

Abstract
Hematopoietic stem cells (HSC) rely on instructive cues from the bone marrow (BM) niche to maintain their quiescence and adapt blood production to the organism’s needs. Alterations in the BM niche are commonly observed in blood malignancies and directly contribute to the aberrant function of disease-initiating leukemic stem cells (LSC). Here, we review recent insights into the cellular and molecular determinants of the normal HSC niche and describe how genetic changes in stromal cells and leukemia-induced BM niche remodeling contribute to blood malignancies. Moreover, we discuss how these findings can be applied to non cell-autonomous therapies targeting the LSC niche.
Introduction
HSCs self-renew and differentiate into all the cells of the hematopoietic system, and are responsible for lifelong blood production (Orkin and Zon, 2008). Under normal conditions, HSCs are found in the BM in specialized niche microenvironments that are critical for their maintenance and functional activity. Stem cell niches were first postulated to exist by Schofield in his pioneering review article on spleen colony-forming units (CFU-S) in the 1970s (Schofield, 1978). Building on these early observations, technical advancements over the past several decades have allowed detailed visualization and mechanistic studies of the key cellular and molecular determinants of the HSC niche. Moreover, the remodeling of the BM microenvironment has emerged as an important event in the development of blood malignancies, involved in controlling the maintenance and activity of disease-initiating LSCs and their progeny. Understanding the differences between normal and malignant BM niches may therefore hold the key to developing non cell-autonomous therapies for a broad range of blood disorders. In this review, we highlight recent work deciphering the normal HSC niche, describe the role of these cellular and molecular niche components in disease settings focusing on myeloid malignancies, review experimental evidence of an active role for the leukemic BM niche in disease development, and discuss therapeutic targeting to abrogate self-reinforcing leukemic niches and restore normal hematopoiesis.
The HSC niche: a puppet master
The HSC niche is now viewed as a complex ecological system found at many locations in different bones, and is composed of a large number of cell types with specialized functions that provide distinct chemical signals and physical interactions essential for HSC maintenance and regulation of blood production (Figure 1). The cellular components of the BM niche can be categorized into two functional types: essential cell types like endothelial cells (EC), mesenchymal stromal cells (MSC) and megakaryocytes (Meg), which provide close proximity signals to HSCs; and accessory cell types like osteoblasts (OB), specialized macrophages and nerve cells, which exert long-range and often indirect influences on HSCs. A number of the signals provided locally by the BM niche cells are known, and their roles in controlling HSC function are now well understood (Pietras et al., 2011; Frenette et al., 2013). Secreted factors like stem cell factor (SCF), transforming growth factor beta-1 (TGF-β1), platelet factor 4 (PF4 or CXCL4), angiopoietin 1 (ANGPT1) and thrombopoietin (TPO) are all critical enforcers of HSC quiescence. Alongside the essential chemokine stromal-derived factor 1 (SDF1α or CXCL12) and its C-X-C chemokine receptor type 4 (CXCR4), adhesion molecules such as vascular cell adhesion protein 1 (VCAM-1), various selectins, and extracellular matrix (ECM) proteins like fibronectin or hyaluronic acid, are all essential regulators of HSC homing and anchoring in the niche. Finally, cell-bound molecules like Notch ligands or locally secreted cytokines like interleukin 7 (IL-7) or erythropoietin (EPO) are important controllers of HSC proliferation and differentiation activity. In adult bones, HSCs are essentially kept in the G0 phase of the cell cycle in a stage of metabolic dormancy or quiescence, which preserves their function by limiting damage associated with cell replication (Bakker and Passegué, 2013). However, quiescent HSCs can quickly respond to a broad range of niche or systemic signals by entering the cell cycle and proliferating (Pietras et al., 2011). These instructive cues are therefore essential for tailoring HSC differentiation and adjusting blood production to the needs of the organism. HSCs can also leave the BM niche upon receiving mobilization signals and enter the bloodstream to ensure immune surveillance of peripheral tissues (Massberg et al., 2007) and engraft distant BM sites (Wright et al., 2001). Thus, HSCs critically depend on short and long-range instructive cues from the BM niche for many aspects of their biology, including cell cycle and trafficking activity, due to the dynamic regulation of the switch between quiescence/proliferation and anchoring/mobilization.
Figure 1. Organization of the HSC niche.
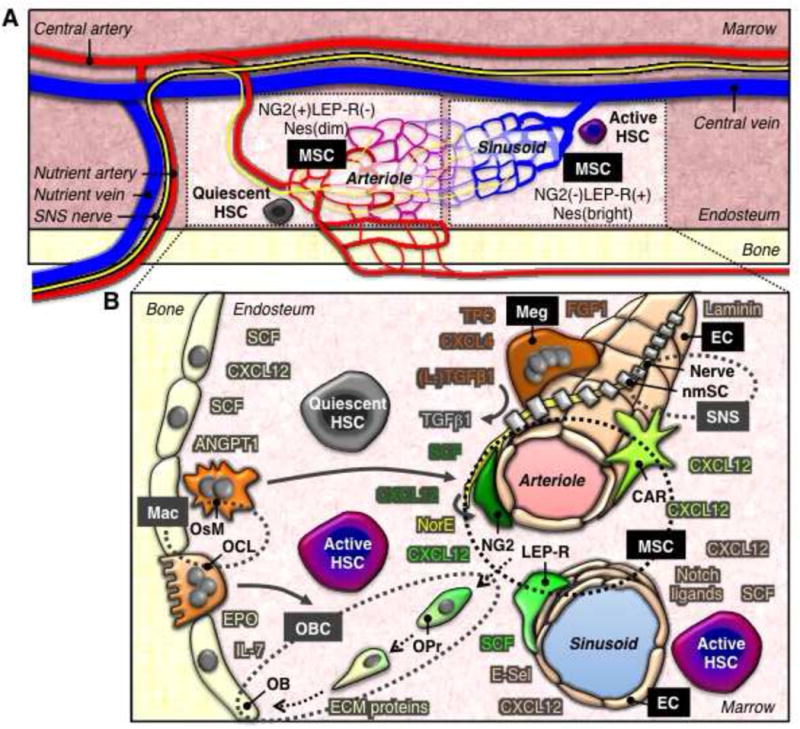
A) Overall anatomy of the marrow cavity depicting the sympathetic innervation and the vasculature, and highlighting the interconnection between arteriole and sinusoid blood vessels. Each of these regions (dotted box) is enriched for a particular subset of perivascular MSCs, which controls a different HSC functional state. Quiescent HSCs are G0 dormant cells. Active HSCs are cells that have just exited quiescence or are already actively cycling or migrating.
B) Blow up of the essential (black) and accessory (grey) HSC niche cells with their respective secreted and/or cell-bound factors (color-coded) that regulate HSC functional states. Doted circles group cells with either similar origin (i.e., perivascular MSC subsets and differentiating OBCs) or similar function (i.e., specialized macrophages (Mac) and SNS components). Black arrows highlight MSC progeny that are differentiating into bone-lining OBs and forming the OBC compartment. Grey arrows indicate the long-range, indirect effects of several accessory HSC niche cells. CAR: CXCL12bright MSCs; E-Sel: E-selectin; LEP-R: NG2−LEP-R+Nesbright MSCs; NG2: NG2+LEP-R−Nesdim MSCs; nmSC: non-myelinating Schwann cells; NorE: norepinephrine; OCL: osteoclasts; OPr: osteoprogenitors; OsM: osteomacs.
The identity of HSC-supportive BM niche cells has recently emerged from technical breakthroughs in imaging HSCs in the marrow cavity, coupled with a series of elegant functional studies in murine models. Immunofluorescence visualization of Lin−CD41−CD48−CD150+ HSCs in their native BM microenvironment shows that while present in the OB-rich trabeculated areas and endosteal bone surfaces, close to 60% of HSCs are directly associated with the vasculature (Figure 1A) (Kiel et al., 2005; Kunisaki et al., 2013). The marrow vasculature is essentially composed of a dense network of small arterioles and sinusoids that are enriched along the bone surface (Nombela-Arrieta et al., 2013). These thin-walled blood vessels are made from a single EC layer and are surrounded by perivascular MSCs and other non-circulating hematopoietic cells like large mature Megs. Arterioles branch up and down the endosteal bone surface from large nutrient arteries and connect to sinusoids, which are then collected into central veins. Both nutrient arteries and central veins enter and exit the long bones via the same nutrient foramina, with sympathetic nerve fibers following a similar path and spreading along the arterioles. The low blood flow of the arteriole/sinusoid network also limits gas exchange and creates a relatively hypoxic microenvironment (Spencer et al., 2014). The marrow cavity is therefore extremely well vascularized and innervated with an immediate access to systemic factors, and a quick ability for HSCs and other hematopoietic cells to enter or leave the bones through the bloodstream. These key features are essential for proper HSC regulation, which explain why ECs and most cell types found in close proximity to the vasculature have HSC-supportive activity (Figure 1B).
Essential components
Essential HSC niche cells have been identified through genetic ablation studies in mice in which their specific deletion or functional inactivation resulted in decreased numbers of HSCs maintained in the BM niche. These cells share several common features including direct contact with HSCs and production of signals important for enforcing HSC quiescence and anchoring HSCs in the niche. The large functional overlap between these cells also contributes to the resilience of the HSC niche, and explains in large part the rather limited impact of most genetic ablation studies.
Vascular ECs
ECs are located at the interface between the bloodstream and the interstitial stromal microenvironment surrounding the blood vessels. ECs are identified using the endothelial-specific markers CD31, MECA-32, V-cadherin and vascular endothelial growth factor receptor 2 (VEGFR2), or by their abundant expression of the ECM basal membrane component laminin. They express many HSC-supportive factors including SCF and CXCL12, various Notch ligands that stimulate HSC self-renewal expansion (Butler et al., 2010), and other cell-bound molecules such as the endothelial-leukocyte adhesion molecule 1 (E-selectin), which promotes HSC proliferation (Winkler et al., 2012). ECs are directly involved in maintaining HSCs in the BM niche as shown by several studies investigating the consequence of Tie2-mediated deletion of Scf or Cxcl12 expression (Ding et al., 2012; Ding and Morrison, 2013; Greenbaum et al., 2013). TIE2 is the receptor for ANGPT1 and is only expressed by the vasculature and some hematopoietic cells. ECs have therefore a widespread role in regulating HSC activity in the adult BM, in addition to their essential function in specifying and educating HSCs during fetal development (Orkin and Zon, 2008; Tamplin et al., 2015)
Perivascular MSCs
MSCs are stem cells for the entire mesenchymal lineage producing bone, cartilage and fat cells, as well as various fibroblast-like stromal cells with essential scaffolding roles in depositing and shaping ECM components. BM MSCs are rare cells that wrap around arteriole and sinusoid vessels, and are in direct contact with the non-luminal side of ECs. Perivascular BM MSCs may, in fact, share functional properties and developmental origins with pericytes that are more broadly found around the smallest blood vessels throughout the body (Crisan et al., 2008; Armulik et al., 2011). They can be visualized with a particular nestin (Nes)-GFP transgene (Méndez-Ferrer et al., 2010) and identified using the classical mesenchymal-specific markers platelet-derived growth factor receptor alpha (PDGFRA or CD140a), CD51 and Sca-1 (Winkler et al., 2010; Pinho et al., 2013), or expression of the leptin receptor (LEP-R) (Zhou et al., 2014) and the pericyte marker neuron/glial antigen 2 (NG2) (Kunisaki et al., 2013). Although there is a significant functional overlap between MSC populations defined with these markers in terms of multipotency and self-renewal capacities, their distribution in the BM cavity and their association with specific blood vessels is distinct (Frenette et al., 2013; Morrison and Scadden, 2014). While NG2+LEP-R−Nesdim pericyte-like MSCs are essentially peri-arteriolar and located close to the endosteal bone surface, NG2−LEP-R+Nesbright reticular-like MSCs are peri-sinusoidal and located more centrally, away from the endosteum (Kunisaki et al., 2013). In addition, quiescent Ki67− HSCs co-localize with peri-arteriolar NG2+ MSCs, while activated Ki67+ HSCs move towards peri-sinusoidal LEP-R+ MSCs, suggesting a differential involvement of MSC subsets in enforcing HSC quiescence and promoting HSC proliferation, respectively. As expected, perivascular MSCs express many HSC-supportive factors including SCF and CXCL12. In fact, high expression of Cxcl12 defines a subset of perivascular MSCs called CXCL12-abundant reticular (CAR) cells, which is important for controlling HSC proliferation and is likely a more mature progenitor subset (Sugiyama et al., 2006; Omatsu et al., 2010). Moreover, perivascular MSCs, like ECs, are directly involved in maintaining HSCs in the BM niche as shown by a series of studies investigating the consequence of either Nes- or Cxcl12-mediated MSC ablation (Méndez-Ferre et al., 2010; Omatsu et al., 2010) or Lep-R- or Prx1-mediated deletion of Scf and Cxcl12 expression in MSCs (Ding et al., 2012; Ding and Morrison, 2013; Greenbaum et al., 2013). Perivascular MSCs therefore have a more specialized function than ECs in controlling HSC cell cycle and trafficking activity, eventually acting through re-localization of activated and/or migrating HSCs to different MSC subsets with different perivascular localizations.
Non-circulating mature Megs
Mature Megs are differentiated hematopoietic cells that are abundant in the BM cavity and other organs in the body, and are found transiently associated with sinusoidal blood vessels for platelet production. They are identified by their unique large and multinucleated morphology and by expression of specific megakaryocytic-markers including CD41, CXCL4 and von Willenbrand factor (vWF). Megs secrete many quiescence-enforcing factors including CXCL4 (Bruns et al., 2014), TGF-β1 (Zhao et al., 2014) and TPO (Nakamura-Ishizu et al., 2014), as well as signals that activate HSC proliferation in regenerative stress conditions such as fibroblast growth factor 1 (FGF1) (Zhao et al., 2014). HSCs are often found in direct contact of Megs, and results from recent Cxcl4-mediated ablation studies demonstrate that Megs are important regulators of HSC cell cycle activity (Kiel et al., 2005; Bruns et al., 2014; Zhao et al., 2014). Mature Megs are therefore an even more specialized component of the HSC niche, which tailor HSC cell cycle activity based on demands and are particularly important in enforcing HSC quiescence.
Accessory components
Accessory HSC niche cells have been identified through similar mouse genetic ablation studies but instead of affecting the number of HSCs maintained in the BM niche, they essentially alter HSC differentiation potential and trafficking ability. These cells can be located at a distance or in close proximity of HSCs, and they usually impact on HSC function by producing factors with long-range effects and by influencing the activity of other BM niche cells.
Differentiating osteoblastic lineage cells
Osteoblastic lineage cells (OBCs) are an imperfectly defined compartment of MSC progeny committed to the osteoblastic lineage that comprises many intermediary stages of differentiation, including immature osteoprogenitors (OPr) and mature bone-lining and bone-forming OBs. OBCs still express some MSC markers like CD51 and the activated leukocyte cell adhesion molecule (ALCAM or CD166), but can be distinguished from MSCs by lack of expression of CD140a and Sca-1 (Nakamura et al., 2010; Winkler et al., 2010). As such, OBCs express transcription factors specific to OPr like Runx2 and Osterix (Sp7), and markers of mature OBs like osteocalcin. In addition, Lin−CD45−CD51+Sca1− OBCs show a significant overlap with CAR MSCs (Schepers et al., 2013), which have more restricted differentiation potential along the adipocytic and osteoblastic lineages and also control the proliferation of lymphoid and erythroid progenitors (Omatsu et al., 2010). OBCs can be genetically manipulated using the Sp7 promoter to target MSCs/OPrs, and the Col1-α1 2.3kb (Col1) promoter-fragment to target mature OBs (Méndez-Ferrer et al., 2015). Differentiating OBCs express many factors important for HSC function including CXCL12, SCF and ANGPT1, as well as ECM proteins like osteocalcin, collagen 1 and osteopontin (Winkler et al., 2010; Schepers et al., 2013). Mature OBs were initially proposed to be essential HSC niche cells based on a positive correlation between OB and immature hematopoietic stem and progenitor cell (HSPC) numbers in two engineered murine models with more OBs (Calvi et al., 2003; Zhang et al., 2003). However, additional studies have since refuted this observation, and showed that mature OBs do not directly affect HSC maintenance in the BM (Frenette et al., 2013). Accordingly, Sp7- or Col1-mediated deletion of Scf or Cxcl12 expression in OBCs does not alter HSC numbers but, instead, reduces the numbers of B lymphoid progenitors (Ding and Morrison, 2013; Greenbaum et al., 2013). In fact, OBs directly secrete IL-7 and are important to control B lymphopoiesis (Wu et al., 2008). OBs also produce EPO and regulate erythropoiesis in response to hypoxic signals (Rankin et al., 2012). Differentiating OBCs are therefore emerging has having a dual function in controlling HSC activity, with the most immature CAR-like OPr subset influencing HSC proliferation, and the most mature OB subset directly tailoring HSC differentiation along the lymphoid and myeloerythroid lineages.
Specialized macrophages
Bone-associated macrophages such as osteomacs (OsM) and bone-resorbing osteoclasts (OCL) are another class of hematopoietic cells that have recently emerged as important niche components (Morrison and Scadden, 2014). OCLs are large multinucleated cells that are positive for tartrate-resistant acid phosphatase (TRAP) activity and resorb bone tissue by secreting both acids and endogenous collagenases. OCLs also regulate through complex cross talk mechanisms MSC differentiation and the production of mature bone-forming OBs, thereby indirectly impacting on HSC function. Blockade of OCL function leads to impaired B lymphopoiesis due to decreased expression of CXCL12 and IL-7 by improperly maturing OBs (Mansour et al., 2011). Moreover, complete OCL ablation in osteopetrotic oc/oc mice results in a dysfunctional HSC niche in which the accumulation of MSCs unable to properly differentiate into OBs directly impair HSC homing (Mansour et al., 2012). Similarly, deletion of OsM, which are a sub-population of bone-associated macrophages expressing F4/80 and CD169, results in HSC mobilization due to indirect effects on other BM niche components (Winkler et al., 2010; Chow et al., 2011). In particular, Nes+ MSCs in OsM-depleted mice have decreased expression of many HSC-supportive factors, including CXCL12 and SCF, which could directly contribute to the observed defect in HSC niche anchoring (Chow et al., 2011). Specialized bone-associated macrophages are therefore controlling HSC differentiation potential and trafficking activity through indirect modulation of MSCs and their OBC progeny.
Sympathetic nervous system
The sympathetic nervous system (SNS) is part of the autonomic nervous system, which acts largely unconsciously and regulates processes such as heart rate, digestion or respiratory rate. However, the SNS is constantly active at a basal level to maintain homeostasis, and it communicates with the body through release of catecholamines such as the neurotransmitter norepinephrine (NorE). NorE binds to β3-adrenergic receptors express by many cells including BM stromal components such as MSCs and OBs (Asada et al., 2013). In fact, the circadian clock and rhythmic secretion of NorE leads to rhythmic down regulation of Cxcl12 expression by BM stromal cells, hence triggering rhythmic release of HSCs from the BM niche and their mobilization to the bloodstream (Méndez-Ferrer et al., 2008). Sympathetic nerve fibers are also sheathed by both myelinating and non-myelinating Schwann cells, which ensure protection and electrical and chemical isolation between the axons. Nonmyelinating Schwann cells (nmSC) are Nes+ similar to some perivascular MSCs, and contribute to keeping HSCs quiescent by activating latent-TGF-β1 found in the surrounding microenvironment (Yamazaki et al., 2011). The SNS is therefore regulating HSC proliferation and trafficking activity through long-range signals and indirect modulation of the activity of other BM niche components.
Advances in understanding the human HSC niche
In contrast to the detailed understanding of the murine HSC niche, the visualization and mechanistic understanding of the key cell types and factors controlling human HSC function in the marrow cavity remains limited mainly due to the difficulty of accurately modeling these complex relationships in the human system. Immunohistochemistry studies indicate that 86% of the primitive CD34+ human HSPCs localize closely to CD271+ alkaline phosphatase (ALP) positive perivascular MSCs that are positive for CXCL12 (Flores-Figueroa et al., 2012), and that CD45+CD34+CD38− human HSCs are enriched in the trabecular areas of the bone (Guezguez et al., 2013). In vitro co-culture studies have provided further information about the identity of the human HSC-supportive BM niche cells and the environmental factors regulating human HSC function. Similar to the mouse, ECs, MSCs and broadly defined OBCs have all been shown to support the maintenance and/or expansion of primitive HSPCs in humans (Taichman, 2005). Two recent studies have highlighted the importance of MSCs by showing that Lin−CD45−CD51+CD140a+CD146+ enriches for a subset of Nes+ MSCs with HSC niche activity in human fetal BM (Pinho et al., 2013), and that Lin−CD45−CD271+CD140a− identifies a population of MSCs in human adult BM that expresses many HSC-supportive factors and are capable of enhancing the in vivo repopulating ability of cultured human HSPCs (Li et al., 2014). To better recapitulate the interactions occurring in human bones, in vitro co-culture models are now moving towards advanced 3D systems using various mixtures of purified BM niche cells (Leisten et al., 2012; Sharma et al., 2012; Raic et al., 2014). Similarly, xenotransplantation approaches are rapidly evolving towards injecting human hematopoietic cells into immunodeficient mice engrafted with ectopic bone-containing human BM microenvironments (Chen et al., 2012; Groen et al., 2012). These new approaches, which also allow genetic manipulation of human niche components, have been successfully used in studying leukemic cell engraftment and will likely help in better understanding the human HSC niche. Current studies hint at many similarities between humans and mice in terms of composition and function of the HSC niche. However, since human HSCs are distinct from mouse HSCs for certain aspects of their biology, the human HSC niche is likely to have unique features that might not be recapitulated in murine models.
Emerging questions
The recent identification of novel bone and fibrosis producing MSC (Kramann et al., 2015) and skeletal stem cell (Worthley et al., 2015; Chan et al., 2015) populations in mice highlight the fact that much remains to be learned about the origin and HSC-supporting function of perivascular MSCs and their derivatives both in mice and humans. In particular, the role of adipocytes still needs to be fully explored given their effect in impairing HSC maintenance (Naveiras et al., 2009). Future work should also refine the purification techniques to better separate the different OBC subsets as well as arterial vs. sinusoidal ECs, and to gain a better understanding of the dynamic nature of the HSC niche and its different cellular constituents. Although some information are currently available about MSC turnover in vivo (Park et al., 2012), the precise kinetics of MSC differentiation and how fast MSCs and their OBC derivatives are at forming and, eventually, resorbing the HSC niche remain entirely unknown. In addition, while many environmental cues are known to trigger a switch from quiescence to proliferation, less is understood about the mechanisms by which quiescence is re-established in HSCs and how BM niche cells contribute to this process. Another largely un-explored aspect of the HSC niche is the biophysical properties of the BM microenvironment including stiffness and ECM composition, and its role in controlling HSC function. Similarly, little is known about potential variability in HSC niche composition depending on anatomical location and bone types, although the overall structure the BM microenvironment is conserved between long bones and the skull, which is often used for intravital live imaging approaches (Lo Celso et al., 2009; Lassailly et al., 2013). Finally, we still need to understand better how these or other BM niche components contribute to the biology of lymphoid and myeloerythroid progenitors, potentially creating similar and/or distinct progenitor niches for the development of these lineages in the marrow cavity.
The LSC niche: a partner in crime
HSCs are constantly exposed to both intrinsic and extrinsic stresses, which can cause DNA damage and lead to mutations if not properly resolved (Bakker and Passegué, 2013). These mutations accumulate with age and can result in malignant transformation (Welch et al., 2012). Many human myeloid malignancies including myeloproliferative neoplasms (MPN) like chronic myelogenous leukemia (CML) and myelodysplastic syndromes (MDS) originate from genetic defects occurring in HSCs (Huntly and Gilliland, 2005; Woll et al., 2014). These transformed HSCs maintain the capability to self-renew and to give rise to various lineages of blood cells, albeit in a deregulated manner, and behave as disease-initiating LSCs (Passegué et al., 2003). Functionally, LSCs are characterized by the ability to initiate and serially propagate diseases upon transplantation, thereby recreating the primary malignancy and its full heterogeneity in recipient mice. In more aggressive diseases, such as acute myeloid leukemia (AML) and blast crisis CML (BC-CML), LSCs are no longer restricted to the HSC compartment and can also emerge from transformed progenitors (Eppert et al., 2011; Goardon et al., 2011). For instance, in both human BC-CML and an MLL-AF9-driven syngeneic murine AML transplantation model, clonal evolution and stabilization of nuclear β-catenin can transform committed granulocyte-macrophage progenitors (GMP) into LSCs that have re-acquired the ability to self-renew and propagate the leukemia (Jamieson et al., 2004; Krivtsov et al., 2006; Wang et al., 2010). Hence, LSCs can have diverse cells of origin and even evolve from HSCs to myeloid progenitors in the course of the disease.
Genetic changes are well-documented in myeloid malignancies and involve mutations in signaling molecules such as JAK2, RAS, FLT3 or KIT (Milosevic and Kralovics, 2013). These genetic lesions provide important cell autonomous growth signals to leukemic cells but also alter how LSCs and their progeny sense environmental signals. Building on the early observations of altered adhesion properties in human CML progenitors (Gordon et al., 1987), more recent intravital imaging approaches have shown that leukemic cells highjack normal BM vascular niche spaces by exploiting signals such as CXCL12 and E-selectin that are important for the homing of healthy HSCs (Sipkins et al., 2005). In addition, LSCs are less dependent than healthy HSCs on certain niche signals for their survival, proliferation and anchoring in the niche. LSCs in murine models of both CML and AML diseases become insensitive to Notch and TGF-β-mediated environmental signals (Santaguida et al., 2009; Krause et al., 2013), which normally limit HSC expansion and myeloid differentiation. LSCs are also more dependent on CD44 extracellular anchoring and on various selectins and their ligands for homing and engraftment in the marrow cavity compared to normal HSCs, as shown both in a syngeneic murine CML transplantation model (Krause et al., 2006; 2014) and with human AML xenograft transplantation (Jin et al., 2006). In addition, LSCs in the syngeneic murine MLL-AF9 AML transplantation model display less dependency on Wnt-derived signals for localization to the marrow compared to normal HSCs (Lane et al., 2011). These examples clearly illustrate the deranged perception of the BM microenvironment by LSCs and their progeny, which is a critical concept in understanding how leukemic hematopoiesis can get a stronghold in the BM niche at the expense of normal hematopoiesis. In addition, it has recently become clear that leukemic hematopoiesis directly remodels the BM niche into a self-reinforcing malignant BM niche that supports disease development at the expense of normal hematopoiesis. Furthermore, striking observations in murine models have shown that genetic lesions directly in BM niche cells could contribute to or even initiate myeloid malignancies. These two modes of disease initiation are not mutually exclusive as demonstrated by the range of crosstalk and common self-reinforcing loops found in both situations, and can also synergize to predispose for additional transforming lesions and more aggressive diseases (Figure 2).
Figure 2. Models of disease initiation.
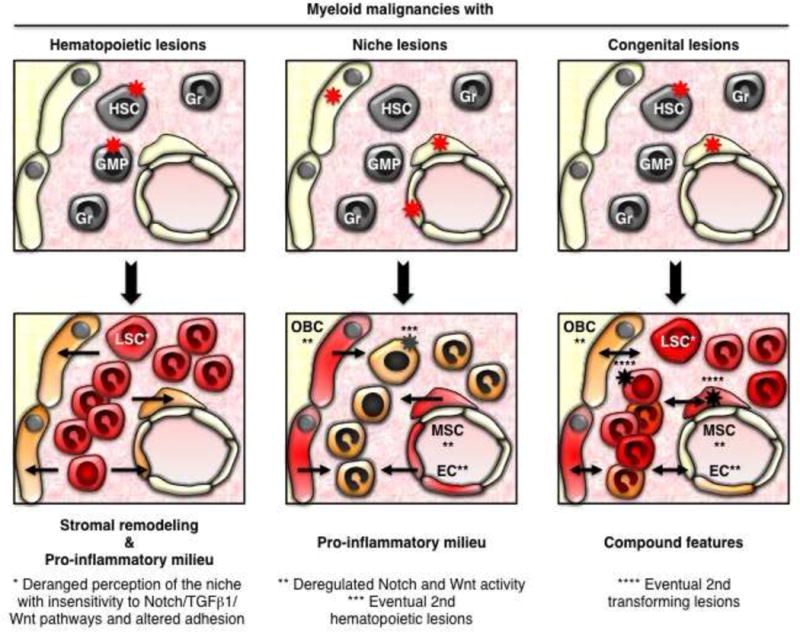
The majority of myeloid malignancies are caused by genetic lesions (stars) occurring in hematopoietic cells, which lead to BM niche remodeling and formation of LSCs with deranged perception of the microenvironment. Genetic lesions can also occur in stromal cells and lead to myeloid malignancies with predisposition to secondary mutations in hematopoietic cells. These two modes of disease initiation are not mutually exclusive as they share several common mechanisms and self-reinforcing loops including deregulated Notch/Wnt signaling and increased production of pro-inflammatory cytokines. Congenital lesions present in both hematopoietic and stromal cells are also observed in myeloid malignancies, and likely synergize as well as predispose for additional transforming lesions and more aggressive diseases. Arrows indicate the directionality of these events.
Niche as initiator of disease
The first evidence supporting the concept that transforming non-cell autonomous genetic changes could contribute to MPN-like diseases came from a murine study showing that deletions of the signaling molecule inhibitor of NF-κB, I kappa B alpha (IκBα), in the liver leads to the development of non-transplantable MPN-like diseases (Rupec et al., 2005). This is due to constitutive expression of the Notch ligand Jagged-1 by IκBα-deficient hepatocytes, which directly drives the aberrant overproduction of myeloid cells. In two subsequent landmark studies published in 2007, Walkley and colleagues directly demonstrated the importance of genetic BM niche changes in the development of myeloid disorders (Walkley et al., 2007a; 2007b). While Mx1-Cre-mediated deletion of the retinoblastoma (Rb) gene in both hematopoietic and stromal elements leads to a widespread MPN-like disease with splenomegaly, mobilization of cells from the BM and eventual loss of HSCs, neither its sole inactivation in hematopoietic cells nor in stromal elements, including BM niche cells, in reciprocal transplantation approaches results in myeloid disorders (Walkley et al., 2007a). In addition, only transplantation of Rb-deleted myeloid cells into Rb-deficient, but not wild type, mice induces the MPN-like disease, thus confirming the dependency of genetically altered myeloid cells on a transformed BM microenvironment. Similarly, while constitutive deletion of the retinoic acid receptor gamma (Rarγ) results in the development of an age-related MPN-like diseases, only transplantation of normal hematopoietic cells into Rarγ-deficient mice, and not of Rarγ-deficient hematopoietic cells into wild type mice, is able to recreate the myeloid condition (Walkley et al., 2007b). Along the same lines, MMTV-mediated deletion of the E3-ubiquitin ligase and canonical Notch ligand regulator mind bomb 1 (Mib1) in stromal elements causes a MPN-like disease that is independent of Mib1 status in the hematopoietic compartment, and can be fully reversed by constitutive activation of Notch signaling in the microenvironment (Kim et al., 2008). Together, these studies highlight the role of non-hematopoietic BM stromal elements in disease initiation, and two subsequent studies hint at MSCs and OBCs as being key players in this process. Sp7-mediated deletion of the miRNA processing Dicer1 gene in MSCs/OPrs is sufficient to drive the development of an MDS-like disease with sporadic transformation to AML, which can be fully reverted upon transplantation into wild type mice (Raaijmakers et al., 2010). Loss of Dicer1 in MSCs/OPrs leads to reduced expression of the ribosome maturation protein Sbds gene, which is mutated in human Shwachman-Bodian-Diamond syndrome, a congenital BM failure disease with known leukemic predisposition. Similarly, Col1-mediated expression of stabilized nuclear β-catenin and constitutive activation of the Wnt pathway in OBCs is sufficient to drive the development of a transplantable AML-like disease with common chromosomal aberrations (Kode et al., 2014). Activated β-catenin stimulates expression of the Notch ligand Jagged-1 by mature OBs, which in turn leads to aberrant activation of Notch signaling in hematopoietic cells and the observed AML-like condition. Another recent study suggests that ECs can also play a direct role in disease initiation. Tie2-mediated deletion of the Rbpj (also known as Cbf1) gene, a non-redundant downstream effector of the canonical Notch signaling pathway, in ECs is sufficient to cause a fatal MPN-like disease (Wang et al., 2014). Loss of Notch signaling in ECs up-regulates miR-155, which directly targets the NF-κB inhibitor κB-Ras1 and activates NF-κB thereby increasing their production of pro-inflammatory cytokines including G-CSF and tumor necrosis factor alpha (TNFα). This pro-inflammatory milieu, in turn, directly drives an MPN-like disease condition. Together, these studies in murine models demonstrate a direct causal role for several HSC niche components (i.e., MSCs, OBCs and ECs) in the development of a broad range of myeloid diseases, and identify deregulated Notch and Wnt signaling pathways, as well as increased production of pro-inflammatory cytokines, as commonly altered mechanisms in BM stromal elements that drive aberrant production of myeloid cells (Figure 2).
These findings beg the question of whether BM niche changes are also important for disease initiation in humans. The evidence is so far limited to correlative data obtained with patient samples. Consistent with a model of increased β-catenin in OBCs driving Notch signaling in human AML cells, 38.3% of a cohort of over 80 patients with MDS, AML, or MDS that progressed to AML had BM biopsies showing increased nuclear β-catenin in Runx2-expressing OBCs associated with increased Notch activity in CD34+ HSPCs (Kode et al., 2014). In support of a role for stromal up-regulation of miR-155 in human MPNs, a significant overexpression of miR-155 was observed in BM aspirates from primary myelofibrosis (PMF) patients (Wang et al., 2014). These data suggest that stromal genetic changes known to drive myeloid disorders in mice are also found in human tissues, and could similarly contribute to disease development. Other correlative and still controversial evidence includes the fact that stromal cell populations isolated from individuals with myeloid malignancies can carry genetic abnormalities that are different from the driver mutation(s) in the leukemic clone (Blau et al., 2007; Kastrinaki et al., 2013). One important caveat with these studies is that they were performed with serially expanded cells, which could therefore have acquired new genetic abnormalities as the direct consequence of the ex vivo culture. However, these results support the idea that genetic changes could independently occur in BM niche cells during the course of the disease. In addition, patients undergoing allogeneic stem cell transplantations (SCT) can develop a rare donor-derived leukemia that is distinct from classical recipient disease relapse (Wiseman, 2011). This phenomenon also supports the idea that an altered BM stromal compartment could directly contribute and/or drive the development of a new leukemia in these patients, although it remains to be determined whether the stromal changes are inherent abnormalities or result from the treatments received before SCT. Nevertheless, it is tempting to speculate that new genetic lesions could be acquired over time in HSC niche components (i.e., MSCs, OBCs and ECs) and directly contribute to the development of human myeloid diseases.
Diseases as initiator of niche changes
Ample experimental evidence in both humans and mice support the opposite concept that malignancies resulting from transforming genetic changes in hematopoietic cells cause a remodeling of the BM niche, which then contributes to disease progression. Many studies in the past several decades have described morphological and functional BM stromal changes in patients with various hematologic conditions, including PMF, MDS and AML. Structural changes in the BM cavity due to impaired angiogenesis and/or bone loss are now well documented in both AML and MDS (Dührsen and Hossfeld, 1996). In PMF, the degree of collagen fiber deposition and ECM remodeling is also directly correlated with overall patient survival (Pereira et al., 1990). In MDS, patient-derived stromal cells appear qualitatively different in their ability to support blast cell colonies and impaired in their ability to maintain long-term cultures of CD34+ HSPCs (Gidali et al., 1996; Aizawa et al., 1999), although these results are still somewhat controversial (Kastrinaki et al., 2013). Reduced contact inhibition in vitro, impaired ability to maintain hematopoietic differentiation and increased osteoblastic lineage gene expression are also aberrant features of stromal cells derived from pediatric MDS patients (Borojevic et al., 2004). In addition, tissue sections of MDS patients show abnormally high numbers of perivascular ALP+CD271+ MSCs expressing CXLC12 (Flores-Figueroa et al., 2012), which could directly contribute to the aberrant BM retention of HSPCs in this disease. In contrast, BM-derived MSCs from AML patients have decreased production of CXCL12 (Ge et al., 2011), which could also contribute to the impaired maintenance of healthy HSCs observed in this disease. Illustrating the crosstalk between leukemic cells and stromal elements, AML cells co-cultured with ECs produce a range of pro-inflammatory cytokines including TNFα, interleukin 6 (IL-6) and interleukin 1 (IL-1), which induce ECs to secrete the myeloid growth factors G-CSF and granulocyte/macrophage colony stimulating factor (GM-CSF). This, in turn, stimulates the growth of the leukemic cells and creates a feed-forward mechanism promoting disease development (Griffin et al., 1987; Oster et al., 1989). The existence of such self-reinforcing mechanisms hints at leukemia-induced BM niche remodeling as a mechanism for disease progression, an idea that is directly supported by recent live imaging and xenograft transplantation data. Key studies from the mid-2000s have visualized the engraftment of human leukemic cells in immunodeficient mice and showed the formation of specialized malignant niches that directly impair healthy HSC function due to overproduction of SCF (Sipkins et al., 2005; Colmone et al., 2008). Recently, human MDS cells were shown to directly rely on deregulated signals provided by their malignant BM niche, including factors such as leukemia inhibitory factor (LIF), VEGF, N-cadherin and other regulators of fibrosis, to efficiently engraft immunodeficient mice (Medyouf et al., 2014). Furthermore, after exposure to MDS cells, normal BM stromal cells adopt a phenotype resembling the malignant niche including LIF up-regulation (Medyouf et al., 2014). Together, these human data argue that malignancies invariably alter the function of the BM niche in ways that favor disease development and impair normal hematopoiesis. Recent studies in murine models have expanded upon these findings, and provide novel mechanistic insights into the formation and function of such self-reinforcing malignant niches (Figure 3).
Figure 3. Self-reinforcing malignant BM niches.
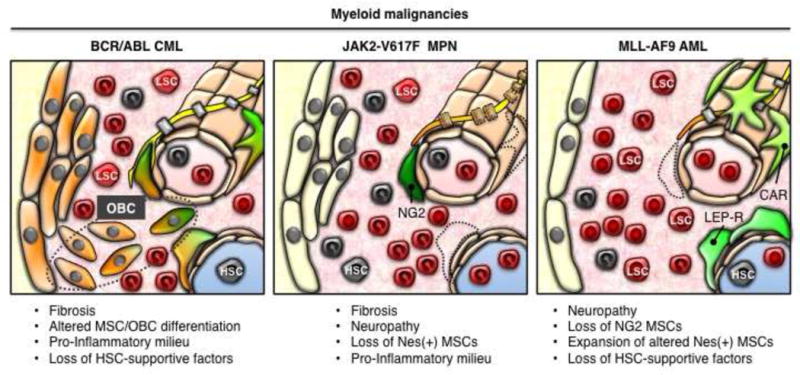
Mechanisms identified in the indicated murine models, which create a self-reinforcing malignant BM niche promoting disease maintenance at the expense of normal hematopoiesis. Several key features have emerged as common themes from these analyses and include (1) altered MSC growth and differentiation with eventual fibrosis, (2) elaboration of pro-inflammatory signals, and (3) decreased production of HSC-supportive factors by stromal cells.
Fibrosis of the malignant niche
CML is the human myeloid malignancy best studied in mice for its remodeling effect on BM stromal cells. CML cells produce G-CSF that decreases expression of CXCL12 by BM stromal cells and directly impairs normal HSC maintenance in an inducible BCR/ABL transgenic model (Zhang et al., 2012). In a syngeneic BCR/ABL transplantation model, CML cells also induce BM stromal cells to secrete platelet growth factor (PLGF), which in turn enhances CML cell proliferation in a self-reinforcing loop (Schmidt et al., 2011). Using the inducible BCR/ABL transgenic model, our group recently identified OBCs as the key HSC niche component directly remodeled by CML cells (Schepers et al., 2013). We show that CML cells stimulate MSCs to proliferate and adopt an abnormal differentiation program resulting in the overproduction of functionally altered OBCs, which accumulate in the BM cavity as inflammatory myelofibrotic cells. We find a role for TPO, the chemokine (C-C motif) ligand 3 (CCL3 or MIP-1α), and direct cell-cell interactions between CML myeloid cells and MSCs in driving OBC expansion, and for changes in TGF-β, Notch and pro-inflammatory signaling activity in remodeling OBCs into myelofibrotic cells. In turn, we show that myelofibrotic OBCs have decreased expression of many HSC retention factors, including CXCL12, and a compromised ability to maintain normal HSCs. In contrast, LSCs are not affected by this malignant remodeling of the niche, likely due to their deranged perception of the BM microenvironment (Krause et al., 2006; 2014). Myelofibrotic OBCs also express pro-inflammatory cytokines (i.e., IL-1 and TNFα), which likely amplify disease development and aberrant myeloid cell production. Together, these studies demonstrate that CML development is associated with the formation a self-reinforcing fibrotic malignant niche that favors LSCs and disease development at the expense of healthy HSCs and normal hematopoiesis. This likely contributes to the clonal dominance of certain LSCs in MPNs that are relatively unfit compared to normal HSCs (Kent et al., 2013), and do not have a strong driver mutation directly providing increased competiveness (Li et al., 2013).
Neuropathy of the malignant niche
Two recent studies using a knock-in Jak2V617F MPN mouse model (Arranz et al., 2014) and the syngeneic MLL-AF9 AML transplantation approach (Hanoun et al., 2014), show that leukemic cells can also damage nerve cells. In both cases, disease development creates neuropathic changes in the BM niche, which affect the activity of perivascular MSCs and alter the function of the HSC niche. Interestingly, IL-1-mediated damage to nerve fibers and nmSCs in Jak2V617F MPN mice leads to a loss of Nes+ MSCs, and an accelerated MPN phenotype that can be reversed by treatment with 4-methycatechol, a neuroprotective drug that signals through β3-adrenergic receptors (Arranz et al., 2014). Interestingly, Jak2V617F MPN mice develop BM fibrosis despite a reduction in size of the MSC compartment and the loss of Nes+ MSCs. In the MLL-AF9 AML transplantation model, in contrast, leukemic cells cause a loss of peri-arteriolar NG2+ MSCs and nerve fibers, which then leads to the expansion of an abnormal population of Nes+ MSCs with skewed osteoblastic differentiation and down-regulated expression of many HSC retention factors, including CXCL12 and SCF, but no detectable BM fibrosis (Hanoun et al., 2014). These changes cooperate in promoting AML development and loss of normal HSCs though their mobilization to the bloodstream. Together, these studies indicate that leukemia-induced neuropathy also promotes the development of a self-reinforcing malignant niche that also favors disease development at the expense of normal HSC maintenance. However, the relation between such leukemia-induced neuropathy and the formation of a fibrotic malignant niche is still unexplored.
Emerging questions
Many aspects of the contribution of the BM microenvironment to myeloid malignancies remain to be investigated. In particular, whether LSCs reside in specific niches that are directly instructing and/or maintaining them is still an open question. It is also currently unknown whether differences in cell of origin (i.e., HSCs vs. progenitors) influence where LSCs home and interact with specific BM niche components. However, what has clearly emerged from current studies in murine models and correlative evidence in patient samples is that BM stromal changes and the formation of a self-reinforcing malignant niche is more than a mere bystander effect of disease development, and can directly contribute to myeloid malignancies. A further understanding of this process in human diseases will require taking advantage of aforementioned 3D co-culture systems and humanized xenograft models to dissect the crosstalk between leukemic cells and the BM microenvironment. These approaches will be particularly useful to investigate the exact contribution to human diseases of genetic changes occurring in BM niche cells, and the translational relevance of the current observations in murine models. Much work also needs to be done in understanding the mechanisms driving the formation and aberrant function of the malignant BM niche in human diseases. Several key features have now emerged as common themes from murine studies and include: (1) altered MSC growth and differentiation as the most widespread consequence of disease development; (2) elaboration of pro-inflammatory signals such as TNFα, IL-1 and IL-6, which help drive myeloid malignancies and BM fibrosis; and (3) decreased production of HSC-supportive factors such as CXCL12, SCF and ANGPT1 by stromal cells, which harms healthy HSCs and favors LSCs. One of the most exciting questions is to address whether these recurrent themes are also important, and potentially targetable, features of malignant niche establishment in human diseases. It will also be interesting to test whether early detection of deranged niche signals, including inflammatory gene activation in BM niche cells and changes in production of niche-associated factors have a prognostic value. Finally, we still need to better understand the connection between the different perivascular MSC subsets and their OBC derivatives in driving BM fibrosis. In this context, the recent identification of Gli1+ MSCs as the general origin of organ fibrosis (Kramann et al., 2015) should help in identifying the source of BM fibrosis in myeloid malignancies.
Targeting the LSC niche: non-cell autonomous treatment strategies
The recent identification of many driver mutations and the development of drugs targeting these deregulated signaling pathways have had dramatic success in treating patients with myeloid malignancies. In the case of CML, BCR/ABL kinase inhibitors have completely changed the landscape of clinical outcomes, leading to longer patient survival and deeper remissions (Jabbour et al., 2013). There has also been success in targeting JAK2 mutations in MPNs as well as FLT3 activation in AMLs (Verstovsek et al., 2012; Jabbour et al., 2013). Even given these recent advancements, significant areas of need remain for the treatment of myeloid malignancies. These include treatment of patients without known driver mutations or for whom no drugs are currently available that target their driver mutations, as well as treatment of elderly patients who are not eligible for intense chemotherapy or curative SCT approaches. The appeal of targeting deranged BM microenvironments fits well with these clinical needs. It represents a non cell-autonomous treatment strategy that could be applied to a broad range of patients with various types of malignancies and underlying driver mutations. It could be a way to preserve normal HSCs and disfavor LSCs, thus deepening clinical remissions, and could easily be used as an adjunct therapy alongside drugs directly targeting driver mutations to aim for curative treatment. Recent discoveries have identified a series of exciting novel features of the LSC niche that could be targeted to abrogate the self-reinforcing malignant BM microenvironment and restore normal hematopoiesis. Some strategies like uncoupling LSCs and leukemic cells from their protective niches have already been tested in the clinic and are at various stages of drug development. Other strategies like targeting the MSC remodeling and inflammatory microenvironment are very promising but remain so far mainly at pre-clinical.
Uncoupling leukemic cells from their protective niches
Targeting factors that maintain minimal residual disease (MRD) in the BM niche to draw remaining LSCs and their progeny out of their protected microenvironment and enhance their killing has caught considerable interest over the past decade (Nair et al., 2010). Leukemic cells co-cultured with BM stromal cells are protected from drug-induced killing via stromal-mediated mechanisms involving prevention of apoptosis and induction of quiescence in LSCs and their progeny. Pre-clinical work in human settings and murine models has demonstrated the effectiveness of blocking the CXCL12-CXCR4 chemokine axis and targeting the adhesion molecules CD44 and VCAM-1 to dislodge leukemic cells from their protective BM niches. Overexpression of CXCR4 on LSCs and their progeny is one of best described mechanisms promoting the BM anchoring and quiescence of leukemic cells via its interaction with CXCL12, hence impairing the cytotoxic effects of various treatment modalities including BCR/ABL inhibitors in CML and Flt3 inhibitors in AML (Jin et al., 2008; Zeng et al., 2009; Nervi et al., 2009). Testing in xenograft models has shown that blocking CXCR4 could mobilize leukemic cells away from their BM stromal niches, leading to better in vivo disease eradication (Nervi et al., 2009; Weisberg et al., 2012). Efficient drugs for manipulating the CXCL12-CXCR4 axis have existed for years, including the FDA-approved Plerixafor (AMD3100) that is routinely used in clinical settings for mobilizing normal HSCs. A phase I/II clinical trial was conducted to test the safety of combining Plerixafor with cytotoxic chemotherapy in patients with relapsed AML, and recently reported as a safe strategy with beneficial clinical outcomes (Uy et al., 2012). Additional clinical trials are currently ongoing using Plerixafor, or other drugs targeting the CXCL12-CXCR4 axis, alone or in combination with other mobilizing agents such as G-CSF in order to sensitize LSCs to conventional chemotherapies both as front-line therapy for new AML patients and as second-line therapy for relapsed diseases. LSCs have also been found to be more dependent on the hyaluronic acid receptor CD44 for their anchoring in the BM niche than normal HSCs (Jin et al., 2006; Krause et al., 2006; 2014), which makes CD44 an exciting target to dislodge leukemic cells from their niche. As drugs to block CD44 already exist, including the FDA-approved anti-CD44v6 monoclonal antibody Bivatuzumab currently used for clinical trials in solid tumors, it will be exciting to see the potential of this strategy in myeloid malignancies especially for patients with MRD or relapsed disease. Finally, VCAM-1 engagement of the α4β1-integrin very large antigen 4 complex (VLA4) is an emerging mechanism mediating the crosstalk between leukemic cells and BM stromal cells that promotes chemoresistance (Jacamo et al., 2014). This interaction can be blocked with the FDA-approved monoclonal antibody Natalizumab, which targets both α4 and αE integrins and is currently used for the treatment of relapsing multiple sclerosis and inflammatory bowel disease. A small molecule specifically targeting VCAM-1 has recently been developed (Hsieh et al., 2014), and would be the ideal drug for testing in myeloid malignancies.
Targeting MSC remodeling and the inflammatory microenvironment
BM stromal remodeling through deregulated growth and differentiation of MSCs has emerged as a key theme in the development of myeloid malignancies. In human MPNs, this result in myelofibrosis, which ultimately culminates in the development of PMF diseases but also contributes to the pathogenesis of other MPNs as exemplified by work in murine models (Abdel-Wahab and Levine, 2009). Strategies aimed at preventing the development of a fibrotic niche and its deleterious consequence for the maintenance of normal HSCs (Schepers et al., 2013) could help restore the marrow’s ability to support healthy HSCs and production of normal blood cells. Currently there are no FDA-approved therapies specifically targeting the fibrotic BM niche, although reversal of myelofibrosis is a clinically achievable goal as shown in CML patients upon BCR/ABL inhibition (Thiele and Kvasnicka, 2006) or in PMF patients upon SCT (Daly et al., 2014). Several agents proposed to target the microenvironment, including immunomodulatory drugs such as Thalidomide and Lenalidomide, proteasome inhibitors such as Bortezomib, and VEGF-targeting drugs such as Sunitinib and Becacizumab, have been tested in subsets of PMF patients. However, these clinical trials have had mixed results and severe tolerability issues (Rambaldi et al., 2008). Considering the importance of TNFα, IL-1 and IL-6 in driving the fibrotic BM niche remodeling (Schepers et al., 2013) and CML development (Reynaud et al., 2011) in murine models, it will be very interesting to test the effect of blocking these specific pro-inflammatory cytokines with the various suppressing, neutralizing and/or antagonizing antibodies that are currently available for clinical use. Their efficacy at treating autoinflammatory conditions such as secondary hemophagocytic lymphohistiocytosis and rheumatoid arthritis show that targeting a single cytokine in a complex inflammatory process can provide significant clinical benefit. It will also be exciting to test whether any of the various FDA-approved bone remodeling agents such as bisphosphonates currently used for osteoporosis and as an adjunct in multiple myeloma treatment are efficacious at blocking the fibrotic remodeling of the MSC/OBC compartment. In murine models of MPN and AML, disease development is also accompanied by SNS neuropathy, which directly alters MSC growth and differentiation and thereby remodels the BM niche. Strategies aimed at either directly protecting these nerve cells or at preserving their function on perivascular MSCs (Arranz et al., 2014; Hanoun et al., 2014), could also help support the function of healthy HSCs and maintain production of normal blood cells. The fact that the clinically approved β3-adrenergic agonist Mirabegron delays disease onset and improve several MPN-associated features several normal features in the Jak2V617F murine MPN model (Arranz et al., 2014), reflects the potential of such therapies for human treatment and provides another exciting path forward.
Emerging questions
The lack of reliance on a particular driver mutation and the wide applicability to various disease types makes targeting the malignant BM niche a worthy endeavor. Despite this, many questions remain regarding the therapeutic benefits of such non-cell autonomous strategies. Further testing of drugs targeting the aberrant homing and adhesion of leukemic cells will demonstrate whether pushing LSCs and their progeny out of hiding in protected BM niches is a promising approach to prevent disease relapse. Time will also tell whether this strategy has any unwanted long-term effects on the maintenance of healthy HSCs. Developing novel approaches to antagonize the soluble factors that promote malignant niche formation also has therapeutic potential. Strategies include IL-1 and IL-6 blocking therapies, which are available in the clinic but not yet used in myeloid malignancies, as well as blockade of novel targets that are important for BM stromal remodeling and myelofibrosis development, such as TPO and MIP-1α. Moreover, establishing new modalities to directly block the cellular changes occurring in the malignant BM niche, including aberrant MSC differentiation into fibrotic tissue and neuropathy-driven MSC alterations, also has great therapeutic potential. Strategies will include using established bone remodeling agents and beta-adrenergic modulators that are available but have not yet been tested in myeloid malignancies. Altogether, this will build an arsenal of new therapeutic compounds for targeting the malignant BM niche and restoring normal hematopoiesis. Finally, while transformed HSCs and progenitors cells can be efficiently replaced through SCT approaches, defective BM stromal function is usually not corrected and can potentially impair the activity of newly transplanted HSCs. The same strategies could therefore be used as preconditioning regimens for SCT in order to restore the marrow’s ability to support healthy HSC function, and ensure that the BM microenvironment preferentially supports normal hematopoiesis over potential remaining LSCs. This could help achieve and maintain deeper clinical remissions in pre- and post-transplantation settings. These exciting new clinical directions illustrate how complementary basic and translational research in deciphering the features of the normal HSC niche, investigating the deregulated properties of the malignant niches and testing the efficacy of new drugs targeting these deregulated microenvironments are bringing us closer to the goal of achieving curative treatments for a broad range of human blood malignancies.
Acknowledgments
We thank members of the Passegué laboratory for critical reading and insights on the manuscript. K.S. is supported by a Dutch NIRM FES0908, T.C. by NIH T32 CA108462 and EP by an LLS Scholar award, NIH R01 HL092471 and an MF Challenge Concept Grant from the MPN Research Foundation. E.P. is a member of Geron scientific advisory board. The other authors have no competing financial interests to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
KS and TBC researched each section. KS, TBC and EP wrote the manuscript.
References
- Abdel-Wahab OI, Levine RL. Primary myelofibrosis: update on definition, pathogenesis, and treatment. Annu Rev Med. 2009;60:23–245. doi: 10.1146/annurev.med.60.041707.160528. [DOI] [PubMed] [Google Scholar]
- Aizawa S, Nakano M, Iwase O, Yaguchi M, Hiramoto M, Hoshi H, Nabeshima R, Shima D, Handa H, Toyama K. Bone marrow stroma from refractory anemia of myelodysplastic syndrome is defective in its ability to support normal CD34-positive cell proliferation and differentiation in vitro. Leukemia Research. 1999;23:239–246. doi: 10.1016/s0145-2126(98)00163-5. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, Lundberg P, Muntión S, Tzeng YS, Lai DM, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512:78–81. doi: 10.1038/nature13383. [DOI] [PubMed] [Google Scholar]
- Asada N, Katayama Y, Sato M, Minagawa K, Wakahashi K, Kawano H, Kawano Y, Sada A, Ikeda K, Matsui T, et al. Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell. 2013;12:737–747. doi: 10.1016/j.stem.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Bakker ST, Passegué E. Resilient and resourceful: genome maintenance strategies in hematopoietic stem cells. Exp Hematol. 2013;41:915–923. doi: 10.1016/j.exphem.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau O, Hofmann WK, Baldus CD, Thiel G, Serbent V, Schümann E, Thiel E, Blau IW. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol. 2007;35:221–229. doi: 10.1016/j.exphem.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Borojevic R, Roela RA, Rodarte RS, Thiago LS, Pasini FS, Conti FM, Rossi MID, Reis LFL, Lopes LF, Brentani MM. Bone marrow stroma in childhood myelodysplastic syndrome: composition, ability to sustain hematopoiesis in vitro, and altered gene expression. Leuk Res. 2004;28:831–844. doi: 10.1016/j.leukres.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20:1315–1320. doi: 10.1038/nm.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, Seandel M, Shido K, White IA, Kobayashi M, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- Chan CKF, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T, et al. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–298. doi: 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Jacamo R, Shi Y, Wang R, Battula VL, Konoplev S, Strunk D, Hofmann NA, Reinisch A, Konopleva M, et al. Human extramedullary bone marrow in mice: a novel in vivo model of genetically controlled hematopoietic microenvironment. Blood. 2012;119:4971–4980. doi: 10.1182/blood-2011-11-389957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208:261–271. doi: 10.1084/jem.20101688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–1865. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Daly A, Song K, Nevill T, Nantel S, Toze C, Hogge D, Forrest D, Lavoie J, Sutherland H, Shepherd J, et al. Stem cell transplantation for myelofibrosis: a report from two Canadian centers. Bone Marrow Transplantation. 2003;32:35–40. doi: 10.1038/sj.bmt.1704075. [DOI] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dührsen U, Hossfeld DK. Stromal abnormalities in neoplastic bone marrow diseases. Ann Hematol. 1996;73:53–70. doi: 10.1007/s002770050203. [DOI] [PubMed] [Google Scholar]
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- Flores-Figueroa E, Varma S, Montgomery K, Greenberg PL, Gratzinger D. Distinctive contact between CD34+ hematopoietic progenitors and CXCL12+ CD271+ mesenchymal stromal cells in benign and myelodysplastic bone marrow. Lab Invest. 2012;92:1330–1341. doi: 10.1038/labinvest.2012.93. [DOI] [PubMed] [Google Scholar]
- Frenette PS, Pinho S, Lucas D, Scheiermann C. Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu Rev Immunol. 2013;31:285–316. doi: 10.1146/annurev-immunol-032712-095919. [DOI] [PubMed] [Google Scholar]
- Ge J, Hou R, Liu Q, Zhu R, Liu K. Stromal-derived factor-1 deficiency in the bone marrow of acute myeloid leukemia. Int J Hematol. 2011;93:750–759. doi: 10.1007/s12185-011-0869-9. [DOI] [PubMed] [Google Scholar]
- Gidali J, Feher I, Hollan SR. Blast colony forming cell-binding capacity of bone marrow stroma from myelodysplastic patients. Stem Cells. 1996;14:577–583. doi: 10.1002/stem.140577. [DOI] [PubMed] [Google Scholar]
- Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, Woll P, Mead A, Alford KA, Rout R, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19:138–152. doi: 10.1016/j.ccr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Gordon MY, Dowding CR, Riley GP, Goldman JM, Greaves MF. Altered adhesive interactions with marrow stroma of haematopoietic progenitor cells in chronic myeloid leukaemia. Nature. 1987;328:342–344. doi: 10.1038/328342a0. [DOI] [PubMed] [Google Scholar]
- Greenbaum A, Hsu YMS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–230. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JD, Rambaldi A, Vellenga E, Young DC, Ostapovicz D, Cannistra SA. Secretion of interleukin-1 by acute myeloblastic leukemia cells in vitro induces endothelial cells to secrete colony stimulating factors. Blood. 1987;70:1218–1221. [PubMed] [Google Scholar]
- Groen RWJ, Noort WA, Raymakers RA, Prins HJ, Aalders L, Hofhuis FM, Moerer P, van Velzen JF, Bloem AC, van Kessel B, et al. Reconstructing the human hematopoietic niche in immunodeficient mice: opportunities for studying primary multiple myeloma. Blood. 2012;120:e9–e16. doi: 10.1182/blood-2012-03-414920. [DOI] [PubMed] [Google Scholar]
- Guezguez B, Campbell CJV, Boyd AL, Karanu F, Casado FL, Di Cresce C, Collins TJ, Shapovalova Z, Xenocostas A, Bhatia M. Regional localization within the bone marrow influences the functional capacity of human HSCs. Cell Stem Cell. 2013;13:175–189. doi: 10.1016/j.stem.2013.06.015. [DOI] [PubMed] [Google Scholar]
- Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Dührsen U, Frenette PS. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15:365–375. doi: 10.1016/j.stem.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YT, Gang EJ, Shishido SN, Kim HN, Pham J, Khazal S, Osborne A, Esguerra ZA, Kwok E, Jang J, et al. Effects of the small-molecule inhibitor of integrin α4, TBC3486, on pre-B-ALL cells. Leukemia. 2014;28:2101–2104. doi: 10.1038/leu.2014.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntly BJP, Gilliland DG. Cancer biology: summing up cancer stem cells. Nature. 2005;435:1169–1170. doi: 10.1038/4351169a. [DOI] [PubMed] [Google Scholar]
- Jabbour E, Cortes J, Ravandi F, O’Brien S, Kantarjian H. Targeted therapies in hematology and their impact on patient care: chronic and acute myeloid leukemia. Semin Hematol. 2013;50:271–283. doi: 10.1053/j.seminhematol.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, Wang Y, Battula VL, Mak PY, Schallmoser K, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood. 2014;123:2691–2702. doi: 10.1182/blood-2013-06-511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CHM, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- Jin L, Tabe Y, Konoplev S, Xu Y, Leysath CE, Lu H, Kimura S, Ohsaka A, Rios M, Calvert L, et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Molecular Cancer Therapeutics. 2008;7:48–58. doi: 10.1158/1535-7163.MCT-07-0042. [DOI] [PubMed] [Google Scholar]
- Kastrinaki MC, Pavlaki K, Batsali AK, Kouvidi E, Mavroudi I, Pontikoglou C, Papadaki HA. Mesenchymal stem cells in immune-mediated bone marrow failure syndromes. Clin Dev Immunol. 2013;2013:265608. doi: 10.1155/2013/265608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent DG, Li J, Tanna H, Fink J, Kirschner K, Pask DC, Silber Y, Hamilton TL, Sneade R, Simons BD, et al. Self-renewal of single mouse hematopoietic stem cells is reduced by JAK2V617F without compromising progenitor cell expansion. PLoS Biol. 2013;11:e1001576. doi: 10.1371/journal.pbio.1001576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kim YW, Koo BK, Jeong HW, Yoon MJ, Song R, Shin J, Jeong DC, Kim SH, Kong YY. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood. 2008;112:4628–4638. doi: 10.1182/blood-2008-03-148999. [DOI] [PubMed] [Google Scholar]
- Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–1180. doi: 10.1038/nm1489. [DOI] [PubMed] [Google Scholar]
- Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, Lezeau S, Attar E, Wu JY, Lin HY, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19:1513–1517. doi: 10.1038/nm.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DS, Lazarides K, Lewis JB, von Andrian UH, Van Etten RA. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood. 2014;123:1361–1371. doi: 10.1182/blood-2013-11-538694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SW, Wang YJ, Lo Celso C, Ragu C, Bullinger L, Sykes SM, Ferraro F, Shterental S, Lin CP, Gilliland DG, et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood. 2011;118:2849–2856. doi: 10.1182/blood-2011-03-345165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassailly F, Foster K, Lopez-Onieva L, Currie E, Bonnet D. Multimodal imaging reveals structural and functional heterogeneity in different bone marrow compartments: functional implications on hematopoietic stem cells. Blood. 2013;122:1730–1740. doi: 10.1182/blood-2012-11-467498. [DOI] [PubMed] [Google Scholar]
- Leisten I, Kramann R, Ventura Ferreira MS, Bovi M, Neuss S, Ziegler P, Wagner W, Knüchel R, Schneider RK. 3D co-culture of hematopoietic stem and progenitor cells and mesenchymal stem cells in collagen scaffolds as a model of the hematopoietic niche. Biomaterials. 2012;33:1736–1747. doi: 10.1016/j.biomaterials.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Li Q, Bohin N, Wen T, Ng V, Magee J, Chen SC, Shannon K, Morrison SJ. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature. 2013;504:143–147. doi: 10.1038/nature12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ghazanfari R, Zacharaki D, Ditzel N, Isern J, Ekblom M, Méndez-Ferrer S, Kassem M, Scheding S. Low/Negative Expression of PDGFR-α Identifies the Candidate Primary Mesenchymal Stromal Cells in Adult Human Bone Marrow. Stem Cell Reports. 2014;3:965–974. doi: 10.1016/j.stemcr.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, Côté D, Rowe DW, Lin CP, Scadden DT. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Anginot A, Mancini SJC, Schiff C, Carle GF, Wakkach A, Blin-Wakkach C. Osteoclast activity modulates B-cell development in the bone marrow. Cell Res. 2011;21:1102–1115. doi: 10.1038/cr.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SEW, Wakkach A, Blin-Wakkach C. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med. 2012;209:537–549. doi: 10.1084/jem.20110994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, Lier A, Eisen C, Nowak V, Zens B, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14:824–837. doi: 10.1016/j.stem.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Scadden DT, Sánchez-Aguilera A. Bone marrow stem cells: current and emerging concepts. Ann N Y Acad Sci. 2015;1335:32–44. doi: 10.1111/nyas.12641. [DOI] [PubMed] [Google Scholar]
- Merzaban JS, Burdick MM, Gadhoum SZ, Dagia NM, Chu JT, Fuhlbrigge RC, Sackstein R. Analysis of glycoprotein E-selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cells. Blood. 2011;118:1774–1783. doi: 10.1182/blood-2010-11-320705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic JD, Kralovics R. Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol. 2013;97:183–197. doi: 10.1007/s12185-012-1235-2. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair RR, Tolentino J, Hazlehurst LA. The bone marrow microenvironment as a sanctuary for minimal residual disease in CML. Biochem Pharmacol. 2010;80:602–612. doi: 10.1016/j.bcp.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Arai F, Iwasaki H, Hosokawa K, Kobayashi I, Gomei Y, Matsumoto Y, Yoshihara H, Suda T. Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood. 2010;116:1422–1432. doi: 10.1182/blood-2009-08-239194. [DOI] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Takubo K, Fujioka M, Suda T. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem Biophys Res Commun. 2014;454:353–357. doi: 10.1016/j.bbrc.2014.10.095. [DOI] [PubMed] [Google Scholar]
- Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460:259–263. doi: 10.1038/nature08099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, Prior JL, Piwnica-Worms D, Bridger G, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 anatagonist AMD3100. Blood. 2009;113:6206–6214. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, Park SY, Lu J, Protopopov A, Silberstein LE. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15:533–543. doi: 10.1038/ncb2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, Nagasawa T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–399. doi: 10.1016/j.immuni.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster W, Cicco NA, Klein H, Hirano T, Kishimoto T, Lindemann A, Mertelsmann RH, Herrmann F. Participation of the cytokines interleukin 6, tumor necrosis factor-alpha, and interleukin 1-beta secreted by acute myelogenous leukemia blasts in autocrine and paracrine leukemia growth control. J Clin Invest. 1989;84:451–457. doi: 10.1172/JCI114186. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, Scadden DT. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10:259–272. doi: 10.1016/j.stem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegué E, Jamieson CHM, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A. 2003;100(Suppl):11842–11849. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira A, Cervantes F, Brugues R, Rozman C. Bone marrow histopathology in primary myelofibrosis: clinical and haematologic correlations and prognostic evaluation. Eur J Haematol. 1990;44:94–98. doi: 10.1111/j.1600-0609.1990.tb00357.x. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195:709–720. doi: 10.1083/jcb.201102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, Frenette PS. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med. 2013;210:1351–1367. doi: 10.1084/jem.20122252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raic A, Rödling L, Kalbacher H, Lee-Thedieck C. Biomimetic macroporous PEG hydrogels as 3D scaffolds for the multiplication of human hematopoietic stem and progenitor cells. Biomaterials. 2014;35:929–940. doi: 10.1016/j.biomaterials.2013.10.038. [DOI] [PubMed] [Google Scholar]
- Rambaldi A, Barbui T, Barosi G. From palliation to epigenetic therapy in myelofibrosis. Hematology Am Soc Hematol Educ Program. 2008:83–91. doi: 10.1182/asheducation-2008.1.83. [DOI] [PubMed] [Google Scholar]
- Rankin EB, Wu C, Khatri R, Wilson TL, Andersen R, Araldi E, Rankin AL, Yuan J, Kuo CJ, Schipani E, Giaccia AJ. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell. 2012;149:63–74. doi: 10.1016/j.cell.2012.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, Sala-Torra O, Radich JP, Passegué E. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20:661–673. doi: 10.1016/j.ccr.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupec RA, Jundt F, Rebholz B, Eckelt B, Weindl G, Herzinger T, Flaig MJ, Moosmann S, Plewig G, Dörken B, et al. Stroma-mediated dysregulation of myelopoiesis in mice lacking I kappa B alpha. Immunity. 2005;22:479–491. doi: 10.1016/j.immuni.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL, Braun BS, Passegue E. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell. 2009;15:341–352. doi: 10.1016/j.ccr.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, Passegué E. Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell. 2013;13:285–299. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Kharabi Masouleh B, Loges S, Cauwenberghs S, Fraisl P, Maes C, Jonckx B, De Keersmaecker K, Kleppe M, Tjwa M, et al. Loss or inhibition of stromal-derived PlGF prolongs survival of mice with imatinib-resistant Bcr-Abl1 (+) leukemia. Cancer Cell. 2011;19:740–753. doi: 10.1016/j.ccr.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25. [PubMed] [Google Scholar]
- Sharma MB, Limaye LS, Kale VP. Mimicking the functional hematopoietic stem cell niche in vitro: recapitulation of marrow physiology by hydrogel-based three-dimensional cultures of mesenchymal stromal cells. Haematologica. 2012;97:651–660. doi: 10.3324/haematol.2011.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, Luster AD, Scadden DT, Lin CP. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508:269–273. doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Tamplin OJ, Durand EM, Carr LA, Childs SJ, Hagedorn EJ, Li P, Yzaguirre AD, Speck NA, Zon LI. Hematopoietic stem cell arrival triggers dynamic remodeling of the perivascular niche. Cell. 2015;160:241–252. doi: 10.1016/j.cell.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105:2631–2639. doi: 10.1182/blood-2004-06-2480. [DOI] [PubMed] [Google Scholar]
- Thiele J, Kvasnicka HM. Myelofibrosis in chronic myeloproliferative disorders–dynamics and clinical impact. Histol Histopathol. 2006;21:1367–1378. doi: 10.14670/HH-21.1367. [DOI] [PubMed] [Google Scholar]
- Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, Kulkarni S, Abboud CN, Cashen AF, Stockerl-Goldstein KE, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–3924. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007a;129:1081–1095. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007b;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang H, Rodriguez S, Cao L, Parish J, Mumaw C, Zollman A, Kamoka MM, Mu J, Chen DZ, et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-κB-dependent manner. Cell Stem Cell. 2014;15:51–65. doi: 10.1016/j.stem.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg E, Azab AK, Manley PW, Kung AL, Christie AL, Bronson R, Ghobrial IM, Griffin JD. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia. 2012;26:985–990. doi: 10.1038/leu.2011.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSC. Blood. 2010 doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, Magnani JL, Lévesque JP. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. 2012;18:1651–1657. doi: 10.1038/nm.2969. [DOI] [PubMed] [Google Scholar]
- Wiseman DH. Donor cell leukemia: a review. Biol Blood Marrow Transplant. 2011;17:771–789. doi: 10.1016/j.bbmt.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Woll PS, Kjällquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, Erlandsson R, Ngara M, Anderson K, Deng Q, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014;25:794–808. doi: 10.1016/j.ccr.2014.03.036. [DOI] [PubMed] [Google Scholar]
- Worthley DL, Churchill M, Comptom JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y, et al. Gremlin 1 identifies a skeletal stem cell with cartilage and reticular stromal potential. Cell. 2015;160:269–284. doi: 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, Scadden DT, Kronenberg HM. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A. 2008;105:16976–16981. doi: 10.1073/pnas.0802898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, Taketo MM, Karlsson S, Iwama A, Nakauchi H. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–1158. doi: 10.1016/j.cell.2011.09.053. [DOI] [PubMed] [Google Scholar]
- Zeng Z, Shi YX, Samudio IJ, Wang R, Ling X, Frolova O, Levis M, Rubin JB, Negrin RR, Estey EH, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–6224. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee S, Hair A, Holyoake TL, Huettner C, Bhatia R. Altered Microenvironmental Regulation of Leukemic and Normal Stem Cells in Chronic Myelogenous Leukemia. Cancer Cell. 2012;21:577–592. doi: 10.1016/j.ccr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, Ahamed J, Li L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20:1321–1326. doi: 10.1038/nm.3706. [DOI] [PubMed] [Google Scholar]
- Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–168. doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]