

Background: The polysaccharide capsule is a major virulence factor of Streptococcus agalactiae.
Results: Mutations of the genes cpsABCD result in aberrant capsule length and localization.
Conclusion: The CpsABCD proteins form a system that modulates termination of capsule elongation.
Significance: This work proposes a model for the unified action of CpsABCD.
Keywords: Bacterial Protein Kinase, Carbohydrate Biosynthesis, Glycobiology, Gram-positive Bacteria, Mutagenesis, Streptococcus agalactiae, Capsular Polysaccharide
Abstract
The production of capsular polysaccharides (CPS) or secreted exopolysaccharides is ubiquitous in bacteria, and the Wzy pathway constitutes a prototypical mechanism to produce these structures. Despite the differences in polysaccharide composition among species, a group of proteins involved in this pathway is well conserved. Streptococcus agalactiae (group B Streptococcus; GBS) produces a CPS that represents the main virulence factor of the bacterium and is a prime target in current vaccine development. We used this human pathogen to investigate the roles and potential interdependencies of the conserved proteins CpsABCD encoded in the cps operon, by developing knock-out and functional mutant strains. The mutant strains were examined for CPS quantity, size, and attachment to the cell surface as well as CpsD phosphorylation. We observed that CpsB, -C, and -D compose a phosphoregulatory system where the CpsD autokinase phosphorylates its C-terminal tyrosines in a CpsC-dependent manner. These Tyr residues are also the target of the cognate CpsB phosphatase. An interaction between CpsD and CpsC was observed, and the phosphorylation state of CpsD influenced the subsequent action of CpsC. The CpsC extracellular domain appeared necessary for the production of high molecular weight polysaccharides by influencing CpsA-mediated attachment of the CPS to the bacterial cell surface. In conclusion, although having no impact on cps transcription or the synthesis of the basal repeating unit, we suggest that these proteins are fine-tuning the last steps of CPS biosynthesis (i.e. the balance between polymerization and attachment to the cell wall).
Introduction
Group B Streptococcus (GBS,2 also known as Streptococcus agalactiae) is the major cause of life-threatening bacterial infections in newborns (1). GBS colonizes the anogenital tract of 10–30% of healthy women, and vertical transmission may cause bacterial septicemia and meningitis in the neonate (2–5). GBS produces a capsule that surrounds and protects the bacterium by preventing complement deposition and opsonophagocytosis (6). Capsular polysaccharides (CPS) conjugated to carrier proteins are currently used as vaccines against several pathogens (7, 8). CPS biosynthesis in GBS has not been investigated in detail but is presumably similar to the better characterized pathway of S. pneumoniae (see Ref. 9 for a detailed review). Briefly, the basic repeating units of the CPS of S. pneumoniae are synthesized at the cytosolic side of the membrane by the sequential action of glycosyltransferases. The unit is anchored to a membrane lipid, and it is transferred to the outer side of the membrane, where it is polymerized into the full-length CPS. The CPS is then covalently bound to the cell wall peptidoglycan, thus creating the mucoid capsule layer covering the bacterial surface (9). Also in GBS, the CPS has been demonstrated to be covalently bound to the cell wall peptidoglycan (10). The CPS from GBS comprises repeating units constituted of 5–7 monosaccharides, depending on the serotype. The enzymes required for the transport and the assembly of the CPS are encoded in the cps operon (11, 12), consisting of 16–18 genes transcribed in a single transcript (12). The cps operon is divided into three main regions (13). The central part of the operon (cpsE–L) determines the capsule serotype and comprises genes encoding glycosyltransferases and a polymerase. The last four genes of the operon (neuA–neuD) encode enzymes that synthesize sialic acid. Finally, the first genes of the operon (cpsA–D) are not directly involved in the biosynthesis of the CPS repeating units (14).
The genes cpsA–D are conserved among all the GBS capsule serotypes and have orthologues in other encapsulated streptococci, such as S. pneumoniae (12). Their function in GBS has been previously investigated through the construction and characterization of knock-out mutants, suggesting that CpsA is required for transcription of the cps operon, whereas CpsC and CpsD have a more undefined role in polymerization/export of CPS (14). Moreover, recombinant CpsA was shown to bind the cps operon promoter in vitro (15). CpsA is a 485-aa membrane protein with a major extracellular portion. The extracellular domains of the homologous proteins in S. pneumoniae and B. subtilis have recently been crystallized and were proposed to be responsible for hydrolysis of the pyrophosphate linkage between the CPS and the membrane lipid anchor (16) and subsequent attachment of CPS to the peptidoglycan (17). Interestingly, these functions appear different from those suggested for CpsA in GBS. Concerning CpsBCD, orthologous proteins in S. pneumoniae (46–64% aa identity) were described to constitute a phosphoregulatory system, where CpsD is an autokinase and CpsB is the cognate phosphatase (18, 19).
By analogy, it can be argued that CPS biosynthesis in GBS is similar to S. pneumoniae overall. However, experimental studies investigating the role of CpsABCD in GBS are limited and present potential discrepancies compared with S. pneumoniae and other related species. Moreover, it is still unclear whether and how the GBS CpsABCD proteins and their counterparts in other species act in concert as a system. In the present work, we attempted to elucidate the molecular details of the final steps in the biosynthesis of the GBS CPS, with a focus on the role of CpsABCD. To that end, we developed a panel of knock-out and functional mutant strains and analyzed the effects on cps operon transcription; CPS quantity, size, and attachment to the cell surface; and CpsD phosphorylation. In vivo molecular interactions between the CpsABCD proteins were also investigated. The resulting data provided novel insights into the role of each individual protein as well as their interdependencies. We concluded that these proteins are not involved in cps transcription or the synthesis of the basal repeating unit, but they are responsible for balancing the processes of polymerization and attachment to the cell wall of the CPS.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
GBS strains 515 and 515ΔcpsE were provided by Dr. Dennis Kasper (Harvard Medical School, Boston, MA) and Dr. Michael Cieslewicz (14), respectively. GBS strains were grown in Todd-Hewitt broth (THB) at 37 °C, 5% CO2. Tryptic soy broth, 15 g/liter agar (TSA) was used as solid medium. Strains were stored at −80 °C in THB medium, 15% glycerol. MAX Efficiency® DH5αTM competent cells (Life Technologies) and chemically competent HK100 and BTH101 Escherichia coli cells were prepared in-house and used for transformation, propagation, and preparation of plasmids. E. coli was grown at 37 °C with agitation (180 rpm) in Luria-Bertani broth (LB) or on LB + 15 g/liter agar plates. Erythromycin was used for selection of GBS (1 μg/ml) or E. coli (100 μg/ml) containing the pJRS233-derived plasmids (20) used for mutagenesis. Kanamycin was used for selection of E. coli (50 μg/ml) containing the pET24b-derived plasmids (Novagen, Pretoria, South Africa) and the pKT25-derived plasmids (Euromedex, Souffelweyersheim, France). Ampicillin was used for selection of E. coli (100 μg/ml) containing the pUT18C-derived plasmids (Euromedex) and the pET15-derived plasmids (Millipore).
Construction of GBS Mutant Strains
To prepare each mutant strain, the shuttle vector pJRS233 (20) containing the gene locus with an in-frame deletion or a codon substitution was constructed. Mutant strains obtained are described in Table 2, and primers used for the development of constructs are listed in Table 1. Constructs for genes with codon substitutions were prepared using a splicing by overlap extension PCR (SOEing-PCR) strategy (21). Briefly, amplicons up- and downstream of the codon substitution were amplified from GBS 515 genomic DNA using the PfuUltra II Fusion HS DNA polymerase (Agilent Technologies). Internal primers used to amplify the two parts of the genes have 15-bp overlapping tails and introduce the codon substitution, and amplicons are then joined together by SOEing-PCR. The resulting fragment was ligated into pJRS233 using BamHI and XhoI restriction sites.
TABLE 2.
GBS strains used in this work
| Strain name | Description | Mutated protein (full-length aa) |
|---|---|---|
| GBS 515 | Wild type strain | |
| ΔcpsE | cpsE deletion | Details in Ref. 14 |
| ΔcpsA | cpsA deletion | Deletion of aa 11–452 (458 aa) |
| CpsA(Δext) | Deletion of the CpsA extracellular domain | Deletion of aa 96–458 (458 aa) |
| CpsA(ΔLytR) | Deletion of the CpsA LytR domain | Deletion of aa 236–458 (458 aa) |
| ΔcpsB | cpsB deletion | Deletion of aa 4–240 (243 aa) |
| CpsB(R139A) | Point mutation in the phosphatase active site | Arginine to alanine in position 139 (243 aa) |
| CpsB(R206A) | Point mutation in the phosphatase active site | Arginine to alanine in position 206 (243 aa) |
| ΔcpsC | cpsC deletion | Deletion of aa 1–222 (230 aa) |
| CpsC(ΔC-term) | Deletion of the CpsC intracellular C-terminal portion | Deletion of aa 198–230 (230 aa) |
| CpsC(Δext) | Deletion of the CpsC extracellular domain | Deletion of aa 53–153 (230 aa) |
| ΔcpsD | cpsD deletion | Deletion of aa 11–225 (232 aa) |
| CpsD(K49A) | Point mutation in the autokinase active site | Lysine to alanine in position 49 (232 aa) |
| CpsD(ΔP-Tyr) | Phosphoacceptor site C-terminal deletion | Deletion of aa 213–224 (232 aa) |
TABLE 1.
Oligonucleotides
Restriction sites are marked in boldface type, overlapping regions used for mutagenesis are underlined, and nucleotide substitutions resulting in amino acid substitutions are marked in boldface type and underlined. For, forward; Rev, reverse; Ampl., amplification.
| Name | Sequence | Description |
|---|---|---|
| NotA5F | TAAAGCGGCCGCCTCTATCACTGACAACAATGG | Ampl. of cpsA + flanking regions, For, 894 bp upstream cpsA start, NotI |
| XhA3R | TATCCTCGAGGAAGAAGTATATTGTGGCGTA | Ampl. of cpsA + flanking regions, Rev, 916 bp downstream cpsA end, XhoI |
| KOA3F | TCGCGCCGTCAACAAAAGAACACAATGGAGGAATAAC | ΔcpsA mutagenesis, For, overlap KOA5R |
| KOA5R | TTGTTGACGGCGCGAATGATTAGACATTGTAA | ΔcpsA mutagenesis, Rev, overlap KOA3F |
| M1A3F | ACTACTTTATATGGATAACAAGAATGATTGATATTCATTC | CpsA(Δext) mutagenesis, For, overlap M1A5R |
| M1A5R | TCCATATAAAGTAGTAGCAACGAAAATAGAAGC | CpsA(Δext) mutagenesis, Rev, overlap M1A3F |
| M2A3F | TCTATATTAGCGGTTAACAAGAATGATTGATATTCATTCTC | CpsA(ΔLyt-R) mutagenesis, For, overlap M2A5R |
| M2A5R | ACCGCTAATATAGATATTAAATACCCCTTCTTTATG | CpsA(ΔLyt-R) mutagenesis, Rev, overlap M2A3F |
| BaB5F | TAAAGGATCCTTATGTTAGCTTAATTGAACTTAGCA | Ampl. of cpsB + flanking regions, For, 904 bp upstream cpsB start, BamHI |
| XhB3R | AAAGCTCGAGGACATAACAGAGTTCCTAGTA | Ampl. of cpsB + flanking regions, Rev, 960 bp downstream cpsB end, XhoI |
| KOB3F | ATTCATTCTCATATCCATTACATTTAGGAGATTTCATGAA | ΔcpsB mutagenesis, For, overlap KOB5R |
| KOB5R | GATATGAGAATGAATATCAATCATTCTTGTTATTCCTC | ΔcpsB mutagenesis, Rev, overlap KOB3F |
| M1BF | GTTGCGCATATAGAGGCGTATAACGCTTTAGA | CpsB(R139A) mutagenesis, For, overlap M1BR |
| M1BR | TCTAAAGCGTTATACGCCTCTATATGCGCAAC | CpsB(R139A) mutagenesis, Rev, overlap M1BF |
| M2BF | CATAACCTTGATGTTGCACCGCCATTTTTAGC | CpsB(R206A) mutagenesis, For, overlap M2BR |
| M2BR | GCTAAAAATGGCGGTGCAACATCAAGGTTATG | CpsB(R206A) mutagenesis, Rev, overlap M2BF |
| BaC5F | ACAAGGATCCACTGTCGAGTCACAAGCATTA | Ampl. of cpsC + flanking regions, For, 905 bp upstream cpsC start, BamHI |
| XhC3R | CAATCTCGAGTTAAACTCTTCAAGATAGCCACG | Ampl. of cpsC + flanking regions, Rev, 943 bp downstream cpsC end, XhoI |
| KOC3F | ATGAATAAAATAGCTATAGTACCAGATTTGAATAAACTT | ΔcpsC mutagenesis, For, overlap KOC5R |
| KOC5R | AGCTATTTTATTCATGAAATCTCCTAAATGTAATGGT | ΔcpsC mutagenesis, Rev, overlap KOC3F |
| M1C3F | ATTATGGGTATTTTGTAAGGAGAATATAATGACTCGTT | CpsC(ΔC-term) mutagenesis, For, overlap M1C5R |
| M1C5R | CAAAATACCCATAATAACTAAAACAATAGTTGATAATCC | CpsC(ΔC-term) mutagenesis, Rev, overlap M1C3F |
| M2C3F | TCAACAAGGATATATGTTACTCAAGTAGAGGATATC | CpsC(Δext) mutagenesis, For, overlap M2C5R |
| M2C5R | ATATATCCTTGTTGAAGAAGTATATTGTGGCGTAA | CpsC(Δext) mutagenesis, Rev, overlap M2C3F |
| BaD5F | TTTAGGATCCCAAAAAGAACGGGTGAAGGAA | Ampl. of cpsD + flanking regions, For, 1018 bp upstream cpsD start, BamHI |
| XhD3R | TCTACTCGAGCTACCATTACGACCTACTCTA | Ampl. of cpsD + flanking regions, Rev, 966 bp downstream cpsD end, XhoI |
| KOD3F | GAAATAGTTGATAGCAAAAGGGATAGAAAAAGGAAGTAA | ΔcpsD mutagenesis, For, overlap KOD5R |
| KOD5R | GCTATCAACTATTTCTAAACGAGTCATTATATTCTC | ΔcpsD mutagenesis, Rev, overlap KOD3F |
| M1DF | GGAAGGGGAAGGAGCATCCACTACTTCA | CpsD(K49A) mutagenesis, Rev, overlap M1DR |
| M1DR | TGAAGTAGTGGATGCTCCTTCCCCTTCC | CpsD(K49A) mutagenesis, For, overlap M1DF |
| M2D3F | GTTAGTGAATCTGTTGGAAAAAGGGATAGAAAAAGG | CpsD(ΔP-Tyr) mutagenesis, For, overlap M2D5R |
| M2D5R | AACAGATTCACTAACTTTATTAAGAATAATACCTAAGAAC | CpsD(ΔP-Tyr) mutagenesis, Rev, overlap M2D3F |
| 1015F | AGGTTTACTTGTGGCGCTTG | qRT-PCR, For, annealing to gyrA |
| 1015R | TCTGCTTGAGCAATGGTGTC | qRT-PCR, Rev, annealing to gyrA |
| 1292F | TCAACTGGACAACGCTTCAC | qRT-PCR, For, annealing to cpsA |
| 1292R | AAGTTGAGCTCCTGGCATTG | qRT-PCR, Rev, annealing to cpsA |
| 1288F | TGCTCATATGTGGCATTGTG | qRT-PCR, For, annealing to cpsE |
| 1288R | AGAAAAGATAGCCGGTCCAC | qRT-PCR, Rev, annealing to cpsE |
| 1289F | TCAATGCGATCCGTACAAAC | qRT-PCR, For, annealing to cpsD |
| 1289R | GTGGATTTTCCTTCCCCTTC | qRT-PCR, Rev, annealing to cpsD |
| CF | TAGGGGATCCCATGAATAAAATAGCTAATACAG | BACTH, For, ampl. of cpsC, BamHI |
| CR | CATTAGAATTCGATTAAAGTTTATTCAAATCTGG | BACTH, Rev, ampl. of cpsC, EcoRI |
| CMutR | CATTAGAATTCGATTACAAAATACCCATAATAAC | BACTH, Rev, ampl. of CpsC(ΔC-term), EcoRI |
| DF | AGGAGGATCCCATGACTCGTTTAGAAATAG | BACTH, For, ampl. of cpsD, BamHI |
| DR | TACAGAATTCGATTACTTCCTTTTTCTATC | BACTH, Rev, ampl. of cpsD, EcoRI |
| AF | GGAGGGATCCAATGTCTAATCATTCGCGCCG | BACTH, For, ampl. of cpsA, BamHI |
| AR | ATCAGGTACCCTTGTTATTCCTCCATTGTGTTC | BACTH, Rev, ampl. of cpsA, KpnI |
| 1289f | CTGTACTTCCAGGGCATGACTCGTTTAGAAATAGTTGATAGC | Recombinant CpsD, For, ampl. of cpsD, 15-bp overlap with pET-TEV |
| 1289r | AATTAAGTCGCGTTACTTCCTTTTTCTATCCCTTTTTCCGTAA | Recombinant CpsD, Rev, ampl. of cpsD, 15-bp overlap with pET-TEV |
Constructs for genes with in-frame deletions were prepared using the polymerase incomplete primer extension method (22). Briefly, the gene and 900–1,000 bp up- and downstream of the coding sequence were amplified from GBS 515 genomic DNA and cloned into pET24b using NotI and XhoI (cpsA inserts) or BamHI and XhoI (cpsB-C-D inserts) restriction sites. In-frame deletions were developed by amplifying the plasmid using primers with 15-bp overlapping tails annealing at the two sides of the region to delete. Linear plasmids were transformed into HK100 competent cells able to recircularize the plasmid. Following propagation and purification of the plasmid, the inserts containing the in-frame deletions were transferred into pJRS233 plasmid by restriction digestion, ligation, and transformation of E. coli DH5α (Life Technologies). Constructs for chromosomal complementation were prepared by cloning the respective WT loci into pJRS233. The various pJRS233 constructs were used for insertion/duplication and excision mutagenesis (14, 20). Briefly, pJRS233-derived plasmids were used to transform electrocompetent GBS 515 cells by electroporation (23). Transformants were selected by growth on TSA + erythromycin at 30 °C for 48 h. Integration was performed by growth of transformants at 37 °C (non-permissive temperature for the suicide shuttle vector) with erythromycin selection. Excision of the integrated plasmid was performed by serial passages in THB at 30 °C and parallel screening for erythromycin-sensitive colonies on the plate. Mutants were verified by PCR sequencing of the loci.
Quantitative RT-PCR Analysis
RNA extracts were prepared as described (24). Bacteria were harvested at two time points, at A600 = 0.4 (log phase) and A600 = 1.7 (early stationary phase). cDNA was prepared using the reverse transcription system (Promega) by using 500 ng of RNA/reaction. Real-time quantitative RT-PCR was performed on 50 ng of cDNA that was amplified using LightCycler® 480 DNA SYBR Green I Master (Roche Applied Science). Reactions were monitored using a LightCycler® 480 instrument and software (Roche Applied Science). Three technical replicates were monitored for each strain/condition analyzed. To quantify cps operon transcription level, primers annealing on cpsA and cpsE were used for all of the strains with the exception of cpsA mutants, where primers for cpsD and cpsE were used. The transcript amounts in each condition were standardized to an internal control gene (gyrA) and compared with standardized expression in the WT strain (ΔΔCT method). The primers used are listed in Table 1.
Quantification of the Capsular Polysaccharide Attached to the Cell Surface
Alkaline extraction of CPS from GBS bacteria was performed as described previously (25). Briefly, bacteria were grown in 50 ml of THB at 37 °C for 8 h (stationary phase). Viable counts were performed and confirmed that cfu numbers between the strains were comparable. GBS cells were collected by centrifugation for 15 min at 3,220 × g at 4 °C; resuspended in PBS, 0.8 n NaOH; and incubated at 37 °C for 36 h. Samples were neutralized by the addition of HCl and pelleted by centrifugation for 10 min at 10,000 × g at 4 °C. The supernatant was diluted 1:10 in distilled H2O, loaded on a Vivaspin 10 tube (Sigma), and centrifuged at 3,220 × g until most of the solution passed through the membrane. After two washes with distilled H2O, the CPS extract was recovered from the membrane by resuspension in water. The amount of CPS present in the extract was estimated by measuring the sialic acid content using the colorimetric resorcinol-hydrochloric acid method (26). Briefly, 120 μl of extract were mixed with 380 μl of water and 500 μl of resorcinol solution (0.2% resorcinol, 0.3 mm copper sulfate, 30% (v/v) HCl). Samples were boiled for 20 min and cooled to room temperature, and absorbance was measured at 564 nm. The sialic acid content of the samples was then determined by comparison with a concomitantly prepared standard curve using serial dilutions of purified sialic acid.
Quantification of CPS in the Growth Medium
Bacteria were grown in 10 ml of THB at 37 °C for 8 h. GBS cells were pelleted by centrifugation for 15 min at 3,220 × g at 4 °C, and the growth medium was collected and filtered using a 0.22-μm Nalgene syringe filter (Thermo Scientific). The amount of capsular polysaccharide in the medium was estimated by dot blot. Serial dilutions (1:2) were prepared in PBS. Two μl of each serial dilution were spotted onto a nitrocellulose membrane. The membrane was dried for 20 min and blocked by soaking in 5% skim milk in PBS, Tween 20 (0.05%) (PBST). Detection by immunoblotting was performed as described below (see “Immunoblotting Experiments”).
Cell Wall Extracts
Bacteria were grown in 10 ml of THB at 37 °C for 8 h. GBS cells were pelleted by centrifugation for 15 min at 3,220 × g at 4 °C and washed in PBS. Extracts of CPS attached to the peptidoglycan were prepared by incubating the bacterial pellet with 200 units of mutanolysin (Sigma) diluted in 50 μl of protoplasting buffer (0.1 m potassium phosphate, 40% sucrose, 10 mm MgCl2) for 1 h at 37 °C. 20 μl of Proteinase K solution (Life Technologies) were added, and samples were incubated at 56 °C for 30 min. After centrifugation for 5 min at 10,000 × g at 4 °C, the supernatant was collected.
Production of α-CpsD Mouse Serum
The cpsD gene was amplified from GBS 515 genomic DNA using the primers in Table 1 and inserted into a modified pET-15 vector (Novagen) by polymerase incomplete primer extension (22). The plasmid was transformed into E. coli BL21(DE3) cells (Novagen), and expression of the His6-tagged fusion protein was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. The bacterial pellet was resuspended in 50 mm Tris-HCl (pH 7.5), 250 mm NaCl, 10 mm imidazole and was lysed by sonication. Extracts were pelleted, and the supernatant was purified using an FF-Crude His-Trap HP nickel chelating column (GE Healthcare). The recombinant CpsD was eluted with 300 mm imidazole, and the buffer was exchanged to PBS using an Amicon Ultra 3K centrifugal filter (Millipore, Cork, Ireland). Antisera specific for CpsD were produced by immunizing (prime and two boosts) eight CD1 mice with 20 μg of purified recombinant protein formulated with 400 μg of alum.
Protein Extracts
Bacteria were grown in 30 ml of THB at 37 °C until exponential growth phase was reached (A600 = 0.4). Cells were pelleted, washed in PBS, and resuspended in 800 μl of Tris-HCl (50 mm, pH 7.5) with Complete protease inhibitor and PhosSTOP phosphatase inhibitor mixture tablets (Roche Applied Science), transferred into Lysing Matrix B 2-ml tubes (MP Biomedicals, Santa Ana, CA), and lysed using the FastPrep-24TM automated homogenizer (6 cycles at 6.5 m/s for 30 s). Tubes were centrifuged at 500 × g for 5 min to eliminate beads and cellular debris.
Immunoblotting Experiments
CPS or protein extracts were separated by electrophoresis on NuPage 4–12% BisTris gels (Life Technologies) according to the manufacturer's instructions. Western blot was performed using the iBlot® blotting system (Life Technologies) according to the manufacturer's instructions. Nitrocellulose membranes were blocked by soaking in 5% (w/v) skim milk in PBST with the exception of membranes probed with the α-Tyr(P) mAb, which were blocked in 3% (w/v) BSA in PBST. Primary mouse α-CPSIa mAb (30E9/B11), were obtained by immunization with Ia glycoconjugate. Mouse α-Tyr(P) mAb (Sigma, clone PT-66), mouse α-RNA polymerase β mAb (Thermo Scientific, clone 8RB13), and mouse α-CpsD polyclonal serum were also used as primary antibodies. All of the primary antibodies and sera were diluted 1:2,000 in 1% (w/v) BSA in PBST, and membranes were incubated for 1 h at room temperature. Membranes were then incubated in 1:15,000 of secondary goat anti-mouse antibody conjugated to horseradish peroxidase. Immunoreactive bands were visualized using the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
Flow Cytometry
Flow cytometry using α-CPSIa mAb (1:10,000) was performed essentially as described elsewhere (27). The secondary antibody was goat anti-mouse allophycocyanin-conjugated F(ab′)2 fragment IgG (Jackson ImmunoResearch, West Grove, PA) diluted 1:200 in PBST. All data were collected using a FACSCanto II (BD Biosciences) by acquiring 10,000 events, and data analysis was performed with Flow-Jo software (version 8.6; TreeStar Inc., Ashland, OR).
Bacterial Two-hybrid Assay
A bacterial two-hybrid assay was employed to test potential protein-protein interactions (28). The cpsC, cpsD, and cpsA coding sequences were amplified using the primers described in Table 1 and cloned into pUT18C and pKT25 plasmids (Euromedex). CpsC was expressed both in a full-length version and with the C-terminal 33-aa tail deleted. Interactions between proteins were tested by introducing the plasmids into the adenylate cyclase-deficient E. coli strain BTH101 (Euromedex). Empty plasmids were tested together with all of the fusion proteins as negative control. Positive control plasmids pKT25-zip and pUT18C-zip were provided by the manufacturer (28). Colonies containing both plasmids were selected by plating on LB + kanamycin (50 μg/ml) + ampicillin (100 μg/ml) agar plates and growing them overnight at 37 °C. Four colonies were selected for each transformation and independently inoculated into 1 ml of LB, 50 μg/ml kanamycin, 100 μg/ml ampicillin, 1 mm isopropyl 1-thio-β-d-galactopyranoside and grown overnight at 30 °C. Two μl from each culture were spotted onto LB agar plates with additives as seen above and 80 μg/ml X-Gal. After incubation overnight at 30 °C, the plates were examined for the formation of blue colonies, indicative of a protein-protein interaction.
Immunogold Labeling and Electron Microscopy
Immunogold electron microscopy of the CPS in WT and mutant strains was performed as described previously (29). Briefly, bacteria were grown in THB until exponential growth phase was reached (A600 = 0.4). Cells were pelleted, washed in PBS, and fixed in PBS, 2% paraformaldehyde for 20 min at room temperature. Five μl of sample were added to 200-square mesh Formvar copper grids coated with a thin carbon film (Ted Pella, Redding, CA) and incubated at room temperature for 5 min. The grids were than incubated in blocking buffer (1% normal rabbit serum, 1% BSA, 1× PBS) and subsequently incubated with 1:1,000 α-CPSIa mAb in blocking buffer. Samples were washed five times in blocking buffer and incubated with secondary gold-conjugated antibodies at 1:40 (goat anti-mouse IgG, 10 nm (Agar Scientific, UK)). Grids were washed in distilled water five times and stained for 45 s with aqueous 1% uranyl acetate, pH 4.5, blotted, air-dried, and finally observed in an FEI Tecnai G2 Spirit system operating at a voltage of 80 kV and at a magnification of 87,000. Images were collected with a CCD Olympus.SIS Morada 2K*4K* camera.
Purification of Capsular Polysaccharide from Bacterial Pellets and Spent Media
The 515 strain and the ΔcpsA, CpsC(Δext), and CpsD(K49A) mutant strains were grown in 1 liter of THB at 37 °C for 8 h. The pellet and the medium obtained from the cultures were separated by centrifugation for 30 min at 8,600 × g at 4 °C. The purification process was based on procedures described previously (25, 27). Briefly, pellets were washed in PBS and successively inactivated by incubation with PBS, 0.8 n NaOH at 37 °C for 36 h. After centrifugation at 4,000 rpm for 20 min, 1 m Tris buffer (1:9, v/v) was added to the supernatant and diluted with 1:1 (v/v) HCl to reach a neutral pH. Spent growth media were inactivated by filtration (0.22 μm). For both bacterial pellets and media, the same purification process was applied. Briefly, 2 m CaCl2 (0.1 m final concentration) and ethanol (30% (v/v) final concentration) were added to the solution. After centrifugation at 4,000 × g for 20 min, the supernatants were subjected to a tangential flow filtration on a 30,000 molecular weight cut-off (Hydrosart Sartorius, 50-cm2 surface) against 16 volumes of 50 mm Tris, 500 mm NaCl, pH 8.8, and 8 volumes of 10 mm sodium phosphate, pH 7.2. Then the samples were loaded onto a preparative size exclusion column (Sephacryl S500 column, GE Healthcare), using 10 mm sodium phosphate, 500 mm NaCl, pH 7.2, as eluent buffer. The CPS samples were subjected to full N-acetylation. After complete drying, samples were solubilized in 300 mm Na2CO3, 300 mm NaCl, pH 8.8. A 1:1 diluted solution of 4.15 μl/ml acetic anhydride in ethanol was added, and the reaction was incubated at room temperature for 2 h. Samples were then purified in a preparative size exclusion column (Sephadex G15 column, GE Healthcare) by using MilliQ water. The polysaccharide content was determined using the colorimetric resorcinol-hydrochloric acid assay (26). The purity of the polysaccharide preparation was assessed by colorimetric assays, which indicated a content of residual proteins below 3% (w/w) and nucleic acids below 1% (w/w). Endotoxin content was <30 endotoxin units/μg of saccharide, measured by the Limulus amebocyte lysate test.
NMR Spectroscopy
1H NMR experiments were recorded by a Bruker Avance III 400 spectrometer, equipped with a high precision temperature controller, using a 5-mm broadband probe (Bruker). TopSpin software (version 3.2; Bruker) was used for data acquisition and processing. 1H NMR spectra were collected at 25 ± 0.1 °C with 32,000 data points over a 10-ppm spectral width, accumulating an appropriate number of scans for high signal/noise ratio. The spectra were weighted with 0.2-Hz line broadening and Fourier-transformed. The transmitter was set at the water frequency, which was used as the reference signal (4.79 ppm). All monodimensional proton NMR spectra were obtained in a quantitative manner using a total recycle time to ensure a full recovery of each signal (5× longitudinal relaxation time T1).
HPLC-SEC
CPS samples were eluted on a TSK gel 6000PW (30 × 7.5 mm) column (particle size, 17 μm; Sigma 8-05765) with a TSK gel PWH guard column (7.5-mm inner diameter × 7.5-cm length; particle size, 13 μm; Sigma 8-06732) (Tosoh Bioscience) and calibrated with a series of defined pullulan standards (Polymer) of average molecular masses ranging from 20,000 to 1,330,000 Da. Void and bed volume calibration was performed with λ-DNA (λ-DNA Molecular Weight Marker III, 0.12–21.2 kbp; Roche Applied Science) and sodium azide (NaN3) (Merck), respectively. The mobile phase was 10 mm sodium phosphate, pH 7.2, at a flow rate of 0.5 ml/min (isocratic method for 50 min). The polysaccharide samples were analyzed at a concentration of 0.2–0.4 mg/ml, using 10 mm sodium phosphate buffer, pH 7.2, as the mobile phase, at a flow rate of 0.5 ml/min.
RESULTS
Generation of Isogenic CpsABCD Mutant Strains in GBS
To investigate the function of cpsABCD, 12 isogenic mutant strains were obtained in the GBS 515 (serotype Ia) genetic background (Table 2). The knock-out (KO) mutants ΔcpsA, ΔcpsB, ΔcpsC, and ΔcpsD contained in-frame deletions where a large part of the gene sequence was removed. Furthermore, we designed ad hoc functional mutant strains to investigate the role of specific domains and/or enzymatic activities of the proteins. Topology and subcellular localization of the CpsABCD proteins were predicted using PSORTb version 3.0 (30) and Octopus (31), and they were consistent with previous literature on S. agalactiae and S. pneumoniae (32, 33). The entire extracellular portion of CpsA, or the LytR domain only, were deleted, generating strains CpsA(Δext) and CpsA(ΔLytR). Through comparison of CpsB with the orthologous gene in S. pneumoniae, we identified two conserved amino acids (Arg-139 and Arg-206) reported to be important for phosphatase activity (34). We generated the two mutant strains CpsB(R139A) and CpsB(R206A), containing alanine substitutions in those positions. Little is known about CpsC and its orthologues, although the predicted C-terminal intracellular tail was suggested to be essential for CpsD activity in Staphylococcus aureus and S. pneumoniae (33, 35). We created the mutant strains CpsC(ΔC-term) and CpsC(Δext), where the predicted intracellular tail (33 aa) and extracellular domain (101 aa) of CpsC were deleted. CpsD is potentially a kinase, by virtue of comparison with the orthologous proteins CapB in S. aureus (36) and Wzd in S. pneumoniae (37). The conserved catalytic residue Lys-49 was mutated into alanine, generating strain CpsD(K49A). In parallel, we identified the repeated motif YGX in the C-terminal tail of CpsD, potentially constituting a phosphoacceptor region (38). This region was truncated (12 aa), generating strain CpsD(ΔP-Tyr). The obtained mutant strains were grown in liquid cultures to examine growth kinetics, which in all cases were similar to the parent strain (data not shown).
Analysis of the cps Operon Transcription in CpsABCD Mutant Strains
CpsA has previously been reported to be involved in cps operon transcription (14, 15). We used real-time quantitative RT-PCR to analyze the transcription of the operon in all of the mutant strains. Bacteria were harvested in logarithmic and early stationary phase, and the transcription was measured using primers annealing to cpsA (with the exception of CpsA mutants, where we used primers annealing to cpsD) (Fig. 1). Compared with the wild type strain 515, mutant strains showed no prominent differences in relative expression of the cps operon, suggesting that none of the cpsABCD genes is involved in transcriptional regulation of the cps operon in the conditions tested. We also noted that the transcription of the cps operon was reduced in early stationary phase compared with exponential phase of growth. The cps operon transcription was also quantified using primers annealing to cpsE, the first gene downstream of cpsABCD. We did not observe any significant differences between the strains analyzed and thus exclude the presence of polar effects on the transcription of the cps operon due to the mutations introduced (Fig. 1).
FIGURE 1.
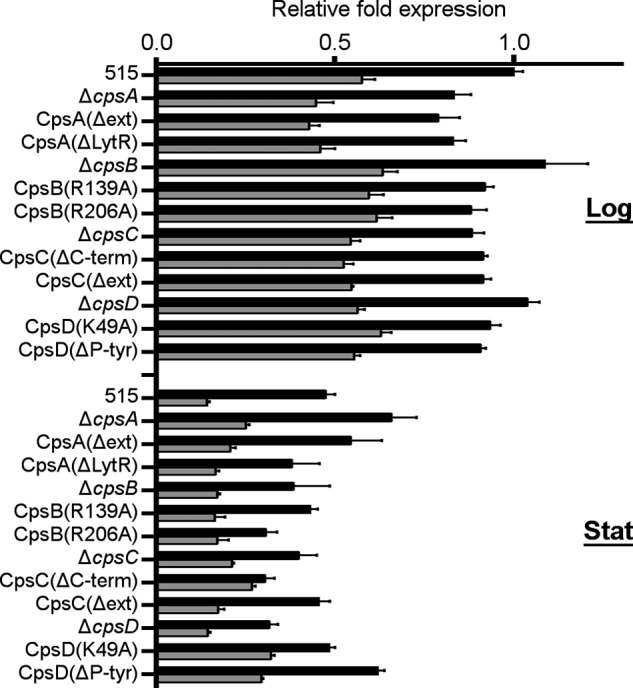
Quantification of the cps operon transcription. cps operon transcription of the WT strain 515 and of its derivative cps mutant strains was measured by real-time quantitative RT-PCR using primers annealing to cpsA/D (black bars) and to cpsE (gray bars). Bacteria were harvested in logarithmic (Log) and early stationary phase (Stat). The relative -fold expression for each strain was calculated in comparison with the WT strain 515 in log phase. Bars, means of three independent experiments performed with triplicate samples. Error bars, S.D.
CpsBCD Forms an Interdependent Kinase/Phosphatase System
In S. pneumoniae the orthologous CpsBCD have been shown to constitute a phosphoregulatory system. The autokinase CpsD phosphorylates its C-terminally located tyrosines (38). CpsB is the cognate phosphatase and CpsC is a membrane protein required for CpsD autokinase activity (19, 33). We hypothesized that these proteins have similar functions in S. agalactiae. The presence of putative phosphoproteins in total bacterial extracts was examined by Western blot using an α-Tyr(P) mAb (α-P-Tyr mAb in Fig. 2A). CpsD was identified using an α-CpsD antibody and appeared phosphorylated in the wild type strain, albeit showing only a faint band. The three cpsB mutants showed an increased level of phosphorylation of CpsD, consistent with absent/reduced phosphatase activity of CpsB, and confirming that the amino acids Arg-139 and Arg-206 are necessary for phosphatase activity. All of the mutations in CpsD resulted in undetectable phosphorylation of this protein. We conclude that CpsD is an autokinase, and when non-functional, it cannot be phosphorylated by other bacterial kinases. Moreover, we showed that one or more of the four tyrosines at the C-terminal of CpsD constitute the phosphoacceptor site and that the lysine in position 49 is necessary for autokinase activity. In addition, if CpsC is absent or lacks the C-terminal intracellular tail, then CpsD is not phosphorylated, whereas, if only the extracellular portion of the protein is deleted, autophosphorylation activity of CpsD is preserved. Thus, the CpsC intracellular 33-aa tail is necessary for CpsD phosphorylation, whereas the CpsC extracellular domain is dispensable.
FIGURE 2.

CpsD and CpsB form an autokinase/phosphatase pair. A, Western blots showing CpsD (α-CpsD), tyrosine phosphorylation of CpsD (α-P-Tyr mAb), and the loading control RNA polymerase subunit β (α-RNApolβ mAb) in total protein extracts from 515 WT and the cps mutant strains. The band corresponding to the phosphorylated CpsD is indicated by an arrow. B, bacterial two-hybrid analysis of CpsC and CpsD. T25-CpsD was tested for interaction with T18-CpsC and T18-CpsC(ΔC-term). Control plasmids (T18 and T25) were tested together with fusion proteins as negative controls. The positive control used was the leucine zipper GCN4 fused to the T25 and T18 fragments (28). The formation of blue colonies indicates a protein-protein interaction.
A protein-protein interaction between the two homologous protein Wzd (CpsC homologue) and Wze (CpsD homologue) in S. pneumoniae has been shown previously (37). Having observed that CpsD autokinase activity required the presence of CpsC, we investigated a putative interaction between CpsC and CpsD of GBS using a bacterial two-hybrid system (28) (see “Experimental Procedures” for details). Indeed, we observed that heterologously expressed CpsC and CpsD interacted (Fig. 2B). Interestingly, we observed that this interaction was abrogated when the CpsC C-terminal 33-aa tail was removed, suggesting that the tail directly interacts with CpsD.
Aberrant CPS Production and Localization in CpsABCD Mutant Strains
CPS production in WT and cps mutant strains was verified by flow cytometry using an α-CPSIa mAb. All of the mutant strains exhibited a clear shift in mean fluorescence compared with the negative control (Fig. 3A), confirming that the strains produce a CPS possessing epitopes recognized by the monoclonal antibodies raised against the wild type CPS. The amount of CPS present on the surface of the different strains was quantified in bacterial extracts obtained by alkaline treatment (25). We detected lower amounts of surface-associated CPS in all of the KO mutant strains compared with the wild type (Fig. 3B), consistent with data published previously (14). Most of the functional mutants also showed a significant capsule reduction, with the exception of mutants with point mutations in CpsB (CpsB(R139A) and CpsB(R206A)) and the CpsD(ΔP-Tyr) strain, which lacks the putative tyrosine phosphoacceptor tail (Fig. 3B).
FIGURE 3.
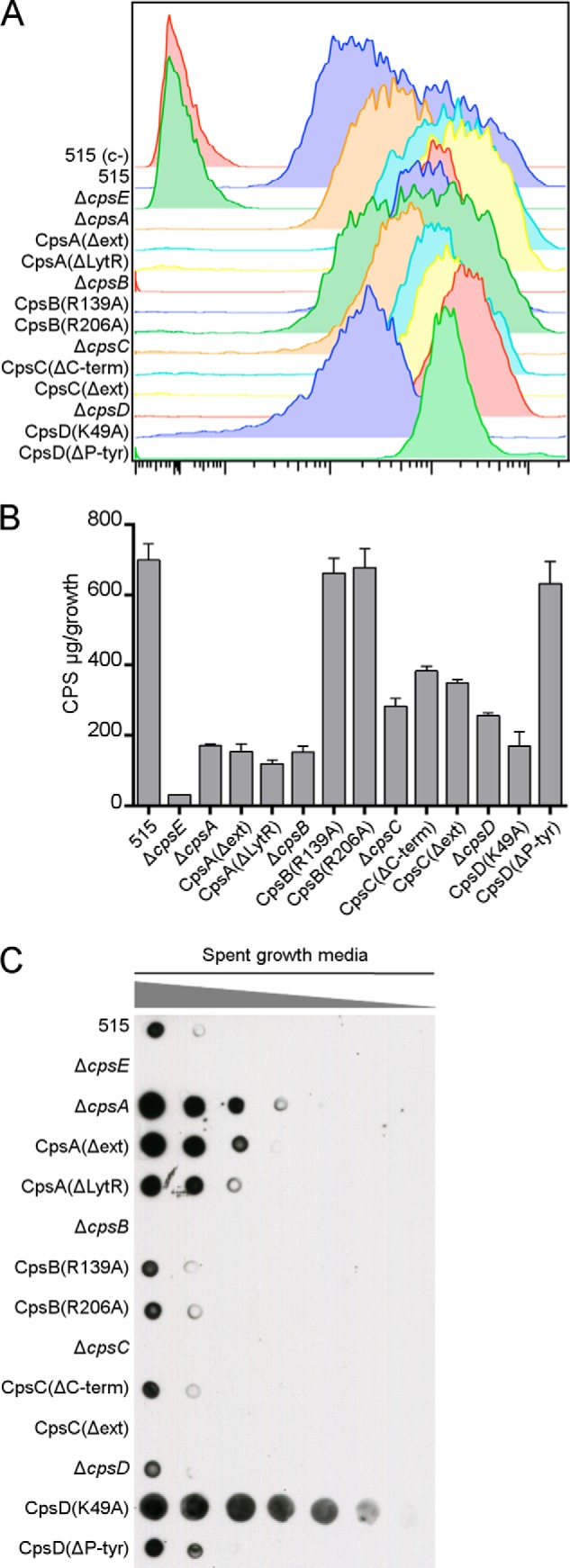
Differences in CPS localization in mutant strains. A, WT and cps mutant strains were incubated with a primary α-CPSIa mAb followed by a secondary goat α-mouse antibody conjugated to allophycocyanin and analyzed by flow cytometry. The unencapsulated ΔcpsE strain was included as a negative control. B, CPS from bacterial pellets was quantified by a resorcinol assay. Bars, means of three independent experiments performed with triplicate samples. Error bars, S.D. C, dot blot showing serial dilutions (1:2) of spent growth media spotted on a nitrocellulose membrane and probed with an α-CPSIa mAb.
In S. agalactiae, the CPS is covalently attached to the cell wall peptidoglycan (10). Certain mutant strains had little CPS attached to the bacterial cell surface (Fig. 3B) despite having normal cps operon transcription. We hypothesized that some of the CPS produced may be shed. To examine this possibility, serial dilutions of spent growth media were spotted on a nitrocellulose membrane and probed with an α-CPSIa mAb. All of the cpsA mutants showed an increased amount of CPS in the medium compared with the WT (Fig. 3C). The same phenotype was observed for the mutant strain with impaired autokinase activity (CpsD(K49A)) but not for the other cpsD mutants. The mutants ΔcpsB, ΔcpsC, and CpsC(Δext) showed no detectable CPS in the culture supernatant, comparable with the negative control ΔcpsE, which does not produce any CPS whatsoever. The ΔcpsA and CpsD(K49A) strains were chromosomally complemented, and the amount of CPS released in the media by the complemented strains was restored to wild type levels (data not shown). The mutant strains with increased CPS in the medium concomitantly showed a significant reduction in the amount of CPS attached to the bacterial surface (Fig. 3B), suggesting a defective attachment of CPS to the cell wall. These data suggest that CpsA could be the enzyme responsible for attachment of CPS to the cell wall and that CpsD autokinase activity is required. Specifically, the LytR domain of CpsA seems to be necessary for CPS attachment to the surface because CPS is shed when this domain is removed.
CPS Length Anomalies Are Observed in Selected Mutant Strains
CPS from bacteria was extracted by mutanolysin treatment, separated on a polyacrylamide gel, and examined by immunoblot with an α-CPSIa mAb (Fig. 4A). Aberrant CPS length was observed in some mutant strains when compared with CPS extracted from the WT. We observed that strains where CpsD is absent/non-functional (the three cpsD mutant strains and the CpsC(ΔC-term) mutant) displayed CPS with an unusually high molecular weight. Interestingly, these mutant strains are those where phosphorylation of CpsD was absent, suggesting that phosphorylation of CpsD may influence CPS chain length. In contrast, a very short CPS was produced by the strains lacking the extracellular domain of CpsC. Surprisingly, we could not detect any CPS in samples from ΔcpsB mutant by Western blot, despite previously having confirmed CPS production by FACS analysis and CPS quantification (Fig. 3, A and B).
FIGURE 4.
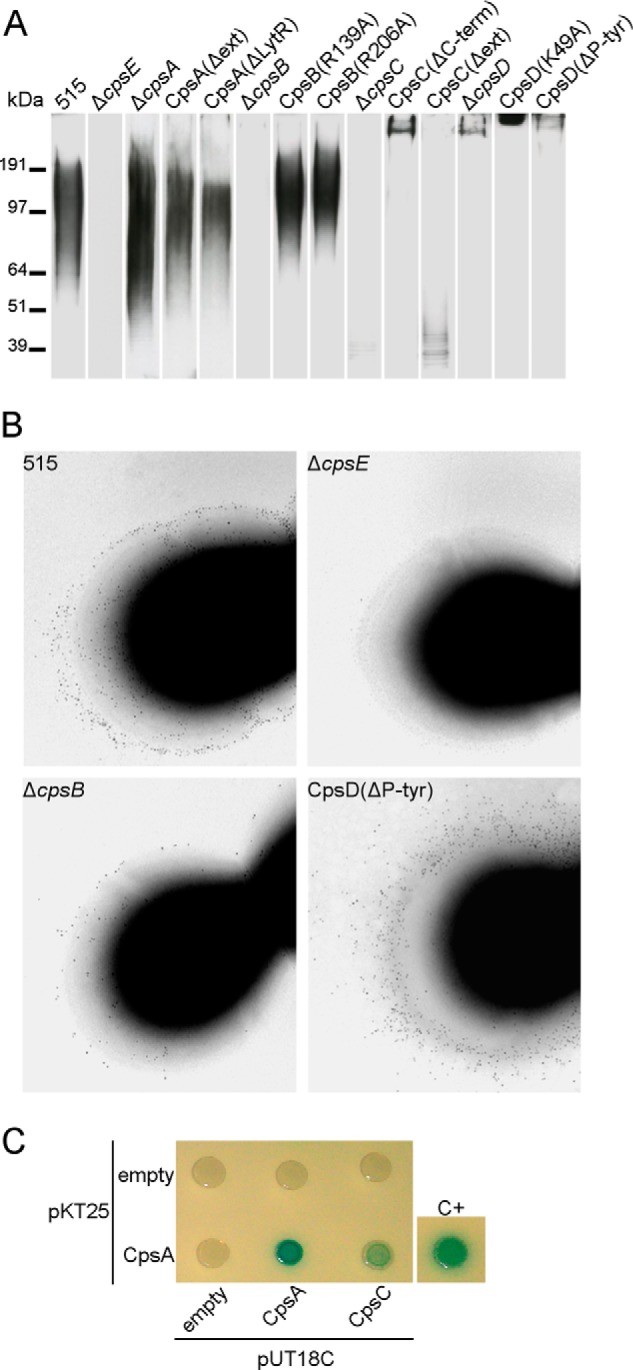
CPS length differences in the WT and cps mutant strains. A, Western blot of CPS bacterial surface extracts (top). CPS was detected with an α-CPSIa mAb. A protein molecular weight marker is included for approximate comparison. Lanes were exposed for different times, in order to permit visualization of CPS from all strains. B, immunogold transmission electron microscopy on whole bacteria using an α-CPSIa mAb as primary antibody and a secondary gold bead-conjugated antibody. Bacterial strains are indicated. C, bacterial two-hybrid analysis. T25-CpsA was tested for interaction with T18-CpsA and T18-CpsC. T18 plasmids were used as negative control. The positive control (C+) is the GCN4 leucine zipper protein provided by the manufacturer in plasmids pKT25-zip and pUT18C-zip (28). Formation of blue colonies indicates that a protein-protein interaction occurred.
Mutant strains that exhibited aberrant CPS phenotypes (i.e. ΔcpsB and all of the cpsC and cpsD mutant strains) were chromosomally complemented. CPS extracts were prepared and analyzed by Western blot (data not shown), and all of the complemented strains appeared indistinguishable from the wild type, confirming that the phenotypes observed are solely due to the specific mutations introduced.
Immunogold transmission electron microscopy was performed in an attempt to visualize the CPS at the bacterial surface (Fig. 4B). Wild type bacteria were observed both as electron-dense diplococci or chains of cocci. Gold beads linked to α-CPSIa were uniformly distributed in a thin layer peripheral to the bacterial surface. The negative control (ΔcpsE), devoid of CPS, had very few if any beads associated with the bacteria. In the CpsD(ΔP-Tyr) mutant, beads formed a wider layer around the bacterial periphery with very few beads in close proximity to the bacteria. In addition, scattered beads were also observed at a large distance from the bacterial surface, suggesting a more extended CPS. The ΔcpsB mutant, in comparison, showed a distribution of beads similar to the WT but with much fewer gold beads. This is consistent with results shown in Fig. 3B indicating a lower CPS amount in this mutant.
As seen in Western blot experiments (Fig. 4A), mutant strains lacking CpsC or the extracellular domain of CpsC produced a short CPS, suggesting that the extracellular part of CpsC is in some way assisting polymerization. We hypothesized that CpsC interferes with CpsA termination of CPS polymerization. This implied a possible interaction between CpsC and CpsA, which was investigated using a bacterial two-hybrid assay. We observed a CpsA self-interaction, suggesting a possible oligomerization of CpsA. Interestingly, we also observed an association between heterologously expressed CpsC and CpsA (Fig. 4C). Such a direct interaction has not been reported previously, and it suggests that CpsC may be forming a transient or stable complex with CpsA, thereby modulating the attachment of CPS to the cell wall.
Biochemical Characterization of the CPS in Selected Mutant Strains
On the basis of the Western blot data, we selected a panel of strains exhibiting different CPS properties for further analytical characterization of the capsular polysaccharides. The ΔcpsA and CpsD(K49A) strains were selected because they partially release the CPS in the growth medium. CpsC(Δext) was chosen because it produces a very short polysaccharide. One-liter cultures were used to obtain bacterial pellets that underwent alkaline treatment, and the CPS was subsequently purified by ethanol precipitation and diafiltration. A simplified procedure was set up to purify the CPS from the growth media. The NMR analysis confirmed the saccharide structural identity of purified CPS obtained from wild type and mutant strains, irrespective of whether they were derived from media or bacteria (data not shown). In the ΔcpsA and CpsD(K49A) strains, the amount of CPS collected from the media represented the majority of the total CPS produced (70 and 77%, respectively) (Table 3). In comparison, very little CPS was purified from the medium of the wild type strain 515. The relative molecular weight distribution of purified CPS was determined by HPLC using a pullulan reference standard to build a calibration curve. The average chain length of the CPS purified from the media of the two mutant strains was significantly higher than that of the CPS of the wild type strain (Table 3 and Fig. 5). The ΔcpsA mutant released a CPS ∼5–10 times longer than the wild type. Interestingly, the CPS purified from the bacterial surface of ΔcpsA strain was instead comparable with that of the wild type.
TABLE 3.
Biochemical characterization of the purified CPS from selected mutant strains
Quantification of the CPS purified from bacterial pellets and from spent growth media from 1-liter cultures. The average molecular mass of CPS was estimated by HPLC-SEC. The mass range represents 95% of the area of the molecular size distribution. NA, not available.
| Strain | Fraction | CPS | Mass | Mass range |
|---|---|---|---|---|
| mg | kDa | kDa | ||
| 515 | Bacteria | 4.1 | 167 | 49–616 |
| Medium | 0.5 | NA | NA | |
| ΔcpsA | Bacteria | 2.8 | 237 | 62–1075 |
| Medium | 6.7 | >1,330 | 115 to >1,330 | |
| CpsC(Δext) | Bacteria | 3.3 | 55 | 23–136 |
| CpsD(K49A) | Bacteria | 1.7 | >1,330 | 1013 to >1,330 |
| Medium | 5.6 | >1,330 | >1,330 |
FIGURE 5.
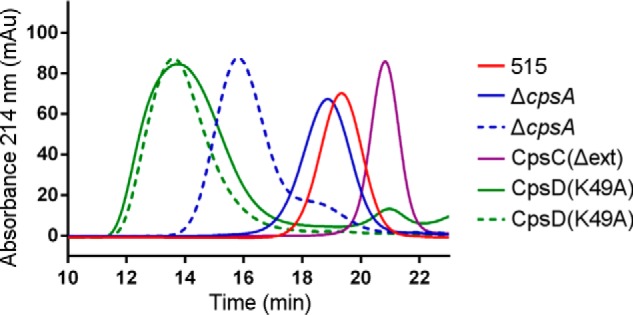
Molecular size analysis of CPS from selected strains. Shown are elution profiles for CPS extracts purified from bacterial pellets (solid lines) and from spent growth media (dotted lines) analyzed by SEC-HPLC. The molecular size of the polysaccharide was calculated by using a calibration curve with pullulan standards.
Using the CpsD(K49A) mutant, we observed that the purified CPS from both fractions exhibits an extremely high molecular weight distribution (more than 10 times longer than the WT), to the point where further size estimation was impossible. We also investigated the CPS purified from the bacterial surface of the mutant strain CpsC(Δext), which showed shorter surface CPS in the immunoblot experiments (Fig. 4A). The amount of CPS extracted from the bacterial pellet of this mutant was higher than in the other mutant strains, and the molecular weight distribution of the purified CPS determined by HPLC indicated a 6-fold smaller CPS compared with the WT (Table 3 and Fig. 5). The growth medium of this strain was not analyzed because previous experiments suggested a minimal release of CPS (Fig. 3C). In conclusion, if CpsA is disrupted, the amount of shed CPS increases dramatically, and that CPS is very long, whereas retained CPS is shorter than that of the wild type. Concomitantly, if CpsD is unable to autophosphorylate, then attached and released CPS are both longer than the wild type, and increased shedding is again observed. Finally, it is noteworthy that shed CPS purified from medium was consistently much longer than WT CPS from the bacterial surface.
DISCUSSION
The cpsABCD genes are relatively well conserved intra- and interspecies (12, 13). In this work, we focused on these four conserved genes and their role in S. agalactiae CPS biosynthesis. Previous studies on homologous proteins from S. pneumoniae have provided molecular details on the phosphorylation and dephosphorylation involving CpsB, -C, and -D (18, 19, 33), but the role of this phosphoregulatory system in the context of the CPS biosynthesis is not completely understood. Moreover, the notion that similar events may be occurring in S. agalactiae is merely an argument by analogy. We attempted mutational studies to experimentally elucidate the role of CpsABCD in S. agalactiae CPS biosynthesis, including mutations that could shed light on the potential interdependencies between these proteins. Our data suggest that CpsA, -B, -C, and -D proteins are not essential for the biosynthesis of the capsular polysaccharide repeating unit because all of the mutant strains retained the ability to produce a CPS recognizable by monoclonal antibodies against the wild type CPS. However, we observed differences in CPS length and localization in our mutant strains, suggesting that these proteins are involved in controlling CPS elongation and attachment to the cell wall. Following is a step-by-step discussion of the working model we propose for CpsABCD (Fig. 6).
FIGURE 6.
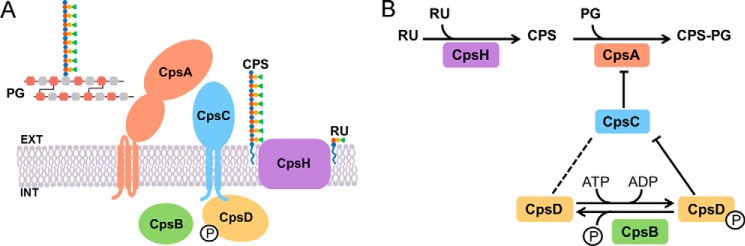
Model of CpsABCD involvement in CPS biosynthesis. A, topology and subcellular localization of the CpsABCD proteins and of the CpsH polymerase was predicted using PSORTb and Octopus. The repeating unit (RU), the capsular polysaccharide (CPS), and the cell wall peptidoglycan (PG) are also represented. B, schematic representation of the working model proposed for the CpsABCD proteins. Arrows, enzymatic reactions; bar-headed line, an inhibitory effect; dotted line, an interdependency.
In the final steps of CPS biosynthesis, the newly synthesized repeating unit (RU) anchored to a polyisoprenoid phosphate lipid is flipped to the outer side of the bacterial membrane, where CpsH is presumably responsible for the polymerization of the repeating units (Fig. 6A). By analogy with other Wzy-dependent systems, such polymerization occurs bottom-up. The nascent CPS is removed from the lipid through a phosphotransferase reaction and subsequently linked to a single membrane-anchored repeating unit (9). The final product is a CPS that is removed from the membrane lipid and covalently attached to GlcNAc in the peptidoglycan backbone (CPS-PG) (10). This linkage effectively renders further polymerization impossible. Our results suggest that the enzyme responsible for this activity is CpsA. In fact, we observed that all of the cpsA mutants showed defects in CPS attachment to the bacterial surface, and, as a corollary, increased amounts of CPS were shed into the growth medium. CpsA belongs to the LCP (LytR-CpsA-Psr) protein family, together with two paralogues, and it was suggested that these enzymes are involved in the final steps of cell wall assembly (39). A study of the homologous Cps2A protein in S. pneumoniae proposed that it may be responsible for transfer of CPS from the membrane lipid to the cell wall peptidoglycan (16). However, the deletion of cps2A in S. pneumoniae was not sufficient to obtain a clear CPS release phenotype, possibly due to redundancy of LCP protein activities (17). In comparison, we observed increased CPS release for all of the cpsA mutant strains, although attachment of CPS to the bacterial surface was not abolished completely. Thus, a possible redundancy between LCP proteins remains a possibility. It is noteworthy that we find no evidence for CpsA involvement in the transcriptional regulation of the cps operon, in contrast to previous reports in the literature (14, 15).
We speculate that the CpsH polymerase and CpsA compete for the same substrate (the nascent CPS). Whereas the enzymatic reaction involving CpsH results in elongation of the CPS by one repeating unit at a time, the CpsA reaction instead terminates elongation by securing CPS to the cell wall. We suggest that the activity of CpsA is moderated by CpsC (Fig. 6B). This notion is supported by the direct interaction of CpsC and CpsA in a bacterial two-hybrid system (Fig. 4C). Moreover, deletion of the extracellular part of CpsC results in abnormally short CPS, suggesting a premature termination of CPS synthesis by CpsA (Fig. 4A).
An interaction between CpsC and CpsD was also observed, strictly dependent on the presence of the short 33-aa intracellular C-terminal tail of CpsC (Fig. 2B). Homologues of CpsC and CpsD in Gram-negative bacteria are found as a single multidomain protein (36, 40). Taken together, this suggests that CpsC and CpsD form a heterodimer or more complex multimers and act in concert. We show that CpsD is an autokinase and phosphorylates tyrosines in its C terminus. In our model, we propose that the phosphorylation state of CpsD directs the conformation of the CpsC extracellular domain through interaction with the C-terminal tail (Fig. 6B). The notion of CpsCD acting in concert is supported by a model of the homologous proteins CapAB in S. aureus, suggesting an octamer complex with conformational changes induced in response to the phosphorylation state of CapB (36).
Among our mutant strains, we observed both very long and very short CPS. We believe that these phenotypes are a result of CpsCD exerting control over the action of CpsA (i.e. a very short CPS suggests unchecked CpsA action and premature termination of CPS biosynthesis). CpsCD can be considered to have entered a “permissive” state, which coincides with CpsD being hyperphosphorylated. In contrast, when CpsD is absent or non-phosphorylated (CpsC(ΔC-term) and all of the cpsD mutants), long CPS are produced, implying that the termination of CPS biosynthesis is impeded. We also observed that when the CpsD protein is in full-length form but non-functional (CpsD(K49A) mutant), the CPS is not only longer than in the wild type; it is also released into the medium. This phenotype is similar to those observed for the cpsA mutants and further supports the notion that CpsD dephosphorylation is directly or indirectly inhibiting CpsA activity. Admittedly, the data on mutants ΔcpsD, Cps(ΔP-Tyr), and CpsC(ΔC-term) are not immediately consistent with this model because the CPS is still attached to the bacterial surface despite having an increased size. However, these mutants all have in common structural truncations that may affect the integrity of the CpsCD complex, resulting in an anomalous configuration with unpredictable consequences. Theoretically possible alternatives to our model include a direct inhibition of polymerization by CpsA or facilitation by CpsC of the polymerization process. In our view, both possibilities are less consistent with the data presented here, compared with the model we propose.
A comparison between the phenotypes of mutant strains ΔcpsA and CpsD(K49A) presents an interesting enigma. In both cases, a majority of the CPS is shed into the medium and is also unusually long. Prior to PG attachment by CpsA, the CPS is tethered to the membrane through the lipid moiety. If the CPS is not transferred from the lipid moiety to the PG, polymerization continues unhindered and results in CPS polymers that are 10-fold longer or more compared with the wild type size. We speculate that shedding occurs because the lipid moiety alone is insufficient to keep such a large molecule tethered in the membrane through hydrophobic interaction. In the ΔcpsA strain, only normal sized CPS is found attached, whereas in the CpsD(K49A) mutant, the attached CPS is as long as the shed CPS. A difference between the mutants is that the CpsD(K49A) has a fully functional CpsA, although the CpsCD is in a permanent “non-permissive” state in relation to CpsA. On the other hand, in the case of the ΔcpsA mutant, CpsCD undergoes normal phosphorylation cycling and will thus periodically enter a “permissive” state, where the CPS is subjective to hydrolysis and may become anchored to PG by other LCP proteins, as suggested previously for S. pneumoniae (17). At present, we do not have a clear understanding of how these mutations result in two somewhat different phenotypes.
In summary, this work examines the concerted action of CpsABCD in the Gram-positive bacterium S. agalactiae. Through the use of multiple functional and structural mutations, the resulting phenotypes allowed us to approach the proteins as a system and define interdependencies. CpsABCD sit at the finishing line of CPS biosynthesis, and the cyclic phosphorylation of CpsD is a main switch that ensures secure attachment to the cell wall, indirectly determining the average length of CPS. A steady state is obtained through autophosphorylation of CpsD and dephosphorylation by CpsB. Perturbances in this system lead to distinct anomalies in capsular localization and polymer length. Apparently, the bacteria are employing a sweet spot in the CpsABCD system, where the current equilibrium results in CPS of a “suitable” length that is securely anchored to the cell surface. The raison d'ètre for that particular design of CPS remains a fascinating subject for future studies.
Acknowledgments
We are grateful to Vittoria Pinto (Novartis Vaccines) for support with NMR spectroscopy and the resorcinol-hydrochloric acid assay. We thank Maria Giuliani and Silvia Mancianti (Novartis Vaccines) for support with protein expression, purification, and formulation for mouse immunization. We thank Fabiola Giusti for assistance with electron microscopy. Finally, we thank Isabel Delany (Novartis Vaccines) for helpful discussions.
Drs. Guido Grandi and Immaculada Margarit Y Ros hold stock options in Novartis Vaccines.
- GBS
- group B Streptococcus
- CPS
- capsular polysaccharide(s)
- THB
- Todd-Hewitt broth
- aa
- amino acid(s)
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- SEC
- size exclusion chromatography
- PG
- peptidoglycan.
REFERENCES
- 1. Schuchat A. (1998) Epidemiology of group B streptococcal disease in the United States: shifting paradigms. Clin. Microbiol. Rev. 11, 497–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barton L. L., Feigin R. D., Lins R. (1973) Group B β hemolytic streptococcal meningitis in infants. J. Pediatr. 82, 719–723 [DOI] [PubMed] [Google Scholar]
- 3. Franciosi R. A., Knostman J. D., Zimmerman R. A. (1973) Group B streptococcal neonatal and infant infections. J. Pediatr. 82, 707–718 [DOI] [PubMed] [Google Scholar]
- 4. Baker C. J., Barrett F. F. (1973) Transmission of group B streptococci among parturient women and their neonates. J. Pediatr. 83, 919–925 [DOI] [PubMed] [Google Scholar]
- 5. Tudela C. M., Stewart R. D., Roberts S. W., Wendel G. D., Jr., Stafford I. A., McIntire D. D., Sheffield J. S. (2012) Intrapartum evidence of early-onset group B streptococcus. Obstet. Gynecol. 119, 626–629 [DOI] [PubMed] [Google Scholar]
- 6. Marques M. B., Kasper D. L., Pangburn M. K., Wessels M. R. (1992) Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect. Immun. 60, 3986–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van den Biggelaar A. H., Pomat W. S. (2013) Immunization of newborns with bacterial conjugate vaccines. Vaccine 31, 2525–2530 [DOI] [PubMed] [Google Scholar]
- 8. Avci F. Y., Kasper D. L. (2010) How bacterial carbohydrates influence the adaptive immune system. Annu. Rev. Immunol. 28, 107–130 [DOI] [PubMed] [Google Scholar]
- 9. Yother J. (2011) Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu. Rev. Microbiol. 65, 563–581 [DOI] [PubMed] [Google Scholar]
- 10. Deng L., Kasper D. L., Krick T. P., Wessels M. R. (2000) Characterization of the linkage between the type III capsular polysaccharide and the bacterial cell wall of group B Streptococcus. J. Biol. Chem. 275, 7497–7504 [DOI] [PubMed] [Google Scholar]
- 11. Rubens C. E., Heggen L. M., Haft R. F., Wessels M. R. (1993) Identification of cpsD, a gene essential for type III capsule expression in group B streptococci. Mol. Microbiol. 8, 843–855 [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto S., Miyake K., Koike Y., Watanabe M., Machida Y., Ohta M., Iijima S. (1999) Molecular characterization of type-specific capsular polysaccharide biosynthesis genes of Streptococcus agalactiae type Ia. J. Bacteriol. 181, 5176–5184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cieslewicz M. J., Chaffin D., Glusman G., Kasper D., Madan A., Rodrigues S., Fahey J., Wessels M. R., Rubens C. E. (2005) Structural and genetic diversity of group B streptococcus capsular polysaccharides. Infect. Immun. 73, 3096–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cieslewicz M. J., Kasper D. L., Wang Y., Wessels M. R. (2001) Functional analysis in type Ia group B Streptococcus of a cluster of genes involved in extracellular polysaccharide production by diverse species of streptococci. J. Biol. Chem. 276, 139–146 [DOI] [PubMed] [Google Scholar]
- 15. Hanson B. R., Runft D. L., Streeter C., Kumar A., Carion T. W., Neely M. N. (2012) Functional analysis of the CpsA protein of Streptococcus agalactiae. J. Bacteriol. 194, 1668–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawai Y., Marles-Wright J., Cleverley R. M., Emmins R., Ishikawa S., Kuwano M., Heinz N., Bui N. K., Hoyland C. N., Ogasawara N., Lewis R. J., Vollmer W., Daniel R. A., Errington J. (2011) A widespread family of bacterial cell wall assembly proteins. EMBO J. 30, 4931–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eberhardt A., Hoyland C. N., Vollmer D., Bisle S., Cleverley R. M., Johnsborg O., Håvarstein L. S., Lewis R. J., Vollmer W. (2012) Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb. Drug Resist. 18, 240–255 [DOI] [PubMed] [Google Scholar]
- 18. Morona J. K., Paton J. C., Miller D. C., Morona R. (2000) Tyrosine phosphorylation of CpsD negatively regulates capsular polysaccharide biosynthesis in Streptococcus pneumoniae. Mol. Microbiol. 35, 1431–1442 [DOI] [PubMed] [Google Scholar]
- 19. Bender M. H., Yother J. (2001) CpsB is a modulator of capsule-associated tyrosine kinase activity in Streptococcus pneumoniae. J. Biol. Chem. 276, 47966–47974 [DOI] [PubMed] [Google Scholar]
- 20. Perez-Casal J., Price J. A., Maguin E., Scott J. R. (1993) An M protein with a single C repeat prevents phagocytosis of Streptococcus pyogenes: use of a temperature-sensitive shuttle vector to deliver homologous sequences to the chromosome of S. pyogenes. Mol. Microbiol. 8, 809–819 [DOI] [PubMed] [Google Scholar]
- 21. Horton R. M., Hunt H. D., Ho S. N., Pullen J. K., Pease L. R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68 [DOI] [PubMed] [Google Scholar]
- 22. Olsen D. B., Eckstein F. (1989) Incomplete primer extension during in vitro DNA amplification catalyzed by Taq polymerase: exploitation for DNA sequencing. Nucleic Acids Res. 17, 9613–9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Framson P. E., Nittayajarn A., Merry J., Youngman P., Rubens C. E. (1997) New genetic techniques for group B streptococci: high-efficiency transformation, maintenance of temperature-sensitive pWV01 plasmids, and mutagenesis with Tn917. Appl. Environ. Microbiol. 63, 3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Faralla C., Metruccio M. M., De Chiara M., Mu R., Patras K. A., Muzzi A., Grandi G., Margarit I., Doran K. S., Janulczyk R. (2014) Analysis of two-component systems in group B Streptococcus shows that RgfAC and the novel FspSR modulate virulence and bacterial fitness. MBio 5, e00870–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wessels M. R., Paoletti L. C., Kasper D. L., DiFabio J. L., Michon F., Holme K., Jennings H. J. (1990) Immunogenicity in animals of a polysaccharide-protein conjugate vaccine against type III group B Streptococcus. J. Clin. Invest. 86, 1428–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Svennerholm L. (1957) Quantitative estimation of sialic acids: II. A colorimetric resorcinol-hydrochloric acid method. Biochim. Biophys. Acta 24, 604–611 [DOI] [PubMed] [Google Scholar]
- 27. Berti F., Campisi E., Toniolo C., Morelli L., Crotti S., Rosini R., Romano M. R., Pinto V., Brogioni B., Torricelli G., Janulczyk R., Grandi G., Margarit I. (2014) Structure of the type IX group B Streptococcus capsular polysaccharide and its evolutionary relationship with types V and VII. J. Biol. Chem. 289, 23437–23448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karimova G., Pidoux J., Ullmann A., Ladant D. (1998) A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barocchi M. A., Ries J., Zogaj X., Hemsley C., Albiger B., Kanth A., Dahlberg S., Fernebro J., Moschioni M., Masignani V., Hultenby K., Taddei A. R., Beiter K., Wartha F., von Euler A., Covacci A., Holden D. W., Normark S., Rappuoli R., Henriques-Normark B. (2006) A pneumococcal pilus influences virulence and host inflammatory responses. Proc. Natl. Acad. Sci. U.S.A. 103, 2857–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu N. Y., Wagner J. R., Laird M. R., Melli G., Rey S., Lo R., Dao P., Sahinalp S. C., Ester M., Foster L. J., Brinkman F. S. (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Viklund H., Elofsson A. (2008) OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24, 1662–1668 [DOI] [PubMed] [Google Scholar]
- 32. Hanson B. R., Lowe B. A., Neely M. N. (2011) Membrane topology and DNA-binding ability of the streptococcal CpsA protein. J. Bacteriol. 193, 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Byrne J. P., Morona J. K., Paton J. C., Morona R. (2011) Identification of Streptococcus pneumoniae Cps2C residues that affect capsular polysaccharide polymerization, cell wall ligation, and Cps2D phosphorylation. J. Bacteriol. 193, 2341–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hagelueken G., Huang H., Mainprize I. L., Whitfield C., Naismith J. H. (2009) Crystal structures of Wzb of Escherichia coli and CpsB of Streptococcus pneumoniae, representatives of two families of tyrosine phosphatases that regulate capsule assembly. J. Mol. Biol. 392, 678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soulat D., Jault J. M., Duclos B., Geourjon C., Cozzone A. J., Grangeasse C. (2006) Staphylococcus aureus operates protein-tyrosine phosphorylation through a specific mechanism. J. Biol. Chem. 281, 14048–14056 [DOI] [PubMed] [Google Scholar]
- 36. Olivares-Illana V., Meyer P., Bechet E., Gueguen-Chaignon V., Soulat D., Lazereg-Riquier S., Mijakovic I., Deutscher J., Cozzone A. J., Laprévote O., Morera S., Grangeasse C., Nessler S. (2008) Structural basis for the regulation mechanism of the tyrosine kinase CapB from Staphylococcus aureus. PLoS Biol. 6, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Henriques M. X., Rodrigues T., Carido M., Ferreira L., Filipe S. R. (2011) Synthesis of capsular polysaccharide at the division septum of Streptococcus pneumoniae is dependent on a bacterial tyrosine kinase. Mol. Microbiol. 82, 515–534 [DOI] [PubMed] [Google Scholar]
- 38. Morona J. K., Morona R., Miller D. C., Paton J. C. (2003) Mutational analysis of the carboxy-terminal (YGX)4 repeat domain of CpsD, an autophosphorylating tyrosine kinase required for capsule biosynthesis in Streptococcus pneumoniae. J. Bacteriol. 185, 3009–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hübscher J., Lüthy L., Berger-Bächi B., Stutzmann Meier P. (2008) Phylogenetic distribution and membrane topology of the LytR-CpsA-Psr protein family. BMC Genomics 9, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whitfield C., Paiment A. (2003) Biosynthesis and assembly of Group 1 capsular polysaccharides in Escherichia coli and related extracellular polysaccharides in other bacteria. Carbohydr. Res. 338, 2491–2502 [DOI] [PubMed] [Google Scholar]