

Background: RppH initiates mRNA degradation in bacteria by removing pyrophosphate from the triphosphorylated 5′ terminus.
Results: X-ray structures of E. coli RppH have revealed molecular features important for RNA recognition, discrimination against mononucleotides, and catalysis.
Conclusion: RppH balances specificity and promiscuity.
Significance: Our findings explain the broad influence of RppH on E. coli mRNA decay and suggest a catalytic mechanism for Nudix hydrolases.
Keywords: Crystal Structure, Hydrolase, RNA-binding Protein, RNA Degradation, RNA Structure, RNA-Protein Interaction
Abstract
5′-End-dependent RNA degradation impacts virulence, stress responses, and DNA repair in bacteria by controlling the decay of hundreds of mRNAs. The RNA pyrophosphohydrolase RppH, a member of the Nudix hydrolase superfamily, triggers this degradation pathway by removing pyrophosphate from the triphosphorylated RNA 5′ terminus. Here, we report the x-ray structures of Escherichia coli RppH (EcRppH) in apo- and RNA-bound forms. These structures show distinct conformations of EcRppH·RNA complexes on the catalytic pathway and suggest a common catalytic mechanism for Nudix hydrolases. EcRppH interacts with RNA by a bipartite mechanism involving specific recognition of the 5′-terminal triphosphate and the second nucleotide, thus enabling discrimination against mononucleotides as substrates. The structures also reveal the molecular basis for the preference of the enzyme for RNA substrates bearing guanine in the second position by identifying a protein cleft in which guanine interacts with EcRppH side chains via cation-π contacts and hydrogen bonds. These interactions explain the modest specificity of EcRppH at the 5′ terminus and distinguish the enzyme from the highly selective RppH present in Bacillus subtilis. The divergent means by which RNA is recognized by these two functionally and structurally analogous enzymes have important implications for mRNA decay and the regulation of protein biosynthesis in bacteria.
Introduction
Messenger RNA degradation is essential for all living cells and is especially critical for bacteria, which live in a rapidly changing environment and require quick changes in protein synthesis to maximize their competitive advantage. An important mRNA decay pathway in bacteria involves endonucleolytic cleavage followed by 3′ to 5′-exonucleolytic digestion (1). Recent studies have demonstrated that bacterial mRNA degradation can also be initiated at the 5′-end (2, 3). This degradation route requires initial conversion of the triphosphorylated 5′ terminus to a monophosphate by the RNA pyrophosphohydrolase RppH (4, 5), followed by 5′ to 3′-exonucleolytic digestion by RNase J in Bacillus subtilis (6) or internal cleavage by the 5′-monophosphate-dependent endonuclease RNase E in Escherichia coli (4, 5). The protective influence of a triphosphate on the 5′-end of bacterial mRNA resembles that of a eukaryotic cap structure; therefore, pyrophosphate removal by RppH activity is both structurally and functionally analogous to mRNA decapping by eukaryotic enzymes. Similarity between the RppH-mediated reaction and eukaryotic decapping is further highlighted by commonalities in the cleavage mechanism. RppH and some eukaryotic decapping enzymes, such as Dcp2, belong to the superfamily of Nudix hydrolyses that share the metal-coordinating Nudix box GX5EX7REXXEEXG, the motif essential for the chemistry of the reaction (7). Despite the different mechanisms and proteins involved in the 5′-end-dependent mRNA decay pathway in E. coli and B. subtilis, RppH is common to both species. Nevertheless, E. coli RppH (EcRppH)2 and B. subtilis RppH (BsRppH) have low (∼20%) sequence identity (6) and share little similarity outside of the Nudix box. How these two rather distinct RppHs bind substrate RNA and perform an analogous catalytic reaction remains puzzling.
Most Nudix hydrolases are active on mononucleotide substrates, and only a few, such as RppH, can act on polynucleotide substrates (7, 8). RppH appears to be a ubiquitous bacterial enzyme and one of the most widespread Nudix hydrolases affecting several critical cellular processes. The loss of RppH activity causes diminished virulence (9, 10), sensitivity to stress (11), abnormal mutagenic repair of DNA breaks (12), and other deficiencies, even though the protein destabilizes only a fraction of mRNAs (hundreds in E. coli) (5, 13). Such unexpected selectivity of RppH-dependent degradation can be explained, at least in part, by the recently discovered 5′-terminal requirements of these enzymes. BsRppH needs two but prefers three or more unpaired 5′-terminal nucleotides for activity and strictly requires guanine at the second position of its substrates (Fig. 1A) (14, 15), suggesting that it triggers the decay of a distinct set of mRNAs. This sequence preference does not correlate with the nucleotide frequency at the 5′-end of B. subtilis transcripts (14, 15). Thus, RppH can be considered a master regulator of protein biosynthesis that selectively controls mRNA translation through different rates of the 5′-end-dependent mRNA decay.
FIGURE 1.
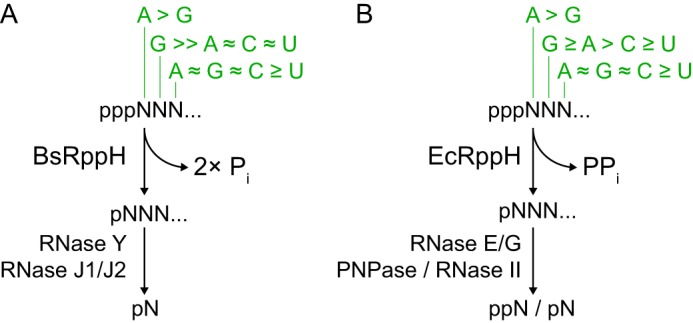
5′-End-dependent mRNA degradation pathways triggered by RppH. A and B, schematics depict sequence- and length-specific removal of pyrophosphate from triphosphorylated 5′ mRNA termini in B. subtilis (A) and E. coli (B). Nucleotide preferences at the first three RNA positions (13, 14, 32) are shown in green. PPi, pyrophosphate; Pi, orthophosphate.
Although vitally important for all organisms, mRNA degradation, especially RppH-mediated 5′-end-dependent mRNA decay in microbes, is among the least understood mechanisms of gene regulation. The biological significance of RppH prompted determination of the NMR structure of free EcRppH (16), x-ray structures of Bdellovibrio bacteriovorus RppH (BdRppH) in the free form and bound to GTP (17), and x-ray structures of BsRppH bound to GTP and RNA (15). However, these RppH structures do not reveal whether EcRppH retains the same specificity as BsRppH or how EcRppH binds mRNA because the putative RNA-binding regions of EcRppH and BsRppH are strikingly different. Moreover, the determined structures do not feature RNA·RppH complexes in catalytically active conformations and do not provide sufficient details to unambiguously explain why BsRppH cleaves off two orthophosphates sequentially, although EcRppH normally removes pyrophosphate in one step (although it too sometimes catalyzes consecutive cleavage reactions) (5, 6, 15).
To reveal the molecular basis of mRNA recognition and catalysis by EcRppH, we have identified minimal RNA substrates, determined x-ray structures of EcRppH in the apo- and RNA-bound forms, and conducted mutational studies. Our structural and biochemical data demonstrate novel structural principles of low specificity mRNA recognition that are characteristic of innumerable proteobacterial RppHs and distinct from those used by more specific RNA pyrophosphohydrolases like BsRppH. Our results also suggest a common catalytic mechanism for many hydrolases of the Nudix superfamily and therefore will have an impact on numerous biological systems.
EXPERIMENTAL PROCEDURES
Protein Production and Purification
The E. coli rppH gene and several variants truncated at the 3′-end were cloned into pSUMOH10 vector (a gift of Dr. H. Li) and expressed with an N-terminal His10-Sumo tag from a T7 RNA promoter in E. coli BL21(DE3)CodonPlusRIL cells using auto-induction (18). Cells from a 2-liter culture were collected by centrifugation and resuspended in 20 ml of lysis buffer containing 50 mm sodium phosphate, pH 7.5, 1 m NaCl, 20 mm imidazole-HCl, pH 8.0, 0.05% (v/v) β-mercaptoethanol, and EDTA-free Protease Inhibitor Mixture (Roche Applied Science). Cells were lysed by EmulsiFlex C-5 (Avestin), and debris was removed by centrifugation at 20,000 × g and at 4 °C for 1 h. Clarified lysate was applied onto a 5-ml HisTrap FF column (GE Healthcare) and washed with the lysis buffer supplemented by 80 mm imidazole-HCl, pH 8.0. Recombinant protein was eluted by the lysis buffer that contained 0.5 m imidazole-HCl, pH 8.0. Peak fractions were combined, and the tag was cleaved off by ULP1 protease at 4 °C for 14–16 h. Digested EcRppH samples were dialyzed against a buffer composed of 50 mm sodium acetate, pH 5.0, 0.3 m NaCl, and 0.05% (v/v) β-mercaptoethanol, applied onto 5-ml HiTrap SP column (GE Healthcare), washed with the same buffer, and eluted with 0.3–1.0 m NaCl gradient in a buffer containing 50 mm sodium acetate, pH 5.0, and 0.05% (v/v) β-mercaptoethanol. Peak fractions were combined and dialyzed against a buffer prepared with 20 mm sodium acetate, pH 5.0, 150 mm NaCl, and 0.05% (v/v) β-mercaptoethanol. At the final step, dialyzed protein samples were concentrated to ≈5 ml and purified on a 2.6 × 60-cm Superdex 75 column (GE Healthcare) equilibrated with the storage buffer that contained 20 mm sodium acetate, pH 5.0, 100 mm NaCl, and 1 mm DTT. Peak fractions were combined, concentrated, and stored at −80 °C.
To re-engineer crystal packing interactions, we predicted mutations on the surface of EcRppH using the surface entropy reduction algorithm (19, 20) and introduced double Ala mutations into the EcRppH(1–160)-coding construct by mega-primer PCR (21) and single mutations in the EcRppH-coding construct by inverse PCR (22).
Preparation of RNA Oligonucleotides
Short RNA oligonucleotides were prepared by in vitro transcription with T7 RNA polymerase and DNA templates that comprise a double-stranded T7 promoter and a single-stranded extension encoding for RNA (23).
DNA templates were prepared by annealing two oligonucleotides in a 100-μl volume at 10 μm concentration. Transcription reactions were performed in 1 ml of buffer containing 100 mm Tris-HCl, pH 8.0, 20 mm MgCl2, 2 mm spermidine, 40 mm DTT, 1 μm DNA template, and 0.1 mg/ml T7 RNA polymerase at 37 °C for 4 h and quenched by 0.1 ml of 0.5 m EDTA. The total concentration of NTPs was kept within 15–16 mm, and concentrations of each NTP were adjusted according to oligoribonucleotide sequence. For example, to prepare pppAGU, we used 5 mm each of ATP, GTP, and UTP, and we used 8 mm each of ATP and GTP to transcribe pppAG. RNAs were purified on a 5-ml HiTrap Q column (GE Healthcare), precipitated by ethanol, washed with 80% ethanol, dried, and dissolved in water.
Crystallization of EcRppH and EcRppH Complexes
Crystals of EcRppH(1–156) were prepared by mixing 2 μl of 0.5 mm protein and 2 μl of reservoir solution containing 0.1 m Tris-HCl, pH 8.0, 1.5 m potassium acetate, 5% (v/v) glycerol, and 20 mm CaCl2. Crystals of EcRppH(1–158)AA were grown from a solution typically prepared by mixing 1 μl of 1 mm protein solution and 2 μl of reservoir solution composed of 0.4 m (NH4)2SO4, 12% (v/v) PEG3350, and 12% (v/v) glycerol. All crystals were grown at 295 K for several days. To obtain crystals of EcRppH·RNA complexes, RNA was added at 1 mm to a drop of the reservoir solution prior to adding EcRppH(1–158)AA.
For structure phasing, a KI soak was performed by transferring the EcRppH(1–156) crystals into a 2 μl drop of the cryoprotecting solution that contained 100 mm Tris-HCl, pH 8.0, 20 mm CaCl2, 25% (v/v) glycerol, 1 m potassium acetate, and 0.5 m KI. After 30 min of incubation at room temperature, the crystal was mounted and frozen in a cryostream at 100 K. EcRppH(1–158)AA crystals were cryoprotected in a solution composed of 0.4 m (NH4)2SO4, 15% (v/v) PEG 3350, and 25% (v/v) glycerol and frozen prior to data collection. To introduce Mg2+ cations, a co-crystal of EcRppH(1–158)AA-ppcpAGU (letter “c” indicates the nonhydrolyzable methylene group) was transferred into a 2-μl drop of the stabilizing solution containing 50 mm MOPS, pH 7.0, 50 mm MgCl2, 50 mm (NH4)2SO4, 15% (v/v) PEG 3350, and 25% (v/v) pentaerythritol propoxylate 5/4 PO/OH. After 30 min of incubation at room temperature, the crystal was frozen without additional cryoprotection.
Cross-linking of EcRppH(1–158)AA·RNA Crystals
To remove sulfate ions from the EcRppH·RNA crystals, the crystals were stabilized by cross-linking with glutaraldehyde. First, to avoid inhibition of cross-linking, NH4+ cations were substituted by Na+ cations by transferring a crystal into a 1-μl droplet of the stabilizing solution containing 20 mm sodium acetate, pH 5.0, 200 mm Na2SO4, 15% (v/v) PEG 3350, 15% (v/v) glycerol, and supplemented by 0.5 mm ppcpAGU. After 30 min of incubation, the crystal was transferred to a fresh 1-μl hanging drop of the same solution and incubated for 1 h over a 2-μl droplet of 50% (w/w) glutaraldehyde placed on a sitting drop bridge in the same well (24). During all procedures, drops were kept in the closed well with 400 μl of stabilizing solution. After cross-linking, the crystal was transferred into a 2-μl drop containing 50 mm MOPS, pH 7.0, 25 mm MgCl2, 15% (v/v) PEG 3350, and 25% (v/v) pentaerythritol propoxylate 5/4 PO/OH for 30 min. Crystals were frozen in cryostream prior to data collection.
Data Collection and Structure Determination
Diffraction data were collected at 100 K on an in-house Rigaku x-ray source and beamline X25 of the National Synchrotron Light Source, Brookhaven National Laboratory. Data were processed using XDS suite (Table 1) (25). The crystal structure for EcRppH(1–156) was solved using single wavelength anomalous diffraction method implemented in PHENIX.AUTOSOL (26). The initial model was built automatically by PHENIX.AUTOBUILD (26) and then adjusted manually in COOT (27). Structure refinement was conducted by PHENIX.REFINE (26) and REFMAC 5 (28) within CCP4 (29). Structures of EcRppH(1–158)AA in the apo-state and with RNA were solved by molecular replacement using PHENIX.AUTOMR (26) with EcRppH(1–156) as a search model. RNA and ions were added at the late stages of refinement based on the Fo − Fc and 2Fo − Fc electron density maps. Ca2+ and Cl− ions were visualized by anomalous electron density maps.
TABLE 1.
Data collection and refinement statistics
| Dataset | EcRppH(1–156) KI soak | EcRppH(1–158)AA | EcRppH·2Mg·ppcpAGU complex | EcRppH·3Mg·ppcpAGU complex |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P 3221 | C 2 | C 2 | C 2 |
| Cell dimensions | ||||
| a, b, c (Å) | 59.70, 59.70, 81.78 | 78.39, 38.85, 56.81 | 78.50, 38.87, 57.20 | 79.62, 36.48, 58.07 |
| α, β, γ (°) | 90, 90, 120 | 90.00, 98.95, 90.00 | 90.00, 100.13, 90.00 | 90.00, 102.30, 90.00 |
| Wavelength (Å) | 1.5418 | 1.5418 | 1.1 | 1.5418 |
| Resolution (Å)a | 20-1.7 (1.80-1.70) | 20-2.0 (2.11-2.00) | 20-1.5 (1.59-1.50) | 20-1.6 (1.70-1.60) |
| Rmerge (%)a | 7.4 (66.1) | 9.0 (52.3) | 4.1 (59.6) | 5.0 (63.2) |
| 〈I〉/σ〈I〉a | 19.58 (3.54) | 10.7 (2.87) | 21.93 (2.65) | 22.6 (2.71) |
| Completeness (%)a | 98.4 (96.3) | 93.2 (84.7) | 95.5 (93.0) | 96.7 (90.6) |
| Unique reflectionsa | 35,292 (5,563) | 10,978 (1,578) | 26,199 (4,066) | 21,065 (3,134) |
| Redundancya | 8.8 (8.5) | 4.9 (4.8) | 6.6 (6.5) | 5.4 (5.0) |
| Phasing | ||||
| Resolution (Å) | 20-1.8 | 20-2.0 | 20-1.5 | 20-1.6 |
| Method | SAD | Molecular replacement | Molecular replacement | Molecular replacement |
| No. of sitesb | 27 | |||
| Figure of merit before/after density modificationb | 0.39/0.68 | |||
| Phaser LLG/TFZc | 105/2.9 | 7,004/4.4 | 1,949/31.3 | |
| Refinement | ||||
| Resolution (Å) | 20.0-1.7 | 20-2.0 | 20-1.5 | 20-1.6 |
| No. of reflections | ||||
| Working set | 35,287 | 10,975 | 26,189 | 20,011 |
| Test set | 1,758 | 549 | 1,310 | 1,054 |
| Rwork/Rfree (%)d | 16.1/19.9 | 17.0/21.6 | 18.9/21.6 | 17.2/19.9 |
| No. of atoms | ||||
| Protein | 1,204 | 1,270 | 1,326 | 1,340 |
| RNA | 54 | 44 | ||
| Anions | 27 | 30 | 10 | 6 |
| Cations | 1 | 2 | 3 | |
| Water | 113 | 91 | 109 | 119 |
| Average B-factors (Å2) | ||||
| Protein | 23.1 | 38.8 | 29.5 | 29.6 |
| RNA | 42.5 | 60.0 | ||
| Anions | 44.7 | 59.4 | 28.6 | 25.0 |
| Cations | 26.2 | 17.9 | 16.1 | |
| Water | 34.2 | 38.3 | 36.5 | 35.4 |
| Root mean square deviation from ideal geometry | ||||
| Bond length (Å)d | 0.004 | 0.007 | 0.005 | 0.007 |
| Bond angles (°)d | 0.972 | 1.098 | 0.998 | 1.016 |
| Ramachandran | ||||
| Favored (%)d | 98 | 97 | 99 | 99 |
| Allowed (%)d | 2 | 3 | 1 | 1 |
| Outliers (%)d | 0 | 0 | 0 | 0 |
a Values in parentheses are for highest resolution shell.
b Values were reported by PHENIX.AUTOSOL.
c Values were reported by PHENIX.PHASER-MR.
d Values were reported by PHENIX.REFINE.
In the EcRppH·3Mg·ppcpAGU complex structure, the tetrahedrally shaped density map was assigned to a noncleaved γ phosphate because of the following: (i) the ≈2.9 Å distance from the β phosphate is somewhat short for positioning of a cleaved-off phosphate; (ii) a water molecule was not sufficient to account for the size of the map; (iii) the α phosphate in this position would require at least the sugar atoms C5′ and O5′ to be shifted and generate a new density map, which was not observed at reasonable σ levels. The γ phosphate was refined at 50% occupancy to reflect the residual density map.
In Vitro Assay of EcRppH Activity
Pyrophosphohydrolytic assays were performed in 10–50-μl reactions containing 50 mm HEPES-Na, pH 7.5, 10 mm MgCl2, 0.1% (v/v) Triton X-100, 100 μm to 1 mm triphosphorylated mono- or oligoribonucleotides, and 100 nm to 10 μm RppH. In control reactions, dilution buffer or alkaline phosphatase at 0.001–0.1 units/μl was added to the reaction mixtures. Reactions were incubated at 37 °C for 10–60 min and quenched by a 10-fold dilution with 50 mm sodium acetate, pH 5.0, for chromatographic and pyrophosphate analysis, or a 2-fold dilution with 40 mm EDTA for thin layer chromatography.
Chromatographic separation of reaction products was performed on 5 × 50-mm Mono Q column (GE Healthcare) by 10 column volumes of a 0–0.5 m NaCl gradient in 10 mm Tris-HCl, pH 8. Elution profiles were recorded at 260 nm, and peaks corresponding to monophosphorylated products or triphosphorylated substrates were integrated using UNICORN software (GE Healthcare). Absolute amounts of the reaction products were calculated based on the values of integrated peaks for substrates and products.
Inorganic pyrophosphate assay was performed colorimetrically according to the published method (30, 31). Briefly, inorganic pyrophosphate from the quenched reactions was precipitated in the presence of 1 mm CaCl2 and 100 mm NaF. Precipitate collected by centrifugation at 20,000 × g for 10 min was washed with acetone, dried, and re-dissolved in solution containing 1.25 n H2SO4, 10 mm ammonium heptamolybdate, and 40 mm β-mercaptoethanol. Intensity of green color developed after 1 h of incubation at room temperature was measured at 700 nm, and the amount of the pyrophosphate liberated during the reaction was calculated from the calibration curve prepared with standard solutions of sodium pyrophosphate.
Thin layer chromatography was performed on PEI-cellulose plates and 0.3 m potassium phosphate buffer, pH 7.5, at room temperature. Chromatography samples were EDTA-quenched EcRppH reaction products that contained 5 nmol of RNA oligonucleotides in 10-μl volumes. RNAs were visualized by UV254 shadowing.
RESULTS
EcRppH Is Active on Short RNAs
Our x-ray crystallography experiments were driven by the unexpected finding, presented in the accompanying article (32), that EcRppH and BsRppH have similar length requirements but distinct sequence preferences for RNA 5′-ends. Like BsRppH, EcRppH favors guanine at the second position but, unlike BsRppH, is also active on RNAs that have adenine or a pyrimidine there (Fig. 1B). The different specificities of EcRppH and BsRppH have important functional consequences for gene regulation mechanisms in bacteria. For instance, BsRppH, which is highly specific, appears to be supplemented by another RNA pyrophosphohydrolase that may target a distinct subset of mRNAs (14, 15), whereas EcRppH, which is less selective, appears to be the only enzyme of its kind in E. coli. Because EcRppH orthologs are present in innumerable bacterial species and flowering plants, although BsRppH orthologs are much less common (32), we set out to investigate the selectivity of EcRppH-like RNA pyrophosphohydrolases by crystallizing EcRppH bound to target RNAs.
To define the minimal RNA substrate sufficient for target-specific EcRppH activity, we compared pyrophosphate removal from 5′-triphosphorylated mononucleotides and RNAs of various lengths. In these RNAs, the first nucleoside was adenosine, preferred by EcRppH in long mRNAs (32); the second nucleoside was varied, and the third nucleoside, less important for selectivity, was uridine for convenience of RNA preparation. The reaction of these substrates with purified EcRppH was monitored by analyzing the products by anion-exchange (Fig. 2, A and B) and thin layer chromatography (Fig. 2C) and by colorimetric measurements of the liberated pyrophosphate (Fig. 2D). With triphosphorylated mononucleotide substrates at physiological pH, EcRppH slowly cleaves off just the γ phosphate and removes both the γ and β phosphates only at elevated enzyme concentrations (Fig. 2B). Under the same conditions, EcRppH is 10–50-fold more active on di- and trinucleotide substrates (Fig. 2, B and D), from which it predominantly removes pyrophosphate in a single step (Fig. 2C). The highest activity was observed with the oligonucleotides pppAG and pppAGU, both of which bear guanosine at the second position (Fig. 2, B and D). Adenosine was slightly less preferred at that position, whereas uridine and cytidine decreased activity by a factor of 1.6–2.3 and 3.4–3.7, respectively. Because the selectivity of EcRppH with oligonucleotides resembled its sequence preferences with long RNA substrates (32), trinucleotides were used as mimics of natural mRNAs in crystallization trials with EcRppH.
FIGURE 2.

EcRppH activity on various substrates. A, representative chromatographic separation of the pyrophosphohydrolysis products of pppAGU RNA. Red, no EcRppH; blue, reaction products with 0.1 μm EcRppH; black, reference compounds. Reactions were carried out with 100 μm substrate for 10 min. B, EcRppH activity on the 5′-triphosphates of mononucleotides and oligonucleotides, as determined by chromatography (A) and graphed as the concentration of hydrolyzed substrates (mean ± S.D., n = 3). C, TLC of 5′-triphosphorylated and modified RNAs treated with 1 μm EcRppH (R lanes) or 0.1 unit/μl alkaline phosphatase (P lanes). pcppAGU and ppcpAGU contain a nonhydrolyzable methylene group (letter “c”) that replaces a bridging oxygen atom. RNAs at 1 mm concentration were incubated with or without EcRppH for 1 h and visualized by UV254 shadowing. D, EcRppH activity determined by colorimetric measurement of liberated inorganic pyrophosphate. Reactions were carried out as in A and B. E, Mg2+ dependence of EcRppH activity with pppAG. pppAG hydrolysis by EcRppH is expressed as a percent of maximal activity observed for the enzyme. Reactions were performed at 100 nm EcRppH and 100 μm pppAG at 37 °C for 10 min. AC50 is concentration of MgCl2, at which half-maximum of enzyme activity is achieved; n is Hill coefficient. Presented values are mean ± S.D., n = 3. F, inhibition of EcRppH activity with (NH4)2SO4. Activity measurements and determination of IC50, at which half-maximum of enzyme activity is achieved, were conducted at 10 mm MgCl2 as in E.
Crystal Structure of EcRppH in the Apo-form
Because initial efforts to co-crystallize EcRppH with various RNA ligands were unsuccessful, we focused on crystallizing EcRppH in the free form and soaking short RNAs into the crystal. Because full-length EcRppH did not crystallize, we progressively removed unstructured C-terminal amino acid residues (16) dispensable for enzymatic activity and identified two constructs, EcRppH(1–156) and EcRppH(1–160), that yielded crystals and crystalline needles. The structure of EcRppH(1–156) was determined at a 1.7-Å resolution (Table 1). Because a symmetry-related molecule blocked access of RNA in these crystals, we re-engineered crystal contacts by replacing residues that have flexible surface-exposed side chains with alanines (20). One truncated mutant, bearing two alanine residues at the C terminus and hereafter referred as EcRppH(1–158)AA, crystallized with the front surface opened for RNA binding (Fig. 3).
FIGURE 3.

Crystal structure of EcRppH in the apo-form. A, structure of EcRppH(1–158)AA. The Nudix box region is highlighted in orange. Sulfate ions are shown as sticks. The dashed green line depicts a disordered region. B, comparison of EcRppH (green) and BsRppH (magenta) structures aligned on the Nudix motif. C, binding of sulfate anions to the catalytic surface of EcRppH. Hydrogen bonds are shown as dashed lines. D, ClustalX sequence alignment of EcRppH and BsRppH. Highlighted amino acids are as follows: yellow, Nudix motif residues; red, residues involved in Mg2+ coordination; blue, residues interacting with RNA or sulfate ions in the RNA·EcRppH and RNA·BsRppH complexes. Asterisks mark identical residues in the protein sequences. Secondary structure elements from the crystal structures of EcRppH (current study) and BsRppH (Protein Data Bank code 4JZV) are shown along the sequences.
Unlike BdRppH (17), EcRppH crystallized with one molecule in the asymmetric unit. EcRppH comprises a Nudix hydrolase domain composed of a mixed β-sheet (β3, β1, β6, β4, and β5) attached to an antiparallel β-sheet (β2 and β7) (Fig. 3A). The β-sheets are sandwiched between α-helices on the left (α1) and right (α4 and α5). The x-ray structure agrees well with the EcRppH NMR structure (16) and is reminiscent of the BdRppH and BsRppH structures overall (Fig. 3B) (15, 17), although it is dissimilar in many important details, consistent with the low sequence identity between the proteins (Fig. 3D). The catalytic site is primarily composed of α1, the α1-β3 loop, and β3, the three structural elements that contain amino acid residues characteristic of the Nudix motif (Fig. 3A). Positively charged surfaces that might participate in RNA binding are located below and to the right of the catalytic site. Two sulfate anions bind in this area, possibly mimicking bound RNA phosphates (Fig. 3C).
EcRppH·2Mg·ppcpAGU Structure Reveals Specifically Bound RNA
To determine a biologically relevant structure of EcRppH bearing an RNA ligand, we preserved the integrity of the enzyme and crystallized catalytically active EcRppH(1–158)AA bound to the trinucleotide ppcpAGU. This RNA substrate contains a nonhydrolysable methylene group (designated “c”) substituted for an oxygen atom between the α and β phosphorus atoms of the 5′-triphosphate moiety (Fig. 2C). In an attempt to further reduce EcRppH activity and prevent minor cleavage between the β and γ phosphates (Fig. 2C) (5), the crystals were grown in the absence of Mg2+ cations, which are essential for catalysis. Enzymatic activity measurements revealed that EcRppH reaches almost full activity at ∼10 mm Mg2+ (Fig. 2E). To obtain the catalytically relevant structure, the concentration of (NH4)2SO4 was lowered to 50 mm to reduce inhibition of catalysis by the high concentration of this salt, and the crystals were stabilized by pentaerythritol propoxylate and soaked at neutral pH in a solution containing 50 mm Mg2+ so as to allow Mg2+ cations to interact with amino acids in the catalytic site. At this lower (NH4)2SO4 concentration, EcRppH retains ∼40% of its maximal activity determined in the absence of (NH4)2SO4 at 10 mm Mg2+ (Fig. 2F).
The resulting 1.5-Å crystal structure of the complex revealed the majority of the protein chain, two Mg2+ cations, and a large part of RNA, only missing the γ phosphate and the third RNA nucleoside (Fig. 4A and Table 1). The 5′-end of the RNA is positioned in the catalytic site, whereas the remainder of the RNA binds EcRppH in the region to the right of the catalytic site. The first nucleobase, Ade1, defined by a fragmented electron density map (Fig. 4B), is rotated outward toward solution, whereas the well defined nucleobase of Gua2 sits in a cleft formed mostly by the β2-β3 loop and α5.
FIGURE 4.

Structure of the EcRppH·2Mg·ppcpAGU complex. A, structure of the complex with the protein depicted by green cartoon, RNA, and a sulfate anion in beige sticks, and Mg2+ cations by magenta spheres. Nudix box is in orange color. B, composite omit 2Fo − Fc map (gray mesh) contoured at 1 σ level around ligands. Mg2+ cations and water molecules are shown as green and red spheres. C, detailed view of the catalytic site. Gray sticks depict amino acid conformations that correspond to the apo-state, and green sticks indicate Mg2+- and RNA-bound conformations. Arrows show movement of glutamates upon binding to Mg2+ cations. Dashed lines depict hydrogen and coordination bonds between the protein, Mg2+ cations (magenta spheres), water molecules (red spheres), and 5′-terminal phosphates (stick representation with atomic colors). D, specific recognition of the second RNA nucleotide base by the protein. Protein residues that participate in RNA binding are shown as green sticks. Blue and red dashed lines depict hydrogen bonds and cation-π interactions, respectively.
The structure revealed two Mg2+ cations bound to several glutamates and a glutamine in the catalytic site. To coordinate the Mg2+ cations, the side chains of these glutamates change their conformation, and we tracked these changes in a single crystal. The 5′-end of the RNA is bound by direct coordination of its β phosphate to Mg1 and Mg2 (Fig. 4C). In addition, both the α and β phosphates form hydrogen bonds with Arg-8 and Gly-39. The γ phosphate was not modeled in the structure because a tetrahedrally shaped electron density adjacent to the β phosphate (Fig. 4B) was ∼0.6 Å further away than expected for a covalently linked γ phosphate. Although this density might belong to the cleaved off γ phosphate, the density most likely corresponds to a SO42− anion capable of binding in the same region in the apo-structure and present in the crystals at the much higher concentration. Despite the loss of the γ phosphate, the diphosphorylated RNA remains a substrate for EcRppH in solution (Fig. 2C). Because none of the ordered water molecules were aligned well for cleavage between α and β phosphates, we hypothesize that this structure reflects one of the states on the reaction pathway either before or after the catalytic step.
Distinct Multiple Interactions Contribute to Binding of the Second Nucleobase
The EcRppH·2Mg·ppcpAGU structure showed that the base of Ade1 apparently rotates freely along the glycosidic bond and does not form specific hydrogen bonds with EcRppH. Contacts between the protein and Ura3 are limited to a nonspecific hydrogen bond between a phosphate and Arg-27 (Fig. 4D). By contrast, Gua2 forms multiple interactions with the protein. First, the sugar-phosphate moiety is hydrogen-bonded to side chains of Gln-37 and Tyr-77. Second, the guanine base is sandwiched between a side chain of Arg-27 and the hydrophobic platform formed by Val-137 and Phe-139. In this position, the base forms cation-π interactions with the guanidinium group of Arg-27. Third, atoms on the Hoogsteen edge are hydrogen-bonded with the side chain of Lys-140. Fourth, on the Watson-Crick edge, N2 makes a direct hydrogen bond to Ser-32 in one of its alternative conformations, whereas N1 forms water-mediated bonds with Ser-32, Trp-3, and Val-137. These multiple contacts of EcRppH with Gua2 strongly suggest that Gua2 is critical for RNA binding and explain the observed preference for guanine in the second position.
Although direct hydrogen bonding to the second nucleobase favors guanine, it neither precludes binding of adenine and pyrimidines in a semi-open RNA-binding site nor explains the much stronger preference for adenine over pyrimidines. The structure suggests that the other factors contributing to higher affinity for purines include hydrophobic and cation-π interactions. Thus, the semi-specific substrate preference of EcRppH is not likely to be determined solely by hydrogen bonding and probably involves an unusual combination of different types of interactions (see “Discussion”).
EcRppH·3Mg·ppcpAGU Structure Reveals Changes in Protein and RNA Conformation
We reasoned that the sulfate anion bound in the catalytic site prevented the EcRppH·2Mg·ppcpAGU complex from adopting a catalytically active conformation. Because sulfate anions participate in crystal packing interactions and cannot be replaced during crystallization, we stabilized the crystals by glutaraldehyde cross-linking and removed loosely bound sulfate anions by soaking in solutions that were sulfate-free and contained Mg2+. This procedure resulted in a 1.6-Å crystal structure of the EcRppH·3Mg·ppcpAGU complex, which contained EcRppH in the same global conformation as before, three Mg2+ cations, and RNA positioned in the RNA-binding site slightly differently than in the EcRppH·2Mg·ppcpAGU structure (Fig. 5, A–C).
FIGURE 5.

Structure of the EcRppH·3Mg·ppcpAGU complex. A, comparison of the RNA conformations in the EcRppH·3Mg·ppcpAGU (beige) and EcRppH·2Mg·ppcpAGU (gray) complexes. B, superposition of the EcRppH·2Mg·ppcpAGU (gray) and EcRppH·3Mg·ppcpAGU (beige) structures on the Nudix residues. Arrows show structural changes in the EcRppH·3Mg·ppcpAGU structure. C, composite omit 2Fo − Fc map (gray mesh) contoured at 1 σ level around ligands. Mg2+ cations and water molecules are shown as green and red spheres. D, intermolecular interactions in the catalytic site. Hydrogen and coordination bonds are depicted by blue dashed lines. W1, putative nucleophilic water molecule Wat-1. E, recognition of the second RNA base by the protein. Blue and red dashed lines depict hydrogen bonds and cation-π interactions, respectively.
In addition to Mg1 and Mg2, another Mg2+ cation, Mg3, binds the catalytic site in place of the sulfate anion and is coordinated by the β phosphate and Glu-53 (Fig. 5D). Glu-56 returns to the apo conformation, although the α phosphate is located far from Arg-8 and in the vicinity of Gln-37 and Mg1. The β phosphate remains in the same position but directly coordinates all three magnesium ions. We observed residual density for the γ phosphate linked to β phosphate (Fig. 5C), likely because our crystal treatment decreased EcRppH activity and prevented complete hydrolysis of the γ phosphate. Importantly, Mg3 binding leads to the formation of a binuclear Mg2·Wat-1·Mg3 complex that stabilizes water molecule Wat-1 in position for an in-line nucleophilic attack on the β phosphorus atom (Fig. 5D). Therefore, the EcRppH·3Mg·ppcpAGU structure appears to be closest to the “catalytic” conformation of the complex.
Concurrently with the conformational changes at the 5′-end of the RNA, Ade1 and Gua2 shift their positions in the EcRppH·3Mg·ppcpAGU structure (Fig. 5, A and B). In comparison with the EcRppH·2Mg·ppcpAGU structure, the 5′-phosphate of Gua2 moves toward Lys-140, whereas the guanine base slides slightly out of the cleft and loses direct hydrogen bonds with Lys-140 (Fig. 5, B and E). In this new position, the Gua2 nucleobase forms only water-mediated hydrogen bonds and weakened cation-π interactions with EcRppH and is therefore more mobile and less defined in the electron density map.
EcRppH Mechanism Involves Conformational Rearrangements
On the basis of these structures, we propose the following catalytic cycle for EcRppH. Initially, EcRppH binds Mg2+ cations Mg1 and Mg2 either directly or likely in complex with the triphosphate moiety of an RNA substrate. Although a Ca2+ cation binds between the Mg1 and Mg2 sites in the EcRppH(1–156) structure, so far our studies failed to identify Mg2+ cations bound to the protein without RNA. Protein-bound Mg1 and Mg2 facilitate binding of the RNA 5′ terminus through electrostatic interactions and coordination of the β phosphate. The Gua2 nucleobase binds deeply and specifically in the protein cleft, resulting in the conformation found in the EcRppH·2Mg·ppcpAGU complex (Fig. 5B). Next, binding of Mg3 and positioning of the α phosphate closely to Gln-37 stabilizes water molecule Wat-1 between Mg2 and Mg3 and close to Glu-56 and the β phosphorus atom (Fig. 6A). In this location, Wat-1 forms a nearly ideal angle (170.6°) for in-line attack on the β phosphorus atom. Interestingly, placement of the α phosphate in the catalytically relevant conformation near Gln-37 causes the Gua2 nucleobase to slide out of the cleft, weakening interactions with the protein, as observed in the EcRppH·3Mg·ppcpAGU complex.
FIGURE 6.
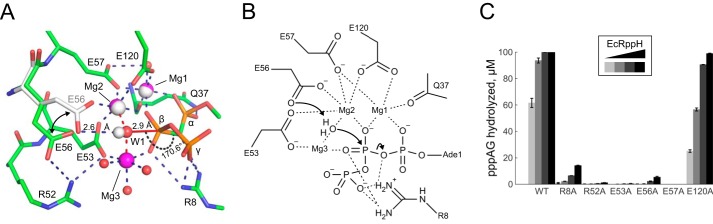
Putative mechanism of EcRppH. A, interactions in the catalytic site. Glu-56, Mg1, Mg2, and water molecule 1 (W1) are depicted as gray sticks and spheres in the EcRppH·2Mg·ppcpAGU structure and as colored sticks and spheres in the EcRppH·3Mg·ppcpAGU structure. Note that Wat-1 (large red sphere) directly coordinates Mg2 and Mg3 (red dashed lines) and is ideally positioned for in-line attack on the β phosphorus. Wat-1 also forms a hydrogen bond with Glu-56 in the EcRppH·2Mg·ppcpAGU structure. Hydrogen bonds are shown as blue dashed lines. B, putative mechanism of RNA pyrophosphohydrolysis by EcRppH. Arrows show electron transfer. Dashed lines depict coordination and hydrogen bonds. C, activity of EcRppH mutants with substitutions in the catalytic center. Reactions of 100 μm pppAG with 0.1 μm (light gray), 0.3 μm (gray), 1 μm (dark gray), or 3 μm (black) enzyme were incubated for 10 min prior to chromatographic analysis. Values are mean ± S.D. with n = 3.
As suggested for other Nudix hydrolases (33), Mg2+ cations can function as Lewis acids and assist in deprotonating Wat-1 by a catalytic residue of RppH. Our structural studies show that Glu-56 is the most plausible candidate for accepting a proton from Wat-1 (Fig. 6, A and B). This residue apparently switches between two conformations during catalysis and may serve as a ligand sensor and reaction trigger. Other adjacent residues and Mg2+ cations hold the substrate in place and stabilize the transition state, although Arg-8 likely assists the departure of the pyrophosphate leaving group, presumably in complex with Mg3. The monophosphorylated RNA product loses β phosphate-mediated interactions with EcRppH and should easily dissociate from the protein if tight interactions with the Gua2 nucleobase have been weakened by the movement of this base out of the cleft.
To evaluate the contribution of Nudix motif residues to catalysis, we replaced the Nudix glutamates with alanines and observed significantly decreased (E53A and E56A) or abolished (E57A) EcRppH activity (Fig. 6D). However, substitution of alanine for Glu-120, which functions as a lid covering Mg1 and Mg2, reduces activity by only ∼2.5-fold. Our data also show that Arg-52 of the Nudix motif holds Glu-53 and Glu-56 in the catalytic site and is absolutely required for catalysis. Arg-8, which coordinates the β phosphate and likely stabilizes the leaving product, is also essential for EcRppH activity. These mutagenesis data demonstrate the important contribution of Nudix amino acids, including a candidate catalytic residue Glu-56, to catalysis and the less critical role of non-Nudix Glu-120 in EcRppH activity.
DISCUSSION
To understand the molecular basis of the bacterial reaction functionally analogous to decapping of eukaryotic mRNAs, we determined the crystal structure of EcRppH in the apo-form and bound to RNA and Mg2+ cations. Although crystallization of low specificity RNA·protein complexes often yields poor electron density maps because of the mobility of the loosely bound RNA and the dissociation of the weak complexes under harsh crystallization conditions, we developed crystal treatment procedures that allowed us to visualize the first two nucleotides of EcRppH-bound RNA almost in their entirety. The EcRppH·RNA structure is more complete than the previously reported BsRppH·RNA structure, which mapped only the 5′-terminal triphosphate moiety, the second nucleotide, and two Mg2+ cations (15).
The EcRppH·RNA structures reveal that EcRppH binds in a bipartite manner to the 5′-phosphates and to a nucleobase at the second position, as well as to adjoined sugar-phosphate moieties (Figs. 7A and 8A). As in the BsRppH·RNA complex (Fig. 8C) (15), the first and third nucleobases are not specifically recognized but must be unpaired to place the second nucleobase into the RNA-binding site (15, 32). Recognition of the second nucleotide by EcRppH and BsRppH allows discrimination between mRNAs and mononucleotides and prevents hydrolysis of cellular NTPs by RppH. Too short to simultaneously reach both the triphosphate and nucleobase two binding sites, NTPs are expected to be poor substrates, as confirmed by the very slow cleavage of the γ phosphate of ATP. This conclusion raises questions about the specificity of Bdellovibrio RppH in vivo because GTP binds to BdRppH at a position equivalent to where the first nucleotide of RNA binds to EcRppH and BsRppH (Fig. 8, A–C) (17). Although the triphosphate of GTP reaches the catalytic center of BdRppH, it was not hydrolyzed in the crystals, suggesting that the published structure of the BdRppH·3Mg·GTP complex may not be representative of how the enzyme functions with an RNA substrate.
FIGURE 7.

Recognition of the second RNA nucleotide by bacterial RppHs. Gray shading depicts the surface of the RNA-binding site visible through a cross-section made above a base plane. A, edge-specific interactions with guanine in the semi-open cleft of EcRppH. B, specific recognition of guanine in the cavity of BsRppH. The preferences of each enzyme for the second RNA nucleotide are shown below the structures.
FIGURE 8.

Comparison of catalytic sites in ternary complexes of EcRppH, BdRppH, and BsRppH with Mg2+ ions and triphosphorylated substrates. RNAs are in stick representation. A–C, surface views of the ligand-binding sites in EcRppH (A), Bd(RppH) (B), and BsRppH (C) colored according to surface potential. Red and blue colors correspond to negatively and positively charged areas, respectively. Mg2+ cations are shown as magenta spheres. D–F, zoomed-in views of catalytic sites in EcRppH (D), BdRppH (E), and BsRppH (F) complex structures. Water molecules (red color) and Mg2+ cations (in colors of corresponding proteins) are shown by spheres. Hydrogen and coordination bonds are depicted by dashed lines.
Our structural studies uncovered markedly dissimilar modes of RNA binding by EcRppH and BsRppH despite their structural and functional similarities. Although both prefer guanine at the second position of their RNA substrates, the enzymes bind the second nucleobase differently, in either a deep cavity (BsRppH) or a cleft (EcRppH), each lined by a distinct set of nonequivalent amino acids (Fig. 7). The most notable sequence-specific contact in the EcRppH·RNA complex is two hydrogen bonds from Lys-140 to the Hoogsteen edge of guanine. These interactions would be expected to play a large part in determining the specificity of mRNA recognition by EcRppH because only guanine in an anti conformation can accept two hydrogen bonds from the lysine side chain, whereas adenine and uracil can accept only one hydrogen bond and cytosine cannot accept a hydrogen bond. Our structural prediction is corroborated by the reduced selectivity of the K140M mutant reported in the accompanying article (32). An additional hydrogen bond between Ser-32 and N2 of guanine might not contribute much to the specificity of EcRppH because only one of two alternative conformations of this residue allows it to contact the RNA. Nevertheless, mutations of Ser-32 can enhance selectivity, suggesting that this residue contributes to the promiscuity of EcRppH (32).
Although the structures with adenosine and pyrimidines in the second position have not been determined yet, our structures with Gua in this position suggest that, in contrast to many other RNA·protein complexes, direct hydrogen bonding alone cannot explain the preference of EcRppH for guanosine. Most likely, hydrophobic and cation-π interactions also contribute to the specificity of RNA binding. Compared with pyrimidines, purines have a larger surface area that promotes stronger interactions with the pedestal formed by the hydrophobic side chains of Val-137 and Phe-139. Purines also form stronger cation-π interactions with the guanidinium group of Arg-27 according to the calculated strength of cation-π interactions in DNA·protein complexes (G > A> T > C) (34). Thus, loss of cation-π interactions explains the reduced ability of the R27A mutant to discriminate between purines and pyrimidines (32). Although cation-π interactions with ligands are not common among Nudix hydrolases, they may be involved in adenine binding by the plant Ap4A hydrolase (35).
The structures illustrate that multiple features of EcRppH that favor binding of purines are compatible with weaker interactions with pyrimidines. Therefore, an unusual combination of low specificity interactions and an open RNA-binding site defines the relaxed specificity of EcRppH. In contrast to EcRppH, BsRppH binds guanine in a tight pocket where the base is surrounded by amino acid side chains that recognize the base through a set of direct hydrogen bonds incompatible with other nucleobases (Fig. 7). Structural comparison reveals that the only sequence-specific contact with nucleobase 2 common to both RNA·protein complexes has remarkably resulted from convergent evolution that positioned nonequivalent lysines to recognize the Hoogsteen edge of guanine.
Our structures of the EcRppH·RNA complexes present two important pieces of information that encourage rethinking the catalytic mechanism of Nudix proteins. The EcRppH·2Mg·ppcpAGU structure demonstrates that Glu-56 makes a hydrogen bond with water molecule Wat-1. The EcRppH·3Mg·ppcpAGU structure reveals that Wat-1 is flanked by two Mg2+ cations and is ideally positioned for the catalytic reaction. These structures hint at the direct involvement of Glu-56 in catalysis, namely in activating Wat-1 for in-line nucleophilic attack on the β phosphorus atom. This hypothesis is supported by the severe effect of the E56A mutation on activity. In addition, in several structures of Nudix hydrolases, the counterpart of Glu-56 is positioned in proximity to a water molecule analogous to Wat-1 (36, 37). Glu-56 is one of the most conserved residues of the Nudix motif and is thought to be essential for coordinating metal cations (33). However, in the EcRppH·3Mg·ppcpAGU and several other structures (36–38), this residue does not coordinate divalent cations. Thus Glu-56 may be more important for activating a water molecule for catalysis than for binding Mg2+ cations involved in catalysis.
Previous studies proposed that another glutamate of the Nudix motif (Glu-53 in EcRppH) sometimes acts as a catalytic residue in Nudix hydrolases (33). However, this residue coordinates two Mg2+ cations in the EcRppH·RNA complex, BdRppH (17), and other Nudix enzyme structures (36, 37) and therefore may have difficulty deprotonating the nucleophilic water molecule. To alleviate this contradiction, it was suggested that Glu-53 could function as a catalytic residue in Nudix hydrolases if coordinated to only a single Mg2+ cation (33). Our structural data and the residual activity of the Glu-53 mutants of EcRppH and BdRppH (17) do not support the idea that Glu-53 is the sole catalytic residue of these RNA pyrophosphohydrolases. Nevertheless, our data do not exclude the possibility that Glu-53 is responsible for the residual activity observed for the E56A mutant at high EcRppH concentrations.
The E57A mutation abolished EcRppH activity entirely, in agreement with the severe effects of Glu-57 mutations on the activity of other Nudix hydrolases, such as MutT (33). Our structures do not reveal any water molecule that could be activated for nucleophilic attack by this residue, but its side chain does undergo a conformational change upon Mg2+ binding. Because Glu-57 coordinates two key Mg2+ cations, this amino acid residue is essential for properly positioning the enzyme-bound metal cations.
The crystal structures of the EcRppH·RNA complexes revealed a conformational change in the side chain of Glu-120 that allows it to coordinate Mg1 and Mg2. Despite the seemingly strategic position of this side chain, which “covers” Mg1 and Mg2 and stabilizes their enzyme-bound positions, Glu-120 has a relatively low impact on catalytic activity, emphasizing the fact that it assists catalysis but does not play an essential role in the reaction mechanism. This glutamate residue not only is absent from many Nudix hydrolases but also is not conserved among RppHs. For example, it is replaced by His in BdRppH.
Mutational analysis showed that replacement of positively charged Arg-8 or Arg-52 significantly reduces EcRppH activity. This effect is easily explained for Arg-52, a conserved residue that orients the side chain of Glu-53 to facilitate its function. The strong reduction in activity observed for the R8A mutant was less expected because Arg-8 is not conserved among RppHs. This residue contacts the γ and β phosphates and likely stabilizes the leaving group. Although Arg-8 is not conserved, BdRppH and BsRppH contain lysines that are situated in proximity to the 5′-terminal phosphates and that may have a function similar to that of Arg-8 in EcRppH (Fig. 8, D–F).
The catalytic mechanism proposed for EcRppH may also pertain to BdRppH (17) and BsRppH (15), which have similarly organized catalytic centers despite the presence of different numbers of bound Mg2+ cations (Fig. 8, D–F). In all of these complexes, Glu-56 adopts the same conformation and therefore has the potential to deprotonate the catalytic water molecule Wat-1. The EcRppH and BdRppH structures show that Wat-1 is aligned for nucleophilic attack on the β phosphorus atom and hydrolysis between the α and β phosphates. Although the BsRppH structure does not reveal Wat-1, such a water molecule would attack the γ phosphorus atom and hydrolyze the bond between the γ and β phosphates. Thus, the preferential initial cleavage between the γ and β phosphates reported for BsRppH can be explained by the differential binding of the triphosphate moiety in the catalytic site so that the γ phosphate in the BsRppH·RNA complex is positioned in the location of the β phosphate in the EcRppH·RNA complex. As suggested in the earlier model of the BsRppH catalysis (15), the flexibility of the first nucleotide may facilitate movement of the β phosphate closer to the metal cations for the second round of catalysis after the γ phosphate has left.
Species ranging from bacteria to mammals can contain over 20 Nudix enzymes involved in distinct cellular activities. Therefore, our mechanistic study to gain insights on the catalytic mechanism of EcRppH will have an impact on many prokaryotic and eukaryotic systems. At the moment, there is no catalytic mechanism accepted as universal for Nudix hydrolases, and several amino acid residues have been proposed to play a catalytic role in different enzymes. However, the Nudix motif is the only set of residues common to all Nudix hydrolases, and because an important catalytic residue is expected to be preserved in evolution, it is likely that such a residue would reside in this motif in the majority of Nudix enzymes. The difficulty in understanding Nudix catalysis likely stems from the abundance of noncatalytic enzyme structures that have been reported and the strong effects on catalysis of various residues that form the catalytic site. The structure of the E. coli ADP-ribose pyrophosphatase (39) indirectly supports our hypothesis that Glu-56 directly participates in catalysis by the majority of Nudix hydrolases. This enzyme has an extra glutamate that can activate the nucleophilic water molecule. However, this glutamate is not conserved, and many Nudix proteins do not have a specialized non-Nudix amino acid residue that could play a similar catalytic role.
An intriguing observation in our study is a slightly different position of the Gua2 nucleobase in the binding cleft in two EcRpH·RNA complexes. Surprisingly, specific hydrogen bonding and deep positioning of this nucleobase in the cleft were observed in the catalytically inactive complex although shallow positioning and loss of specific hydrogen bonds were seen in the catalytically relevant complex. We also noticed that placement of the α phosphate in the catalytically relevant conformation appears to be structurally incompatible with deep binding of the Gua2 nucleobase in the cleft. To explain two distinct means of the Gua2 nucleobase binding, we hypothesize that EcRppH initially forms base-specific interactions with the nucleotide at the second position of RNA, although the triphosphate moiety and Mg2+ cations are not fully bound to the enzyme. Weak initial binding of the triphosphate moiety may constitute a specific adaptation of RppH to reject nucleotides and other phosphate-containing noncognate substrates so that productive interactions are formed only with RNA. Following initial binding, movement of the triphosphate to the catalytic conformation would then result in tighter binding, hydrolysis, and sliding of the second nucleobase partway out of the cleft, thereby facilitating release of the monophosphorylated RNA product from the protein.
The crystal structures of the EcRppH·RNA complexes reveal how a Nudix hydrolase that does not possess an established RNA-binding fold has nevertheless evolved for RNA binding and catalysis. The bipartite mRNA recognition mechanism involving the 5′-terminal phosphates and the second RNA nucleotide distinguishes EcRppH from Argonaute (40), RIG-I (41, 42), and other proteins (43, 44) that recognize RNA ends. The unique combination of molecular features revealed by the EcRppH·RNA complexes expands the known repertoire of principles that govern RNA binding and demonstrates that functionally and structurally analogous enzymes can employ very different structural principles for the recognition of similar substrates.
Acknowledgments
We thank the Belasco laboratory for fruitful discussions and sharing unpublished results. We thank the staff of beamlines X-25 (National Synchrotron Light Source, Brookhaven National Laboratory).
This work was supported by New York University start-up funds (to A. S.).
The atomic coordinates and structure factors (codes 4S2V, 4S2W, 4S2X, and 4S2Y) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- EcRppH
- E. coli RppH
- BsRppH
- B. subtilis RppH
- BdRppH
- B. bacteriovorus RppH
- Ap4A
- P1,P4-di(adenosine-5′) tetraphosphate.
REFERENCES
- 1. Belasco J. G. (2010) All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat. Rev. Mol. Cell Biol. 11, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mathy N., Bénard L., Pellegrini O., Daou R., Wen T., Condon C. (2007) 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 129, 681–692 [DOI] [PubMed] [Google Scholar]
- 3. Even S., Pellegrini O., Zig L., Labas V., Vinh J., Bréchemmier-Baey D., Putzer H. (2005) Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 33, 2141–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Celesnik H., Deana A., Belasco J. G. (2007) Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell 27, 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deana A., Celesnik H., Belasco J. G. (2008) The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451, 355–358 [DOI] [PubMed] [Google Scholar]
- 6. Richards J., Liu Q., Pellegrini O., Celesnik H., Yao S., Bechhofer D. H., Condon C., Belasco J. G. (2011) An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol. Cell 43, 940–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McLennan A. G. (2006) The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 63, 123–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McLennan A. G. (2013) Substrate ambiguity among the nudix hydrolases: biologically significant, evolutionary remnant, or both? Cell. Mol. Life Sci. 70, 373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Badger J. L., Wass C. A., Kim K. S. (2000) Identification of Escherichia coli K1 genes contributing to human brain microvascular endothelial cell invasion by differential fluorescence induction. Mol. Microbiol. 36, 174–182 [DOI] [PubMed] [Google Scholar]
- 10. Bessman M. J., Walsh J. D., Dunn C. A., Swaminathan J., Weldon J. E., Shen J. (2001) The gene ygdP, associated with the invasiveness of Escherichia coli K1, designates a Nudix hydrolase, Orf176, active on adenosine (5′)-pentaphospho-(5′)-adenosine (Ap5A). J. Biol. Chem. 276, 37834–37838 [DOI] [PubMed] [Google Scholar]
- 11. Lundin A., Nilsson C., Gerhard M., Andersson D. I., Krabbe M., Engstrand L. (2003) The NudA protein in the gastric pathogen Helicobacter pylori is an ubiquitous and constitutively expressed dinucleoside polyphosphate hydrolase. J. Biol. Chem. 278, 12574–12578 [DOI] [PubMed] [Google Scholar]
- 12. Al Mamun A. A., Lombardo M. J., Shee C., Lisewski A. M., Gonzalez C., Lin D., Nehring R. B., Saint-Ruf C., Gibson J. L., Frisch R. L., Lichtarge O., Hastings P. J., Rosenberg S. M. (2012) Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338, 1344–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luciano D. J., Hui M. P., Deana A., Foley P. L., Belasco K. J., Belasco J. G. (2012) Differential control of the rate of 5′-end-dependent mRNA degradation in Escherichia coli. J. Bacteriol. 194, 6233–6239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsieh P. K., Richards J., Liu Q., Belasco J. G. (2013) Specificity of RppH-dependent RNA degradation in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 110, 8864–8869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Piton J., Larue V., Thillier Y., Dorléans A., Pellegrini O., Li de la Sierra-Gallay I., Vasseur J. J., Debart F., Tisné C., Condon C. (2013) Bacillus subtilis RNA deprotection enzyme RppH recognizes guanosine in the second position of its substrates. Proc. Natl. Acad. Sci. U.S.A. 110, 8858–8863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bi Y., Li H., Fan S., Xia B., Jin C. (2009) 1H, 13C and 15N resonance assignments of RNA pyrophosphohydrolase RppH from Escherichia coli. Biomol NMR Assign. 3, 149–151 [DOI] [PubMed] [Google Scholar]
- 17. Messing S. A., Gabelli S. B., Liu Q., Celesnik H., Belasco J. G., Piñeiro S. A., Amzel L. M. (2009) Structure and biological function of the RNA pyrophosphohydrolase BdRppH from Bdellovibrio bacteriovorus. Structure 17, 472–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 19. Derewenda Z. S. (2004) Rational protein crystallization by mutational surface engineering. Structure 12, 529–535 [DOI] [PubMed] [Google Scholar]
- 20. Derewenda Z. S., Vekilov P. G. (2006) Entropy and surface engineering in protein crystallization. Acta Crystallogr. D Biol. Crystallogr. 62, 116–124 [DOI] [PubMed] [Google Scholar]
- 21. Miyazaki K., Takenouchi M. (2002) Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. BioTechniques 33, 1033–1034, 1036–1038 [DOI] [PubMed] [Google Scholar]
- 22. Imai Y., Matsushima Y., Sugimura T., Terada M. (1991) A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res. 19, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milligan J. F., Groebe D. R., Witherell G. W., Uhlenbeck O. C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 15, 8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lusty C. J. (1999) A gentle vapor-diffusion technique for cross-linking of protein crystals for cryocrystallography. J. Appl. Crystallogr. 32, 106–112 [Google Scholar]
- 25. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heinonen J. K., Honkasalo S. H., Kukko E. I. (1981) A method for the concentration and for the colorimetric determination of nanomoles of inorganic pyrophosphate. Anal. Biochem. 117, 293–300 [DOI] [PubMed] [Google Scholar]
- 31. Putnins R. F., Yamada E. W. (1975) Colorimetric determination of inorganic pyrophosphate by a manual or automated method. Anal. Biochem. 68, 185–195 [DOI] [PubMed] [Google Scholar]
- 32. Foley P. L., Hsieh P. K., Luciano D., Belasco J. G. (February 5, 2015) Specificity and evolutionary conservation of Escherichia coli RppH. J. Biol. Chem. 290, 10.1074/jbc.M114.634659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mildvan A. S., Xia Z., Azurmendi H. F., Saraswat V., Legler P. M., Massiah M. A., Gabelli S. B., Bianchet M. A., Kang L. W., Amzel L. M. (2005) Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 433, 129–143 [DOI] [PubMed] [Google Scholar]
- 34. Wintjens R., Liévin J., Rooman M., Buisine E. (2000) Contribution of cation-π interactions to the stability of protein-DNA complexes. J. Mol. Biol. 302, 395–410 [DOI] [PubMed] [Google Scholar]
- 35. Fletcher J. I., Swarbrick J. D., Maksel D., Gayler K. R., Gooley P. R. (2002) The structure of Ap4A hydrolase complexed with ATP-MgFx reveals the basis of substrate binding. Structure 10, 205–213 [DOI] [PubMed] [Google Scholar]
- 36. Bailey S., Sedelnikova S. E., Blackburn G. M., Abdelghany H. M., Baker P. J., McLennan A. G., Rafferty J. B. (2002) The crystal structure of diadenosine tetraphosphate hydrolase from Caenorhabditis elegans in free and binary complex forms. Structure 10, 589–600 [DOI] [PubMed] [Google Scholar]
- 37. Kang L. W., Gabelli S. B., Cunningham J. E., O'Handley S. F., Amzel L. M. (2003) Structure and mechanism of MT-ADPRase, a nudix hydrolase from Mycobacterium tuberculosis. Structure 11, 1015–1023 [DOI] [PubMed] [Google Scholar]
- 38. Scarsdale J. N., Peculis B. A., Wright H. T. (2006) Crystal structures of U8 snoRNA decapping nudix hydrolase, X29, and its metal and cap complexes. Structure 14, 331–343 [DOI] [PubMed] [Google Scholar]
- 39. Gabelli S. B., Bianchet M. A., Ohnishi Y., Ichikawa Y., Bessman M. J., Amzel L. M. (2002) Mechanism of the Escherichia coli ADP-ribose pyrophosphatase, a Nudix hydrolase. Biochemistry 41, 9279–9285 [DOI] [PubMed] [Google Scholar]
- 40. Ma J. B., Yuan Y. R., Meister G., Pei Y., Tuschl T., Patel D. J. (2005) Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature 434, 666–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y., Ludwig J., Schuberth C., Goldeck M., Schlee M., Li H., Juranek S., Sheng G., Micura R., Tuschl T., Hartmann G., Patel D. J. (2010) Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 17, 781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu C., Xu H., Ranjith-Kumar C. T., Brooks M. T., Hou T. Y., Hu F., Herr A. B., Strong R. K., Kao C. C., Li P. (2010) The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18, 1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leung D. W., Amarasinghe G. K. (2012) Structural insights into RNA recognition and activation of RIG-I-like receptors. Curr. Opin. Struct. Biol. 22, 297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abbas Y. M., Pichlmair A., Górna M. W., Superti-Furga G., Nagar B. (2013) Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494, 60–64 [DOI] [PMC free article] [PubMed] [Google Scholar]