editor. Plant-associated bacteria. Netherlands: Springer; 2006. pp. 573–644. [Google Scholar]
- 55. Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I. Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics. 2009; 10: 104 10.1186/1471-2164-10-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shyntum DY, Venter SN, Moleleki LN, Toth I, Coutinho TA. Comparative genomics of type VI secretion systems in strains of Pantoea ananatis from different environments. BMC Genomics. 2014; 15: 163 10.1186/1471-2164-15-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol. 2012; 66: 453–472. 10.1146/annurev-micro-121809-151619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hsu F, Schwarz S, Mougous JD. TagR promotes PpkA-catalysed type VI secretion activation in Pseudomonas aeruginosa. Mol Microbiol. 2009; 72: 1111–1125. 10.1111/j.1365-2958.2009.06701.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mougous JD, Gifford CA, Ramsdell TL, Mekalanos JJ. Threonine phosphorylation post-translationally regulates protein secretion in Pseudomonas aeruginosa. Nat Cell Biol. 2007; 9: 797–803. [DOI] [PubMed] [Google Scholar]
- 60. English G, Trunk K, Rao VA, Srikannathasan V, Hunter WN, Coulthurst SJ. New secreted toxins and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens. Mol Microbiol. 2012; 86: 921–936. 10.1111/mmi.12028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sana TG, Hachani A, Bucior I, Soscia C, Garvis S, Termine E, et al. The second type VI secretion system of Pseudomonas aeruginosa strain PAO1 is regulated by quorum sensing and Fur and modulates internalization in epithelial cells. J Biol Chem. 2012; 287: 27095–27105. 10.1074/jbc.M112.376368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe. 2010; 7: 25–37. 10.1016/j.chom.2009.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lesic B, Starkey M, He J, Hazan R, Rahme LG. Quorum sensing differentially regulates Pseudomonas aeruginosa type VI secretion locus I and homologous loci II and III, which are required for pathogenesis. Microbiology. 2009; 155: 2845–2855. 10.1099/mic.0.029082-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Matz C, Deines P, Boenigk J, Arndt H, Eberl L, Kjelleberg S, et al. Impact of violacein-producing bacteria on survival and feeding of bacterivorous nanoflagellates. Appl Environ Microbiol. 2004; 70: 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Demain AL. Why do microorganisms produce antimicrobials Society for general microbiology: Cambridge University Press; 1995. pp. 205. [Google Scholar]
- 66. Proenca DN, Espirito SC, Grass G, Morais PV. Draft genome sequence of Serratia sp. strain M24T3, isolated from pinewood disease nematode Bursaphelenchus xylophilus. J Bacteriol. 2012; 194: 3764 10.1128/JB.00670-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Harris AK, Williamson NR, Slater H, Cox A, Abbasi S, Foulds I, et al. The Serratia gene cluster encoding biosynthesis of the red antibiotic, prodigiosin, shows species- and strain-dependent genome context variation. Microbiology. 2004; 150: 3547–3560. [DOI] [PubMed] [Google Scholar]
- 68. Thomashow LS, Weller DM, Bonsall RF, Pierson LS. Production of the antibiotic phenazine-1-carboxylic Acid by fluorescent pseudomonas species in the rhizosphere of wheat. Appl Environ Microbiol. 1990; 56: 908–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang WL, Liu J, Huo YB, Ling JQ. Bacteriocin immunity proteins play a role in quorum-sensing system regulated antimicrobial sensitivity of Streptococcus mutans UA159. Arch Oral Biol. 2013; 58: 384–390. 10.1016/j.archoralbio.2012.09.001 [DOI] [PubMed] [Google Scholar]
- 70. Dobson A, Cotter PD, Ross RP, Hill C. Bacteriocin production: a probiotic trait?. Appl Environ Microbiol. 2012; 78: 1–6. 10.1128/AEM.05576-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Monreal J, Reese ET. The chitinase of Serratia marcescens. Can J Microbiol. 1969; 15: 689–696. [DOI] [PubMed] [Google Scholar]
- 72. Vaaje-Kolstad G, Horn SJ, Sorlie M, Eijsink VG. The chitinolytic machinery of Serratia marcescens—a model system for enzymatic degradation of recalcitrant polysaccharides. FEBS J. 2013; 280: 3028–3049. 10.1111/febs.12181 [DOI] [PubMed] [Google Scholar]
- 73. Suzuki K, Sugawara N, Suzuki M, Uchiyama T, Katouno F, Nikaidou N, et al. Chitinases A, B, and C1 of Serratia marcescens 2170 produced by recombinant Escherichia coli: enzymatic properties and synergism on chitin degradation. Biosci Biotechnol Biochem. 2002; 66: 1075–1083. [DOI] [PubMed] [Google Scholar]
- 74. Zarei M, Aminzadeh S, Zolgharnein H, Safahieh A, Daliri M, Noghabi KA, et al. Characterization of a chitinase with antifungal activity from a native Serratia marcescens B4A. Braz J Microbiol. 2011; 42: 1017–1029. 10.1590/S1517-838220110003000022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Parani K, Shetty GP, Saha BK. Isolation of Serratia marcescens SR1 as a Source of Chitinase Having Potentiality of Using as a Biocontrol Agent. Indian J Microbiol. 2011; 51: 247–250. 10.1007/s12088-011-0065-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Frankowski J, Lorito M, Scala F, Schmid R, Berg G, Bahl H. Purification and properties of two chitinolytic enzymes of Serratia plymuthica HRO-C48. Arch Microbiol. 2001; 176: 421–426. [DOI] [PubMed] [Google Scholar]
- 77. Monreal J, Reese ET. The chitinase of Serratia marcescens. Can J Microbiol. 1969; 15: 689–696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
From outer to inner layer: (1) nucleotide positions in kilobases (kb) (black); (2) COG database-annotated CDSs (light red); (3) ACLAME database-annotated potential horizontal transferring genes (orange: from plasmids, blue: from prophage and black: from virus); (4) ARDB-annotated potential drug resistant genes (green); (5) prodigiosin biosynthetic gene cluster (red), genes related to biosynthesis of chitinases (black), siderophores (blue); (6) tRNA region (purple), rRNA (red); (7) Predicted genomic islands (yellow); (8) GC density.
(PDF)
Progressive Mauve [33] alignment of S. marcescens FS14, FGI 94, WW4 and Db11 genome sequences with default parameters. Each same color block represents a locally collinear block (LCB) (i.e. homologous region shared by genomes without any rearrangements). Rearrangement of genomic regions was observed in the four genomes, in term of collinearity and their localization on the negative or positive strand (indicated by their genomic position below or above the black horizontal center line in the Mauve alignment, respectively).
(PNG)
Maximum Likelihood Tree based on amino acid sequences of the conserved T3SS ATPase associated with S. marcescens FGI 94 constructed by 44-representive orthologs from each species using MEGA5. 7 different families of T3SS were identified. T3SS in S. marcescens FGI 94 was found in the Hrp1 family, which mainly composed of plant pathogens. The ATPase of the flagellum of E. coli was used as an outgroup. Bootstrap values are shown as percentages of 100 replicates, numbers at nodes represent bootstrap values, and only bootstrap values of >50 are shown.
(TIF)
COG-annotated genes of S. marcescens FS14 were compared to 9 other Serratia genomes: S. marcescens WW4 (CP003959), S. marcescens Db11 (HG326223), S. marcescens FGI 94 (CP003942), S. proteamaculans 568 (CP000826), S. plymuthica 4Rx13 (CP006250), S. plymuthica S13 (CP006566), S. plymuthica AS13 (CP002775), S. liquefaciens ATCC27592 (CP006252), and S. fonticola RB-25 (CP007044).
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
Genome data are deposited into NCBI GenBank (accession number: CP005927) and may be downloaded by following: http://www.ncbi.nlm.nih.gov/nuccore/CP005927.