

Background: Methyl-coenzyme M reductase (MCR) catalyzes the final step in methanogenesis.
Results: MCR forms binary complexes with both substrates, but stabilizes only the productive binary and ternary complexes.
Conclusion: Substrate-induced conformational changes promote correct binding order and chemistry.
Significance: This first rapid kinetics study of MCR with natural substrates describes how an enzyme can enforce a strictly ordered ternary complex reaction mechanism.
Keywords: Electron Paramagnetic Resonance (EPR), Enzyme Inhibitor, Enzyme Kinetics, Enzyme Mechanism, Metalloenzyme, Nickel, Pre-steady-state Kinetics
Abstract
Methyl-coenzyme M reductase (MCR) is a nickel tetrahydrocorphinoid (coenzyme F430) containing enzyme involved in the biological synthesis and anaerobic oxidation of methane. MCR catalyzes the conversion of methyl-2-mercaptoethanesulfonate (methyl-SCoM) and N-7-mercaptoheptanoylthreonine phosphate (CoB7SH) to CH4 and the mixed disulfide CoBS-SCoM. In this study, the reaction of MCR from Methanothermobacter marburgensis, with its native substrates was investigated using static binding, chemical quench, and stopped-flow techniques. Rate constants were measured for each step in this strictly ordered ternary complex catalytic mechanism. Surprisingly, in the absence of the other substrate, MCR can bind either substrate; however, only one binary complex (MCR·methyl-SCoM) is productive whereas the other (MCR·CoB7SH) is inhibitory. Moreover, the kinetic data demonstrate that binding of methyl-SCoM to the inhibitory MCR·CoB7SH complex is highly disfavored (Kd = 56 mm). However, binding of CoB7SH to the productive MCR·methyl-SCoM complex to form the active ternary complex (CoB7SH·MCR(NiI)·CH3SCoM) is highly favored (Kd = 79 μm). Only then can the chemical reaction occur (kobs = 20 s−1 at 25 °C), leading to rapid formation and dissociation of CH4 leaving the binary product complex (MCR(NiII)·CoB7S−·SCoM), which undergoes electron transfer to regenerate Ni(I) and the final product CoBS-SCoM. This first rapid kinetics study of MCR with its natural substrates describes how an enzyme can enforce a strictly ordered ternary complex mechanism and serves as a template for identification of the reaction intermediates.
Introduction
Methyl-coenzyme M reductase (MCR)2 (EC 2.8.4.1) from methanogenic archaea catalyzes the rate-limiting and final step of methane (CH4) production (1) and the first step in anaerobic CH4 oxidation (2, 3). In the direction of CH4 formation, MCR converts methyl-coenzyme M (methyl-SCoM) and coenzyme B (CoB7SH) to CH4 and the heterodisulfide of coenzyme M and coenzyme B (CoBS-SCoM) (4). CH4 is not only a potential source of renewable energy but also a potent greenhouse gas; thus, understanding the basis of CH4 production and oxidation has environmental impacts.
The crystal structures show that MCR is a dimer of heterotrimers (α2β2γ2) with a molecular mass of 270 kDa (5). The three subunits (αβγ) tightly associate to form two 50-Å hydrophobic channels (one in each heterotrimer) (6) ending in a pocket that accommodates a redox-sensitive nickel tetrapyrrole cofactor (coenzyme F430), which plays an essential role in catalysis (7, 8). Currently, 16 distinct enzymatic and complexed states of MCR have been spectroscopically characterized, including EPR-active (MCRred1 (Ni(I)-F430), MCRred2 (rhombic Ni(I)-F430), MCRox1 (high spin Ni(II) thiyl-radical), MCRox2 (high spin rhombic Ni(II) thiyl-radical), MCRMe (methyl-Ni(III)), and related alkyl-Ni(III)) (9–14) and MCRsilent (meaning that the enzyme is in an EPR-silent Ni(II) form, which includes MCRred1-silent (five coordinate Ni(II)-F430), and MCRox1-silent (Ni(II) thiolate) (6, 15, 16). To initiate catalysis, the enzyme must be in the MCRred1 state, which contains the redox-active nickel as Ni(I) (7, 8). Only when cells are harvested strictly anaerobically and exposed to special reductive activation conditions and the enzyme purified by demanding anaerobic procedures is active enzyme (containing >70–80% Ni(I)) obtained (17, 18).
Although x-ray structures are only available from one Ni(III) (methyl-Ni) and inactive Ni(II) states, all of them show that both substrates access the active site through the same long narrow channel, which opens into the hydrophobic cavity above the hydrocorphinoid plane of F430 (6, 10, 15, 16, 19). The phosphate group of CoB7SH is positioned by ionic interactions with MCR residues located halfway down the channel with its thiol group located 8.7 Å from the nickel. The binding site of HSCoM (presumably methyl-SCoM) is more deeply buried within the enzyme indicating that this substrate may enter the active site before the CoB7SH substrate for productive chemistry to occur. This has been borne out by steady-state (12, 20–23) and single turnover (21) kinetic studies. It has been proposed that, once both substrates are bound in the active site, CoB7SH induces a conformational change that brings the methyl-SCoM into closer proximity to the nickel and this promotes C-S bond cleavage (6, 15, 16, 19, 24).
The role of nickel in the MCR catalytic cycle is controversial and two competing catalytic mechanisms for MCR have been proposed (Fig. 1). These two mechanisms differ in the nature of the first intermediate in the mechanism (25). Mechanism I involves attack of the Ni(I) nucleophile on the methyl group of methyl-SCoM to generate a methyl-Ni(III) intermediate (16, 26). This proposed mechanism I is based on mechanistic work with F430 model complexes (27), on the location of substrates in the active site of inactive Ni(II) MCR structures (6), and on mechanistic and crystallographic studies of the active Ni(I) enzyme with 3-bromopropanesulfonate and methyl halide (10, 11, 13, 28). Mechanism II starts with Ni(I) attack on the sulfur atom of methyl-SCoM, promoting the homolytic cleavage of the methyl-sulfur bond and generating a methyl radical (•CH3) and a Ni(II)·thiolate complex. This mechanism is based on density functional theory calculations (29–31) and on isotope effects studies of the reaction of MCR with methyl-SCoM and homologous substrate ethyl-SCoM (32–34).
FIGURE 1.
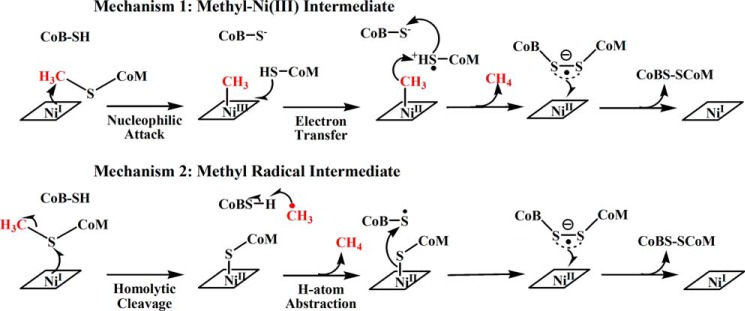
Two of the proposed catalytic mechanisms for methyl-coenzyme M reductase.
This article represents the first transient kinetic study of active MCR with its native substrates (methyl-SCoM and CoB7SH). Thus, many mechanistic details of the MCR reaction had been lacking. For example, the rate constants for formation of the various binary and ternary MCR·substrate complexes were unknown and no intermediates have been observed. In this report, the reaction of active MCR with the native substrates was explored using chemical quench and stopped-flow spectrophotometric methods. Conversion of MCR among the various intermediate nickel states was monitored to obtain rate constants for the individual steps in the reaction. Our results provide a kinetic and thermodynamic explanation for the strictly ordered binding mechanism, in which methyl-SCoM must enter the MCR active site prior to CoB7SH.
EXPERIMENTAL PROCEDURES
Organism and Materials
Methanothermobacter marburgensis (formally M. thermoautotrophicum strain Marburg) was obtained from the Oregon Collection of Methanogens (Portland, OR) catalog as OCM82. All buffers, media ingredients, and other reagents were acquired from Sigma. The N2 (99.98%), CO (99.99%), H2/CO2 (80%/20%), and Ultra High Purity (UHP) H2 (99.999%) gasses were obtained from Cryogenic Gases (Grand Rapids, MI). A stock solution of 200 mm Ti(III) citrate was synthesized by adding sodium citrate to Ti(III) trichloride (30 weight % solution in 2 n hydrochloric acid) under anaerobic conditions and adjusting the pH to 7.0 with sodium bicarbonate (35). The concentration of Ti(III) citrate was determined by titrating against a solution of methyl viologen. Methyl-SCoM and [14C]methyl-SCoM were prepared from HSCoM and methyl iodide and [14C]methyl iodide, respectively (36). The homodisulfide CoBS-SCoB was prepared from 7-bromoheptanoic acid (37, 38). The free thiol forms of CoB7SH was generated by the reduction of the homodisulfide as previously described (14).
Cell Growth and Purification
Solutions were prepared and all steps of purification were performed under anaerobic conditions in a Vacuum Atmospheres (Hawthorne, CA) anaerobic chamber maintained under nitrogen gas at an oxygen level below 1 ppm. M. marburgensis was cultured on H2/CO2 (80/20%) at 65 °C in a 14-liter fermentor (New Brunswick Scientific Co., Inc., New Brunswick, NJ) to an optical density of 5–6 at 578 nm. Culture media were prepared as previously described (39) with a slight modification of the sulfur and reducing source, by adding 50 mm sodium sulfide (instead of H2S gas) at a flow rate of 1 ml/min during the entire growth period. Before harvest, the cells were treated with 100% H2 for 30 min in the fermenter. Then, the cells were harvested, transferred to the anaerobic chamber, and resuspended in 50 mm Tris-HCl, pH 7.6, containing 10 mm HSCoM and 0.1 mm Ti(III) citrate, and transferred into a 1-liter serum-stopped anaerobic high pressure bottle. The headspace of the bottle containing the resuspended cells was then purged with CO for 10 min or at timed intervals to generate the active MCRred1 state as previously described (17). The purification of MCRred1 (Ni(I) state) was performed as described earlier (39). All steps were performed in the presence of 10 mm HSCoM and 0.1 mm Ti(III) citrate. This purification procedure generates about 60–70% MCRred1 and this is the form of the enzyme that was used in all experiments unless otherwise stated. The concentration of MCRred1 was determined by UV-visible spectroscopy using extinction coefficients of 27.0 and 9.15 mm−1 cm−1 at 385 and 420 nm, respectively, using a multiple wavelength calculation as previously described (39). The concentration of MCRsilent, which contains the inactive Ni(II) form of F430, was calculated using extinction coefficients of 22.0 and 12.7 mm−1 cm−1 at 420 and 385 nm, respectively (39).
UV-visible and EPR Spectroscopic Studies
Absorbance spectra were recorded in the anaerobic chamber using a diode array spectrophotometer (model DT 1000A, Analytical Instrument Systems, Inc., Flemington, NJ). EPR spectra were recorded on a Bruker EMX spectrometer (Bruker Biospin Corp., Billerica, MA), equipped with an Oxford ITC4 temperature controller, a Hewlett-Packard model 5340 automatic frequency counter, and Bruker gaussmeter. The EPR spectroscopic parameters included the following: temperature, 70 K; microwave power, 10 milliwatt; microwave frequency, 9.43 GHz; receiver gain, 2 × 104; modulation amplitude, 10.0 G; modulation frequency, 100 kHz. Spin concentration was determined by double integration of the sample spectrum obtained under nonsaturating conditions and comparison to that of 1 mm copper perchlorate standard. All samples for EPR spectroscopy were prepared in 50 mm Tris-HCl, pH 7.6, in a Vacuum Atmospheres anaerobic chamber.
Determination of Dissociation Constants
The interaction of substrates with MCR was determined by EPR and fluorescence methods. The enzyme used in these equilibrium binding (or dissociation) experiments was prepared by removing HSCoM and Ti(III) citrate from MCR by buffer exchanging into 50 mm Tris-HCl, pH 7.6, using Amicon Ultra15 centrifuge filter units with a 50-kDa cut-off (Millipore).
Binding of HSCoM and methyl-SCoM causes a significant change in the EPR spectrum of the active MCRred1 state in which the hyperfine lines due to the interaction between Ni(I) and the equatorial tetrapyrrole nitrogen ligands are enhanced. To determine the interaction between MCR and methyl-SCoM, a 30 μm solution of MCR was mixed with various concentrations of methyl-SCoM (2.5–500 μm, final) and anaerobically transferred into the EPR tube before freezing the mixtures in liquid nitrogen. All these manipulations were performed in the anaerobic chamber. The EPR spectrum was then measured and the change in the intensity of the hyperfine lines was plotted against the concentration of methyl-SCoM. The dissociation constant was calculated according to Equation 1 where EL is the concentration of the MCR·ligand complex, E0 is the total MCR concentration, L0 is the total ligand concentration, and Kd is the dissociation constant (40).
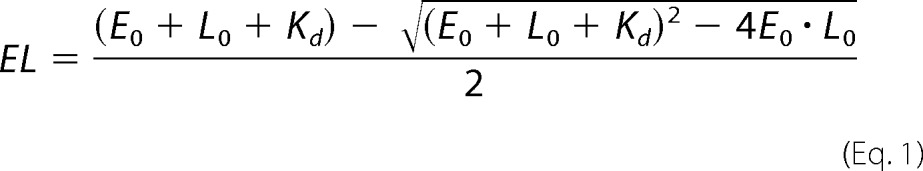 |
To measure the dissociation constants of the MCR·methyl-SCoM and MCR·CoB7SH complexes, we also monitored the changes in the fluorescence spectrum when MCR was mixed with various concentrations of each substrate (methyl-SCoM or CoB7SH). In brief, either substrate (methyl-SCoM or CoB7SH) was anaerobically titrated into an anaerobic fluorescence cuvette containing a solution of MCRred1 (∼30 μm) in 50 mm Tris-HCl, pH 7.6. For each titration, the mixture was incubated for 10 min before the fluorescence spectrum was recorded. The final concentrations of methyl-SCoM and CoB7SH used were 2.5–500 and 30–960 μm, respectively. The change in fluorescence was plotted against the concentration of substrates and the dissociation constant was calculated as described above.
Steady-State Experiments
MCR activity was measured by following the time-dependent loss of radioactivity from the methyl group of 14CH3-SCoM, which is converted to highly volatile and insoluble [14C]methane. The assay was performed at 25 °C under anaerobic conditions in 50 mm Tris-HCl, pH 7.6, containing 20 mm 14CH3-SCoM, 2 mm hydroxocobalamin (to reduce and recycle the heterodisulfide product), 20 mm Ti(III) citrate, and 0.05–1 mm CoB7SH in a final volume of 0.2 ml. The reaction was started by adding 10 μm MCRred1. 1 m Perchloric acid was used for quenching the reaction at each time point before monitoring the decrease in radioactivity of 14CH3-SCoM. This assay was also performed without hydroxocobalamin to study the MCR reaction without recycling of the heterodisulfide product. The enzyme activity was calculated from the initial slopes of 14CH3-SCoM decay in the linear portion of the time course. One unit of MCR activity is equivalent to the decay of 1 μmol of 14CH3-SCoM per minute at pH 7.6, 25 °C.
Rapid Reaction Experiments
Reactions were carried out in 50 mm Tris-HCl, pH 7.6, 25 °C. Stopped-flow measurements were performed with an Applied Photophysics (Leatherhead, United Kingdom) spectrophotometer (SX.MV18 with the Pro-Data upgrade) in photodiode array mode. The optical path length of the observation cell was 1 cm. The sample handing unit (drive syringes and mixing chamber) of the stopped-flow is located in a Vacuum Atmospheres anaerobic chamber. MCRred1 used in the rapid kinetic studies was prepared by removing HSCoM and Ti(III) citrate by buffer exchange into 50 mm Tris-HCl, pH 7.6, as described above. For studying the reaction of the MCR·methyl-SCoM complex with CoB7SH, MCRred1, and methyl-SCoM were preincubated in the first syringe and mixed with CoB7SH, at various concentrations, in the second syringe. Similarly, for studying reactions of MCR·CoB7SH complex with methyl-SCoM, the order of incubation was reversed with MCRred1 and CoB7SH mixed against various concentrations of methyl-SCoM substrate. The reaction was monitored in the photodiode array mode by following the change of the Ni(I) state of MCR at 385 nm and the Ni(II) or Ni(III) states at 420 nm. Apparent rate constants (kobs) from kinetic traces were calculated from exponential fits using the software Kinetic Studio (Tgk Scientific, Wiltshire, United Kingdom) or program A (written at the University of Michigan by Rong Chang, Jung-yen Chiu, Joel Dinverno, and D. P. Ballou). Rate constants were determined from plots of kobs versus methyl-SCoM or CoB7SH concentrations using a Marquardt-Levenberg nonlinear fit algorithm that is included in Kaleida Graph (Synergy Software). Simulations were performed by numerical methods using the Runge-Kutta algorithm implemented in Berkeley Madonna 8.3 with time step of 10−3 s. Simulations according to the model proposed (Schemes 1 and 2) were conducted, and the results were compared with the stopped-flow data.
SCHEME 1.
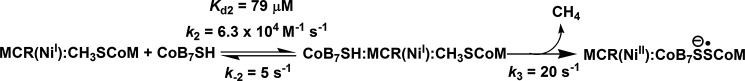
Reaction of MCR·methyl-SCoM complex with CoB7SH.
SCHEME 2.

Reaction of MCR·CoB7SH complex with methyl-SCoM.
Rapid Quench-flow Experiments
All experiments used perchloric acid as the quenching solution and were performed at 25 °C using a Bio-Logic (Claix, France) model SFM-400 rapid quench-flow system in an anaerobic chamber. The rapid quench-flow system employed three syringes. An anaerobic solution of MCR·methyl-SCoM (nonradioactive) or MCR·14CH3-SCoM in 50 mm Tris-HCl, pH 7.6, from the first syringe was mixed with CoB7SH from the second syringe. The reaction mixture was incubated for various time periods before quenching with a 0.2 m perchloric acid solution from a third syringe. The quenched samples were collected from the sample loop, and injected into 10-ml serum bottles sealed with rubber stoppers. The decrease in radioactivity of 14CH3-SCoM was monitored from the reaction of MCR with radioactive methyl-SCoM using liquid scintillation counting. A similar reaction was performed using nonradioactive CH3-SCoM and the amount of CH4 produced was monitored using gas chromatography.
RESULTS
Measurement of Interactions between MCR and Its Substrates
The interactions of MCR with methyl-SCoM and CoB7SH were examined by EPR and fluorescence spectroscopy to determine the dissociation constants of the binary complexes between this enzyme and each substrate. Changes in the fluorescence properties of activated MCRred1 upon binding of methyl-SCoM and CoB7SH were monitored using a spectrofluorometer at 25 °C. A solution of the MCRred1 (∼30 μm) was mixed with various concentrations of methyl-SCoM (Fig. 2A) and CoB7SH (Fig. 2B). MCRred1 was excited at its wavelength maximum at 385 nm and binding was monitored via following the change in fluorescence emission at 470 nm (Fig. 2, A and B). The binding of methyl-SCoM substrate to MCRred1 is very tight (Kd = 13 ± 4 μm) (inset in Fig. 2A, Table 1).
FIGURE 2.

Changes in the fluorescence spectra of MCR upon titration with methyl-SCoM (A) or CoB7SH (B). The insets in A and B show plots of the fluorescence changes at the wavelength where maximum signal change was observed (470 nm) versus substrate concentration. Superhyperfine splitting in the EPR spectra of MCR upon titration with 500 μm methyl-SCoM is shown in C. The upper inset in C represents the intensity differences at the g-value from 2.08 to 2.05 where the superhyperfine splitting was observed. The lower inset in C shows plots of the maximum intensity differences versus methyl-SCoM concentration. The dissociation constants were summarized in Table 1.
TABLE 1.
Rate constants obtained from kinetic analysis and simulations
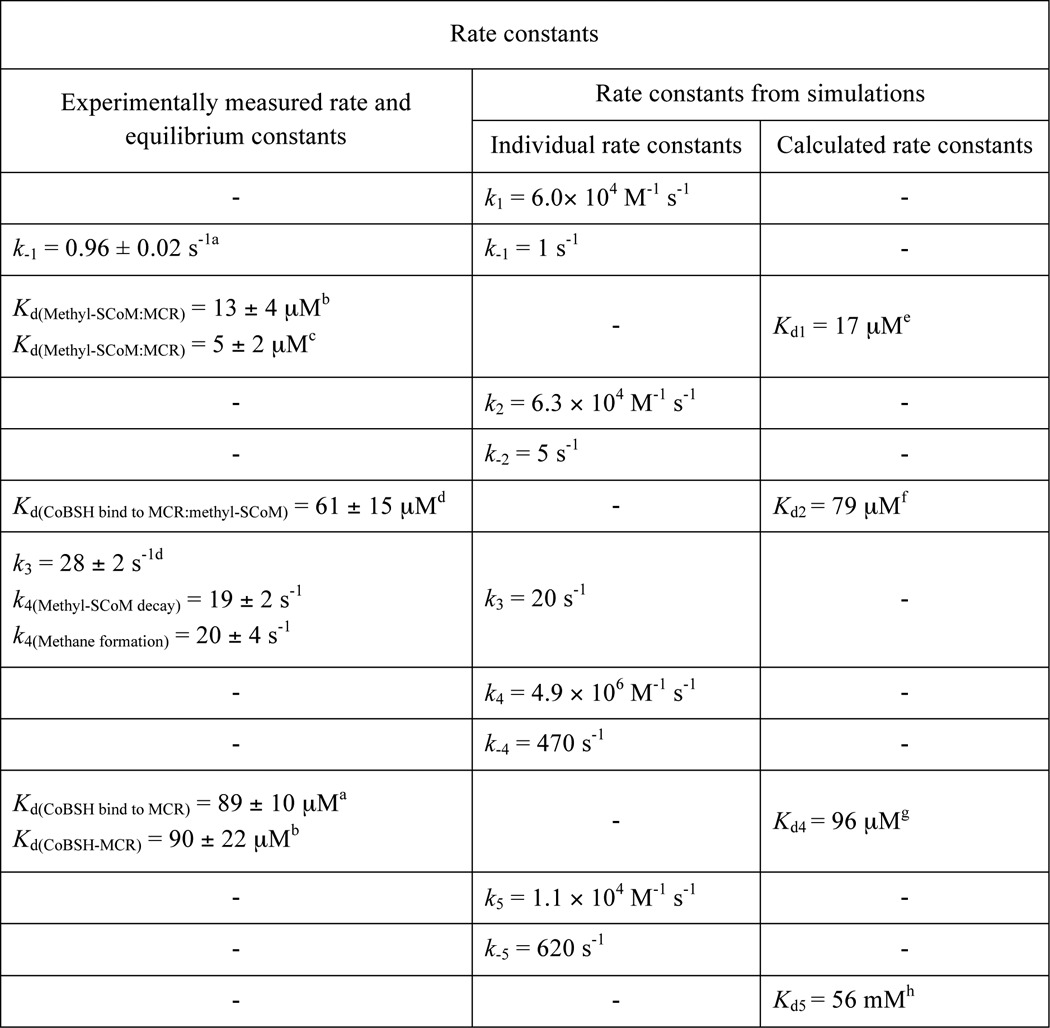
a Observed rate constants and equilibrium constants measured from the reaction of MCR·CoB7SH complex with methyl-SCoM.
b Equilibrium constants measured from the fluorescence binding experiment.
c Equilibrium constants measured from the EPR binding experiment.
d Observed rate constants and equilibrium constants measured from the reaction of MCR·methyl-SCoM complex with CoB7SH.
eKd1 = k−1/k1, the dissociation constant for methyl-SCoM binding to MCR.
f Kd2 = k−2/k2, the dissociation constant for CoB7SH binding to MCR·methyl-SCoM complex.
g Kd4 = k−4/k4; the dissociation constant for CoB7SH binding to MCR.
h Kd5 = k−5/k5; the dissociation constant for methyl-SCoM binding to MCR·CoB7SH complex.
Similar fluorescence experiments were performed to measure binding of CoB7SH. The Kd value (90 ± 22 μm) (inset in Fig. 2B) for the MCR-CoB7SH complex was 7 times higher than that for the complex with methyl-SCoM (13 ± 4 μm) (Table 1), suggesting that MCR binds methyl-SCoM more tightly than CoB7SH.
We also measured the Kd values for the binary complexes of MCRsilent with methyl-SCoM and CoB7SH and found that the Ni(II) form of the enzyme binds methyl-SCoM 3-fold more weakly (Kd = 43 ± 6 μm, data not shown) than the Ni(I) state. However, MCRsilent binds CoB7SH with a similar affinity (105 ± 17 μm, data not shown) as MCRred1. These experiments with the Ni(II) enzyme were performed as with the Ni(I) state except that MCRsilent was excited at its wavelength maximum at 420 nm and fluorescence emission was measured at 470 nm.
Binding of methyl-SCoM was also measured by monitoring the changes in the superhyperfine splitting patterns in the MCRred1 EPR spectrum as the enzyme was titrated with methyl-SCoM (Fig. 2C). This effect of methyl-SCoM on the EPR spectrum of MCR has been observed earlier (9) and is likely due to the effect of this substrate in decreasing the microheterogeneity of the F430 cofactor in the active site. Plotting the maximum intensity differences (upper inset in Fig. 2C) at each concentration of methyl-SCoM (lower inset in Fig. 2C) fits to a Kd value of 5 ± 2 μm, which is compatible with the value from the fluorescence measurements (Table 1). Because the inactive Ni(II) form of MCR is not observed in the EPR spectrum, this EPR-derived measurement monitors binding of methyl-SCoM substrate to the active Ni(I) state, whereas the fluorescence experiments could reflect binding to the Ni(I) as well as the inactive Ni(II) form of the enzyme. Importantly, we do not observe any effect of CoB7SH on the EPR spectrum of MCRred1 (data not shown).
Monitoring Kinetics of the Reaction of the MCR·methyl-SCoM Complex with CoB7SH by Stopped-flow
To help interpret the pre-steady-state data, we performed steady-state experiments at 25 °C in the presence and absence of hydroxocobalamin, which catalyzes reduction of the heterodisulfide product. Thus, the steady-state reaction with hydroxocobalamin is similar to conditions in the cell, where heterodisulfide reductase catalyzes heterodisulfide reduction. Under these conditions, the v versus S data nicely fit a hyperbolic curve according to the following Michaelis parameters: kcat = 18 ± 3 s−1; Vmax = 7.8 ± 1.2 units/mg; Km(CoBSH) = 169 ± 79 μm. For the steady-state condition in which the heterodisulfide product is not recycled (reaction without hydroxocobalamin), the rate (kcat = 0.96 ± 0.03 s−1; Vmax = 0.41 ± 0.01 units/mg; Km(CoBSH) = 42 ± 7 μm) is significantly lower, suggesting that product (CoBS-SCoM) release may limit regeneration of the free active state of the enzyme for the next catalytic cycle.
To follow the MCR-catalyzed reaction of methyl-SCoM with CoB7SH by pre-steady-state kinetics, we monitored absorbance changes associated with the F430 cofactor. A premixed solution containing MCRred1 (18 μm) and methyl-SCoM (18 μm) was anaerobically mixed with various concentrations of CoB7SH using a stopped-flow spectrophotometer. Thus, these conditions are single turnover in methyl-SCoM. To monitor as much of the MCR reaction time course as possible, the pre-steady-state kinetic experiments were performed at 25 °C, instead of at the optimum growth temperature (60 °C), where kcat is 250 s−1 (7) and where most steady-state reactions have been performed (7, 20–22).
The MCR reaction has been shown to follow an ordered bi-bi ternary complex mechanism (12, 20–23). Correspondingly, the pre-steady-state reaction of the binary MCR·methyl-SCoM complex with CoB7SH is strictly dependent on the presence of both substrates. No change in the Ni(I) absorption spectrum was observed in the reaction of MCR with either methyl-SCoM or CoB7SH alone (data not shown). The decay of Ni(I) and the formation of Ni(II)/Ni(III) were monitored using a diode array detector set at a wavelength range from 350 to 700 nm. We observed two dominant spectral changes: a maximum absorbance decrease due to the decay of Ni(I) at 385 nm (Fig. 3A, and inset in B), and an absorbance increase at 420 nm due to Ni(II) or Ni(III) formation (Fig. 3B, and inset in B). Therefore, kinetic traces at both 385 and 420 nm were used for data analysis (Fig. 3).
FIGURE 3.

Kinetic traces of the reaction of a pre-mixed solution containing MCRred1 (18 μm) and methyl-SCoM (18 μm) with CoB7SH at varying concentrations (0.05, 0.125, 0.25, 0.5, and 1 mm, final concentrations after mixing) from top to bottom (A) and from bottom to top (B), respectively, in 50 mm Tris-HCl, pH 7.6. The reactions were performed under anaerobic conditions using the stopped-flow spectrophotometer at 25 °C and monitored by following the decay of Ni(I) at 385 nm (A), and the formation of Ni(II)/Ni(III) at 420 nm (B). The reactions showed four kinetic phases as shown in C. The first phase (0.002–0.5 s) was the phase with a large decrease in absorbance at 385 nm (decay of Ni(I)). The first phase of the multiexponential trace was fit to a single exponential equation. The dashed lines represent simulations based on the model presented in Scheme 1 with the values of the kinetic parameters listed in Table 1 and following extinction coefficients: ϵ385(MCR(NiI)·CH3SCoM) = 35,040 m−1 cm−1; ϵ385(CoB7SH·MCR(NiI)·CH3SCoM) = 30,010 m−1 cm−1; ϵ385(MCR(NiII)·CoB7S−·SCoM) = 25,490 m−1 cm−1; ϵ420(MCR(NiI):CH3SCoM) = 20,620 m−1 cm−1; ϵ420(CoB7SH·MCR(NiI)·CH3SCoM) = 20,550 m−1 cm−1; and ϵ420(MCR(NiII)·CoB7S−·SCoM) = 20,100 m−1 cm−1. The inset in B shows the spectral changes when the MCR·methyl-SCoM complex was reacted with 1 mm CoB7SH from 0.001 s (solid line) to 1 s (dashed line). Arrows indicate the direction of change over time at 385 and 420 nm. C, the reaction of MCR·methyl-SCoM complex with 1 mm CoB7SH (final) over a 1000-s log time scale at 385 nm.
As shown in Fig. 3C, this reaction showed four distinct kinetic phases. The first phase, occurring over the first 0.5 s of the reaction, exhibits a maximum decrease in absorbance at 385 nm of 0.1, which equals 28% of the amount of the enzyme present (as well as substrate converted) and coincides with a consistent amplitude increase at 420 nm. This phase exhibits a kobs value (kmax = 28 s−1), which is greater than the steady-state kcat (18 s−1, determined in the presence of hydroxocobalamin) and is analyzed in more detail below. The second phase (0.5–10 s) occurs with a smaller decrease in absorbance at 385 nm (a maximum absorbance amplitude of 0.03, equaling another 10% of the MCRred1). For the second phase of the reaction, although the amplitude is dependent on the concentration of CoB7SH, the value of kobs2 (kmax = 0.40 ± 0.02 s−1) is CoB7SH independent and is similar to the steady-state kcat for the reaction performed in the absence of hydroxocobalamin (0.96 s−1). This indicates that, under these single-turnover conditions, as the first 28% of the bound methyl-SCoM is depleted, an inhibitory binary complex with CoB7SH begins to accumulate. The nature of this complex and its reactivity with methyl-SCoM is described in more detail below. The third phase (10–70 s) occurs without a noticeable absorbance change, suggesting that it corresponds to the steady-state reaction, where MCR does not undergo a net redox change. The fourth phase (70–1000 s) occurs with an absorbance increase at 385 nm (a maximum absorbance amplitude of 0.07), as the Ni(II) state is converted back to Ni(I). The change of this phase is independent of CoB7SH concentrations. Using 70 s as a starting point for this phase, the kobs4 is 0.12 ± 0.01 s−1. Presumably, the final 62% of the bound methyl-SCoM is converted to methane during these last two phases of the reaction.
We focused our analysis on the first phase because the other phases are slower than kcat and, thus, would not provide information on intermediates in the catalytic mechanism. The data collected at each wavelength for the decay of Ni(I) and the formation of Ni(II)/Ni(III) in the first phase of the reaction of MCR·methyl-SCoM complex with CoB7SH gave similar observed rate constants (Fig. 3). A maximum absorbance amplitude (at the highest concentration of CoB7SH, 1 mm) of 10.3 ± 0.4 × 10−2 at 385 nm and 1.4 ± 0.1 × 10−2 at 420 nm (Figs. 3 and 4A) is consistent with a net conversion of 5 μm Ni(I) to Ni(II)/Ni(III). The decrease in absorbance at 385 nm (Fig. 3A, and inset in B, circles in Fig. 4A) is synchronized with the amplitude increase at 420 nm (Fig. 3B, and inset in B, diamonds in Fig. 4A), indicating that the Ni(I) state of MCRred1 is converted directly (without another intermediate) to a Ni(II)/Ni(III) intermediate (Fig. 4A). The observed rate constants (kobs) from kinetic traces at both wavelengths (385 and 420 nm) showed a hyperbolic dependence on the CoB7SH concentrations that approached a limiting value of 28 ± 2 s−1 and a half-saturation CoB7SH value of 61 ± 15 μm (Fig. 4B, Table 1).
FIGURE 4.

Plot of the amplitude change at 385 (○) and 420 nm (♢) (A) and the observed rate constant (kobs) of the reaction of the MCR·methyl-SCoM complex (B) with CoB7SH based on the kinetic traces in Fig. 3. The plot of kobs versus CoB7SH concentration was hyperbolically dependent on the CoB7SH concentration and analyzed according to Equation 2. The vertical line at each point indicates the standard deviation of the measurement.
The results shown in Figs. 3 and 4 can be described by a two-step mechanism shown in Scheme 1. The observed rate constant (kobs) of the reaction is hyperbolically dependent on the CoB7SH concentration (Fig. 4B) indicating that the first step in the mechanism is rapid binding of CoB7SH substrate to the MCR·methyl-SCoM complex to form the ternary CoB7SH·MCR(NiI)·CH3SCoM complex (Scheme 1). This rapid equilibrium step must precede the chemical reaction (involving k3), which is accompanied by a large decrease in absorbance at 385 nm (Ni(I) decay) coincident with a large increase in absorbance at 420 nm (Ni(II)/Ni(III) formation) (Figs. 3 and 4A, Scheme 1).
 |
Simulations according to the model described in Scheme 1 generated data that agree well with the experimental data (Fig. 3, solid versus dashed lines). Observed rate constants from the experimental data and apparent rate constants from the simulations are compared in Table 1. When the observed rate constants were plotted as a function of CoB7SH (S in Equation 2) concentration according to Equation 2, the Kd was derived to be 61 ± 15 μm and the limiting value of kobs at saturating CoB7SH concentrations was 28 ± 2 s−1 (Fig. 4B, Table 1). Based on simulations according to Scheme 1, the Kd2 (k−2/k2) for interaction of CoB7SH with the MCR·methyl-SCoM complex was 79 μm and k3, the rate constant for the chemical step, which is conversion of the ternary complex into the products, CH4 and CoBS-SCoM anion radical, and Ni(I) to Ni(II)/Ni(III), was derived to be 20 s−1 (Table 1). These values are in excellent agreement with the experimental Kd and limiting kobs values determined according to Equation 2 (Table 1), providing support for the kinetic model described in Scheme 1.
One limitation of Scheme 1 (and Scheme 2, shown below) is that we have not included the final step in the mechanism, reformation of the Ni(I) enzyme. This is because of the multiexponential nature of the reaction, where recycling of the Ni(I) enzyme occurs in the fourth phase (the absorbance increase at the end, Fig. 3C). This phase of the reaction is currently being studied by following the reverse reaction of MCRred1 with CoBS-SCoM and CH4.
Rapid Chemical Quench Measurement of Kinetics of Methyl-SCoM Conversion to CH4 by Reaction of the MCR·methyl-SCoM Complex with CoB7SH
To further test the model described in Scheme 1, the conversion of methyl-SCoM to CH4 was followed by rapid chemical quench methods under similar conditions to the stopped-flow experiments. A solution containing MCRred1 (36 μm) and [14C]methyl-SCoM (36 μm) was rapidly mixed with 2 mm CoB7SH, and incubated for various time periods between 0.01 and 0.97 s. The reaction was quenched by mixing with 0.2 m perchloric acid and analyzed by liquid scintillation counting. The amount of remaining [14C]methyl-SCoM was plotted versus time (Fig. 5A) and fit to a single-exponential curve, revealing a limiting rate constant of 19 ± 2 s−1 (Table 1).
FIGURE 5.

Rapid chemical-quench study of the reaction of the MCR·methyl-SCoM complex with CoB7SH. Reactions of a pre-mixed solution containing equimolar MCRred1 (36 μm) and [14C]methyl-SCoM/nonradioactive methyl-SCoM (36 μm) with 2 mm CoB7SH were quenched with 0.2 m perchloric acid at various times using the rapid-quench apparatus. The percentage conversion of [14C]methyl-SCoM was plotted versus time (A). The percentage conversion was determined by comparing the remaining concentration of [14C]methyl-SCoM to the initial concentration. The percentage CH4 product formed was plotted versus time (B). The percentage conversion was determined by comparing the amount of CH4 formed at a particular quench time versus that formed at the end of the reaction (10 min). Both plots yielded a single exponential curve with a rate constant of 19 ± 2 and 20 ± 4 s−1, respectively (Table 1). A vertical line at each point indicates a standard deviation of the measurement.
CH4 formation was analyzed by gas chromatography in an experiment performed as just described but using nonradioactive methyl-SCoM as a substrate. A plot of formed CH4 versus time at 1 mm CoB7SH (Fig. 5B) also fit to a single-exponential curve with an observed limiting rate constant of 20 ± 4 s−1 (Table 1). Thus, decay of methyl-SCoM and formation of CH4 are limited by the same rate constant (∼20 s−1), which also agrees with the rate constant for conversion of Ni(I) to Ni(II)/Ni(III) in the stopped-flow experiment (20 s−1) (Table 1). These results imply that the MCR chemical reaction occurred in the same time frame as methyl-SCoM substrate decay and CH4 product formation. Therefore, the overall reaction of the MCR·methyl-SCoM complex with CoB7SH can be summarized as shown in Scheme 1, with a rapid equilibrium binding of CoB7SH to the MCR·methyl-SCoM complex to generate the active ternary complex (CoB7SH·MCR(NiI)·CH3SCoM) that undergoes chemical conversion of methyl-SCoM to CH4 at a rate constant of 20 s−1 (Scheme 1, Table 1).
Monitoring Kinetics of the Reaction of the MCR·CoB7SH Complex with Methyl-SCoM by Stopped-flow
A equimolar mixture of MCRred1 (18 μm) and CoB7SH (18 μm) was mixed with methyl-SCoM at various concentrations using a stopped-flow spectrophotometer under anaerobic conditions at 25 °C. Interestingly, under these conditions, the absorbance 385 nm increased (Fig. 6A, circles in Fig. 7A) as the absorbance at 420 nm decreased (Fig. 6B, diamonds in Fig. 7A), indicating conversion of a Ni(II)/Ni(III) state to the active Ni(I) state. The limiting amplitude (at the highest concentration of methyl-SCoM; 2 mm) is 3.1 ± 0.2 × 10−2 at 385 nm and 1.2 ± 0.7 × 10−4 at 420 nm, which corresponds to a conversion of only 1 μm Ni(II)/III to Ni(I), which is 5% of the amount of MCRred1 in the reaction. As described in Scheme 2, our results indicate that this reaction involves dissociation of CoB7SH from the unproductive binary MCR·CoB7SH complex (step 4), followed by binding of methyl-SCoM to generate the productive MCR·methyl-SCoM complex (step 1). Then, CoB7SH rebinds to form the active ternary complex (CoB7SH·MCR(NiI)·CH3SCoM) before the chemical reaction occurs (step 2–3). The kinetic traces at both 385 and 420 nm were used for data analysis (Fig. 6). The regeneration of Ni(I) and the decay of Ni(II)/Ni(III) by the reaction of MCR·CoB7SH complex with methyl-SCoM resulted in one kinetic phase (Fig. 6). The observed rate constants (kobs) determined from kinetic traces at both wavelengths (385 and 420 nm) showed a hyperbolic dependence on the methyl-SCoM concentrations (Fig. 7B). At high substrate concentrations, the observed rate constants approached a limiting value of 0.96 ± 0.02 s−1 (Fig. 7B, Table 1), which is similar to the value of kcat for the steady-state reaction performed in the absence of hydroxocobalamin (0.96 s−1). The methyl-SCoM concentration that gives a half-saturation value of the plot is 89 ± 10 μm (Fig. 7B, Table 1). The rapid chemical quench measurement performed under similar conditions gave a kmax ∼1 s−1 (data not shown), which agrees well with the value obtained by stopped-flow.
FIGURE 6.
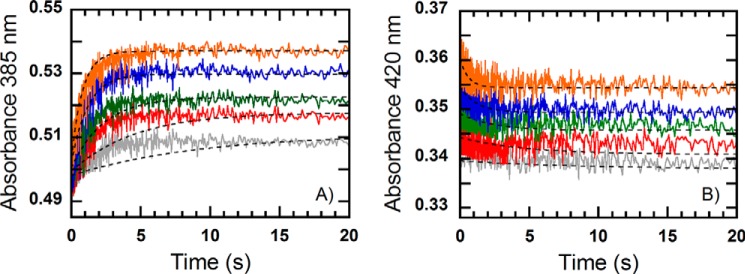
Kinetics of the reaction of a pre-mixed solution of MCRred1 (18 μm) and CoB7SH (18 μm) with methyl-SCoM at concentrations of 0.125, 0.25, 0.5, 1, and 2 mm (final concentrations) in 50 mm Tris-HCl pH 7.6. The reactions were performed under anaerobic conditions using the stopped-flow spectrophotometer at 25 °C and monitored at 385 (A) and 420 nm (B). The traces were fit to a single exponential equation. The dashed lines represent simulations based on the model presented in Scheme 2 with kinetic parameters listed in Table 1 and the following extinction coefficients: ϵ385(MCR(NiI)) = 37,340 m−1 cm−1; ϵ385(MCR(NiI)·CH3SCoM) = 35,040 m−1 cm−1; ϵ385(CoB7SH·MCR(NiI)·CH3SCoM) = 30,010 m−1 cm−1; ϵ385(MCR(NiII)·CoB7S−·SCoM) = 25,490 m−1 cm−1; ϵ385(MCR(NiI)·CoB7SH) = 35,790 m−1 cm−1; ϵ385(CoB7SH·MCR(NiI)·CH3SCoM*) = 35,550 m−1 cm−1; ϵ420(MCR(NiI)) = 18,060 m−1 cm−1; ϵ420(MCR(NiI)·CH3SCoM) = 20,620 m−1 cm−1; ϵ420(CoB7SH·MCR(NiI)·CH3SCoM) = 20,550 m−1 cm−1; and ϵ420(MCR(NiII)·CoB7S−·SCoM) = 20,100 m−1 cm−1; ϵ420(MCR(NiI)·CoB7SH) = 21,070 m−1 cm−1; and ϵ420(CoB7SH·MCR(NiI)·CH3SCoM*) = 22,920 m−1 cm−1.
FIGURE 7.
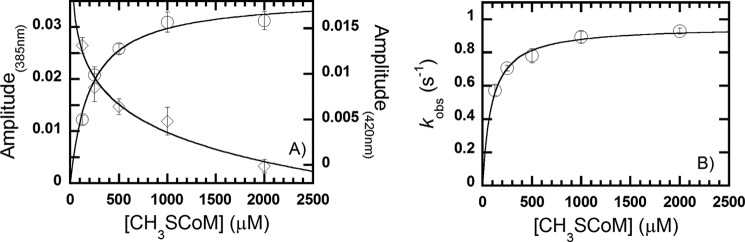
Plot of the amplitude change at 385 (○) and 420 (♢) nm (A) and the observed rate constant (kobs) of the reaction of MCR·CoB7SH complex with methyl-SCoM (B) based on the kinetic traces in Fig. 6. The plot of kobs versus methyl-SCoM concentration was hyperbolically dependent on methyl-SCoM concentration and analyzed according to Equation 2. The vertical lines at each point indicate the standard deviation of the measurement.
The results shown in Figs. 6 and 7 are described by the five-step reaction shown in Scheme 2. The rate constants for the chemical steps (step 2–3) are based on the results from the reaction of the productive MCR·methyl-SCoM complex with CoB7SH described in Scheme 1. That the observed rate constants (kobs) are hyperbolically dependent on methyl-SCoM concentrations (Fig. 7B) indicates that the first step is a dissociation of CoB7SH substrate from the unproductive MCR·CoB7SH complex to generate the active Ni(I)-MCRred1 state (step 4), as indicated by an increase in absorbance at 385 (Fig. 6A) and a decrease in absorbance at 420 nm (Fig. 6B). This step is required to permit binding of methyl-SCoM to the free enzyme to generate the productive MCR·methyl-SCoM complex (step 1) prior to CoB7SH binding to form active ternary complex (CoB7SH·MCR(NiI)·CH3SCoM) (step 2), which reacts as described in Scheme 1. Moreover, binding of methyl-SCoM to an inhibitory MCR·CoB7SH complex might occur to generate a non-productive CoB7SH·MCR(NiI)·CH3SCoM* complex (step 5). These results are a striking demonstration of a strictly ordered substrate binding mechanism in which productive chemistry only occurs when the substrates bind in the correct order.
Simulations according to the model described in Scheme 2 generated data that agree well with the experimental data (Fig. 6, solid versus dashed lines) and the observed rate constants and equilibrium constants from the experimental data closely match those obtained from the simulations, as shown in Table 1. The plot of the observed rate constants (Fig. 7B) was analyzed according to Equation 2. The Kd4 (k−4/k4) for the dissociation of CoB7SH from the MCR·CoB7SH complex, based on simulations is 96 μm, which agree well with the Kd derived from kinetic constants (89 ± 10 μm; Fig. 7B, Table 1). The observed rate constant from the experimental data at the saturating concentration of methyl-SCoM was determined to be 0.96 ± 0.02 s−1 from the plot shown in Fig. 7B, which is a rate constant for dissociation of methyl-SCoM from the MCR·methyl-SCoM complex (k−1 in Scheme 2). On the basis of kinetic constants obtained from simulations, the apparent rate constant at this step was calculated as 1 s−1 (k−1 in Table 1), which agrees well with the experimental data. All kinetic constants derived from the experimental data and simulations agree well, indicating that the model and kinetic parameters shown in Scheme 2 and Table 1 accurately represent the MCR mechanism.
Order of Substrate Binding
The results from reaction of the MCR·CoB7SH complex with methyl-SCoM suggested that the methyl-SCoM substrate must enter the MCR active site before the CoB7SH substrate for a productive catalysis (Scheme 2). We then conducted stopped-flow experiments to better understand how MCR enforces the order of binding of methyl-SCoM and CoB7SH. A solution of 25 μm MCRred1 was preincubated with various concentrations of CoB7SH (0.025, 0.1, 0.25, 0.5, and 1 mm, final) and rapidly mixed with methyl-SCoM (1 mm, final). The reaction was monitored at 385 nm (Fig. 8A). Interestingly, the kinetic traces shows significantly increased amplitude change as the CoB7SH concentration decreases (Fig. 8). As indicted in Scheme 2, these results show that the active Ni(I) state of MCR was regenerated in high amounts at low concentrations of CoB7SH. Therefore, all data confirm the order of binding of the substrate for MCR is sequential, which is that the methyl-SCoM substrate must bind before the CoB7SH substrate (Scheme 2).
FIGURE 8.
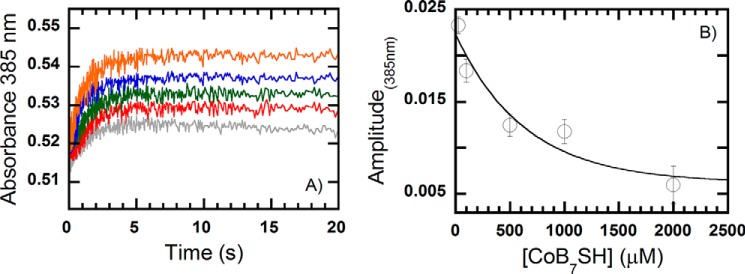
Kinetics of the reaction of a pre-mixed solution of MCRred1 (25 μm) and various concentrations of CoB7SH (0.025, 0.1, 0.25, 0.5, and 1 mm, from top to bottom) with 1 mm methyl-SCoM in 50 mm Tris-HCl, pH 7.6. All concentrations quoted are those after mixing. A, the reactions were performed under anaerobic conditions using the stopped-flow spectrophotometer at 25 °C and monitored by following the formation of Ni(I) at 385 nm. B, amplitude changes from the traces in A. The amplitude values at low to high CoB7SH concentrations are 2.3 ± 0.1 × 10−2, 1.8 ± 0.1 × 10−2, 1.2 ± 0.1 × 10−2, 1.1 ± 0.1 × 10−2, and 6.0 ± 0.2 × 10−3, respectively. Thus, the amplitude of MCRred1 formation (increase in 385 nm) decreases when the concentration of CoB7SH increases.
DISCUSSION
This article describes the first rapid kinetic study of the reaction of MCR with its native substrates. We have derived the overall kinetic scheme and the individual rate constants associated with each step in the mechanism, as described in Scheme 2 and Table 1. The results of equilibrium binding (Figs. 2, 4, and 7, Table 1) and pre-steady-state experiments combined with kinetic simulations (Figs. 3, 6, and 8, Table 1, Schemes 1 and 2) revealed important information about MCR specifically and in general about how an enzyme can dictate strict order in substrate binding.
The proposed reaction mechanism in Scheme 2 was tested for its validity by comparing kinetic constants obtained from pre-steady-state kinetic and substrate binding experiments with simulations. For example, based on the simulations according to Scheme 2, the Kd1 for methyl-SCoM interactions with free MCR was calculated to be 17 μm (k−1/k1), which is in agreement with the values of the Kd measured from fluorescence (13 ± 4 μm) and EPR (5 ± 2 μm) binding experiments (Table 1). Furthermore, the Kd2 for CoB7SH binding to the MCR·methyl-SCoM complex, which is based on simulations of the kinetic mechanism in Schemes 1 and 2 (k−2/k2 = 79 μm), agrees well with the values obtained by the pre-steady-state reaction of the MCR·methyl-SCoM complex with CoB7SH (61 ± 15 μm) (Table 1). Moreover, the Kd4 for CoB7SH dissociation from the inhibitory MCR·CoB7SH complex, which is based on simulations of the kinetic mechanism in Scheme 2 (k−4/k4 = 96 μm), agrees well with the values obtained by the pre-steady-state reaction of the MCR·CoB7SH complex with methyl-SCoM (89 ± 10 μm) and the fluorescence studies of CoB7SH binding to MCR (90 ± 22 μm) (Table 1). That the experimental and simulated values are in such excellent agreement indicates that the kinetic mechanism in Scheme 2 is a realistic model for the MCR reaction.
One of the most interesting results relates to how MCR enforces a strictly ordered substrate binding mechanism in which the chemical conversion of substrates to products can only occur when methyl-SCoM enters the active site prior to CoB7SH (Figs. 3–5). Previous studies have also indicated that the MCR reaction proceeds via an ordered bi-bi ternary complex mechanism (12, 20–23); however, surprisingly, our fluorescence and EPR results clearly demonstrate that either substrate can bind tightly to the enzyme in the absence of the other substrate, with the Kd value for the MCR·CoB7SH complex (90 ± 22 μm) being only 7–18-fold weaker than that for the MCR·methyl-SCoM complex (5–13 μm) (Fig. 2, Table 1). This result on its own might suggest a random binding mechanism. However, our kinetic studies clearly show that only one binary complex is productive, the MCR·methyl-SCoM complex, whereas the MCR·CoB7SH complex is inhibitory. This selectivity seems intuitive upon examination of the crystal structure of MCR (6) because it is difficult to imagine how methyl-SCoM could reach its binding site near the F430 cofactor if CoB7SH, a large molecule that occupies most of the substrate binding channel, is already present.
How does the enzyme promote formation of the productive complex while preventing formation of the inhibitory nonproductive complex? Our simulations demonstrate that formation of the unproductive ternary complex (CoB7SH·MCR(NiI)·CH3SCoM*), formed from methyl-SCoM binding to the inhibitory MCR·CoB7SH complex, is highly disfavored (Kd5 = 56 mm; Table 1), whereas formation of the productive CoB7SH·MCR(NiI)·CH3SCoM complex from CoB7SH binding to the MCR·methyl-SCoM complex is highly favored (Kd2 = 79 μm; Table 1). Thus, MCR is highly effective in preventing formation of the unproductive ternary complex and ensuring that methyl-SCoM is in place near the Ni center before CoB7SH enters. For example, if both substrates are present in the cell at 1 mm concentrations, MCR·methyl-SCoM would be the highly predominant binary complex at the MCR active site.
Besides being able to block formation of the unproductive ternary complex (CoB7SH·MCR(NiI)·CH3SCoM*), MCR promotes binding of CoB7SH to the MCR·methyl-SCoM complex (Kd2 = 79 μm; Table 1) to form the active ternary complex (CoB7SH·MCR(NiI)·CH3SCoM). Formation of the stable MCRred1·methyl-SCoM complex (Kd1 = 17 μm; Table 1) most likely elicits a conformational change in the enzyme as indicated by the striking effect of methyl-SCoM binding on the superhyperfine splitting pattern (Fig. 2C). This is likely due to an increase in organization (lowering the microheterogeneity) at the active site and narrowing the conformational distribution of various groups that interact with the F430 cofactor (9). Similarly, comparison of the crystal structures of the MCRred1-silent state before and after reacting with HSCoM revealed that the binding of HSCoM induces a conformational change that affects the conformation of the tunnel in which CoB7SH binds (16), likely increasing the affinity of MCR·methyl-SCoM complex for CoB7SH. This selectivity for methyl-SCoM in organizing the active site is specific; we do not observe any effect of CoB7SH on the EPR spectrum of MCRred1.
Besides the positive effect of methyl-SCoM on preparing the active site for binding CoB7SH, the enzyme sets up roadblocks against CoB7SH binding first to generate an inhibitory complex. This is reflected also in structural studies of the inactive Ni(II) state of MCR (6, 16) because, in the absence of substrates, the active site exhibits a significantly higher degree of flexibility than when both HSCoM and CoB7SH are bound. This finding was interpreted to indicate that MCR prevents inhibitory binding of CoB7SH to active sites without the first substrate methyl-SCoM being bound (16). Our kinetic data demonstrate that one mechanism for avoiding inhibition is that methyl-SCoM promotes the dissociation of CoB7SH from the MCR active site (steps 1 and 4 in Scheme 2). For example, when the MCR·CoB7SH complex is reacted with various concentrations of methyl-SCoM (Figs. 6–8), an increase in the active Ni(I) state is observed. This reactivation is likely to be due to the dissociation of inhibitory CoB7SH from the MCR active site (step 4 in Scheme 2) to generate active Ni(I)-MCRred1.
These organizing effects are likely to also promote the chemical reaction between methyl-SCoM and CoB7SH and to explain the synergy between the two substrates shown in Scheme 2 and in Table 1 that prevents any chemical reaction between methyl-SCoM and MCR until CoB7SH binds. The results showing dissociation of methyl-SCoM from the unproductive ternary complex (CoB7SH·MCR(NiI)·CH3SCoM*; Kd5 = 56 mm) and the similar dissociation constant for interactions between CoB7SH and free MCR(NiI) (Kd4 = 96 μm), and for re-binding of CoB7SH to the MCR·methyl-SCoM complex to generate the productive ternary complex (CoB7SH·MCR(NiI)·CH3SCoM; Kd3 = 79 μm), support the hypothesis that methyl-SCoM binding triggers a conformational change that promotes binding of the second substrate (CoB7SH). Dey et al. (12) proposed the existence of two populations of MCR enzyme (substrate-bound and substrate-free) forms that slowly interconvert, with the methyl-SCoM-bound state able to undergo a conformational change to rapidly react with CoB7SH. It is possible that this conformational change drives the end of the 7-thioheptanoyl chain of CoB7SH into closer contact with the nickel center (and the methyl group) in the MCR·methyl-SCoM complex. Such a mechanism is indicated by 19F-ENDOR data for MCRred1 reacted with HSCoM and the coenzyme B analog, F3C-S-S-coenzyme B in which it was shown that the 7-thioheptanoyl chain of the CoBSH analog moves more than 2 Å closer to the nickel center of F430 (24). Perhaps a conformational change of the glycine-rich loop in the β subunit of the inactive MCR (Ni(II)) crystal structure might couple the start of the reaction with binding of the second substrate, CoB7SH (16). In addition, in the reaction of the MCR·methyl-SCoM complex with CoB7SH, the kobs depends on the CoB7SH concentration (Fig. 4B), indicating that CoB7SH must bind before the irreversible chemical step occurs.
Step 3 of Scheme 2 describes the chemical reaction of MCR. After formation of active ternary complex (CoB7SH·MCR(NiI)·CH3SCoM), the chemical reaction occurs with a rate constant of 20 s−1 (Table 1). The rate constant of Ni(I) decay matches that of Ni(II)/Ni(III) formation (Figs. 3 and 4A), suggesting that no intermediate is formed between the states. Alternative explanations are that 1) any intermediate that forms does not accumulate to detectable levels because its formation rate is too slow relative to a relatively rapid decay rate, or 2) an intermediate with a nearly identical absorption spectrum at 420 nm (e.g. alkyl-Ni(III) species) is formed. Rapid-quench techniques allowed us to measure the rate constant of methyl-SCoM substrate decay and CH4 product formation (Fig. 5), which can be directly compared with the rate constant from the stopped-flow method (Fig. 4B, Table 1). The data also provided independent measurements of rate constants in addition to the values based on nickel absorbance change. The rate constants for methyl-SCoM substrate decay and CH4 product formation are 19 ± 2 and 20 ± 4 s−1, respectively (Fig. 5, Table 1). These values agree well with the rate constant for conversion of Ni(I) to Ni(II)/Ni(III) (20 s−1) determined by stopped-flow methods (Figs. 3 and 4B, Table 1). These results indicate that decay of the methyl-SCoM substrate and the formation of the CH4 product occur via a direct transformation of Ni(I) to the Ni(II)/Ni(III) state. Therefore, the formation of enzyme substrate complex (CoB7SH·MCR(NiI)·CH3SCoM) was followed by the chemical reaction step to form MCR(NiII)·CoB7S−·SCoM with the rapid dissociation of CH4 product (step 3 in Scheme 2).
Several experimental and theoretical studies have been performed in an effort to understand the catalytic mechanism of MCR and the geometric and electronic structures of the intermediates in the catalytic cycle (6, 10, 13, 16, 28, 30, 31, 33, 41–43). The major distinction between the two competing mechanisms lies in the proposed intermediate generated in the first step of catalysis. The first mechanism involves formation of an organometallic methyl-Ni(III) intermediate starting from nucleophilic attack of the MCR-Ni(I) center at the carbon atom of methyl-SCoM in SN2 fashion (6, 42, 43). This is followed by protonolysis to generate CH4 and the heterodisulfide product. The putative methyl-Ni(III) intermediate has been generated independently by adding methyl halides to the active MCR-Ni(I) and is characterized by EPR spectroscopy (13, 28) and x-ray crystal structure (10). The second mechanism involves a direct attack of the Ni(I) on the S of methyl-SCoM, resulting in a homolytic cleavage of the thioether bond and formation of a Ni(II)-thiolate species and a •CH3. This mechanism is based on density functional theory calculations (30, 31, 41) and kinetic isotope effect (33). From our kinetic results, we cannot rule out either of these mechanisms; thus, the nature of the Ni(II) or Ni(III) intermediates that are formed during the reaction with the natural substrates (methyl-SCoM and CoB7SH) requires future investigation. However, the kinetic data demonstrate that any intermediate that forms at 25 °C should have a decay rate greater than 20 s−1.
In conclusion, this article describes the first rapid kinetic study of MCR with its native substrates and has elucidated its kinetic mechanism (Scheme 2) and revealed the rate constants for each of these steps (Table 1). Our results suggest that the reaction mechanism of MCR is a complex process comprising several reaction steps starting with the binding of methyl-SCoM to the MCR active site leading to the formation of a reactive MCR·methyl-SCoM complex. This complex may trigger movement of CoB7SH toward the nickel and perhaps motion of methyl-SCoM toward CoB7SH to generate the active enzyme substrate complex (CoB7SH·MCR(NiI)·CH3SCoM), which is poised for the chemical reaction to occur and generate CH4 and the MCR(NiII)·CoB7S−·SCoM. Our mechanistic results will serve as the grounds for future studies identifying the intermediates in the MCR reaction mechanism. Our results also may explain how other enzymes enforce ordered mechanisms. Although some enzymes may prevent binding of the “incorrect/second” substrate to the free enzyme and recognize only the “correct/first” substrate, MCR can form a binary complex with either substrate. It appears to couple binding of methyl-SCoM to expulsion of any pre-bound CoB7SH from its nonproductive binding mode. It also uses conformational energy to promote CoB7SH binding to the productive binary complex and to effect the synergetic movement of substrates in position for the chemical reaction. Further studies are required to identify the protein-associated groups that drive these conformational changes and to identify the nature of the key intermediates in the mechanism.
Acknowledgments
We thank Dr. Dariusz Sliwa and Nathan Sheskey for preparation of coenzyme B and Dr. Dariusz Sliwa for preparation of the [14C]methyl-SCoM used during this study.
This work was supported by the U.S. Dept. of Energy, Office of Science, Office of Basic Energy Sciences Award DE-FG02–08ER15931.
- MCR
- methyl-coenzyme M reductase
- methyl-SCoM
- methyl-coenzyme M
- HSCoM
- coenzyme M
- CoB7SH
- N-7-mercaptoheptanolyl-threonine phosphate or coenzyme B
- CoBS-SCoM
- the heterodisulfide product of the MCR reaction.
REFERENCES
- 1. Thauer R. K. (1998) Biochemistry of methanogenesis: a tribute to Marjory Stephenson. 1998 Marjory Stephenson Prize Lecture. Microbiology 144, 2377–2406 [DOI] [PubMed] [Google Scholar]
- 2. Scheller S., Goenrich M., Boecher R., Thauer R. K., Jaun B. (2010) The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 465, 606–608 [DOI] [PubMed] [Google Scholar]
- 3. Shima S., Thauer R. K. (2005) Methyl-coenzyme M reductase and the anaerobic oxidation of methane in methanotrophic Archaea. Curr. Opin. Microbiol. 8, 643–648 [DOI] [PubMed] [Google Scholar]
- 4. DiMarco A. A., Bobik T. A., Wolfe R. S. (1990) Unusual coenzymes of methanogenesis. Annu. Rev. Biochem. 59, 355–394 [DOI] [PubMed] [Google Scholar]
- 5. Ellefson W. L., Wolfe R. S. (1981) Component C of the methylreductase system of Methanobacterium. J. Biol. Chem. 256, 4259–4262 [PubMed] [Google Scholar]
- 6. Ermler U., Grabarse W., Shima S., Goubeaud M., Thauer R. K. (1997) Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 278, 1457–1462 [DOI] [PubMed] [Google Scholar]
- 7. Goubeaud M., Schreiner G., Thauer R. K. (1997) Purified methyl-coenzyme-M reductase is activated when the enzyme-bound coenzyme F430 is reduced to the nickel(I) oxidation state by titanium(III) citrate. Eur. J. Biochem. 243, 110–114 [DOI] [PubMed] [Google Scholar]
- 8. Becker D. F., Ragsdale S. W. (1998) Activation of methyl-SCoM reductase to high specific activity after treatment of whole cells with sodium sulfide. Biochemistry 37, 2639–2647 [DOI] [PubMed] [Google Scholar]
- 9. Mahlert F., Bauer C., Jaun B., Thauer R. K., Duin E. C. (2002) The nickel enzyme methyl-coenzyme M reductase from methanogenic archaea: in vitro induction of the nickel-based MCR-ox EPR signals from MCR-red2. J. Biol. Inorg. Chem. 7, 500–513 [DOI] [PubMed] [Google Scholar]
- 10. Cedervall P. E., Dey M., Li X., Sarangi R., Hedman B., Ragsdale S. W., Wilmot C. M. (2011) Structural analysis of a Ni-methyl species in methyl-coenzyme M reductase from methanothermobacter marburgensis. J. Am. Chem. Soc. 133, 5626–5628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dey M., Kunz R. C., Lyons D. M., Ragsdale S. W. (2007) Characterization of alkyl-nickel adducts generated by reaction of methyl-coenzyme M reductase with brominated acids. Biochemistry 46, 11969–11978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dey M., Li X., Kunz R. C., Ragsdale S. W. (2010) Detection of organometallic and radical intermediates in the catalytic mechanism of methyl-coenzyme M reductase using the natural substrate methyl-coenzyme M and a coenzyme B substrate analogue. Biochemistry 49, 10902–10911 [DOI] [PubMed] [Google Scholar]
- 13. Dey M., Telser J., Kunz R. C., Lees N. S., Ragsdale S. W., Hoffman B. M. (2007) Biochemical and spectroscopic studies of the electronic structure and reactivity of a methyl-Ni species formed on methyl-coenzyme M reductase. J. Am. Chem. Soc. 129, 11030–11032 [DOI] [PubMed] [Google Scholar]
- 14. Kunz R. C., Dey M., Ragsdale S. W. (2008) Characterization of the thioether product formed from the thiolytic cleavage of the alkyl-nickel bond in methyl-coenzyme M reductase. Biochemistry 47, 2661–2667 [DOI] [PubMed] [Google Scholar]
- 15. Grabarse W., Mahlert F., Shima S., Thauer R. K., Ermler U. (2000) Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: unusual amino acid modification, conservation and adaptation. J. Mol. Biol. 303, 329–344 [DOI] [PubMed] [Google Scholar]
- 16. Grabarse W., Mahlert F., Duin E. C., Goubeaud M., Shima S., Thauer R. K., Lamzin V., Ermler U. (2001) On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J. Mol. Biol. 309, 315–330 [DOI] [PubMed] [Google Scholar]
- 17. Zhou Y., Dorchak A. E., Ragsdale S. W. (2013) In vivo activation of methyl-coenzyme M reductase by carbon monoxide. Front. Microbiol. 4, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prakash D., Wu Y., Suh S. J., Duin E. C. (2014) Elucidating the process of activation of methyl-coenzyme M reductase. J. Bacteriol. 196, 2491–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cedervall P. E., Dey M., Pearson A. R., Ragsdale S. W., Wilmot C. M. (2010) Structural insight into methyl-coenzyme M reductase chemistry using coenzyme B analogues. Biochemistry 49, 7683–7693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ellermann J., Hedderich R., Böcher R., Thauer R. K. (1988) The final step in methane formation. Investigations with highly purified methyl-CoM reductase (component C) from Methanobacterium thermoautotrophicum (strain Marburg). Eur. J. Biochem. 172, 669–677 [DOI] [PubMed] [Google Scholar]
- 21. Horng Y. C., Becker D. F., Ragsdale S. W. (2001) Mechanistic studies of methane biogenesis by methyl-coenzyme M reductase: evidence that coenzyme B participates in cleaving the C - S bond of methyl-coenzyme M. Biochemistry 40, 12875–12885 [DOI] [PubMed] [Google Scholar]
- 22. Bonacker L. G., Baudner S., Mörschel E., Böcher R., Thauer R. K. (1993) Properties of the two isoenzymes of methyl-coenzyme M reductase in Methanobacterium thermoautotrophicum. Eur. J. Biochem. 217, 587–595 [DOI] [PubMed] [Google Scholar]
- 23. Goenrich M., Duin E. C., Mahlert F., Thauer R. K. (2005) Temperature dependence of methyl-coenzyme M reductase activity and of the formation of the methyl-coenzyme M reductase red2 state induced by coenzyme B. J. Biol. Inorg. Chem. 10, 333–342 [DOI] [PubMed] [Google Scholar]
- 24. Ebner S., Jaun B., Goenrich M., Thauer R. K., Harmer J. (2010) Binding of coenzyme B induces a major conformational change in the active site of methyl-coenzyme M reductase. J. Am. Chem. Soc. 132, 567–575 [DOI] [PubMed] [Google Scholar]
- 25. Ermler U. (2005) On the mechanism of methyl-coenzyme M reductase. Dalton Trans. 3451–3458 [DOI] [PubMed] [Google Scholar]
- 26. Signor L., Knuppe C., Hug R., Schweizer B., Pfaltz A., Jaun B. (2000) Methane formation by reaction of a methyl thioether with a photo-excited nickel thiolate: a process mimicking methanogenesis in archaea. Chem. Eur. J. 6, 3508–3516 [DOI] [PubMed] [Google Scholar]
- 27. Lahiri G. K., Stolzenberg A. M. (1993) F430 model chemistry: evidence for alkyl- and hydrido-nickel intermediates in the reactions of the nickel(I) octaethylisobacteriochlorin anion. Inorg. Chem. 32, 4409–4413 [Google Scholar]
- 28. Yang N., Reiher M., Wang M., Harmer J., Duin E. C. (2007) Formation of a nickel-methyl species in methyl-coenzyme M reductase, an enzyme catalyzing methane formation. J. Am. Chem. Soc. 129, 11028–11029 [DOI] [PubMed] [Google Scholar]
- 29. Chen S. L., Pelmenschikov V., Blomberg M. R., Siegbahn P. E. (2009) Is there a Ni-methyl intermediate in the mechanism of methyl-coenzyme M reductase? J. Am. Chem. Soc. 131, 9912–9913 [DOI] [PubMed] [Google Scholar]
- 30. Pelmenschikov V., Blomberg M. R., Siegbahn P. E., Crabtree R. H. (2002) A mechanism from quantum chemical studies for methane formation in methanogenesis. J. Am. Chem. Soc. 124, 4039–4049 [DOI] [PubMed] [Google Scholar]
- 31. Chen S. L., Blomberg M. R., Siegbahn P. E. (2012) How is methane formed and oxidized reversibly when catalyzed by Ni-containing methyl-coenzyme M reductase? Chem. Eur. J. 18, 6309–6315 [DOI] [PubMed] [Google Scholar]
- 32. Scheller S., Goenrich M., Mayr S., Thauer R. K., Jaun B. (2010) Intermediates in the catalytic cycle of methyl coenzyme M reductase: isotope exchange is consistent with formation of a σ-alkane-nickel complex. Angew. Chem. Int. Ed. Engl. 49, 8112–8115 [DOI] [PubMed] [Google Scholar]
- 33. Scheller S., Goenrich M., Thauer R. K., Jaun B. (2013) Methyl-coenzyme M reductase from methanogenic archaea: Isotope effects on the formation and anaerobic oxidation of methane. J. Am. Chem. Soc. 135, 14975–14984 [DOI] [PubMed] [Google Scholar]
- 34. Scheller S., Goenrich M., Thauer R. K., Jaun B. (2013) Methyl-coenzyme M reductase from methanogenic archaea: isotope effects on label exchange and ethane formation with the homologous substrate ethyl-coenzyme M. J. Am. Chem. Soc. 135, 14985–14995 [DOI] [PubMed] [Google Scholar]
- 35. Zehnder A. J., Wuhrmann K. (1976) Titanium(III) citrate as a nontoxic oxidation reduction buffering system for the culture of obligate anaerobes. Science 194, 1165–1166 [DOI] [PubMed] [Google Scholar]
- 36. Gunsalus R. P., Romesser J. A., Wolfe R. S. (1978) Preparation of coenzyme M analogues and their activity in the methyl coenzyme M reductase system of Methanobacterium thermoautotrophicum. Biochemistry 17, 2374–2377 [DOI] [PubMed] [Google Scholar]
- 37. Bobik T. A., Wolfe R. S. (1988) Physiological importance of the heterodisulfide of coenzyme M and 7-mercaptoheptanoylthreonine phosphate in the reduction of carbon dioxide to methane in Methanobacterium. Proc. Natl. Acad. Sci. U.S.A. 85, 60–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Noll K. M., Donnelly M. I., Wolfe R. S. (1987) Synthesis of 7-mercaptoheptanoylthreonine phosphate and its activity in the methylcoenzyme M methylreductase system. J. Biol. Chem. 262, 513–515 [PubMed] [Google Scholar]
- 39. Kunz R. C., Horng Y. C., Ragsdale S. W. (2006) Spectroscopic and kinetic studies of the reaction of bromopropanesulfonate with methyl-coenzyme M reductase. J. Biol. Chem. 281, 34663–34676 [DOI] [PubMed] [Google Scholar]
- 40. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochem. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 41. Pelmenschikov V., Siegbahn P. E. (2003) Catalysis by methyl-coenzyme M reductase: a theoretical study for heterodisulfide product formation. J. Biol. Inorg. Chem. 8, 653–662 [DOI] [PubMed] [Google Scholar]
- 42. Goenrich M., Mahlert F., Duin E. C., Bauer C., Jaun B., Thauer R. K. (2004) Probing the reactivity of Ni in the active site of methyl-coenzyme M reductase with substrate analogues. J. Biol. Inorg. Chem. 9, 691–705 [DOI] [PubMed] [Google Scholar]
- 43. Li X., Telser J., Kunz R. C., Hoffman B. M., Gerfen G., Ragsdale S. W. (2010) Observation of organometallic and radical intermediates formed during the reaction of methyl-coenzyme M reductase with bromoethanesulfonate. Biochemistry 49, 6866–6876 [DOI] [PMC free article] [PubMed] [Google Scholar]