

Background: Rac GTPase is essential for the survival of primary cerebellar granule neurons (CGNs).
Results: NSC23766-mediated Rac inhibition induces CGN apoptosis through deactivation of prosurvival MEK5/ERK5 signaling.
Conclusion: Targeted inhibition of Rac versus global inhibition of Rho GTPases induces apoptosis via disruption of unique MAP kinase signaling pathways.
Significance: Elucidating the pathways that mediate neuronal apoptosis is critical for understanding the pathology of neurodegenerative diseases.
Keywords: Apoptosis, B Cell Lymphoma 2 (Bcl-2), ERK, Neurodegeneration, Rac (Rac GTPase), Bad, MEK, NSC23766
Abstract
Rho family GTPases play integral roles in neuronal differentiation and survival. We have shown previously that Clostridium difficile toxin B (ToxB), an inhibitor of RhoA, Rac1, and Cdc42, induces apoptosis of cerebellar granule neurons (CGNs). In this study, we compared the effects of ToxB to a selective inhibitor of the Rac-specific guanine nucleotide exchange factors Tiam1 and Trio (NSC23766). In a manner similar to ToxB, selective inhibition of Rac induces CGN apoptosis associated with enhanced caspase-3 activation and reduced phosphorylation of the Rac effector p21-activated kinase. In contrast to ToxB, caspase inhibitors do not protect CGNs from targeted inhibition of Rac. Also dissimilar to ToxB, selective inhibition of Rac does not inhibit MEK1/2/ERK1/2 or activate JNK/c-Jun. Instead, targeted inhibition of Rac suppresses distinct MEK5/ERK5, p90Rsk, and Akt-dependent signaling cascades known to regulate the localization and expression of the Bcl-2 homology 3 domain-only protein Bad. Adenoviral expression of a constitutively active mutant of MEK5 is sufficient to attenuate neuronal cell death induced by selective inhibition of Rac with NSC23766 but not apoptosis induced by global inhibition of Rho GTPases with ToxB. Collectively, these data demonstrate that global suppression of Rho family GTPases with ToxB causes a loss of MEK1/2/ERK1/2 signaling and activation of JNK/c-Jun, resulting in diminished degradation and enhanced transcription of the Bcl-2 homology 3 domain-only protein Bim. In contrast, selective inhibition of Rac induces CGN apoptosis by repressing unique MEK5/ERK5, p90Rsk, and Akt-dependent prosurvival pathways, ultimately leading to enhanced expression, dephosphorylation, and mitochondrial localization of proapoptotic Bad.
Introduction
Rho family GTPases belong to the Ras superfamily of monomeric G-proteins and include the well described proteins Rac, Rho, and Cdc42. Although Rho GTPases are best recognized for their prominent roles in regulating actin cytoskeletal dynamics, more recent studies have demonstrated an important role for Rho GTPases in regulating neuronal survival (1). Indeed, apoptosis induced by inhibitors of 3-HMG-CoA reductase (statins) was correlated with loss of plasma membrane-associated Rho GTPase family members in rat cortical neurons (2), and down-regulation of Rho family GTPases is closely associated with an increase in neuronal apoptosis (3). More recently, Rac has been highlighted as a specific member of the Rho GTPase family critical for maintaining neuronal survival. For example, prior studies demonstrated that either dominant negative Rac expression or siRNA-mediated knockdown of the Rac GEF Alsin (ALS2) induces apoptosis of cultured motor neurons (4, 5). The critical involvement of Rac in neurodegeneration is further evidenced by studies demonstrating that loss-of-function mutations in ALS2 are causative in juvenile-onset amyotrophic lateral sclerosis (6). Collectively, these data highlight Rac as a crucial modulator of neuronal survival and suggest that Rac dysregulation may underlie some forms of neurodegenerative disease.
In previous studies, we examined the neurotoxic effects of the large cytotoxin Clostridium difficile toxin B (ToxB)2 in CGNs. ToxB monoglucosylates a key threonine residue in the switch 1 region of Rho GTPases, preventing Rho, Rac, and Cdc42 from interacting with their downstream effectors (7). Global inhibition of Rho GTPases with ToxB induces CGN apoptosis through dysregulation of critical prosurvival and proapoptotic signaling cascades (8, 9). Specifically, ToxB induces down-regulation of Rac1 GTPase as well as elements of a Rac-dependent mitogen-activated protein (MAP) kinase pathway, including the p21-activated kinase (PAK), MEK1/2, and ERK1/2 signaling cascade. Inhibition of this pathway in CGNs induces apoptosis in part through reduced degradation of the proapoptotic BH3-only protein Bim. In addition to repression of a prosurvival MEK1/2/ERK1/2 signaling cascade, we have reported previously that broad Rho GTPase inhibition with ToxB induces CGN death through activation of a JNK/c-Jun pathway that stimulates transcription of Bim (10). Therefore, ToxB globally suppresses Rho GTPase function and induces CGN apoptosis through dysregulation of specific MAP kinase signaling cascades, leading to enhanced expression and diminished degradation of the proapoptotic BH3-only protein Bim.
The focus of this study was to compare the effects of ToxB on neuronal survival to those of a more targeted inhibitor of Rac GTPase. Although ToxB has been shown to inhibit Rac, Rho, and Cdc42, NSC23766 suppresses a more discrete pool of Rho family GTPases through inhibition of the Rac-specific GEFs Tiam1 and Trio, two of the most prominent regulators of Rac in the brain (11). The application of this targeted Rac inhibitor allows for a refined understanding of the Rho family GTPase-regulated signaling pathways essential to neuronal survival. Contrary to our results with ToxB, we demonstrate that targeted inhibition of Rac with NSC23766 does not turn off prosurvival MEK1/2/ERK1/2 signaling in CGNs, nor does it activate the JNK/c-Jun cascade. Instead, NSC23766 induces apoptosis via repression of the distinct MAP kinase pathway MEK5/ERK5. Further establishing these findings, adenoviral expression of constitutively active MEK5 significantly protects CGNs from NSC23766-mediated Rac inhibition but is unable to protect CGNs from ToxB-mediated global Rho GTPase inhibition. We report that loss of MEK5/ERK5 signaling in NSC23766-treated CGNs results in deactivation of the downstream effectors p90Rsk and Akt, leading to induction, dephosphorylation, and mitochondrial localization of the BH3-only, proapoptotic protein Bad. These findings are novel in that they are the first to distinguish the precise MAP kinase signaling pathways that regulate neuronal apoptosis in response to selective inhibition of Rac versus global suppression of Rho family GTPases.
EXPERIMENTAL PROCEDURES
Materials
NSC23766, JNK inhibitor II (SP600125), and the caspase inhibitors BOC, LEHD, and QVD (non-O-methylated) were obtained from Calbiochem. ZVAD was purchased from Enzo Lifesciences (Farmingdale, NY). C. difficile toxin B was prepared using a method published previously (7). The monoclonal antibody for active Rac1 was procured from NewEast Biosciences (Malvern, PA). Hoechst dye 33258 and DAPI were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Polyclonal antibody to active caspase-3 was from Promega (Madison, WI). Cy3- and FITC-conjugated secondary antibodies for immunofluorescence were from Jackson ImmunoResearch Laboratories (West Grove, PA). The caspase activation assay was purchased from Life Technologies. The monoclonal β-tubulin antibody conjugated to Alexa Fluor 555, polyclonal antibodies for pPAK1 (Ser-144)/pPAK2 (Ser-141), PAK1/2/3, pERK1/2 (Thr-202/Tyr-204), total ERK1/2, pMEK1/2 (Ser-217/221), total MEK1/2, pERK5 (Thr-218/Tyr-220), total ERK5, pMEK5, total MEK5, pAkt (Ser-473), and the monoclonal antibodies against Actin, c-Jun, pBad (Ser-136), and pP90Rsk (Ser-380) were purchased from Cell Signaling Technology (Beverly, MA). Polyclonal antibodies for pMEK5 (Ser-311/Thr-315) and MAP2 monoclonal antibody were obtained from Abcam (Cambridge, MA). The monoclonal antibody used to detect β-tubulin was purchased from Sigma. The rabbit monoclonal antibody for Bad (N-term) was purchased from Millipore (Billerica, MA). Horseradish peroxidase-linked secondary antibodies and reagents for enhanced chemiluminescence detection were from Amersham Biosciences.
CGN Culture
CGNs were isolated from 7-day-old Sprague-Dawley rat pups of both sexes (15–19 g) as described previously (12). Briefly, neurons were plated on 35-mm-diameter plastic dishes coated with poly-l-lysine at a density of ∼2.0 × 106 cells/ml in basal modified Eagle's medium containing 10% fetal bovine serum, 25 mm KCl, 2 mm l-glutamine, and penicillin (100 units/ml)/streptomycin (100 μg/ml) (Life Technologies). 24 h after plating the CGNs, cytosine arabinoside (10 μm) was added to the culture medium to limit the growth of non-neuronal cells. In general, experiments were performed after 6–7 days in culture.
Cell Lysis and Immunoblotting
Following treatment as described under “Results,” cell lysates were prepared for Western blotting as described previously (8). The protein concentration of the samples was determined using a protein assay kit (BCA, Thermo Scientific), and SDS-polyacrylamide gel electrophoresis was performed using equal amounts of protein followed by transfer to PVDF membranes (Amersham Biosciences). The entire PVDF membranes, when processed, were blocked in PBS (pH 7.4) containing 0.1% Tween 20 (PBS-T) and 1% BSA for 1 h at room temperature (25 °C). Subsequently, membranes were incubated for 1 h with primary antibodies diluted in blocking solution. Excess primary antibody was removed by washing the membranes with PBS-T five times over 25 min. The corresponding horseradish peroxidase-conjugated secondary antibodies diluted in PBS-T were incubated with the membranes for 1 h following the washes. Excess secondary antibody was removed by washing with PBS-T five times over 25 min. Immunoreactive proteins were detected by enhanced chemiluminescence.
Densitometric Analysis of Western Blots
Quantitative changes in the phosphorylation or expression levels of individual proteins were assessed by densitometry of Western blots. When appropriate, expression levels of assessed proteins were normalized to a loading control. In general, phosphospecific bands were normalized to the total expression level for a given protein. All quantitative data are on the basis of an analysis of blots obtained from at least three independent experiments. The protein expression level or phosphorylation level of the control cell lysates was set at 100%, and all other values are reported relative to the control value. Statistical differences in phosphorylation or protein expression between control cell lysates and lysates from cells treated with the Rac inhibitor were analyzed by paired Student's t test. All statistical analyses were performed by comparing raw densitometric values.
Immunofluorescence Microscopy
After treatment, CGNs were washed once with PBS (1 ml/well) and then fixed in 4% paraformaldehyde (1 ml/well) for 1 h at room temperature (25 °C). Cells were washed once with PBS (1 ml/well) and then permeabilized and blocked in 0.2% Triton X-100 and 5% BSA in PBS (pH 7.4). Primary antibodies were diluted in 2% BSA and 0.2% Triton X-100 in PBS. Cells were incubated in the primary antibody for 1 h at room temperature or overnight at 4 °C depending on the antibody. After incubation, cells were washed five times in PBS (5 min per wash) and then incubated for 1 h at 25 °C with Cy3- or FITC-conjugated secondary antibodies and DAPI-diluted in 2% BSA and 0.2% Triton X-100 in PBS. Following the second incubation, neurons were washed an additional five times with PBS (5 min per wash) before the addition of anti-quench (0.1% p-phenylenediamine in 75% glycerol) in PBS. Fluorescent images were captured by using a ×63 oil immersion objective on a Zeiss Axioplan 2 microscope equipped with a Cooke Sensicam deep-cooled charge-coupled device camera and a Slidebook software analysis program for digital deconvolution (Intelligent Imaging Innovations, Inc., Denver, CO).
Quantification of Apoptosis
Following treatment, Hoechst dye (8 μg/ml final concentration) was added directly to each well and incubated at 25 °C for 30 min to stain CGN nuclei. Cells were then washed with PBS (1 ml/well) and fixed by incubation in 4% paraformaldehyde (in PBS) for 30 min at 25 °C. Cells were subsequently washed twice with PBS (1 ml/well). To quantify apoptosis, CGNs displaying condensed chromatin or nuclear fragmentation were scored as apoptotic. Approximately 300 neurons were counted in a minimum of three ×63 fields of a 35-mm dish, and final counts represent data collected from a minimum of three independent experiments performed in duplicate.
Adenovirus Preparation and Infection
Adenoviral constitutively active MEK5 was purchased from Cell BioLabs (San Diego, CA), and adenoviral GFP was a gift from Marja T. Nevalainen (Thomas Jefferson University) and was prepared as described previously (13). CGNs were infected on day 5 in vitro with adenovirus with a multiplicity of infection of 35 carrying GFP or constitutively active MEK5. 48 h post-infection, some cells were treated with 175 μm NSC23766 for an additional 48 h. 72 h post-infection, another group of cells was treated with 60 ng/ml ToxB for an additional 24 h. 96 h post-infection, cells were fixed for fluorescent imaging and quantification of apoptosis as described above.
Data Analysis
In general, qualitative Western blotting observations were made on the basis of three independent experiments. Graphical data represent the mean ± S.E. for the number of independent experiments performed (n). Statistical differences between the means of unpaired sets of data were assessed by one-way analysis of variance and further analyzed using a post hoc Tukey's test. p < 0.05 was considered statistically significant.
RESULTS
The Specific Rac Inhibitor NSC23766 Induces Inactivation of Rac-GTP, Resulting in CGN Apoptosis
The targeted Rac inhibitor NSC23766 selectively and reversibly hinders GDP/GTP exchange carried out by the Rac-specific GEFs Tiam1 and Trio. Specifically, this cell-permeable pyrimidine compound targets a tryptophan within the GEF binding site, prohibiting Tiam1 and Trio from binding and activating Rac (14). Consistent with this mechanism of inhibition, CGNs incubated for 48 h with NSC23766 (200 μm) exhibited significantly decreased immunoreactivity for active Rac (Rac-GTP) compared with control CGNs (Fig. 1). Consistent with the involvement of Rac in regulating actin assembly, inhibition of Rac for 48 h resulted in marked fragmentation of the actin cytoskeletal network (Fig. 2A). The maintenance of an intact microtubule network, visualized by β-tubulin and MAP2 immunoreactivity, confirmed that the relative lack of Rac-GTP staining observed in NSC23766-treated cells was not due to the global absence or disruption of cellular components (Fig. 2B, top row). Moreover, examination of nuclei by Hoechst staining revealed significantly increased apoptotic cell death in NSC23766-treated CGNs compared with untreated controls, as evidenced by nuclear fragmentation and/or condensation (Fig. 2, B, bottom row, and C). Collectively, these data indicate that the targeted inhibition of a select pool of Rac GTPase activated by Tiam1 and Trio is sufficient to induce CGN apoptosis.
FIGURE 1.

The specific Rac inhibitor NSC23766 reduces activation of Rac1, resulting in CGN apoptosis. CGNs were incubated for 48 h in either control medium alone (Con) or medium containing NSC23766 (NSC, 200 μm). A, cells were then fixed, and their nuclei were stained with DAPI. Active Rac1 was visualized using a monoclonal antibody that specifically detects Rac1-GTP and a FITC-conjugated secondary antibody. Scale bar = 10 μm.
FIGURE 2.
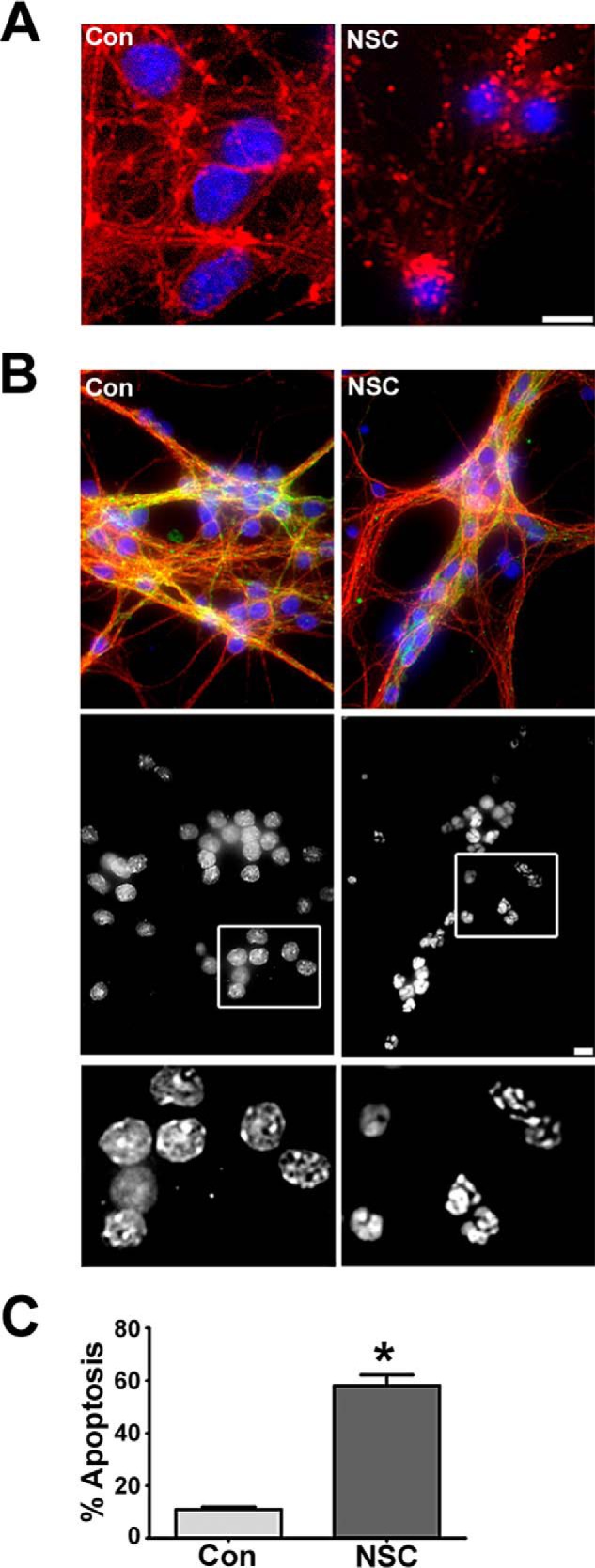
NSC23766 disrupts the actin cytoskeleton and induces apoptosis. A, cells were incubated for 48 h in either control medium alone (Con) or medium containing NSC23766 (NSC, 200 μm). Cells were fixed, nuclei were stained with DAPI, and the actin cytoskeletal was visualized with an antibody against actin. Scale bar = 10 μm. B, cells were incubated for 48 h in either control medium alone or medium containing NSC23766 (200 μm). Cells were fixed, and their nuclei were stained with DAPI. The microtubule network was visualized using a monoclonal antibody for β-tubulin conjugated with Cy3 Alexa Fluor 555 and a mouse monoclonal MAP2 antibody followed by a FITC-conjugated secondary antibody. Although the microtubule network remains largely intact, CGNs display condensed or fragmented nuclei characteristic of apoptosis following treatment with NSC23766. Bottom row, black and white images of DAPI-stained nuclei. Scale bar = 10 μm. C, quantification of CGN apoptosis. Hoechst-stained CGNs displaying condensed and/or fragmented nuclei were scored as apoptotic. Data represent the mean ± S.E. of four independent experiments. *, p < 0.05.
CGN Death Induced by NSC23766 Is Largely Caspase-independent
To determine whether NSC23766-mediated CGN death occurs via a mechanism that is dependent upon activation of caspases, we examined activation of caspase-3 because its proteolytic cleavage signifies commitment to apoptotic cell death (15). In agreement with this mechanism of cell death, CGNs treated with NSC23766 displayed increased immunoreactivity for active (cleaved) caspase-3 (Fig. 3A) and demonstrated enhanced activation of caspases that were blocked by inclusion of pan-caspase inhibitors in a caspase-3/7 activation assay (data not shown). To definitively establish the involvement of caspase-3 activation in NSC23766-mediated CGN death, we next examined the potential protective effects of caspase inhibitors. CGN viability assessed by nuclear staining was notably reduced in NSC23766-treated cells compared with untreated controls. However, inclusion of the pan-caspase inhibitors BOC, QVD, or ZVAD had no apparent protective effects against this Rac inhibitor (Fig. 3B). In marked contrast, incubation with either BOC or LEHD (a caspase-9 inhibitor) alone significantly protected CGNs treated with the global Rho GTPase inhibitor ToxB (Fig. 3C). These data suggest that CGN death induced by targeted inhibition of Rac GTPase occurs via a mechanism that involves a dispensable activation of caspase-3.
FIGURE 3.

CGN death induced by NSC23766 is largely caspase-independent, unlike Toxin B-induced apoptosis. A, cells were incubated for 48 h in either control medium alone (Con) or medium containing NSC23766 (NSC, 200 μm). Cells were then fixed, and their nuclei were stained with DAPI. Active caspase-3 was visualized using a polyclonal antibody and a Cy3-conjugated secondary antibody. NSC23766 treatment causes increased activation of caspase-3 in CGNs (arrows). Scale bar = 10 μm. B, cells were incubated for 48 h in either control medium or medium containing NSC23766 (200 μm) with or without BOC (50 μm), QVD (20 μm), or ZVAD (10 μm). The percentage of cell viability was measured by nuclear morphology. Data represent the mean ± S.E. of three independently performed experiments. ***, p < 0.001; NS, no significant difference between NSC and NSC cotreated with caspase inhibitors individually (BOC, QVD, or ZVAD). C, cells were incubated for 24 h in either control medium or medium containing ToxB (60 ng/ml) with or without BOC (50 μm) or LEHD (100 μm, a caspase-9 inhibitor). Data represent the mean ± S.E. of four independently performed experiments. *, significantly different from control (p < 0.05); ††, significantly different from ToxB (p < 0.01).
NSC23766 Reduces the Phosphorylation of PAK but Does Not Suppress the Downstream MEK1/2 and ERK1/2 Signaling Cascade in CGNs
We have reported previously that broad Rho GTPase inhibition with ToxB deactivates a prosurvival MAP kinase signaling cascade involving PAK1/2/3, MEK1/2, and ERK1/2. Therefore, we examined whether this signaling cascade was similarly suppressed in NSC23766-treated CGNs. Although decreased levels of pPAK1/2/3 were detected in CGNs incubated with NSC23766 for 48 h (Fig. 4, A and D), specific Rac inhibition did not result in significantly decreased phosphorylation of MEK1/2 or ERK1/2 (Fig. 4, B, C, and E). These data suggest that targeted inhibition of a select pool of Rac activated by Tiam1 and Trio in CGNs induces cell death through a mechanism that is distinct from global inhibition of Rho GTPases with ToxB.
FIGURE 4.

NSC23766 reduces the phosphorylation of PAK but does not suppress the downstream MEK1/2 and ERK1/2 signaling cascade in CGNs. A, CGNs were incubated for 48 h in either control medium alone (Con) or medium containing NSC23766 (NSC, 200 μm). Cells were then lysed, proteins were resolved using SDS-PAGE, and PVDF membranes were immunoblotted with antibodies to pPAK1/2 and total PAK1/2/3. The blots shown are representative of three independent experiments. B, cells were treated as described in A and subsequently immunoblotted with antibodies against pMEK1/2 and total MEK1/2. The blots shown are representative of three independent experiments. C, cells were treated as described in A and subsequently immunoblotted with antibodies against pERK1/2 and total ERK1/2. The blots shown are representative of three independent experiments. D, quantitative analysis of pPAK densitometry. *, p < 0.05 versus control. E, quantitative analysis of pMEK1/2 and pERK1/2 densitometry.
NSC23766, but Not ToxB, Decreases MEK5/ERK5 Phosphorylation in CGNs
The above results suggest the possibility that the more targeted Rac inhibitor may work through suppressing MEK5 and ERK5, components of an alternative MAP kinase signaling cascade, to repress prosurvival signaling downstream of Rac and PAK. Indeed, an immunoblot analysis indicated that NSC23766 treatment caused significantly decreased phosphorylation of both MEK5 and ERK5 in CGNs (Fig. 5, A–C). MEK5 is also cleaved in response to NSC23766 treatment in CGNs, resulting in reduced levels of total MEK5 (Fig. 5A). Conversely, ToxB treatment had little to no effect on the phosphorylation of either MEK5 or ERK5 in CGNs (Fig. 5, A–C). These data indicate that the targeted inhibition of Rac GTPase may elicit cell death via deactivation of an alternative prosurvival MAP kinase pathway downstream of PAK and MEK5/ERK5 compared with ToxB-treated CGNs.
FIGURE 5.
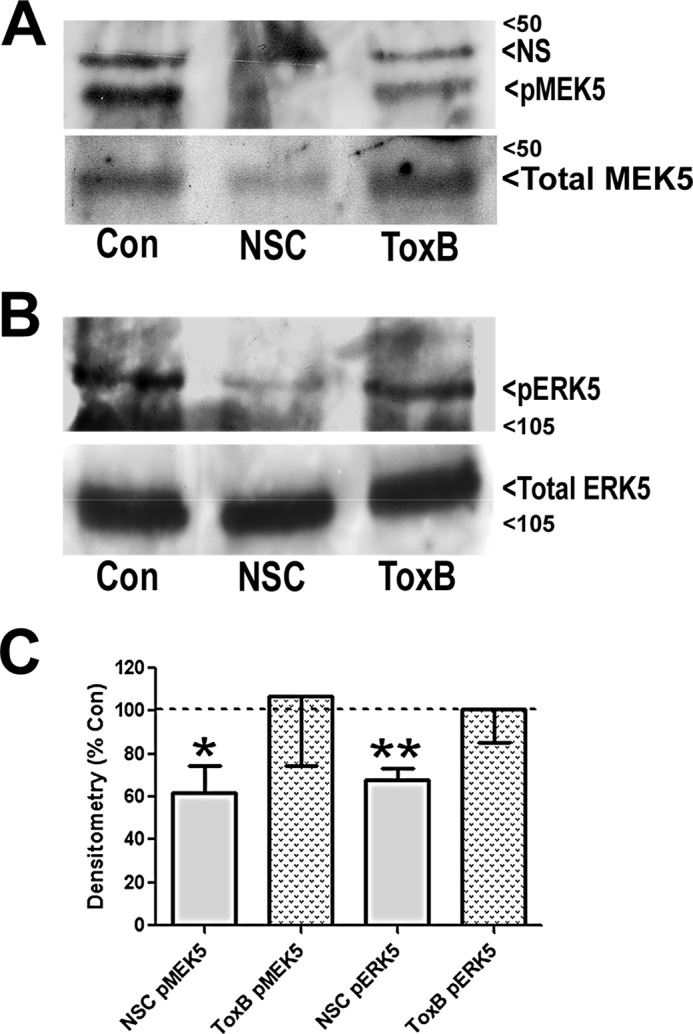
NSC23766, but not ToxB, decreases MEK5/ERK5 phosphorylation in CGNs. A, CGNs were incubated in either control medium alone (Con), medium containing NSC23766 (NSC, 200 μm) for 48 h or medium containing ToxB (60 ng/ml) for 24 h. Cells were then lysed, proteins were resolved using SDS-PAGE, and PVDF membranes were immunoblotted with antibodies to pMEK5 and total MEK5. The blots shown are representative of three independent experiments. NS, not significant. B, cells were treated as described in A and subsequently immunoblotted with antibodies against pERK5 and total ERK5. The blots shown are representative of three independent experiments. C, quantitative analysis of pMEK5 and pERK5 densitometry. *, p < 0.05; **, p < 0.01 versus control).
NSC23766 Treatment in CGNs Induces Dephosphorylation of p90Rsk and Akt, Leading to the Up-regulation and Mitochondrial Localization of the Proapoptotic BH3-only Protein Bad
A recent study has demonstrated that neuronal cell death induced by deletion of ERK5 is correlated with a marked decrease in phosphorylation of p90Rsk and Akt (16). To determine whether the loss of ERK5 phosphorylation induced by selective inhibition of Rac has a similar effect in CGNs, we examined the phosphorylation of p90Rsk and Akt in NSC23766-treated CGNs. Consistent with this previous study, we report distinct decreases in phosphorylated p90Rsk and Akt in NSC23766-treated CGNs compared with controls (Figs. 6, A, B, and E). One of the shared prosurvival functions of p90Rsk and Akt is to suppress the expression of Bad via repression of Bad transcription by the cAMP response element-binding protein (CREB) (16, 17). Bad is a member of the Bcl-2 family of proteins that promotes apoptosis through its ability to heterodimerize with, and therefore inhibit, prosurvival members of this family, such as Bcl-xL (18, 19). We report that treatment with the targeted Rac inhibitor NSC23766 resulted in up-regulation of proapoptotic Bad, an effect that was not observed with ToxB (Fig. 6C). Interestingly, both of the inhibitors also triggered the cleavage of Bad (Fig. 6C), producing an ∼15-kDa truncated form of the protein that is reportedly a more potent inducer of apoptosis (20).
FIGURE 6.

NSC23766 treatment in CGNs deactivates p90Rsk and Akt leading to increased expression, dephosphorylation, and mitochondrial localization of the proapoptotic BH3-only protein Bad. A, CGNs were incubated in either control medium alone (Con) or medium containing NSC23766 (NSC, 200 μm) for 48 h. Cells were then lysed, proteins were resolved using SDS-PAGE, and PVDF membranes were immunoblotted with antibodies to phospho-p90Rsk and subsequently stripped and reprobed for β-tubulin as a loading control. B, CGNs were incubated in either control medium alone or medium containing NSC23766 (200 μm) for various periods up to 48 h. Cell lysates were prepared for immunoblotting as described in A, and PVDF membranes were blotted for pAkt (Ser-473). Subsequently, membranes were stripped and reprobed for Actin as loading control. C, CGNs were treated and prepared for immunoblotting as described in A with the addition of a ToxB-treated condition for 24 h. Cell were lysed, and PVDF membranes were blotted for total Bad and subsequently stripped and reprobed for Actin as a loading control. D, CGNs were incubated as described in C. Cell lysates were prepared, and PVDF membranes were blotted for pBad (Ser-136) and subsequently stripped and reprobed for Actin as a loading control. E, quantitative analysis of p-p90Rsk, p-AKT, and p-Bad (Ser-136) densitometry. *, p < 0.05 versus control; **, p < 0.01 versus control. F, CGNs were incubated as described in C. Cells were then fixed, and their nuclei were stained with DAPI. Bad was visualized using a polyclonal antibody and a Cy3-conjugated secondary antibody. Images are representative of three independent experiments. Scale bar = 10 μm. G, CGNs were incubated as described in C, and cells were subfractionated into mitochondrial (Mito) and cytosolic (Cyto) fractions. Subcellular fractions were immunoblotted for Bad and OPA1 (a mitochondrial marker). The blots shown are representative of three independent experiments.
We next examined Bad phosphorylation because it has been demonstrated that phosphorylation at specific serine residues inactivates the proapoptotic function of Bad by sequestering the protein away from the mitochondria to 14-3-3 scaffolding proteins in the cytosol (21). Expression of pBad (Ser-136) was markedly reduced in NSC23766-treated but not ToxB-treated CGNs (Fig. 6, D and E). Prior reports have characterized a critical prosurvival function for Akt-dependent phosphorylation of Bad at Ser-136 (22, 23). In agreement with these studies, NSC23766-treated CGNs also displayed a marked reduction in pAkt (Ser-473) (Fig. 6, B and E). In contrast, we have reported previously that ToxB treatment does not alter the phosphorylation of Akt in CGNs (12). Because the Akt-mediated phosphorylation of Bad targets it to 14-3-3 scaffolding proteins in the cytosol, we next analyzed whether the dephosphorylation of Bad induced by inhibition of Rac was associated with translocation of this BH3-only protein to mitochondria. Immunofluorescent staining for Bad in control CGNs revealed a diffuse localization in the cytosol of the cell bodies with little detectable staining in cell processes (Fig. 6F, left column). In ToxB-treated CGNs, a similar distribution of Bad was observed in cells not actively undergoing apoptosis, whereas clearly apoptotic cells displayed undetectable levels of Bad expression (Fig. 6F, right column). In marked contrast to these results, CGNs incubated with the selective Rac inhibitor NSC23766 showed a striking redistribution of Bad to neuronal processes and an overall punctuate distribution of the protein (Fig. 6F, center column). To establish whether the punctuate appearance of Bad induced by the inhibition of Rac was due to its translocation to mitochondria, CGNs were subfractionated into mitochondrial and cytosolic fractions that were then immunoblotted for Bad. In control CGNs, Bad was exclusively localized to the cytosolic fraction, whereas, in NSC2376-treated CGNs, Bad was exclusively found in the mitochondrial fraction (Fig. 6G). Collectively, these data indicate that targeted inhibition of Rac with NSC23766 induces CGN apoptosis by turning off p90Rsk and Akt, likely downstream of MEK5/ERK5, leading to enhanced expression, dephosphorylation, and mitochondrial localization of proapoptotic Bad. In contrast, neither Bad dephosphorylation nor Bad translocation to mitochondria appears to be induced by ToxB.
Unlike ToxB, NSC23766 Treatment Does Not Activate a Proapoptotic JNK/c-Jun/Bim Pathway in CGNs
We have shown previously that ToxB-mediated repression of MEK1/2/ERK1/2 signaling induces intrinsic apoptosis of CGNs via decreased phosphorylation, and, therefore, diminished degradation, of the proapoptotic BH3-only protein Bim (8). In addition, we revealed that ToxB-treated CGNs displayed increased activation of a JNK/c-Jun signaling cascade that led to enhanced transcription of Bim (10). Therefore, we next examined whether targeted inhibition of Rac in CGNs had a similar effect on c-Jun and/or Bim. NSC23766-treated CGNs did not display increased c-Jun expression at any time point up to 48 h. As expected, within 8 h of ToxB treatment, the expression of both total c-Jun and a higher molecular weight band corresponding to phosphorylated c-Jun was observed in CGNs (Fig. 7, A and E). To investigate the potential involvement of JNK activation in promoting CGN death downstream of Rac inhibition, we examined the neuroprotective effects of the JNK inhibitor SP600125. Although inclusion of the JNK inhibitor did not significantly protect CGNs from selective Rac inhibition with NSC23766, the inhibitor significantly mitigated CGN apoptosis in response to ToxB treatment (Fig. 7, B and C). Consistent with activation of a prodeath JNK/c-Jun signaling pathway acting upstream of the BH3-only protein Bim, the expression of Bimshort was elevated in ToxB-treated CGNs. In contrast, we did not detect an increase in Bim expression in NSC23766-treated CGNs at any time point up to 48 h (Fig. 7, D and E). These results indicate that broad inhibition of Rho GTPases with ToxB elicits CGN apoptosis primarily through the up-regulation of Bim in a JNK/c-Jun-dependent manner, whereas neither JNK/c-Jun signaling nor Bim appear to play a significant role in NSC23766-mediated CGN death.
FIGURE 7.

Unlike ToxB, NSC23766 treatment does not activate a proapoptotic JNK/c-Jun/Bim pathway in CGNs. A, CGNs were incubated in either control medium alone (Con), medium containing ToxB (60 ng/ml), or medium containing NSC23766 (NSC, 200 μm) for various periods up to 8 and 48 h, respectively. Cells were then lysed, proteins were resolved using SDS-PAGE, and PVDF membranes were immunoblotted with an antibody against c-Jun and subsequently stripped and reprobed for Actin as a loading control. B, CGNs were incubated in either control medium alone, medium containing ToxB (40 ng/ml) for 24 h, or medium containing NSC23766 (200 μm) for 48 h. In addition, some cells were exposed to NSC or ToxB in the presence of the JNK inhibitor (JNK Inh, 20 μm). Cells were then fixed, and their nuclei were stained with Hoechst dye. Scale bar = 10 μm. C, quantification of CGN apoptosis from three independent experiments conducted as described in B. ***, significantly different from control (p < 0.001); †††, significantly different from ToxB (p < 0.001); NS, no significant difference from NSC. D, CGNs were incubated in either control medium alone, medium containing ToxB (60 ng/ml) for 8 h, or medium containing NSC23766 (200 μm) for various periods up to 48 h. Cells were then lysed, proteins were resolved using SDS-PAGE, and PVDF membranes were immunoblotted with an antibody against Bim and subsequently stripped and reprobed for β-tubulin as a loading control. The blots shown are representative of three independent experiments. E, quantitative analysis of c-Jun and Bim densitometry. *, p < 0.05; **, p < 0.01 versus control).
Adenoviral Infection with Constitutively Active MEK5 Protects CGNs from NSC23766 Treatment but Not ToxB
To definitively establish the precise MAP kinase signaling pathway that is deactivated in CGNs exposed to NSC23766-targeted Rac inhibition versus ToxB-mediated global Rho GTPase inhibition, we examined the protective effects of an adenoviral constitutively active mutant of MEK5 (Ad-CA MEK5) in which the dual phosphorylation site Ser-311/Ser-315 was changed to Asp-311/Asp-315. Infection with Ad-CA MEK5, but not Ad-GFP, increased the expression of MEK5 in CGNs (Fig. 8A). It is important to note that neither adenoviral construct increased the basal level of apoptosis on its own (data not shown). As we have shown previously, global inhibition of Rho GTPases with ToxB resulted in significant CGN apoptosis (Fig. 8B). Consistent with ToxB inducing death through a mechanism independent of MEK5/ERK5 signaling, preincubation of CGNs with either Ad-GFP or Ad-CA MEK5 did not confer significant protection against ToxB treatment (Fig. 8, B and C). Conversely, although CGNs exposed to selective inactivation of Rac GTPase with NSC23766 showed condensed and fragmented nuclei indicative of apoptotic cell death, CGNs preincubated with Ad-CA MEK5, but not Ad-GFP, displayed a nuclear morphology strikingly more similar to the controls (Fig. 8D). Quantification of these results demonstrated that adenoviral infection with Ad-CA MEK5 significantly protected CGNs from NSC23766 treatment (Fig. 8E). In addition to protecting CGNs from NSC23766-mediated apoptosis, preincubation with Ad-CA MEK5 significantly reduced the appearance of the cleaved form of Bad in the mitochondrial fraction of cells treated with the Rac inhibitor (Fig. 8F). It is worth noting that the protection conferred in CGNs preincubated with Ad-CA MEK5 and subsequently treated with NSC23766 was not complete, presumably because the infection efficiency was ∼40–50%, as estimated by the percentage of CGNs expressing Ad-GFP. Nonetheless, these results further corroborate that selective inactivation of Rac GTPase versus global inhibition of Rho GTPases induces CGN death through deactivation of unique prosurvival MAP kinase signaling pathways.
FIGURE 8.

Adenoviral infection with constitutively active MEK5 protects CGNs from NSC23766 treatment but not ToxB. A, CGNs were incubated in control medium (Con) or infected for 48 h with an adenovirus carrying GFP (Ad-GFP) or constitutively active MEK5 (Ad-CA MEDK5). Cell lysates were separated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted for MEK5 expression. Blots were subsequently stripped and reprobed for actin expression. B, CGNs were infected for 48 h with either Ad-GFP or Ad-CA MEK5. After the 48-h incubation period, CGNs were treated with ToxB (40 ng/ml) for an additional 24 h as indicated. At the end of the incubation, cells were fixed and nuclei were stained with DAPI. Scale bar = 10 μm. C, quantification of apoptosis from cells treated as described in B. NS, not significant. D, CGNs were infected for 48 h with either Ad-GFP or Ad-CA MEK5. After the 48-h incubation period, CGNs were treated with NSC23766 (NSC, 175 μm) for an additional 48 h as indicated. At the end of the incubation, cells were fixed, and nuclei were stained with DAPI. Scale bar = 10 μm. E, quantification of apoptosis from cells treated as described in D. Data represent the mean ± S.E. of three independently performed experiments. *, significantly different from control (p < 0.05); ***, significantly different from control (p < 0.001); †, significantly different from NSC (p < 0.05). F, CGNs were infected for 48 h with either Ad-GFP or Ad-CA MEK5. After the 48-h incubation period, CGNs were treated with NSC23766 (200 μm) for an additional 48 h. Following the incubation period, CGNs were separated into cytosolic and mitochondrial fractions and immunoblotted for Bad. The nonspecific band shown was unique to the mitochondrial fractions and is used to indicate equal loading.
DISCUSSION
Although a number of studies have highlighted a critical prosurvival role for Rho family GTPases in various neuronal cell types (8–10, 12, 24, 25), the mechanisms by which these G-proteins induce their neuroprotective effects remain largely unknown. In previous studies, we utilized C. difficile ToxB as a selective inhibitor of Rac, Rho, and Cdc42 to reveal that the PAK/MEK/ERK and JNK/c-Jun signaling cascades, components of the Rac-activated MAP kinase pathway, critically regulate CGN survival (8, 10). To advance our understanding of Rho family GTPases in neuronal survival, we compared the effects of ToxB to NSC23766, an inhibitor which specifically prevents Rac activation by the GEFs Tiam1 and Trio (14). Initial results confirmed that NSC23766 promotes CGN death through the loss of GTP-bound (active) Rac. However, although both ToxB and NSC23766 induce CGN death in the presence of serum and depolarizing potassium, this study indicates that CGNs succumb to cell death via unique signaling pathways in response to specific Rac inhibition versus global Rho GTPase inhibition.
Similar to ToxB, NSC23766 induces activation of caspase-3 in CGNs. However, although ToxB-induced CGN death was sensitive to caspase inhibitors, the effects of NSC23766 on neuronal death were not mitigated to a significant degree when coincubated with broad-spectrum pan-caspase inhibitors. The finding that caspase-3 is not sufficient to induce NSC23766-mediated CGN death is a key point of divergence between the effects of ToxB and NSC23766 on neuronal survival and indicates that NSC23766 is capable of promoting CGN death through a caspase-independent pathway. Given the antagonistic relationship between Rac and Rho signaling, it is possible that specific inhibition of only Rac via NSC23766 unmasks a Rho-dependent pathway that does not rely on caspase activation for the execution of CGN death. Indeed, Rac directly suppresses Rho activity through binding and activating the Rho GAP, p190RhoGAP, in HeLa cells (26). Moreover, the downstream effector of Rac, PAK, has been shown to regulate the activation of Rho through GEFs in a variety of cell types (27). Consistent with this idea, Rho has been demonstrated to sensitize corticohippocampal neurons to cell death (28), and inhibition of the Rho effector Rho kinase exerts a neuroprotective effect in various in vitro and in vivo models of neuronal cell death, such as cerebral ischemia and excitotoxicity (29, 30). In addition to Rho kinase, Rho GTPase has many other downstream effectors that could contribute to neuronal death via a mechanism that is independent of caspase activation.
The second contrast between the activities of ToxB and NSC23766 was apparent in their divergent effects upon MAP kinase signaling. Although ToxB suppressed MEK1/2/ERK1/2 signaling, the selective Rac inhibitor had no effect of this MAP kinase pathway and, instead, suppressed a unique MEK5/ERK5 cascade. Indeed, adenoviral expression of constitutively active MEK5 was only sufficient to mitigate the toxic effects of the selective Rac inhibitor NSC23766 in CGNs while also significantly reducing the appearance of the cleaved form of Bad in the mitochondrial fraction of cells treated with the Rac inhibitor. In contrast, expression of constitutively active MEK5 displayed no protective effect against global Rho GTPase inhibition with ToxB. It is possible that this stark contrast may be attributed to differential regulation of PAK. Upstream of the MEK/ERK signaling cascades, both Rho family GTPase inhibitors reduced the phosphorylation of PAK in CGNs. However, one limitation of these findings is that, although we have utilized an antibody that detects phosphorylation of PAK1/2/3, there are six known isoforms of PAK. Therefore, regulation of unique MAP kinase pathways in response to Rac inhibition may be a result of differential inactivation of specific PAK isoforms. Furthermore, an additional member of the Rho GTPase family that is inhibited by ToxB, Cdc42, also regulates PAK activity in a manner similar to Rac (31). Inactivation of distinct PAK isoforms in response to targeted inhibition of Rac versus global inhibition of Rho GTPases may represent a key point at which the signaling cascades regulated by these inhibitors deviate.
Although deactivation of specific PAK isoforms is a potential point at which the signaling cascades regulated by distinct Rho family GTPases may diverge, this could also be accomplished downstream of PAK signaling through MEKKs. MEKKs are upstream regulators of MAP kinase pathways such as MEK/ERK, JNK, and p38 (32, 33). As the most well studied MAP kinase signaling pathway, MEK1/2 activation has largely been attributed to MEKKs that belong to the Raf family of proteins (i.e. A-Raf, Raf-1, and B-Raf). This may be a critical point of divergence in MEK/ERK signaling because recent studies have demonstrated that MEKK2 and MEKK3 specifically activate the downstream effector MEK5 via heterodimerization of a PB1 domain that is specific to MEKK2, MEKK3, and MEK5 (33, 34). Furthermore, although both MEKK2 and MEKK3 bind to MEK5 and activate the MEK5/ERK5 pathway, it has been shown that MEKK2 binds MEK5 with higher affinity, stimulating ERK5 activity to a greater extent (33). Collectively, these previous studies suggest that the distinct MAP kinase pathways that are deactivated by either ToxB or NSC23766 in CGNs may diverge downstream of PAK at the level of specific MEKK activation.
Overall, these data suggest that CGN death triggered by the targeted inhibition of a select pool of Rac activated by the GEFs Tiam1 and Trio involves the suppression of a MEK5/ERK5 signaling cascade to induce apoptosis. This is in agreement with previous studies highlighting a critical prosurvival function for ERK5 in dopaminergic neurons and sympathetic neurons (16, 35). Moreover, past studies have indicated that ERK5 is activated by MEK5 but not by MEK1 or MEK2, speaking to the highly specific nature of the MEK5/ERK5 interaction (36, 37). Finally, others have demonstrated that Rac1 can signal to MEK5/ERK5 (38). This Rac effect presumably occurs through PAK-mediated activation of MEKK2/3 and their subsequent activation of MEK5 (34, 39). Therefore, selective inhibition of Rac is capable of suppressing a putative PAK/MEKK2/3/MEK5/ERK5 prosurvival pathway.
Previous experimentation has shown that ERK5 is capable of triggering a phosphorylation cascade that results in the phosphorylation of the MAPK-activated kinase p90Rsk and the subsequent activation of the transcription factor CREB, which regulates transcription of prosurvival and proapoptotic genes in neurons, including repression of the proapoptotic Bad gene (40–43). Accordingly, recent findings in neurons indicate that, in the absence of ERK5, phosphorylation of p90Rsk is impaired and cell death is elicited in a Bad-dependent manner (16). Consistent with the observed reduction in phosphorylated p90Rsk in response to NSC23766, we report that NSC23766 treatment also elicited up-regulation of Bad in CGNs. Given that ToxB did not enhance Bad expression, it appears that deactivation of p90Rsk is unique to NSC23766-treated CGNs. Interestingly, both NSC23766 and ToxB induced the cleavage of Bad, generating elevated levels of a 15-kDa cleavage product. This is consistent with a previous report that revealed that caspase cleavage of Bad at its N terminus produces a 15-kDa truncated form of Bad that is a more effective inducer of apoptosis than the full-length protein (20). However, given that pan-caspase inhibitors do not protect NSC23766-treated CGNs to a significant extent, our data suggest that caspase cleavage of Bad is not the prominent mechanism of death in CGNs exposed to NSC23766. Together, these data suggest an important role for the MEK5/ERK5 pathway in neuronal survival. Specifically, targeted inhibition of a discrete pool of Rac activated by Tiam1 and Trio appears to trigger CGN death through repression of a novel prosurvival pathway involving PAK, possibly MEKK2/3, and the MEK5/ERK5 signaling cascade, including its downstream targets p90Rsk and Bad (Fig. 9).
FIGURE 9.

Comparing CGN death induced by NSC23766 and ToxB. The schematic illustrates the Rac-dependent signaling cascades examined in this study. A, pathway regulated by the Rho GTPases Rac, Rho, and Cdc42. B, pathway regulated by the specific pool of Rac activated by the GEFs Tiam1 and Trio. Dashed lines represent hypothesized effects. Our findings suggest that targeted inhibition of the specific pool of Rac activated by the GEFs Tiam1 and Trio promotes CGN death through an alternative pathway to that employed by the broad-spectrum Rho family inhibitor ToxB. These pathways diverge downstream of PAK, with NSC23766 repressing the MEK5/ERK5 signaling cascade, whereas ToxB inhibits MEK1/2 and ERK1/2. ToxB also induces the activation of JNK/c-Jun, a pathway that is not activated by NSC23766. ToxB ultimately results in decreased degradation and enhanced transcription of Bim, whereas NSC23766 promotes the dephosphorylation, mitochondrial localization, and induction of Bad.
In addition to regulation of Bad at the transcriptional level via CREB, p90Rsk also catalyzes the phosphorylation of Bad at serine 112, suppressing Bad-mediated apoptosis in neurons (17, 44). However, we did not detect any decrease in the phosphorylation of Bad at Ser-112 (data not shown). Instead, we observed that NSC23766-treated CGNs exhibited a marked reduction in levels of pBad (Ser-136). Interestingly, several studies have highlighted that phosphorylation of Bad at Ser-136 may be the more critical 14-3-3 binding site that enhances the association of Bad with cytosolic 14-3-3 scaffolding proteins, effectively sequestering Bad away from mitochondria (21, 45). Several reports have indicated that phosphorylation of Bad at this particular site is largely attributed to Akt activation (22, 23). This is consistent with our data demonstrating reduced phosphorylation (activation) of Akt (Ser-473) in NSC23766-treated CGNs. Because we have reported previously that ToxB does not regulate Akt signaling in CGNs (12), the finding that NSC23766-treated CGNs display a marked reduction in activated Akt highlights an additional signaling pathway that is differentially regulated in response to specific Rac inhibition versus global Rho GTPase inhibition. However, the particular mechanism by which Akt is deactivated in NSC23766-treated CGNs is currently unknown. For instance, although it is well documented that Akt can be phosphorylated and activated by Rac through phosphatidylinositol 3-kinase (23, 25), ERK5 has also been shown to modestly activate Akt to promote survival of non-neuronal cells (46). Therefore, the precise mechanism by which Akt is dephosphorylated following Rac inhibition warrants further investigation. Despite these considerations, our data indicate that inhibition of a specific pool of Rac activated by the GEFs Tiam1 and Trio induces apoptosis through a mechanism consistent with Akt deactivation and subsequent dephosphorylation and translocation of Bad to the mitochondria.
Finally, in addition to the distinct differences in MEK/ERK, p90Rsk, and Akt signaling, ToxB and NSC23766 also appear to dissimilarly regulate JNK/c-Jun signaling in CGNs. Although we have reported previously that ToxB induced mitochondrial-dependent CGN apoptosis through a mechanism consistent with JNK/c-Jun-dependent transcriptional up-regulation of the BH3-only protein Bim (10), we report here that c-Jun was not induced in CGNs exposed to the specific Rac inhibitor NSC23766. Moreover, inclusion of the JNK inhibitor, SP600125, did not exert a protective effect in NSC23766-treated CGNs. Furthermore, although we report an increase in expression of the pro-apoptotic protein Bim in response to ToxB in CGNs, Bim was not elevated above control levels in NSC23766-treated CGNs. Therefore, although ToxB appears to induce CGN death through a mechanism dependent upon JNK/c-Jun-mediated induction of Bim, Bim does not appear to execute cell death in CGNs exposed to the specific Rac inhibitor NSC23766. Many studies have indicated that Rac and Cdc42 can regulate JNK activation. Therefore, this distinction in signaling may be due to the intrinsic differences between inactivation of specifically Rac versus global inhibition of Rho GTPases.
In conclusion, the specificity with which NSC23766 suppresses Rac activity allowed us to further investigate signaling pathways regulated by this Rho family GTPase that are involved in neuronal survival. We revealed that CGNs exposed to ToxB or NSC23766 succumb to neuronal death through inactivation of distinct MAP kinase signaling pathways. Comparison of the mechanisms behind specific Rac inhibition with those associated with broader Rho family GTPase inhibition has been central to elucidating the signaling pathways that regulate survival in CGNs. It is interesting to note that inhibition of the select pool of Rac activated by Tiam1 and Trio is sufficient to induce dysregulation of multiple signaling pathways, including MEK5/ERK5, p90Rsk, and Akt. Furthermore, the critical function of Rac in maintaining neuronal survival is highlighted by the fact that targeted inhibition of Rac is sufficient to induce CGN death in the presence of both serum and depolarizing potassium. Accordingly, this study serves to advance our understanding of the key prosurvival roles of Rho family GTPases in neurons. These findings may ultimately contribute to the discovery of novel signaling molecules that could be targeted therapeutically for debilitating neurodegenerative diseases, such as amyotrophic lateral sclerosis, for which loss of Rac function is thought to play a significant role in neuronal cell death.
This work was supported by a Merit Review award from the Department of Veterans Affairs (to D. A. L.).
- ToxB
- C. difficile toxin B
- CGN
- cerebellar granule neuron
- MAP
- mitogen-activated protein
- PAK
- p21-activated kinase
- GEF
- guanine nucleotide exchange factor
- CREB
- cAMP response-element binding protein.
REFERENCES
- 1. Linseman D. A., Loucks F. A. (2008) Diverse roles of Rho GTPases in neuronal development, survival, and death. Front. Biosci. 13, 657–676 [DOI] [PubMed] [Google Scholar]
- 2. Tanaka T., Tatsuno I., Uchida D., Moroo I., Morio H., Nakamura S., Noguchi Y., Yasuda T., Kitagawa M., Saito Y., Hirai A. (2000) Geranylgeranyl-pyrophosphate, an isoprenoid of mevalonate cascade, is a critical compound for rat primary cultured cortical neurons to protect the cell death induced by 3-hydroxy-3-methylglutaryl-CoA reductase inhibition. J. Neurosci. 20, 2852–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Desagher S., Severac D., Lipkin A., Bernis C., Ritchie W., Le Digarcher A., Journot L. (2005) Genes regulated in neurons undergoing transcription-dependent apoptosis belong to signaling pathways rather than the apoptotic machinery. J. Biol. Chem. 280, 5693–5702 [DOI] [PubMed] [Google Scholar]
- 4. Topp J. D., Gray N. W., Gerard R. D., Horazdovsky B. F. (2004) Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 279, 24612–24623 [DOI] [PubMed] [Google Scholar]
- 5. Jacquier A., Buhler E., Schäfer M. K., Bohl D., Blanchard S., Beclin C., Haase G. (2006) Alsin/Rac1 signaling controls survival and growth of spinal motoneurons. Ann. Neurol. 60, 105–117 [DOI] [PubMed] [Google Scholar]
- 6. Yamanaka K., Vande Velde C., Eymard-Pierre E., Bertini E., Boespflug-Tanguy O., Cleveland D. W. (2003) Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor disease. Proc. Natl. Acad. Sci. U.S.A. 100, 16041–16046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Just I., Selzer J., Wilm M., von Eichel-Streiber C., Mann M., Aktories K. (1995) Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375, 500–503 [DOI] [PubMed] [Google Scholar]
- 8. Loucks F. A., Le S. S., Zimmermann A. K., Ryan K. R., Barth H., Aktories K., Linseman D. A. (2006) Rho family GTPase inhibition reveals opposing effects of mitogen-activated protein kinase kinase/extracellular signal-related kinase and Janus kinase/signal transducer and activator of transcription signaling cascades on neuronal survival. J. Neurochem. 97, 957–967 [DOI] [PubMed] [Google Scholar]
- 9. Stankiewicz T. R., Loucks F. A., Schroeder E. K., Nevalainen M. T., Tyler K. L., Aktories K., Bouchard R. J., Linseman D. A. (2012) Signal transducer and activator of transcription-5 mediates neuronal apoptosis induced by inhibition of Rac GTPase activity. J. Biol. Chem. 287, 16835–16848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Le S. S., Loucks F. A., Udo H., Richardson-Burns S., Phelps R. A., Bouchard R. J., Barth H., Aktories K., Tyler K. L., Kandel E. R., Heidenreich K. A., Linseman D. A. (2005) Inhibition of Rac GTPase triggers a c-Jun and Bim-dependent mitochondrial apoptotic cascade in cerebellar granule neurons. J. Neurochem. 94, 1025–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmidt A., Hall A. (2002) Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16, 1587–1609 [DOI] [PubMed] [Google Scholar]
- 12. Linseman D. A., Laessig T., Meintzer M. K., McClure M., Barth H., Aktories K., Heidenreich K. A. (2001) An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J. Biol. Chem. 276, 39123–39131 [DOI] [PubMed] [Google Scholar]
- 13. Ahonen T. J., Xie J., LeBaron M. J., Zhu J., Nurmi M., Alanen K., Rui H., Nevalainen M. T. (2003) Inhibition of transcription factor STAT5 induces cell death of human prostate cancer cells. J. Biol. Chem. 278, 27287–27292 [DOI] [PubMed] [Google Scholar]
- 14. Gao Y., Dickerson J. B., Guo F., Zheng J., Zheng Y. (2004) Rationale design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. U.S.A. 101, 7618–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Porter A. G., Jänicke R. U. (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99–104 [DOI] [PubMed] [Google Scholar]
- 16. Finegan K. G., Wang X., Lee E. J., Robinson A. C., Tournier C. (2009) Regulation of neuronal survival by the extracellular signal-related protein kinase 5. Cell Death Differ. 16, 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonni A., Brunet A., West A. E., Datta S. R., Takasu M. A., Greenberg M. E. (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286, 1358–1362 [DOI] [PubMed] [Google Scholar]
- 18. Kelekar A., Chang B. S., Harlan J. E., Fesik S. W., Thompson C. B. (1997) Bad is a BH3 domain-containing protein that forms an inactivating dimmer with Bcl-xL. Mol. Cell Biol. 17, 7040–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kitada S., Krajewska M., Zhang X., Scudiero D., Zapata J. M., Wang H. G., Shabaik A., Tudor G., Krajewski S., Myers T. G., Johnson G. S., Sausville E. A., Reed J. C. (1998) Expression and location of pro-apoptotic Bcl-2 family protein BAD in normal human tissues and tumor cell lines. Am. J. Pathol. 152, 51–61 [PMC free article] [PubMed] [Google Scholar]
- 20. Condorelli F., Salomoni P., Cotteret S., Cesi V., Srinivasula S. M., Alnemri E. S., Calabretta B. (2001) Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol. Cell Biol. 21, 3025–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zha J., Harada H., Yang E., Jockel J., Korsmeyer S. J. (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3- not Bcl-X(L). Cell 87, 619–628 [DOI] [PubMed] [Google Scholar]
- 22. Datta S. R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M. E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241 [DOI] [PubMed] [Google Scholar]
- 23. del Peso L., González-García M., Page C., Herrera R., Nuñez G. (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278, 687–689 [DOI] [PubMed] [Google Scholar]
- 24. Kobayashi K., Takahashi M., Matsushita N., Miyazaki J., Koike M., Yaginuma H., Osumi N., Kaibuchi K., Kobayashi K. (2004) Survival of developing motor neurons mediated by Rho GTPase signaling pathway through Rho-kinase. J. Neurosci. 24, 3480–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kanekura K., Hashimoto Y., Kita Y., Sasabe J., Aiso S., Nishimoto I., Matsuoka M. (2005) A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J. Biol. Chem. 280, 4532–4543 [DOI] [PubMed] [Google Scholar]
- 26. Wildenberg G. A., Dohn M. R., Carnahan R. H., Davis M. A., Lobdell N. A., Settleman J., Reynolds A. B. (2006)) p120-catenin and p190Rho Gap regulate cell-cell adhesion by coordinating between Rac and Rho. Cell 127, 1027–1039 [DOI] [PubMed] [Google Scholar]
- 27. Hakoshima T., Shimizu T., Maesaki R. (2003) Structural basis of the Rho GTPase signaling. J. Biochem. 134, 327–331 [DOI] [PubMed] [Google Scholar]
- 28. Barberan S., McNair K., Iqbal K., Smith N. C., Prendergast G. C., Stone T. W., Cobb S. R., Morris B. J. (2011) Altered apoptotic response in neurons lacking RhoB GTPase. Eur. J. Neurosci. 34, 1737–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rikitake Y., Kim H. H., Huang Z., Seto M., Yano K., Asano T., Moskowitz M. A., Liao J. K. (2005) Inhibition of Rho kinase (ROCK) leads to increased cerebral flow and stroke prevention. Stroke 36, 2251–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jeon B. T., Jeong E. A., Park S. Y., Son H., Shin H. J., Lee D. H., Kim H. J., Kang S. S., Cho G. J., Choi W. S., Roh G. S. (2013) The Rho-kinase (ROCK) inhibitor Y-27632 protects against excitotoxicity-induced neuronal death in vivo and in vitro. Neurotox. Res. 23, 238–248 [DOI] [PubMed] [Google Scholar]
- 31. Kreis P., Thévenot E., Rousseau V., Boda B., Muller D., Barnier J. V. (2007) The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc-42 dependent pathway. J. Biol. Chem. 282, 21497–21506 [DOI] [PubMed] [Google Scholar]
- 32. Fanger G. R., Johnson N. L., Johnson G. L. (1997) MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J. 16, 4961–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun W., Kesavan K., Schaefer B. C., Garrington T. P., Ware M., Johnson N. L., Gelfand E. W., Johnson G. L. (2001) MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J. Biol. Chem. 276, 5093–5100 [DOI] [PubMed] [Google Scholar]
- 34. Nakamura K., Johnson G. L. (2003) PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J. Biol. Chem. 278, 36989–36992 [DOI] [PubMed] [Google Scholar]
- 35. Cavanaugh J. E., Jaumotte J. D., Lakoski J. M., Zigmond M. J. (2006) Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J. Neurosci. Res. 84, 1367–1375 [DOI] [PubMed] [Google Scholar]
- 36. English J. M., Vanderbilt C. A., Xu S., Marcus S., Cobb M. H. (1995) Isolation of MEK5 and differential expression of alternatively spliced forms. J. Biol. Chem. 270, 28897–28902 [DOI] [PubMed] [Google Scholar]
- 37. Zhou G., Bao Z. Q., Dixon J. E. (1995) Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 270, 12665–12669 [DOI] [PubMed] [Google Scholar]
- 38. Cheng J., Wang Y., Liang A., Jia L., Du J. (2012) FSP-1 silencing in bone marrow cells suppresses neointima formation in vein graft. Circ. Res. 110, 230–240 [DOI] [PubMed] [Google Scholar]
- 39. Porter A. C., Fanger G. R., Vaillancourt R. R. (1999) Signal transduction pathways regulated by arsenate and arsenite. Oncogene 54, 7794–77802 [DOI] [PubMed] [Google Scholar]
- 40. Watson F. L., Heerssen H. M., Bhattacharyya A., Klesse L., Lin M. Z., Segal R. A. (2001) Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci. 4, 981–988 [DOI] [PubMed] [Google Scholar]
- 41. Park E. M., Cho S. (2006) Enhanced ERK dependent CREB activation reduces apoptosis in staurosporine-treated human neuroblastoma SK-N-BE(2)C cells. Neurosci. Lett. 402, 190–194 [DOI] [PubMed] [Google Scholar]
- 42. Ranganathan A., Pearson G. W., Chrestensen C. A., Sturgill T. W., Cobb M. H. (2006) The MAP kinase ERK5 binds to and phosphorylates p90 RSK. Arch. Biochem. Biophys. 449, 8–16 [DOI] [PubMed] [Google Scholar]
- 43. Ha S., Redmond L. (2008) ERK mediates activity dependent neuronal complexity via sustained activity and CREB-mediated signaling. Dev. Neurobiol. 68, 1565–1579 [DOI] [PubMed] [Google Scholar]
- 44. Bertolotto C., Maulon L., Filippa N., Baier G., Auberger P. (2000) Protein kinase C θ and epsilon promote T-cell survival by a rsk-dependent phosphorylation and inactivation of Bad. J. Biol. Chem. 275, 37246–37250 [DOI] [PubMed] [Google Scholar]
- 45. Masters S. C., Yang H., Datta S. R., Greenberg M. E., Fu H. (2001) 14-3-3 inhibits Bad-induced cell death through interaction with serine-136. Mol. Pharmacol. 60, 1325–1331 [DOI] [PubMed] [Google Scholar]
- 46. Lennartsson J., Burovic F., Witek B., Jurek A., Heldin C. H. (2010) Erk5 is necessary for sustained PDGF-induced Akt phosphorylation and inhibition of apoptosis. Cell Signal. 22, 955–960 [DOI] [PubMed] [Google Scholar]