

Abstract
Purpose
Metabolic liver disease (MLD) often progresses to life-threatening conditions. This study intends to describe the outcomes of liver transplantation (LTx) for MLD at a living donor-dominant transplantation center where potentially heterozygous carrier grafts are employed.
Methods
We retrospectively evaluated the medical records of 54 patients with MLD who underwent LTx between November 1995 and February 2012 at Asan Medical Center in Seoul, Korea. The cumulative graft and patient survival rates were analyzed according to patient age, and living or deceased donor LTx. Recurrence of the original disease was also investigated.
Results
The post-transplant cumulative patient survival rates at one, five, and 10 years were 90.7%, 87.5% and 87.5%, and the graft survival rates were 88.8%, 85.5%, and 85.5%, respectively. There were no differences in the patient survival rates according to the recipient age, human leukocyte antigen matching, and living or deceased donor LTx. There were also no differences in the patient survival rates between the MLD and the non-MLD groups for children. Recurrence of the original metabolic disease was not observed in any patient during the follow-up period.
Conclusion
Our results suggest that the living donor-dominant transplantation program is well-tolerated in MLD without recurrence of the original MLD using all types of transplantation.
Keywords: Metabolic diseases, Liver transplantation, Adult, Child
INTRODUCTION
Liver is an organ with a major role in the human body's metabolic process. Therefore, a serious condition affecting the liver, such as metabolic liver disease (MLD), can cause life-threatening conditions, including acute liver failure, end-stage liver disease, and serious symptoms arising from abnormal liver metabolism, such as hyperammonemia, deteriorating neuropsychological functions, and coagulation factor deficiency. If it is not possible to prevent metabolic decompensation, liver transplantation is an alternative therapeutic option [1]. As it is also now an accepted therapeutic option for non-metabolic, end stage liver diseases in children [2,3]. Favorable outcomes of cadaveric liver transplantation (LTx) for MLD have also been reported [4].
Most MLDs have genetic traits and most of them are autosomal recessive; however, various genetic inheritance factors, including maternal inheritance, can occur. Therefore, living related donor LTx might affect the outcome of the original disease as living related donor LTx may involve a heterozygous carrier. As for Wilson's disease (WD), it has an autosomal recessive genetic trait and is the most common cause of the need for LTx for MLD at our medical center. Studies of the copper metabolism in heterozygotes of WD reported that copper metabolism tends to be abnormal and that recipients of heterozygotic donor grafts probably maintain these abnormalities [5,6]. Asonuma et al. [7] suggested that minor, persistent abnormalities of copper metabolism in a heterozygous donor have no clinical impact, and we also confirmed this result in a previous report [8]. Through our medical center's living donor-dominant transplantation program, we investigated the outcome of LTx in MLD with a potential heterozygous carrier as the donor.
MATERIALS AND METHODS
Between November 1995 and February 2012, 3,319 patients underwent LTx at Asan Medical Center, in Seoul, Korea. Among these patients, 54 with MLD were enrolled in this study and their medical records were reviewed. Indications for LTx for WD at our medical center were acute liver failure or decompensated liver cirrhosis unresponsive to medical treatment for more than two months [5,8,9]. A neurologic complication was not included as an indication for LTx at our medical center. Indications for LTx in other MLDs included liver failure, liver cirrhosis with complication, and medically uncontrollable original disease.
The criteria for living donor selection have been described in another study [10]. As ornithine transcarbamylase deficiency is inherited in an X-linked recessive trait, mothers of patients were excluded from the donor selection in order to eliminate the possibility of co-dominance [11]. Surgical techniques and postoperative management are also described in other studies [2,10].
We analyzed the patient demographics, recipient and donor characteristics, graft type, and patient survival. We analyzed the patient survival and graft survival rates in all of the patient groups. We compared the demographic data and the patient survival rate between the adult and the pediatric groups. The non-MLD patients were those who did not have metabolic etiologies. The patient survival rate of non-MLD patients in the pediatric group was also analyzed and compared with that of the MLD group.
Given the possibility that living donors can be heterozygous carriers, our recipients were classified into three groups according to the donor type, as follows: the haplo-matched group, i.e. those with first degree-related donors; the mixed group, i.e. those with a haplo-matched donor and unrelated donor (dual LTx); and the unrelated groups. Donor grafts were also classified as deceased and living-donor grafts. We compared the patient survival rates according to the donor type.
The cumulative graft and patient survival rates were calculated by using the Kaplan-Meier method according to the patient age and the donor type. The differences of cumulative survival were assessed using the log-rank method. Chi-square testing was used to compare categorical variables. In the pediatric population, the cumulative graft and patient survival rates were analyzed in non-MLD patients who underwent LTx. SPSS commercial statistics software was used for all statistical analyses (PASW Statistics ver. 18.0; IBM Co., Armonk, NY, USA). The p-values less than 0.05 were considered to be significant.
RESULTS
Patient characteristics are summarized in Table 1. Of the 3,076 adult patients, 31 (1.0%) and of the 243 pediatric patients, 23 (9.5%) (under 18 years of age) were included in our study. The median age of the 54 patients was 17.6 years (range, 6 months-59 years). The median follow-up period was 70.9 months. There were 23 men (42.6%) and 31 women (57.4%), with a male-to-female ratio of 1 : 1.3. The etiologies of the 54 patients with MLD were WD (n=44, 81.4%), glycogen storage disease (GSD) type III (n=2, 3.7%), GSD type IV (n=1, 1.8%), ornithine transcarbamylase deficiency (n=2, 3.7%), citrullinemia type II (citrin deficiency) (n=2, 3.7%), citrullinemia type I (n=1, 1.8%), protein C deficiency (n=1, 1.8%), and factor X deficiency (n=1, 1.8%). According to the donor type, the recipients were divided into four groups, i.e., haplo-matched living donor (n=18, 33.3%), unrelated living donor (n=17, 31.5%), mixed-type living donor (n=7, 13.0%), and deceased donor (n=12, 22.2%).
Table 1. Recipient Demographics and Clinical Characteristics (n=54).

Values are presented as the number (%) or mean±standard deviation (range).
The post-transplant cumulative rates of the total patient survival were 90.7%, 87.5%, and 87.5% and those of the graft survival were 90.8%, 87.5%, and 87.5% at one, five, and 10 years respectively (Fig. 1). There were no differences between the two groups, and which shows a high graft survival rate without retransplantation.
Fig. 1. Cumulative post-transplant patient and graft survival rates for 54 patients with metabolic liver diseases.

The survival rates of the pediatric patients were 90.2%, 90.2%, and 90.2% at one, five and 10 years, while the survival rates of the adult patients were 91.3%, 84.3%, and 84.3% (p=0.72). There were no statistically significant differences between the pediatric and the adult patients. This suggests that the outcomes did not differ between adult and pediatric patients when the etiology was the same and the same surgical techniques were used at a single medical center.
The cumulative survival rates in the non-MLD pediatric patients group were 91.9%, 87.2%, and 85.8% at one, five, and 10 years, respectively (Fig. 2A). There were no statistically significant differences between the MLD and non-MLD group and which indicates that the patient survival rates did not differ between the two groups. Therefore, LTx can be an effective therapy option in MLD as well as in non-MLD patients.
Fig. 2. (A) Comparison of post-transplant patient survival between the metabolic liver disease (MLD) group (n=23) and the non-MLD group (n=220) in the pediatric age group. (B) Comparison of the post-transplant patient survival among the living donor types (haplo-matched donors [n=18], unrelated donors [n=17], mixed donors [n=7]). (C) Comparison of the post-transplant patient survival between the living donors (n=42) and the cadaveric donors (n=12).

There were also no differences in the patient survival rates in the living donor types (p=0.62), i.e., the patient survival rates at one, five, and 10 years in the haplo-matched groups were 94.4%, 86.6%, and 86.6%, while in the unrelated groups they were 86.7%, 86.7%, and 86.7%, respectively. The patient survival rates in the mixed groups were 100.0%, 100.0%, and 100.0% at one, five, and 10 years (Fig. 2B), and there were no statistically significant differences between the donor graft types. This indicates that heterozygote carrier graft does not affect the survival rates of liver transplant recipients with MLD. This also suggests that haplo-matched donor graft can be safely used in MLD.
The patient survival rates of the living donors were 92.5%, 88.6%, and 88.6%, while those of the deceased donors were 84.6%, 84.6%, and 84.6% at one, five and 10 years, respectively (p=0.48), and there were no statistically significant differences between the donor graft types (Fig. 2C).This shows that there is no difference between emergency liver transplant with a deceased donor and elective liver transplant with a living donor.
A comparison of demographic data between the pediatric and the adult patients is shown in Table 2. Gender, the indications of LTx, the donor types, and the operation types were compared. Acute liver failure was the most common indication of LTx in both groups (p=0.040). The haplo-matched donors were predominant in the pediatric patients, while the unrelated donors were predominant in the adult group (p=0.006). The gender and the operation types did not show statistically significant differences.
Table 2. Comparison of Pediatric Patient and Adult Patient Demographic Data.
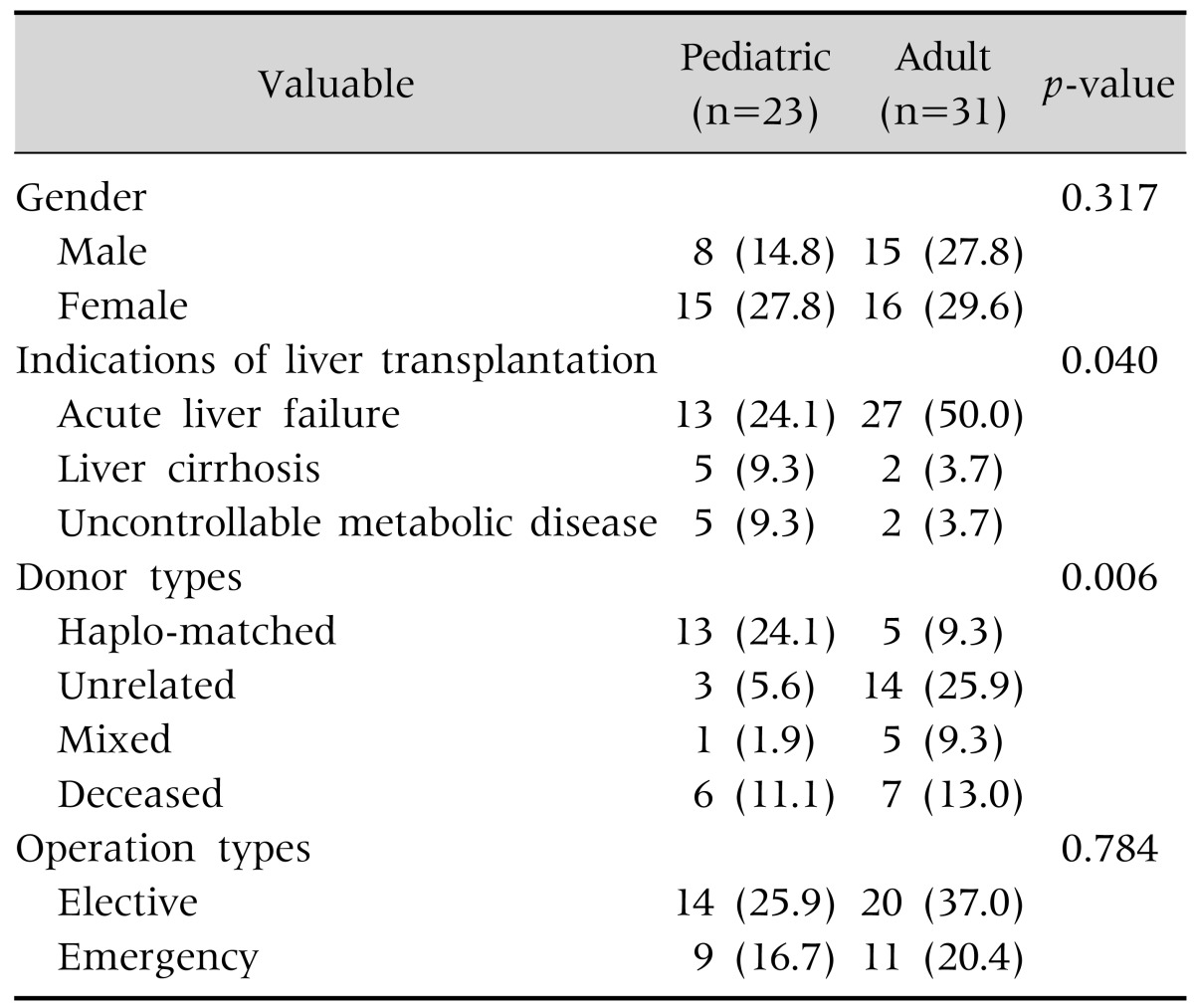
Values are presented as the number (%).
There were six post-transplant deaths during the study period, five of which occurred during hospitalization. One patient died during the long-term follow-up period. Of the 48 surviving patients, none showed any evidence of recurrence of their original disease.
DISCUSSION
Most studies regarding LTx for MLD involve pediatric patients. The one- and five-year patient survival rates reported in the United States [12] were 94% and 92%, respectively, whereas they were 91%, 86%, respectively, in the United Kingdom [13]. These figures were comparable to the 90% and 90%, respectively, seen at our medical center. Therefore, the LTx in MLD performed at our medical center can be deemed as tolerable and consistently used as a treatment option. These results also suggest that the survival rates are similar between cadaveric LTx, the mainstay of liver transplant in the western countries, and living donor liver transplant which is the main option at our medical center. As there were no differences between living-donor and deceased-donor LTx at our medical center, the donor type has little bearing on the liver transplant outcomes. Likewise, there were no differences in the LTx outcome with a potential heterozygous carrier graft. Our study also showed that haplo-matched donor graft could be safely used for MLD with no risk of recurrence of the original disease. Similar results were reported in Japan where only living-donor transplantation is performed [14]. The most common indication for LTx in our study population was acute liver failure. The donor types differed between the pediatric and the adult patients. The haplo-matched donors were predominant in the pediatric patients probably because their parents were eligible and willing to donate their liver to their children.
In our study population, WD was the most common indication for LTx. WD develops from abnormal copper metabolism in the liver due to ATP7B mutation and manifests as various liver diseases and psychiatric symptoms [15]. Acute liver failure and decompensated liver cirrhosis are common indications for LTx [5,9].
However, neurological deterioration alone was not listed as an indication for LTx at our medical center as the need for LTx due to neurological symptoms is still debatable. Improvement following LTx has been reported in some cases of neurological WD [16,17], however, patients with neuropsychiatric signs have a significantly shorter survival than patients with liver disease alone [18]. Therefore, LTx should be contraindicated in WD patients with severe neurological impairment [19]. Further investigation is required on this topic.
Eighteen out of the 44 patients with WD underwent LTx with a haplo-matched donor and no recurrence was reported in our study. Yoshitoshi et al. [20] presented the outcome of living donor LTx in 32 patients with WD, in whom 29 donors were parents (heterozygotes). The overall survival rate for these patients was 90.6% and 83.7% at one and five years, respectively, and without recurrence. This indicates that the use of liver grafts from a heterozygous donor can be safe.
As for GSD, dietary modifications and medical interventions compose the first line of therapy, however, LTx can be considered for patients with very poor metabolic control, the risk of adenocarcinoma with multiple recurrent adenomas, and liver failure [21,22]. At our medical center, as we did not perform LTx for GSD type 1, further studies are needed to evaluate the efficacy of GSD type 1 in our medical center. Three patients with type 3 and 4 who underwent LTx seemed to be at risk for extrahepatic manifestations, including cardiac symptoms, and were placed under close observation. However, we have not observed such symptoms since the transplantation.
As for urea cycle disorders, expert consensus guidelines recommend LTx should be performed between 3 and 12 months of age once the child weighs more than 5 kg to avoid complications such as neurological complications [23]. In our study, only one patient with an age of 8 months underwent LTx. LTx was performed on two patients aged 2-3 years due to late diagnosis. Two citrin deficiency patients were diagnosed in adolescence due to the late onset of symptoms. Although LTx was performed late in these patients, there were no neurological complications. Close observation and early diagnosis is needed when urea cycle disorder is suspected to avoid neurological complications. As for protein C deficiency, the best way to manage protein C deficiency would be to provide an exogenous source of protein C [24]. However, protein C concentrate was not widely available in South Korea, LTx was an applicable treatment option. Our patient has undergone LTx at age of six month. There were no serious complications of life-long immunosuppression such as renal impairment and development of secondary malignancy during the follow-up period.
There are some limitations to our study. First, some MLD patients, such as those with propionic acidemia or methylmalonic acidemia, did not undergo LTx. There has only been one case report of a patient with propionic acidemia who underwent LTx in Korea and with incomplete improvement of metabolic control [25]. Further research is needed regarding safety and outcome of LTx in those patients. Second, as the majority of our MLD patients had WD, selection bias can, therefore, not be ruled out.
In conclusion, as shown in the long-term patient survival rate following transplantation, LTx in MLD is tolerable, regardless of the donor relationship, recipient age, and graft type. LTx may, therefore, help to prevent future life-threatening situations in these patient groups.
References
- 1.Shneider BL. Pediatric liver transplantation in metabolic di'sease: clinical decision making. Pediatr Transplant. 2002;6:25–29. doi: 10.1034/j.1399-3046.2002.1p057.x. [DOI] [PubMed] [Google Scholar]
- 2.Oh SH, Kim KM, Kim DY, Lee YJ, Rhee KW, Jang JY, et al. Long-term outcomes of pediatric living donor liver transplantation at a single institution. Pediatr Transplant. 2010;14:870–878. doi: 10.1111/j.1399-3046.2010.01357.x. [DOI] [PubMed] [Google Scholar]
- 3.Oh SH, Kim KM, Kim DY, Song SM, Kim T, Hwang S, et al. Clinical experience of more than 200 cases of pediatric liver transplantation at a single center: improved patient survival. Transplant Proc. 2012;44:484–486. doi: 10.1016/j.transproceed.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Arnon R, Kerkar N, Davis MK, Anand R, Yin W, González-Peralta RP SPLIT Research Group. Liver transplantation in children with metabolic diseases: the studies of pediatric liver transplantation experience. Pediatr Transplant. 2010;14:796–805. doi: 10.1111/j.1399-3046.2010.01339.x. [DOI] [PubMed] [Google Scholar]
- 5.Bellary S, Hassanein T, Van Thiel DH. Liver transplantation for Wilson's disease. J Hepatol. 1995;23:373–381. doi: 10.1016/0168-8278(95)80194-4. [DOI] [PubMed] [Google Scholar]
- 6.Sternlieb I, Scheinberg IH. The role of radiocopper in the diagnosis of Wilson's disease. Gastroenterology. 1979;77:138–142. [PubMed] [Google Scholar]
- 7.Asonuma K, Inomata Y, Kasahara M, Uemoto S, Egawa H, Fujita S, et al. Living related liver transplantation from heterozygote genetic carriers to children with Wilson's disease. Pediatr Transplant. 1999;3:201–205. doi: 10.1034/j.1399-3046.1999.00014.x. [DOI] [PubMed] [Google Scholar]
- 8.Kim JT, Chang SH, Choi BH, Kim KM, Yoo HW, Lee YJ, et al. Living-related liver transplantation with heterozygote carrier graft in children with wilson disease. Korean J Pediatr Gastroenterol Nutr. 2003;6:161–166. [Google Scholar]
- 9.Roche-Sicot J, Benhamou JP. Acute intravascular hemolysis and acute liver failure associated as a first manifestation of Wilson's disease. Ann Intern Med. 1977;86:301–303. doi: 10.7326/0003-4819-86-3-301. [DOI] [PubMed] [Google Scholar]
- 10.Hwang S, Lee SG, Lee YJ, Sung KB, Park KM, Kim KH, et al. Lessons learned from 1,000 living donor liver transplantations in a single center: how to make living donations safe. Liver Transpl. 2006;12:920–927. doi: 10.1002/lt.20734. [DOI] [PubMed] [Google Scholar]
- 11.Kim BS, Kim KM, Yoo HW, Lee SG. A case of ornithine transcarbamylase deficiency successfully treated with protein restriction and living related liver transplantation. J Korean Pediatr Soc. 1999;42:868–873. [Google Scholar]
- 12.Kayler LK, Rasmussen CS, Dykstra DM, Punch JD, Rudich SM, Magee JC, et al. Liver transplantation in children with metabolic disorders in the United States. Am J Transplant. 2003;3:334–339. doi: 10.1034/j.1600-6143.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 13.Sze YK, Dhawan A, Taylor RM, Bansal S, Mieli-Vergani G, Rela M, et al. Pediatric liver transplantation for metabolic liver disease: experience at King's College Hospital. Transplantation. 2009;87:87–93. doi: 10.1097/TP.0b013e31818bc0c4. [DOI] [PubMed] [Google Scholar]
- 14.Morioka D, Kasahara M, Takada Y, Corrales JP, Yoshizawa A, Sakamoto S, et al. Living donor liver transplantation for pediatric patients with inheritable metabolic disorders. Am J Transplant. 2005;5:2754–2763. doi: 10.1111/j.1600-6143.2005.01084.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee BH, Kim JH, Lee SY, Jin HY, Kim KJ, Lee JJ, et al. Distinct clinical courses according to presenting phenotypes and their correlations to ATP7B mutations in a large Wilson's disease cohort. Liver Int. 2011;31:831–839. doi: 10.1111/j.1478-3231.2011.02503.x. [DOI] [PubMed] [Google Scholar]
- 16.Stracciari A, Tempestini A, Borghi A, Guarino M. Effect of liver transplantation on neurological manifestations in Wilson disease. Arch Neurol. 2000;57:384–386. doi: 10.1001/archneur.57.3.384. [DOI] [PubMed] [Google Scholar]
- 17.Marin C, Robles R, Parrilla G, Ramírez P, Bueno FS, Parrilla P. Liver transplantation in Wilson's disease: are its indications established? Transplant Proc. 2007;39:2300–2301. doi: 10.1016/j.transproceed.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 18.Medici V, Mirante VG, Fassati LR, Pompili M, Forti D, Del Gaudio M, et al. Monotematica AISF 2000 OLT Study Group. Liver transplantation for Wilson's disease: The burden of neurological and psychiatric disorders. Liver Transpl. 2005;11:1056–1063. doi: 10.1002/lt.20486. [DOI] [PubMed] [Google Scholar]
- 19.Kassam N, Witt N, Kneteman N, Bain VG. Liver transplantation for neuropsychiatric Wilson disease. Can J Gastroenterol. 1998;12:65–68. doi: 10.1155/1998/414236. [DOI] [PubMed] [Google Scholar]
- 20.Yoshitoshi EY, Takada Y, Oike F, Sakamoto S, Ogawa K, Kanazawa H, et al. Long-term outcomes for 32 cases of Wilson's disease after living-donor liver transplantation. Transplantation. 2009;87:261–267. doi: 10.1097/TP.0b013e3181919984. [DOI] [PubMed] [Google Scholar]
- 21.Franco LM, Krishnamurthy V, Bali D, Weinstein DA, Arn P, Clary B, et al. Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis. 2005;28:153–162. doi: 10.1007/s10545-005-7500-2. [DOI] [PubMed] [Google Scholar]
- 22.Mazariegos G, Shneider B, Burton B, Fox IJ, Hadzic N, Kishnani P, et al. Liver transplantation for pediatric metabolic disease. Mol Genet Metab. 2014;111:418–427. doi: 10.1016/j.ymgme.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. doi: 10.1186/1750-1172-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angelis M, Pegelow CH, Khan FA, Verzaro R, Tzakis AG. En bloc heterotopic auxiliary liver and bilateral renal transplant in a patient with homozygous protein C deficiency. J Pediatr. 2001;138:120–122. doi: 10.1067/mpd.2001.109199. [DOI] [PubMed] [Google Scholar]
- 25.Ryu J, Shin YH, Ko JS, Gwak MS, Kim GS. Intractable metabolic acidosis in a child with propionic acidemia undergoing liver transplantation -a case report- Korean J Anesthesiol. 2013;65:257–261. doi: 10.4097/kjae.2013.65.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]