

Abstract
Objective
To compare short-term effects of fine particles (PM2.5; aerodynamic diameter <2.5 µm) from different sources on the blood levels of markers of systemic inflammation.
Methods
We followed a panel of 52 ischaemic heart disease patients from 15 November 2005 to 21 April 2006 with clinic visits in every second week in the city of Kotka, Finland, and determined nine inflammatory markers from blood samples. In addition, we monitored outdoor air pollution at a fixed site during the study period and conducted a source apportionment of PM2.5 using the Environmental Protection Agency's model EPA PMF 3.0. We then analysed associations between levels of source-specific PM2.5 and markers of systemic inflammation using linear mixed models.
Results
We identified five source categories: regional and long-range transport (LRT), traffic, biomass combustion, sea salt, and pulp industry. We found most evidence for the relation of air pollution and inflammation in LRT, traffic and biomass combustion; the most relevant inflammation markers were C-reactive protein, interleukin-12 and myeloperoxidase. Sea salt was not positively associated with any of the inflammatory markers.
Conclusions
Results suggest that PM2.5 from several sources, such as biomass combustion and traffic, are promoters of systemic inflammation, a risk factor for cardiovascular diseases.
Keywords: particulate air pollution, inflammation, cardiovascular health
What this paper adds.
Ambient fine particulate matter (PM2.5; aerodynamic diameter <2.5 µm) has adverse effects on cardiovascular health.
Systemic inflammation has been proposed to be one mechanism linking inhaled PM2.5 with cardiovascular events.
Some epidemiological studies suggest that PM2.5 from traffic has an effect on inflammation, but the influence of PM2.5 from other sources is poorly known.
In this study, PM2.5 from several sources was associated with systemic inflammation.
Multitude of harmful sources of particulate air pollution makes abatement of health effects more challenging and more expensive, because legislation and air pollution control methods cannot focus on any particular source type.
Introduction
An association between ambient particulate matter (PM) and cardiovascular health has been established in many studies.1 2 Low-level inflammation plays a key role in the development of cardiovascular diseases, such as atherosclerosis,3 while acute changes in inflammatory status are associated with the vulnerability of atherosclerotic plaque.1 Therefore, it has been hypothesised that increased systemic inflammation due to inhalation of PM may explain the observed associations between daily ambient PM and cardiovascular mortality and hospitalisations. Indeed, daily levels of outdoor particulate air pollution have been reported to be associated with the concentration of inflammatory markers.1
Ambient air fine particles (PM2.5; particles <2.5 µm in aerodynamic diameter) are a mixture of numerous components originating from different sources and atmospheric processes. Association between black carbon particles, an indicator for combustion derived particles, and cardiopulmonary morbidity and mortality has been concluded in a systematic review by Janssen et al.4 However, black carbon per se may not be responsible for the observed health effects, but may merely act as an indicator of harmful PM constituents originating from combustion processes. Many epidemiological studies have reported particles originating from combustion of fossil fuels, including road traffic, to be associated with cardiopulmonary health, but fewer studies have included particles originating from biomass combustion, another source of black carbon.5 One important source category is formed by long-range transport (LRT) of particles. The source is comprised of secondary aerosols, formed over a longer time in the atmosphere, and is typically characterised by sulfate. Sulfate has been associated with cardiovascular and respiratory health effects in short-term epidemiological studies.6 In general, there is a lack of epidemiological studies comparing the strength of association between different sources and health.
We have previously reported that even low daily levels of PM2.5, typical for smaller cities in Europe and North America, may increase systemic inflammation among elderly subjects with coronary heart disease.7 In this paper, we exploit the same health study conducted in Kotka, Finland, and evaluate associations between the daily concentrations of source-specific PM2.5 and the levels of inflammatory markers in blood.
Methods
Study design
We recruited 52 elderly persons with clinically stable ischaemic heart disease (IHD) for the study (see online supplementary figure S1). Participants lived in the city centre of Kotka (55 000 inhabitants) in Finland. We followed the study participants every second week between 15 November 2005 and 12 May 2006. However, a significant air pollution episode, caused by large wildfires in Eastern Europe, occurred in Eastern Finland at the end of April 2006. Because these exceptionally high air pollution levels during the episode did not represent normal exposure, we excluded the end of the measurement period from the data. Therefore the final data comprised measurements conducted between 15 November 2005 and 21 April 2006; the number of repeated visits ranged from 3 to 11 (medium=5) depending on the number of visits cancelled (eg, because of symptoms of influenza). We scheduled clinical visits on the same weekday and at the same time of the day (for 96% of the participants the variability was ± 1h) to minimise potential influence of weekly and circadian variation. The study protocol was based on the AIRGENE study8 and the recruitment process has been described previously by Huttunen et al.7 The main inclusion criteria for the study subjects were IHD diagnosed by a physician, being a non-smoker, and age 35–80 years. We excluded participants with diagnosed chronic diseases with a strong inflammatory component. Inflammatory diseases leading to exclusion were Crohn's disease, colitis ulcerosa, rheumatoid arthritis, haemophilia and HIV. Persons who had had myocardial infarction, surgery, angioplasty, balloon angioplasty, bypass operation or infection within 6 months were also excluded. We received ethics approval from the ethics committee of the Kymenlaakso Hospital District. In addition, all study subjects signed a written informed consent.
Clinical measurements
At the first visit, we collected information on the health status, medication intake and lifestyle factors of the study subjects with a baseline questionnaire. During each clinical visit, we collected venous blood samples in EDTA and citrate plasma tubes for the determination of the inflammatory markers. We processed the blood samples for further analysis within 0.5 h. We centrifuged the samples at 4°C for 15 min and stored plasma at −70°C until measurement of the indicators of systemic inflammation.
We analysed the cytokines interleukin (IL)-1β, IL-6, IL-8 and IL-12, and interferon (IFN) γ from the EDTA-plasma with an immunologic ELISA method (OptEIA, Becton Dickinson, San José, California, USA) at the Department of Environmental Health, National Institute for Health and Welfare (Kuopio, Finland). Cytokine analysis was optimised with in-house titration experiments; the minimum detectable level was determined for each cytokine by defining the concentration above two SDs of the average optical densities of 20 replicates of the zero standard (IL-1β, 14 pg/mL; IL-6, 2 pg/mL; IL-8, 1 pg/mL; IL-12, 5 pg/mL; and IFNγ, 4 pg/mL). For cytokines IL-1β, IL-6 and IFNγ, the proportion of samples below the detection limit was considered too high (42–70%) to conduct reliable statistical analyses. Thus, only the results for IL-8 and IL-12 are presented in this paper.
We analysed high-sensitive C-reactive protein (CRP), fibrinogen (FB), white blood cell (WBC) count and myeloperoxidase (MPO) at the Kymenlaakso Hospital Services (Laboratory of Clinical Chemistry, Carea, Kotka, Finland). CRP was analysed from EDTA-plasma with an immunoturbidimetric method (Sentinel CRP Vario List No. 6K26-02), and FB from citrate-plasma with a chromogenic method. WBC counting was performed by an automatic haematological analyser (CellDyn 4000, Abbott), and levels of MPO were determined by ELISA (MPO Elisa Kit, Immunodiagnostik) using the instrument Multiscan Ex (Thermolabsystems). Detection limits for the CRP, FB and MPO analyses were <0.1 mg/mL, <0.5 g/L and 1.4 ng/mL, respectively. Values below the detection limit were included as such in the data for IL-8 and IL-12, and treated as missing values for CRP, WBC and FB.
Air pollution data and meteorological parameters
Before the beginning of the study period, we set up a measurement station to monitor general air quality prevailing at the study area. The station was located in a schoolyard in the city centre, and was surrounded by three-storey apartment buildings at a distance of 15–30 m. A major road leading from the city centre to the surrounding areas was located 1000 m west from the station. All study participants lived within a distance of 1.6 km from the fixed outdoor air pollution monitoring site. The sample intakes were placed about 5–6 m above the level of the nearest street.
We measured particle number concentration (PNC) with a condensation particle counter (CPC 3022, particles >20 nm in aerodynamic diameter), and nitrogen oxide (NO) and nitrogen dioxide (NO2) with the chemiluminescence method (AC-30M Environment, Poissy, France). We collected 24 h outdoor PM2.5 samples with an EPA-well impactor ninety-six (WINS) sampler,9 which operated at a flow rate of 16.7 l/min. We used prewashed (methanol and deionised) polytetrafluoroethylene (PTFE) filters (diameter 47 mm, pore size 3 µm, type FS, Millipore Ireland, Carrigtwohill, Ireland) to collect the samples. Filters were changed daily at noon (86% of filters were collected between 10:00 and 14:00) and 24 h averages were calculated from the continuous data from noon to noon.
We weighed the PTFE filters with a Mettler M3 microbalance (Mettler Instumente, Zurich, Switzerland) before and after sampling. Temperature and relative humidity in the weighing room ranged from 22.6 to 23.8°C and from 10.1 to 26.3%, respectively. We stabilised the filters inside a laminar flow bench for about 30 min before weighing, and used the Electrical discharger (Mettler Toledo, HAUG, Leinfelden-Achterdingen, Germany) and Po-210 (1U400 static master, NRD, Grand Island, New York, USA) radioactive source to control the static electricity. After weighing, we stored the samples in a freezer (−20°C) until chemical analyses.
We determined the absorption coefficient (ABS), an indicator of combustion derived particles, by measuring reflectance of the PTFE filters from a EPA-WINS sampler with a smoke stain reflectometer (Model M34D, Diffusion Systems, London, UK). Reflectance of PM2.5 filters was transformed into an absorption coefficient (a) according to ISO983510:
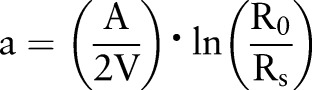 |
1 |
where A=loaded filter area (m2), V=sampled volume (m3), R0=average reflectance of field blank filters and Rs=reflectance of the sampled filter. Absorption coefficients are expressed in 1/m×1/105. We used only one half of each PTFE filter in this ABS measurement.
We used the other half of the filters to analyse a group of selected ions with an ion chromatograph (DX500, Dionex Corporation, Sunnyvale, California, USA). We analysed the anions (Cl−, NO3−, SO42− and oxalate) using an AS11 column and 1–20 mM sodium hydroxide eluent with a flow rate of 1.5 mL/min, and the cations (Na+, NH4+, K+, Mg2+ and Ca2+) using a CS12 column and 20 mM methanesulfonic acid eluent with a flow rate of 1.2 mL/min. In addition, we analysed monosaccharide anhydrides, including levoglucosan, using an anion-exchange chromatograph coupled to an ion trap mass spectrometer (Agilent Technologies SL, Santa Clara, California, USA).11 Chemical analyses are described in more detail by Aurela et al.12
The city of Kotka recorded meteorological parameters (air temperature, relative humidity, barometric pressure and wind direction) with an automatic weather station located at a distance of 1.5 km from the fixed outdoor measurement site.
Source apportionment
We conducted a source apportionment for PM2.5 using the EPA PMF 3.0. model of the US Environmental Protection Agency. Information on the source-apportionment analyses can be found in the online supplementary material.
Statistical analyses
We performed statistical analyses using linear mixed models with a random patient intercept and a compound symmetry covariance structure in SAS V.9.2. Based on the distribution of model residuals, we log transformed IL-12, IL-8, CRP and MPO. Furthermore, we evaluated immediate and lagged air pollution effects (up to 4 days lag). We defined lag 0 concentration as the 24 h average concentration from noon of the previous day to noon of the day of the clinic visit; lag 1 was defined as the preceding 24 h period, and lags 2–4 accordingly.
We built first a confounder model without air pollutants separately for each inflammatory marker. We used penalised splines (P-splines) in the additive mixed model framework to allow for non-linear confounder-response functions,13 and used minimisation of Akaike's information criteria method to select the shape and lag of the confounder into the model. Long-term time trend and apparent temperature (which contains temperature and relative humidity) were included as confounders. In the first step, we added a variable for long-term time trend in the model, in either linear or non-linear form. In the second step, we chose between lag 0 and the average of lags 1–3 for apparent temperature, testing again for the linearity of the effect. We tested both compound symmetry and first-order autocorrelation structures to confirm the applicability of the chosen correlation structure. As sensitivity analyses, we evaluated the effect of extreme values (>3 times the SD) on the results, and matched the lag-time of temperature in the models with the lag-time of air pollution. We present the associations as percentage changes of the outcome mean per 1 µg/m3 increase in air pollutant concentration together with 95% CIs.
Results
Baseline characteristics of the study population are presented in table 1. Fifty-two study participants had at least three valid repeated visits, and the total number of clinical visits during the 5 months study period was 444. Mean concentrations of IL-12, IL-8, CRP, FB, WBC and MPO were 474 pg/mL, 3.1 pg/mL, 2.3 mg/L, 3.1 g/L, 6.7×109/L and 76 mg/L, respectively. Proportions of samples below the detection limit ranged from 1% for CRP to 7% for IL-8.
Table 1.
Baseline characteristics of the study population (52 ischaemic heart disease patients)
| Characteristic | Value |
|---|---|
| Age, mean (years) (range) | 71 (50–80) |
| Body mass index, mean (kg/m2) (range) | 29 (21–49) |
| Sex, n (%) | |
| Male | 32 (62) |
| Female | 20 (38) |
| Self reported history, n (%) | |
| Myocardial infarction | 25 (48) |
| Angina pectoris | 31 (60) |
| Arrhythmia | 26 (50) |
| Congestive heart failure | 13 (25) |
| Hypertension | 25 (48) |
| Stroke | 1 (2) |
| Diabetes | 10 (19) |
| Arthrosis | 11 (21) |
| Asthma | 7 (13) |
| Total cholesterol, mean (mmol/L) (SD) | 4.2 (0.8) |
| High-density lipoprotein cholesterol, mean (mmol/L) (SD) | 2.0 (3.0) |
| Medication, n (%) | |
| Statins | 43 (83) |
| Lipid-lowering medication | 47 (90) |
| Antithrombotic medication | 46 (88) |
| Regular use of β-blockers | 42 (81) |
We identified five source categories in the PM2.5 in source apportionment. Descriptive statistics for source specific PM2.5 and Spearman's correlations (r) for air pollution and temperature are presented in table 2. The main source category was regional and LRT (56% of the measured mass). The average contributions of LRT, traffic, biomass combustion, sea salt and pulp industry were 4.5, 0.6, 1.6, 0.1 and 1.0 µg/m3, respectively. The model explained, on average, 99% of the variation in measured PM2.5 concentration. The levels of PM2.5 from different sources had mainly very weak positive correlations with each other, and the highest correlation (r=0.56) was observed between LRT and biomass combustion. Correlations between sea salt and other sources were negative. Mean temperature during the study period was −4.3°C, and it had moderate negative correlation with PM related to traffic and pulp industry.
Table 2.
Descriptive statistics and Spearman's correlations for air pollution and temperature (T)
| Percentile | Correlations | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean (±SD) | 25th | 50th | 75th | Max | TRAF | BIOM | SALT | IND | PM2.5 | ABS | PNC | T | |
| LRT (µg/m3) | 130 | 4.5 (±3.4) | 1.7 | 4.3 | 6.0 | 17.4 | 0.16 | 0.56 | −0.54 | 0.07 | 0.86 | 0.60 | 0.30 | −0.11 |
| TRAF (µg/m3) | 130 | 0.6 (±0.4) | 0.3 | 0.5 | 0.7 | 1.7 | 0.19 | −0.07 | 0.14 | 0.25 | 0.44 | 0.84 | −0.41 | |
| BIOM (µg/m3) | 130 | 1.6 (±1.5) | 0.5 | 1.1 | 2.2 | 7.4 | −0.16 | 0.11 | 0.76 | 0.88 | 0.30 | −0.15 | ||
| SALT (µg/m3) | 130 | 0.1 (±0.2) | 0.01 | 0.02 | 0.06 | 1.2 | 0.21 | −0.38 | −0.22 | −0.12 | −0.02 | |||
| IND (µg/m3) | 130 | 1.0 (±1.0) | 0.3 | 0.7 | 1.4 | 6.0 | 0.27 | 0.23 | 0.27 | −0.41 | ||||
| PM2.5 (µg/m3) | 160 | 8.1 (±5.0) | 4.5 | 6.8 | 10.0 | 27.6 | 0.82 | 0.40 | −0.19 | |||||
| ABS (m−1×10−5) | 131 | 1.5 (±0.9) | 0.9 | 1.3 | 1.9 | 4.8 | 0.49 | −0.23 | ||||||
| PNC (cm−3) | 155 | 6300 (±5100) | 3100 | 4599 | 7937 | 29 332 | −0.42 | |||||||
| T (°C) | 160 | −4.3 (±6.1) | −7.6 | −2.8 | 0.3 | 7.3 | ||||||||
ABS, absorption coefficient; BIOM, biomass combustion; IND, pulp industry; LRT, regional and long-range transport; PM2.5, particles <2.5 µm in aerodynamic diameter; PNC, particle number concentration; SALT, sea salt; TRAF, traffic emissions.
Associations between the levels of source-specific PM2.5 and the levels of inflammatory markers are presented in table 3. Under the assumption of independent observations, we would have expected six false statistically significant associations (5 outcomes*4 lags*6 sources, α-level 0.05), but observed 11 (and one borderline significant). We found the strongest, statistically significant, association between traffic emissions and IL-12 at a lag of 1 day. Biomass combustion and pulp industry were also positively associated with IL-12; for LRT the effect estimate was borderline significant. Biomass combustion was also associated with CRP and MPO at a 2-day lag, whereas pulp industry was associated with MPO (lag 0), but not with CRP. LRT was associated with both CRP and MPO at 1- and 2-day lags. Sea salt had a strong negative association with IL-12 at lag 1 and with IL-8 at lag 0.
Table 3.
Associations of source-specific PM2.5 (fine particles, particles <2.5 µm in aerodynamic diameter) on inflammatory markers
| Inflammatory | Lag | LRT | TRAF | BIOM | SALT | IND |
|---|---|---|---|---|---|---|
| marker | % change (95% CI) | % change (95% CI) | % change (95% CI) | % change (95% CI) | % change (95% CI) | |
| IL-12 | 0 | 1.60 (0.00 to 3.22) | 0.73 (−12.3 to 15.46) | 3.39 (0.19 to 6.71) | −5.43 (−34.29 to 36.11) | 3.81 (−0.64 to 8.46) |
| 1 | 0.79 (−0.72 to 2.31) | 17.97 (3.19 to 34.86) | 3.46 (0.63 to 6.37) | −29.98 (−48.71 to −4.41) | 4.49 (−0.43 to 9.66) | |
| 2 | 0.26 (−1.14 to 1.68) | 4.33 (−8.91 to 19.49) | 1.24 (−1.83 to 4.42) | 26.32 (−7.11 to 71.77) | −1.16 (−7.60 to 5.72) | |
| 3 | 0.57 (−0.88 to 2.03) | 14.28 (−0.81 to 31.68) | 1.68 (−1.27 to 4.71) | 10.21 (−17.92 to 47.99) | 8.36 (0.57 to 16.75) | |
| IL-8 | 0 | 0.64 (−1.66 to 2.98) | −7.69 (−24.82 to 13.33) | −0.60 (−5.13 to 4.15) | −59.32 (−76.58 to −29.34) | −5.36 (−11.69 to 1.42) |
| 1 | 1.71 (−0.37 to 3.83) | −0.29 (−18.28 to 21.65) | 1.96 (−1.98 to 6.06) | −18.19 (−47.15 to 26.63) | −4.51 (−11.26 to 2.75) | |
| 2 | 0.88 (−1.07 to 2.87) | 6.56 (−13.00 to 30.51) | 2.57 (−1.66 to 6.97) | −11.54 (−44.93 to 42.09) | −4.88 (−13.30 to 4.37) | |
| 3 | 0.66 (−1.38 to 2.75) | 9.27 (−10.59 to 33.53) | 2.09 (−1.89 to 6.23) | −15.93 (−45.22 to 29.03) | 3.71 (−6.54 to 15.09) | |
| CRP | 0 | 0.40 (−2.32 to 3.19) | 9.23 (−12.01 to 35.61) | −1.85 (−6.92 to 3.48) | 17.09 (−36.80 to 116.91) | −1.19 (−7.86 to 5.97) |
| 1 | 3.15 (0.75 to 5.60) | 19.21 (−4.29 to 48.47) | 4.40 (−0.16 to 9.17) | −24.86 (−59.66 to 39.96) | −5.86 (−12.80 to 1.63) | |
| 2 | 2.53 (0.32 to 4.78) | 1.54 (−18.18 to 26.01) | 6.14 (1.12 to 11.41) | −14.74 (−47.69 to 38.99) | −1.98 (−11.35 to 8.37) | |
| 3 | 1.63 (−0.74 to 4.06) | 6.69 (−16.88 to 36.93) | 1.82 (−2.80 to 6.65) | −19.48 (−50.22 to 30.24) | −8.85 (−19.03 to 2.62) | |
| FB | 0 | −0.04 (−0.57 to 0.49) | 0.28 (−4.39 to 4.95) | 0.12 (−0.89 to 1.13) | 8.76 (−1.67 to 19.19) | −0.52 (−1.81 to 0.77) |
| 1 | 0.29 (−0.20 to 0.77) | −1.60 (−5.66 to 2.46) | 0.39 (−0.46 to 1.24) | −2.31 (−10.93 to 6.30) | −0.30 (−1.64 to 1.05) | |
| 2 | −0.04 (−0.51 to 0.42) | 0.92 (−2.87 to 4.72) | 0.01 (−0.93 to 0.94) | −5.98 (−14.73 to 2.77) | −1.32 (−3.01 to 0.38) | |
| 3 | −0.31 (−0.78 to 0.15) | 0.74 (−3.36 to 4.84) | −0.17 (−1.09 to 0.75) | −2.93 (−11.34 to 5.48) | −0.52 (−2.41 to 1.38) | |
| WBC | 0 | 0.11 (−0.40 to 0.62) | −1.19 (−5.08 to 2.70) | 0.14 (−0.82 to 1.09) | −1.34 (−12.34 to 9.66) | 0.08 (−1.20 to 1.37) |
| 1 | 0.16 (−0.27 to 0.58) | −1.52 (−5.46 to 2.41) | 0.02 (−0.82 to 0.85) | 4.24 (−6.94 to 15.41) | −1.17 (−2.61 to 0.27) | |
| 2 | 0.05 (−0.34 to 0.43) | 1.45 (−2.35 to 5.25) | 0.77 (−0.09 to 1.63) | −4.78 (−13.35 to 3.79) | 0.19 (−1.56 to 1.95) | |
| 3 | −0.19 (−0.60 to 0.22) | 2.97 (−1.26 to 7.20) | 0.25 (−0.55 to 1.06) | −2.21 (−10.77 to 6.34) | −0.41 (−2.41 to 1.58) | |
| MPO | 0 | 0.88 (−0.08 to 1.84) | 3.90 (−4.07 to 12.54) | 1.25 (−0.62 to 3.15) | −12.55 (−29.51 to 8.50) | 2.95 (0.45 to 5.51) |
| 1 | 1.08 (0.20 to 1.97) | 3.13 (−4.81 to 11.73) | 1.05 (−0.59 to 2.71) | −8.57 (−26.46 to 13.67) | 1.26 (−1.53 to 4.13) | |
| 2 | 0.80 (0.02 to 1.59) | 3.96 (−3.71 to 12.24) | 2.07 (0.31 to 3.86) | −6.53 (−22.35 to 12.51) | 1.36 (−2.31 to 5.16) | |
| 3 | 0.42 (−0.41 to 1.26) | 3.74 (−5.02 to 13.30) | 0.55 (−1.05 to 2.18) | −13.01 (−26.37 to 2.78) | −2.55 (−6.43 to 1.49) |
Effect estimates calculated for 1 µg/m3 increase in source-specific PM2.5.
BIOM, biomass combustion; CRP, C-reactive protein; FB, fibrinogen; IL, interleukin; IND, pulp industry; LRT, regional and long-range transport; PM2.5, particles <2.5 µm in aerodynamic diameter; MPO, myeloperoxidase; SALT, sea salt; TRAF, traffic emissions; WBC, white blood cell count.
Effect estimates for the 5-day average concentrations of the source-specific PM2.5 are presented in figure 1. Concerning IL-12, we found the highest effect estimate again for traffic, but it was not statistically significant. Also the association between IL-12 and biomass combustion was only suggestive, but LRT and biomass combustion showed a positive association with CRP. For MPO, a suggestive evidence of association was observed for biomass. PM2.5 from pulp industry was positively associated with IL-12, but negatively with CRP and IL-8. For comparison, the effect estimates for the 5-day average concentrations calculated per interquartile increase of the source-specific PM2.5 are presented for IL-12 and CRP in online supplementary figure S4.
Figure 1.

Estimated effect of 5-day average source-specific PM2.5 (particles <2.5 µm in aerodynamic diameter) on inflammatory markers. Effect estimates have been calculated as percentage changes of the outcome mean per 1 µg/m3 increase in air pollutant concentration together with 95% CIs. BIOM, biomass combustion; CRP, C-reactive protein; FB, fibrinogen; IL, interleukin; IND, pulp industry; LRT, regional and long-range transport; MPO, myeloperoxidase; SALT, sea salt; TRAF, traffic emissions; WBC, white blood cell count.
In general, we did not find substantial changes in the results, when we excluded extreme concentrations (at maximum 50 measurements per lag). However, more negative associations were observed for sea salt. Associations between pulp industry and inflammatory markers got mainly weaker, whereas negative associations between sea salt and inflammatory markers strengthened when we matched the lag-time of source-specific PM2.5 with the lag time of temperature. Because of the relatively high correlation (r=0.56) between LRT and biomass combustion, we also run two-pollutant models. Effect estimates for LRT turned out to be more robust for the adjustment (data not shown).
Discussion
To our knowledge, this is the first epidemiological study to evaluate associations between daily variation in source-specific fine particulate air pollution and markers of systemic inflammation. In this panel study of elderly individuals with IHD, we found significant positive associations between source-specific PM2.5 and inflammatory markers CRP, IL-12 and MPO. The strongest associations were observed for LRT, traffic emissions and biomass combustion contributions to PM2.5. We also observed decreased levels of inflammatory markers in association with PM2.5 rich in sea salt.
CRP is the most well-known inflammatory marker which is commonly used in clinical tests to represent systemic inflammation in the human body. An increased level of CRP is predictive of an elevated risk for future cardiovascular morbidity and mortality.14 Particulate air pollution has been reported to be associated with the levels of CRP in several studies.15–17 In this study, we observed consistent associations between CRP and biomass combustion as well as LRT. CRP has been reported to react to changes in air pollution with a few days lag,15–17 which was also true in our study. However, it is not well known how fast other inflammatory markers react to changes in air pollution levels (or in any other stressors). Reaction may occur immediately after exposure and then disappear, but sometimes, for example, acute phase proteins can induce a new delayed reaction of cytokines after a few days. Therefore, we did not have any strong hypothesis on the lag times of inflammation markers other than CRP.
Cytokine IL-12 is produced by macrophages and dendritic cells, and it is suggested to enhance the development of atherosclerosis by stimulating T-cell recruitment into atherosclerotic plaques.18 IL-12 was another marker of systemic inflammation for which we found evidence of an air pollution effect: PM2.5 from LRT, biomass combustion and traffic were associated with increased IL-12 concentrations in the blood of IHD patients.
MPO, a leucocyte enzyme, is stored in neutrophils and monocytes, and is released by leucocyte activation and degranulation. This has been proposed to be a risk marker for coronary artery disease.19 20 In our study MPO was associated with PM2.5 from LRT, biomass combustion and pulp industry.
Our study also included some inflammatory markers which apparently were not affected by air pollution. For FB, IL-8 and WBC we found no consistent associations with PM2.5 originating from any of the five sources. FB is a commonly used marker of inflammation and increased risk of blood coagulation in clinical medicine. Results from previous studies are inconclusive: positive,21 22 negative23 and non-existent15 associations between PM and FB have been reported.
WBC count has been considered to be a stable marker of vascular inflammation, and a predictor of long-term cardiovascular events.24 25 Some previous studies have associated short-term increases in the levels of ambient PM with elevated numbers of WBCs,26 27 whereas some other studies have found no evidence of association.23
In southern Finland more than half of all ambient PM2.5 is long-range transported air pollution. In our study, the proportion of LRT was 56%. In Helsinki, Finland, almost all of sulfate and ammonium and more than half of nitrate are long-range transported.28 In our study LRT was characterised, in addition to sulfate and ammonium, by oxalate—an indicator of biomass combustion, especially during the cold and low-solar radiation wintertime when it is not formed by atmospheric processes. Some of the biomass combustion particles were likely emitted by nearby sources, whereas the rest came via regional and LRT, but separation of these two is not unambiguous. Biomass combustion correlated moderately with LRT.
Small-scale wood combustion as a primary or secondary heating source in residential areas is common throughout Finland. In the Helsinki metropolitan area, where nearly 80% of households are connected to the district heating system, the contribution of biomass combustion derived aerosol in PM2.5 has been estimated by Saarnio et al29 to be about 31–66% in suburban areas and 18–29% in urban areas during the coldest heating season (October–March). The higher proportion of PM from biomass combustion in suburban areas reflected the higher contribution of local residential wood combustion. In our study 20% of the explained PM2.5 was from biomass combustion, which agrees well with the findings from the Helsinki metropolitan area.
It is noteworthy that fine particles from biomass combustion were associated with CRP, IL-12 and MPO in our study. The finding supports the recent review by WHO,6 where evidence was found for an effect of particles from biomass combustion on cardiovascular health. The review concluded that cardiovascular effects of particles from biomass combustion may be comparable to those from traffic. The conclusion was supported by those few short-term studies, which have compared in the same set-up the associations of biomass and traffic-related PM2.5 with daily cardiovascular mortality or hospitalisations.30 31
Vehicular traffic exhausts and stationary combustion processes are major sources of fine particles. It has been suggested that in urban areas traffic is also the most important source of ultrafine particles,32 33 which have been associated with increased levels of inflammation and coagulation markers in some studies.21 34 We did not have a specific indicator component for traffic emissions, but in our source apportionment analysis, the factor describing traffic emissions was characterised by high levels of NO and PNC.
The mass portion of sea salt can vary depending on the season. The proportion of sea salt from all measured fine particles in Kotka was 1% during our field measurements. Sea salt concentrations were associated negatively with the levels of inflammatory markers, and sea salt had a moderate negative correlation with LRT. Based on back trajectories, LRT particles rich in secondary sulfate are typically transported to Kotka from central and Eastern Europe, whereas air masses containing sea salt passed over cleaner sea areas.
In our previous study7 we evaluated associations between different PM size fractions and the concentrations of inflammatory markers in blood. Given as percentage changes of outcome mean per 1 µg/m3 increase in PM2.5 concentration, the strongest associations between ambient PM2.5 and inflammatory markers were found for CRP (2.6%; 95% CI 0.9% to 4.5%) and for IL-12 (1.3%; 95% CI 0.3% to 2.4%). Here we studied the effects of PM2.5 from different sources; the effect estimate found for IL-12 was even higher for traffic-related PM2.5 (18.0%; 95% CI 3.2% to 34.9%) than for total ambient PM2.5. In the case of CRP, the effect estimates for ambient PM2.5 were clearly higher than for source-related PM2.5. Furthermore, MPO was associated in the previous study with total PM2.5 (0.6%; 95% CI 0.05% to 1.2%). For MPO, effect estimates for PM2.5 from LRT, biomass and pulp industry were somewhat higher than those for total PM2.5.
A strength of this study is that exposure assessment is based on data from the central measurement site located only a short distance from the homes of the study participants. Another strength is the substantial number of repeated measurements per participant. In our opinion, these results are also rather generalisable to other types of study populations, as it is unlikely that the differences between sources in harmfulness would depend on, for example, disease status. However, generalisability of the results for the source category industry is more questionable because the composition of emissions from industry depends heavily on the type of industry. This may explain why, in contrast to our study, in a recent German study on the long-term effects of air pollution on CRP an association with industry was observed.35
One limitation of the study is that the source apportionment was based on outdoor measurements instead of measurements of actual exposures, which may cause source-dependent exposure misclassification. Several studies have reported that daily outdoor PM2.5, ABS and sulfate or sulfur (indicating LRT) correlate longitudinally well with indoor concentrations and personal exposures.36–39 There is some evidence that outdoor concentrations of PM2.5 from non-tailpipe emissions may not reflect well the personal exposures.39 In general, there is very little information on correlations between outdoor, indoor or personal source-specific PM2.5 concentrations.40 41 Strengths and limitations of the source apportionment are discussed in the online supplementary material.
An inherent limitation of systemic inflammation as an outcome is caused by the fact that many factors influence short-term changes in inflammation, such as exercise and stress. This will add noise in the results and make associations between air pollution and inflammation more difficult to observe. Moreover, the role of CRP and FB as predictors of acute exacerbations of heart diseases has been established, but the role of other inflammatory markers in this context is less clear.
Conclusion
Our results suggest that particulate air pollution from several sources, including residential wood combustion and traffic, are potential promoters of systemic inflammation, a risk factor for cardiovascular diseases.
Supplementary Material
Acknowledgments
Eija Värri and Eeva Linkola from the City of Kotka Environment Centre are gratefully acknowledged for municipal data on meteorology and air pollutants.
Footnotes
Contributors: TL, TY-T, JP, ROS, IS and HD were involved in the study design. M-RH and KH provided data for the inflammatory markers. IS acted as the responsible researcher of the study. ROS, AP and RH contributed to the exposure assessment. MA and RH were responsible for the chemical analyses of air pollution. TY-T conducted the source apportionment for air pollution. TS conducted all statistical analyses under the supervision of PT and TL. TS drafted this manuscript and all authors read and provided feedback for the manuscript.
Funding: The HIPPU project was funded by the Finnish Funding Agency for Technology and Innovation (TEKES/EAKR 70078/04), the Kymenlaakso Hospital District and the Cities of Kotka and Hamina, Kotka Energy Ltd, Sunila Ltd and Stora Enso Plc. Additional funding for TL came from the Academy of Finland (122783), and for TS as she has been student in Doctoral Program in Environmental Health and received part of her funding from there.
Competing interests: None.
Patient consent: Obtained.
Ethics approval: This study was approved by the ethics committee of the Kymenlaakso Hospital District.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Brook RD, Rajagopalan S, Pope CA III, et al. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation 2010;121:2331–78. [DOI] [PubMed] [Google Scholar]
- 2.Pope CA III, Dockery DW. Health effects of fine particulate air pollution: lines that connect. J Air Waste Manag Assoc 2006;56:709–42. [DOI] [PubMed] [Google Scholar]
- 3.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002;105:1135–43. [DOI] [PubMed] [Google Scholar]
- 4.Janssen NAH, Gerlofs-Nijland ME, Lanki T, et al. Health effects of black carbon. Copenhagen Ø, Denmark: World Health Organization, 2012. [Google Scholar]
- 5.Naeher LP, Brauer M, Lipsett M, et al. Woodsmoke health effects: a review. Inhal Toxicol 2007;19:67–106. [DOI] [PubMed] [Google Scholar]
- 6.WHO. Review of evidence on health aspects ofair pollution—REVIHAAP Project: final technical report. 2013. [PubMed]
- 7.Huttunen K, Siponen T, Salonen I, et al. Low-level exposure to ambient particulate matter is associated with systemic inflammation in ischemic heart disease patients. Environ Res 2012;116:44–51. [DOI] [PubMed] [Google Scholar]
- 8.Peters A, Schneider A, Greven S, et al. Air pollution and inflammatory response in myocardial infarction survivors: gene-environment interactions in a high-risk group. Inhal Toxicol 2007;19:161–75. [DOI] [PubMed] [Google Scholar]
- 9.Peters TM, Vanderpool RW, Wiener RW. Design and calibration of the EPA PM2.5 well impactor ninety-six (WINS). Aerosol Sci Technol 2001;34:389–97. [Google Scholar]
- 10.ISO 9835. Ambient air-determination of a black smoke index 1993.
- 11.Dye C, Yttri KE. Determination of monosaccharide anhydrides in atmospheric aerosols by use of high-performance liquid chromatography combined with high-resolution mass spectrometry. Anal Chem 2005;77:1853–8. [DOI] [PubMed] [Google Scholar]
- 12.Aurela M, Sillanpää M, Pennanen A, et al. Characterization of urban particulate matter for a health-related study in southern Finland. Boreal Env Res 2010;15:513–32. [Google Scholar]
- 13.Greven S, Küchenhoff H, Peters A. Additive mixed models with P-splines. Proceedings of the 21st International workshop on Statistical Modelling 2006;201–7. [Google Scholar]
- 14.Zakynthinos E, Pappa N. Inflammatory biomarkers in coronary artery disease. J Cardiol 2009;53:317–33. [DOI] [PubMed] [Google Scholar]
- 15.Delfino RJ, Staimer N, Tjoa T, et al. Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ Health Perspect 2008;116:898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pope CA III, Hansen ML, Long RW, et al. Ambient particulate air pollution, heart rate variability, and blood markers of inflammation in a panel of elderly subjects. Environ Health Perspect 2004;112:339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruckerl R, Ibald-Mulli A, Koenig W, et al. Air pollution and markers of inflammation and coagulation in patients with coronary heart disease. Am J Respir Crit Care Med 2006;173:432–41. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Niessner A, Nakajima T, et al. Interleukin 12 induces T-cell recruitment into the atherosclerotic plaque. Circ Res 2006;98:524–31. [DOI] [PubMed] [Google Scholar]
- 19.Meuwese MC, Stroes ES, Hazen SL, et al. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol 2007;50:159–65. [DOI] [PubMed] [Google Scholar]
- 20.Zhang R, Brennan M, Fu X, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA 2001;286:2136. [DOI] [PubMed] [Google Scholar]
- 21.Pekkanen J, Brunner EJ, Anderson HR, et al. Daily concentrations of air pollution and plasma fibrinogen in London. Occup Environ Med 2000;57:818–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruckerl R, Greven S, Ljungman P, et al. Air pollution and inflammation (interleukin-6, C-reactive protein, fibrinogen) in myocardial infarction survivors. Environ Health Perspect 2007;115:1072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seaton A, Soutar A, Crawford V, et al. Particulate air pollution and the blood. Thorax 1999;54:1027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JC, Schwartz J. Metabolic syndrome and inflammatory responses to long-term particulate air pollutants. Environ Health Perspect 2008;116:612–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Margolis KL, Manson JE, Greenland P, et al. Leukocyte count as a predictor of cardiovascular events and mortality in postmenopausal women: the Women's Health Initiative Observational Study. Arch Intern Med 2005;165:500–8. [DOI] [PubMed] [Google Scholar]
- 26.Bruske I, Hampel R, Socher MM, et al. Impact of ambient air pollution on the differential white blood cell count in patients with chronic pulmonary disease. Inhal Toxicol 2010;22:245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz J. Air pollution and blood markers of cardiovascular risk. Environ Health Perspect 2001;109:405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ojanen C, Pakkanen T, Aurela M, et al. Hengitettävien hiukkasten kokojakauma, koostumus ja lähteet pääkaupunkiseudulla [In Finnish, abstract in English] 1998;7.
- 29.Saarnio K, Niemi JV, Saarikoski S, et al. Using monosaccharide anhydrides to estimate the impact of wood combustion on fine particles in the Helsinki Metropolitan Area. Boreal Environ Res 2012;17:163. [Google Scholar]
- 30.Mar TF, Ito K, Koenig JQ, et al. PM source apportionment and health effects. 3. Investigation of inter-method variations in associations between estimated source contributions of PM2.5 and daily mortality in Phoenix, AZ. J Expo Sci Environ Epidemiol 2006;16:311–20. [DOI] [PubMed] [Google Scholar]
- 31.Sarnat JA, Marmur A, Klein M, et al. Fine particle sources and cardiorespiratory morbidity: An application of chemical mass balance and factor analytical source-apportionment methods. Environ Health Perspect 2008;116:459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morawska L, Ristovski Z, Jayaratne ER, et al. Ambient nano and ultrafine particles from motor vehicle emissions: Characteristics, ambient processing and implications on human exposure. Atmos Environ 2008;42:8113–38. [Google Scholar]
- 33.Shi JP, Khan AA, Harrison RM. Measurements of ultrafine particle concentration and size distribution in the urban atmosphere. Sci Total Environ 1999;235:51–64. [Google Scholar]
- 34.Hildebrandt K, Ruckerl R, Koenig W, et al. Short-term effects of air pollution: a panel study of blood markers in patients with chronic pulmonary disease. Part Fibre Toxicol 2009;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hennig F, Fuks K, Moebus S, et al. Association between source-specific particulate matter air pollution and hs-CRP: local traffic and industrial emissions. Environ Health Perspect 2014;122:703–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janssen NAH, Hoek G, Harssema H, et al. Personal exposure to fine particles in children correlates closely with ambient fine particles. Arch Environ Health 1999;54:95–101. [DOI] [PubMed] [Google Scholar]
- 37.Janssen NAH. Associations between ambient, personal, and indoor exposure to fine particulate matter constituents in Dutch and Finnish panels of cardiovascular patients. Occup Environ Med 2005;62:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarnat JA, Koutrakis P, Suh HH. Assessing the relationship between personal particulate and gaseous exposures of senior citizens living in Baltimore, MD. J Air Waste Manage Assoc 2000;50:1184–98. [DOI] [PubMed] [Google Scholar]
- 39.Montagne D, Hoek G, Nieuwenhuijsen M, et al. Temporal associations of ambient PM2.5 elemental concentrations with indoor and personal concentrations. Atmos Environ 2014;86:203–11. [Google Scholar]
- 40.Yli-Tuomi T, Lanki T, Hoek G, et al. Determination of the sources of indoor PM(2.5) in Amsterdam and Helsinki. Environ Sci Technol 2008;42:4440–6. [DOI] [PubMed] [Google Scholar]
- 41.Minguillón MC, Schembari A, Triguero-Mas M, et al. Source apportionment of indoor, outdoor and personal PM2.5 exposure of pregnant women in Barcelona, Spain. Atmos Environ 2012;59:426–36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.