

Background: GPCRs can activate selective signaling pathways according to the nature of the bound ligand.
Results: Coupling selectivity of chemokine receptors CCR2, CCR5, and CCR7 with G protein subtypes was measured and compared with data obtained with other functional readouts.
Conclusion: Some signaling bias was detected at CCR2 and CCR5.
Significance: Signaling bias appears relatively subtle for natural ligands such as chemokines.
Keywords: Bioluminescence Resonance Energy Transfer (BRET), Biosensor, Chemokine, G Protein-coupled Receptor (GPCR), Signaling
Abstract
The ability of G protein-coupled receptors (GPCRs) to activate selective signaling pathways according to the conformation stabilized by bound ligands (signaling bias) is a challenging concept in the GPCR field. Signaling bias has been documented for several GPCRs, including chemokine receptors. However, most of these studies examined the global signaling bias between G protein- and arrestin-dependent pathways, leaving unaddressed the potential bias between particular G protein subtypes. Here, we investigated the coupling selectivity of chemokine receptors CCR2, CCR5, and CCR7 in response to various ligands with G protein subtypes by using bioluminescence resonance energy transfer biosensors monitoring directly the activation of G proteins. We also compared data obtained with the G protein biosensors with those obtained with other functional readouts, such as β-arrestin-2 recruitment, cAMP accumulation, and calcium mobilization assays. We showed that the binding of chemokines to CCR2, CCR5, and CCR7 activated the three Gαi subtypes (Gαi1, Gαi2, and Gαi3) and the two Gαo isoforms (Gαoa and Gαob) with potencies that generally correlate to their binding affinities. In addition, we showed that the binding of chemokines to CCR5 and CCR2 also activated Gα12, but not Gα13. For each receptor, we showed that the relative potency of various agonist chemokines was not identical in all assays, supporting the notion that signaling bias exists at chemokine receptors.
Introduction
G protein-coupled receptors (GPCRs)4 constitute one of the largest families of cell surface receptors. They are involved in many physiological processes and are targeted by ∼40% of all modern medicinal drugs (1). Agonist binding to a receptor promotes conformational changes that result in the tight coupling of the receptor to a heterotrimeric G protein. Thereafter, conformation change within the G protein and the exchange of bound GDP for GTP allow the active Gα-GTP and Gβγ subunits to interact with effectors to transduce the signal initiated by agonist binding (2, 3). In humans, genes encoding at least sixteen Gα subunits, five Gβ subunits, and twelve Gγ subunits have been identified, several of which have splice variants (4). Specific combinations of α, β, and γ subunits in G proteins affects which downstream targets are activated, providing the means to integrate the various signals coming into a cell. In addition to the canonical G protein-dependent pathways, G protein-independent signaling pathways have also been reported (5–7). Of particular interest are arrestins, which were originally considered to be solely involved in receptor desensitization and now have been identified as multifunctional scaffolds interacting with a number of signaling proteins such as MAPKs, PI3K, or protein kinase B (8). Over the past few years, a growing number of biased ligands have been reported that preferentially activate G protein- or arrestin-dependent pathways, and several of these biased ligands were shown to possess distinct functional properties when compared with “balanced” ligands (5, 7).
From a therapeutic point of view, molecules that selectively activate the arrestin pathway without affecting G protein-dependent signaling, or the inverse, could have improved efficacy or decreased side effects (6). Biased signaling therefore appears to be a novel and promising concept that challenges our knowledge of GPCR function and raises new opportunities for the use of these receptors as therapeutic targets (9). It is therefore important to identify ligands and receptors that possess biased signaling properties and to understand how agonist binding induces the selective activation of a signaling pathway within the cell. Chemokines are a good model system for the analysis of GPCR signaling bias. Chemokines are small basic proteins that control the homing and migration of immune cells. Approximately 40 chemokines and more than 20 chemokine receptors have been identified so far (10). Each receptor has its own specific repertoire of chemokine ligands, ranging from a one to a half a dozen. There is therefore significant redundancy in the chemokine system, but relatively little is known regarding the selectivity of pathways activated by chemokines downstream of their common receptor. Signaling selectivity has been first described for the two natural ligands of the chemokine receptor CCR7, CCL19, and CCL21. Both CCL19 and CCL21 were reported to activate G protein-dependent pathways, but only CCL19 was proposed to recruit β-arrestin and activate MAPK Erk1/2 (11–13). As a result, CCR7 became a prototypical example of biased signaling, particularly with respect to natural agonists. Based on these first studies, it was anticipated that other chemokine receptors might display a similar bias by preferentially activating either G protein- or arrestin-dependent pathways. Consistent with this hypothesis, Rajagopal et al. (14) recently reported that other chemokine receptors have different tendencies to induce either G protein- or arrestin-dependent signaling. In addition, selectivity might exist in the activation of different G protein subtypes or isoforms. However, until recently, it was difficult to elucidate which G protein subtype was activated. In this paper, we used bioluminescence resonance energy transfer (BRET) biosensors to monitor the conformational changes in G proteins during their activation and determined which G protein subtypes were activated by various chemokines. We combined the use of these sensors with other functional assays to revisit the signaling bias previously identified for CCR7 and to analyze the signaling selectivity of various chemokine ligands through activation of the closely related receptors CCR2 and CCR5.
EXPERIMENTAL PROCEDURES
Reagents, Plasmids, and Cell Lines
Chemokines and Maraviroc were purchased from R & D Systems. TAK-779 was purchased from the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, NIAID. Plasmids encoding G protein and arrestin constructs were described previously (16). The plasmid encoding the cAMP sensor YFP-Epac-RLuc (CAMYEL) was purchased from ATCC. Human embryonic kidney cells (HEK293T) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). CHO-K1 cells expressing apoaequorin were cultured in Ham's F-12 medium supplemented with 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Cells stably expressing apoaequorin and chemokine receptors were cultured in the presence of 10 μg/ml Zeocin and G418 (Invitrogen).
Binding Assays
Binding experiments were performed as previously described (15). CHO-K1 cells were incubated for 45 min at 25 °C in the assay buffer (50 mm Hepes, pH 7.4, 1 mm CaCl2, 5 mm MgCl2, 250 mm sucrose, 0.5% BSA) with 0.1 nm [125I]CCL2, [125I]CCL4, or [125I]CCL19 as tracers and variable concentrations of unlabeled competitors. Tubes were incubated for 1 h at room temperature, and bound tracer was separated by filtration through GF/B filters presoaked in 1% polyethyleneimine. Filters were counted in a γ-scintillation counter. Binding parameters were determined with the PRISM software (GraphPad Software) using nonlinear regression applied to a single site model. Ki values were calculated from IC50 values based on the Cheng-Prusoff equation.
G-protein BRET Assay
G protein activation was assayed by BRET as previously described (16, 17). Briefly, plasmids encoding G protein biosensors and receptors of interest were cotransfected into HEK293T cells by using the calcium phosphate method. Forty-eight hours after transfection, cells were washed twice with PBS, detached, and resuspended in PBS plus 0.1% (w/v) glucose at room temperature. The cells were then distributed (80 μg of proteins/well) in a 96-well microplate (Optiplate; PerkinElmer Life Sciences). BRET2 between RLuc8 and GFP10 was measured 1 min after addition of coelenterazine 400a/deep blue C (5 μm, Gentaur). BRET readings were collected using an Infinite F200 reader (Tecan Group Ltd). The BRET signal was calculated as the ratio of emission of GFP10 (510–540 nm) to RLuc8 (370–450 nm).
cAMP BRET Assay
cAMP inhibition was assayed by BRET as previously described (18). Briefly, plasmids encoding YFP-Epac-RLuc biosensor and receptors of interest were cotransfected into HEK293T cells by using the calcium phosphate method. Twenty-four hours post-transfection, cells were collected and seeded in 96-well microplates (catalog no. 165306; Nunc) and cultured for an additional 24 h. Cells were then rinsed once with PBS and resuspended in PBS plus 0.1% (w/v) glucose. BRET1 between RLuc and YFP was measured at 20 °C in presence of the RLuc substrate coelenterazine h (5 μm; Promega) and 40 μm of the nonspecific phosphodiesterase inhibitor isobutylmethylxanthine. Chemokines were added 5 min after coelenterazine h, and 5 μm forskolin was added 5 min later. After 5 min of incubation, BRET readings were collected using a Mithras LB940 multilabel reader (Berthold Technologies). The BRET signal was calculated as the ratio of emission of YFP (520–570 nm) to RLuc (370–480 nm).
β-Arrestin BRET Assay
β-Arrestin recruitment was measured by a BRET proximity assay as previously described (17). Briefly, plasmids encoding Rluc-β-arrestin 2 and receptors fused to Venus were cotransfected into HEK293T cells by using the calcium phosphate method. Twenty-four hours post-transfection, cells were collected and seeded in 96-well microplates (catalog no. 165306; Nunc) and cultured for an additional 24 h. Cells were then rinsed once with PBS and incubated in PBS plus 0.1% (w/v) glucose at 25 °C to slow down kinetics of arrestin recruitment and improve temporal resolution. BRET1 between RLuc and YFP was measured after the addition of coelenterazine h (5 μm; Promega). Chemokines were added 5 min after coelenterazine h, and BRET readings were collected using a Mithras LB940 Multilabel Reader (Berthold Technologies). The BRET signal was calculated as the ratio of emission of YFP (520–570 nm) to RLuc (370–480 nm).
Intracellular Calcium Mobilization Assay
Calcium mobilization was measured in HEK293 cells expressing CCR2 or CCR7 with an aequorin-based assay as previously described (15). Because signals induced by chemokines were barely detectable in HEK293 cells expressing CCR5, calcium mobilization was performed in CHO-K1 cells stably expressing CCR5. Briefly, cells expressing apoaequorin and the receptor of interest were incubated for 4 h in the dark in the presence of 5 μm coelenterazine h (Promega) and then diluted before use to reach the appropriate cell density. The cell suspension (25,000 cells/well) was added to wells containing various concentrations of chemokines, and luminescence was recorded for 30 s in an EG&G Berthold luminometer (PerkinElmer Life Sciences).
MAPK Assays
HEK293 cells expressing CCR7 were starved for 12–18 h in serum-free medium prior to stimulation. After stimulation, cells were collected by centrifugation and heated to 100 °C for 5 min in 2× Laemmli sample buffer. Whole cell lysates were resolved on 10% Tris/Tricine polyacrylamide gels and transferred to nitrocellulose membranes. Phosphorylated ERK1/2, total ERK1/2, and β-arrestins were detected by using rabbit polyclonal anti-phospho-ERK1/2 (catalog no. 4370; Cell Signaling; 1:1,000) and anti-ERK1/2 (catalog no. 4695S; Cell Signaling; 1:2,000) antibodies. Chemiluminescent detection was performed using ECL Plus reagent (PerkinElmer Life Sciences).
Bias Analysis
The bias index (β) was estimated by using the following equation, where RA denotes the relative efficacy of a ligand (Lig) through pathway a and pathway b relative to a reference agonist (ref) chosen arbitrary (15).
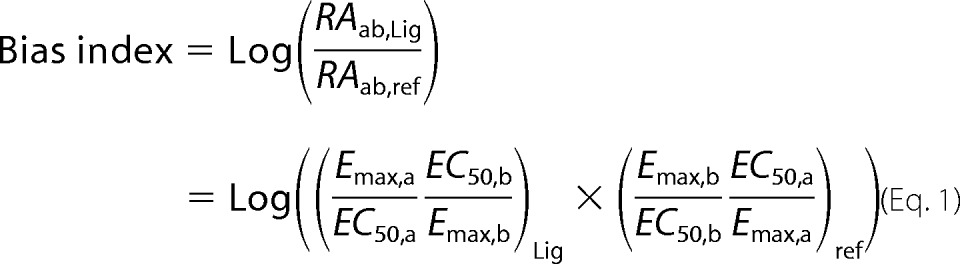 |
Statistical analyses were performed where appropriate using one-way analysis of variance with Dunnett's post-test, and statistical significance related to the reference balanced ligand was taken as p < 0.05.
RESULTS
Signaling Selectivity at Chemokine Receptor CCR7
In a competition binding assay, we first confirmed that CCL19 and CCL21 compete with radiolabeled CCL19 for CCR7 binding with nearly equivalent pIC50 values (Fig. 1A and Table 1). We next investigated the panel of G proteins activated by CCR7 upon binding of CCL19 or CCL21 by using BRET-based biosensors directly monitoring the activation of G proteins. This technology relies on the large interdomain movement that occurs within heterotrimeric G proteins upon GDP/GTP exchange, resulting in a decrease of the BRET signal between probes inserted at specific locations within Gα and Gγ subunits (16, 17). This technology detects early signaling events that occur shortly after receptor stimulation, enabling us to identify the G protein subtypes activated by a receptor. CCR7 was transiently coexpressed in HEK-293T cells with ten different biosensors belonging to the four major classes of G proteins (Gαs, Gαi/o, Gαq/11, and Gα12/13), and stimulated with either CCL19 or CCL21. Among the ten G protein isoforms tested, neither chemokine significantly activated the Gαq, Gα11, Gαs, Gα12, or Gα13 proteins at 100 nm (data not shown). CCL19 binding triggered a significant activation of the three Gαi subtypes (Gαi1, Gαi2, and Gαi3) and the two Gαo isoforms (Gαoa and Gαob) (Fig. 1B). In contrast, CCL21 binding led to less efficient activation of the Gαi/o proteins. A dose-response curve indicated that CCL21 activates Gαi and Gαo at least ten times less potently than CCL19 (Fig. 1, C–E, and Table 1). We are, however, uncertain of the maximal efficacy of Gα activation by CCL21 because we did not saturate the BRET signal at 100 nm. We next measured the inhibition of cAMP production as a representative readout of Gαi/o proteins activation and showed that CCL21 inhibited cAMP production triggered by forskolin at lower potency but similar efficacy compare with CCL19, in complete agreement with data generated with the G protein biosensors (Fig. 1F). We also investigated the ability of CCR7 to recruit β-arrestin by a BRET proximity assay, which measures the energy transfer between β-arrestin-2-Rluc and CCR7 fused to the yellow fluorescent protein Venus. Because of the rapid recruitment of arrestin at 37 °C, our assays were performed at 25 °C to decrease the reaction kinetics and gain temporal resolution. CCL21 (at 100 nm) induced the recruitment of β-arrestin-2 slower than CCL19 (Tau values of 4.6 and 2.4 min, respectively), in perfect agreement with previously published data (11, 12) (Fig. 1G). It should be noted that the maximal BRET value (BRETmax) reached with CCL21 stimulation is also 30% lower than the BRETmax with CCL19 stimulation. This might reflect distinct conformations of receptor-arrestin complexes (12) but is most likely the consequence of decreased arrestin recruitment at the tested concentrations. Indeed, we showed that CCL21 activates β-arrestin-2 ten times less potently than CCL19 (Fig. 1H and Table 1). Finally, we showed that CCL21 also triggered calcium mobilization and ERK1/2 phosphorylation less potently than CCL19 (Fig. 1, I and J). Collectively, our data indicate that CCL21 binds CCR7 as efficiently as CCL19 but is less potent in activating all the assays including G proteins and β-arrestin-2, thus challenging the notion of signaling bias as previously claimed between these two pathways.
FIGURE 1.

A, competition binding assays performed on CHO-K1 cells expressing CCR7. Cells were incubated with 0.1 nm 125I-CCL19 as tracer and unlabeled CCL19 (●) or CCL21 (○) as competitors. The data were normalized for nonspecific binding (0%) in the presence of 300 nm of competitor (CCL19) and specific binding in the absence of competitor (100%). All points were run in triplicate (error bars indicate S.E.). B–E, G protein activation by CCR7. Real time measurement of BRET signal in HEK293T cells coexpressing G protein biosensors and CCR7 and stimulated for 1 min with 50 nm CCL19 (black bars and circles) or CCL21 (open bars and circles) is shown. The results are expressed as the differences in BRET signals measured in the presence and absence of stimulation. The data represent the means ± S.E. of three to six independent experiments. Statistical significance between stimulated and unstimulated cells was assessed using Tukey's test. ***, p < 0.001. F, inhibition of cAMP by CCR7. Measurement of BRET signal in HEK293T cells coexpressing cAMP biosensor and CCR7 and stimulated sequentially by CCL19 (●) or CCL21 (○) and forskolin. The results were normalized for the basal signal in absence of stimulation (0%), and the maximal response was obtained with forskolin only (100%). The data represent the means ± S.E. of three independent experiments. G and H, recruitment of β-arrestin 2 by CCR7. Real time measurement of BRET signal in HEK293T cells expressing β-arrestine2-RLuc8 and CCR7-Venus and stimulated with CCL19 (black circles) or CCL21 (open circles) is shown. Cells were stimulated by 100 nm chemokines in E and for 30 min in F. The results are expressed as the difference in BRET signals measured in the presence and absence of stimulation. The data represent the means ± S.E. of three independent experiments. I, calcium mobilization by CCR7. Calcium mobilization was measured in HEK293 cells using the aequorin-based functional assay. Cells expressing CCR7 were stimulated with increasing concentrations of CCL19 (●) or CCL21 (○), and luminescence was recorded for 30 s. The results were normalized for the basal luminescence in absence of agonist (0%), and the maximal response was obtained with 50 μm acetylcholine (100%). The data represent the means ± S.E. of three independent experiments. J, phosphorylation of ERK1/2 by CCR7. HEK293 cells stably expressing CCR7 were stimulated with 100 nm CCL19 or CCL21 for 2 min. The results are expressed as the ratio between the amount of phospho-ERK1/2 and total ERK1/2 following quantification on Western blots. The data represent the means ± S.E. of three independent experiments.
TABLE 1.
Binding and signaling properties of CCR7
Binding and signaling parameters were measured on HEK293 or CHO-K1 cells expressing CCR7. The pIC50, pEC50 and Emax values were obtained from experiments displayed in Fig. 1. The values represent the means ± S.E. of at least three independent experiments. ND, not determined.
| Ligands | [125I]CCL19 |
Gαi2 |
Gαi3 |
Gαoa |
β-Arr2 |
cAMP |
Ca2+ |
MAPK |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pIC50 | pKi | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | |
| CCL19 | −8.5 ± 0.1 | −8.8 ± 0.1 | −8.1 ± 0.2 | −0.11 ± 0.01 | −8.3 ± 0.1 | −0.19 ± 0.01 | −8.2 ± 0.1 | −0.11 ± 0.01 | −8.6 ± 0.1 | 0.17 ± 0.01 | −9.7 ± 0.2 | 47 ± 3 | −8.1 ± 0.3 | 25 ± 4 | −8.8 ± 0.3 | 1.2 ± 0.2 |
| CCL21 | −8.1 ± 0.1 | −8.4 ± 0.1 | ND | ND | −7.3 ± 0.2 | −0.15 ± 0.03 | −7.6 ± 0.1 | −0.07 ± 0.01 | ND | ND | −8.8 ± 0.3 | 56 ± 8 | ND | ND | −7.4 ± 0.5 | 1.2 ± 0.4 |
Activation of G Protein Subtypes by CCR5
To further analyze the signaling selectivity of chemokine receptors, we also investigated the activation of G proteins through CCR5, a receptor that binds multiple chemokines. We first measured the binding affinities of some of these chemokines (CCL-3, -4, -5, -8, and -13) and showed that they compete with radiolabeled CCL4 for CCR5 binding with calculated pIC50 values consistent with previous estimates (Fig. 2 and Table 2) (19). We next compared the ability of these chemokines to trigger G protein activation by using BRET-based G protein biosensors. CCR5 was coexpressed with the various biosensors and stimulated with the agonist chemokines CCL3, CCL4, CCL5, CCL8, and CCL13. As controls, cells were also exposed to the antagonist chemokine CCL7 and to TAK779 and Maraviroc, two small molecules displaying inverse agonist properties. Stimulation of CCR5 by CCL3, CCL4, CCL5, CCL8, and CCL13 at 100 nm significantly activated all Gαi and Gαo isoforms and also Gα12, with CCL13 activating each G protein the least (Fig. 3). Stimulation of cells expressing the biosensors without CCR5 was used as negative control and generated signals of much weaker magnitude (Fig. 3). No significant BRET signal was detected for the Gαq, Gα11, Gαs, or Gα13 proteins (Fig. 3 and data not shown). The chemokine antagonist CCL7 had no significant effect, whereas TAK779 and Maraviroc displayed a tendency to increase the BRET signal for some biosensors (Gαi2, Gαi3, and Gαoa) in the presence of CCR5, in agreement with their inverse agonist properties. Dose-response curves performed with some representative G protein biosensors also indicated that the calculated pEC50 values are similar for the different Gα proteins. CCL5 was the most potent agonist in these assays, despite having a similar CCR5 binding affinity than CCL3 and CCL4. CCL13 was the least potent agonist, in agreement with its lower affinity for CCR5 (Fig. 4 and Table 2). However, the calculated Emax values were more variable for specific chemokines and Gα proteins, with CCL13 being the agonist with the lowest efficacy. Collectively, our data did not reveal major activation bias among chemokines and between the Gαi, Gαo, and Gα12 protein subtypes, although CCL5 displayed higher potency in most assays, and CCL8 tended to be less effective in activating Gαo proteins. We next used cAMP accumulation as a representative readout of Gαi/o protein activation. As expected, all chemokine agonists inhibited the cAMP production triggered by forskolin with relative potencies compatible with our binding data. The calculated pEC50 values are lower than those estimated with the G protein biosensors, likely as a consequence of the higher sensitivity of this assay caused by signal amplification along the cascade (Fig. 5A and Table 2). CCL13 inhibited cAMP accumulation with a lower potency, but with a similar efficiency compared with the other agonist chemokines. Finally, we showed that agonist chemokines activated calcium mobilization with similar pEC50 and Emax values, with the exception of CCL13, which was again less potent in this assay (Fig. 5B and Table 2). In contrast to the stimulation of CCR7, our data showed that stimulation of CCR5 triggered the activation of G12, indicating that chemokine receptors can activate distinct G proteins. Moreover, our results also showed that CCR5 activated Gα12 but not Gα13, although they belong to the same family (Fig. 3).
FIGURE 2.
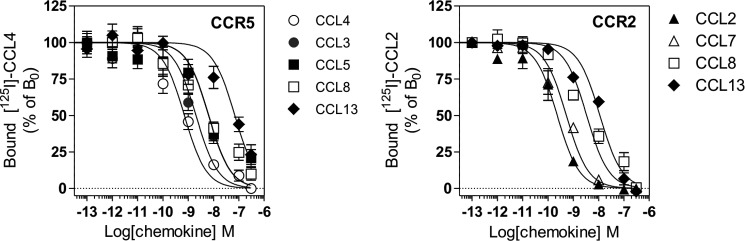
A, competition binding assays performed on cells expressing CCR5. CHO-K1 cells expressing CCR5 were incubated with 0.1 nm 125I-CCL4 as tracer and unlabeled CCL3, CCL4, CCL5, CCL8, and CCL13 as competitors. The data were normalized for nonspecific binding (0%) in the presence of 300 nm CCL4 and specific binding in the absence of competitor (100%). All points were run in triplicate (error bars indicate S.E.). B, competition binding assays performed on cells expressing CCR2. CHO-K1 cells expressing CCR2 were incubated with 0.05 nm 125I-CCL2 as tracer and unlabeled CCL5, CCL7, CCL8, and CCL13 as competitors. The data were normalized for nonspecific binding (0%) in the presence of 300 nm CCL2 and specific binding in the absence of competitor (100%). All points were run in triplicate (error bars indicate S.E.).
TABLE 2.
Binding and signaling parameters of CCR5
Binding and functional parameters were measured on HEK293 or CHO-K1 cells expressing CCR5. The pIC50, pEC50, and Emax values were obtained from experiments as displayed in Figs. 2, 4, 5, and 9. The values represent the means ± S.E. of at least three independent experiments. ND, not determined.
| Ligands | [125I]CCL4 |
Gαi1 |
Gαob |
Gα12 |
β-Arr2 |
cAMP |
Ca2+ |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pIC50 | pKi | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | |
| CCL3 | −8.7 ± 0.1 | −8.9 ± 0.1 | −8.1 ± 0.4 | −0.09 ± 0.02 | −8.1 ± 0.4 | −0.08 ± 0.02 | −8.6 ± 0.3 | −0.017 ± 0.002 | −8.6 ± 0.2 | 0.08 ± 0.01 | −10.6 ± 0.2 | 43 ± 3 | −7.2 ± 0.1 | 119 ± 12 |
| CCL4 | −9.2 ± 0.1 | −9.4 ± 0.1 | −8.5 ± 0.2 | −0.07 ± 0.02 | −8.3 ± 0.2 | −0.10 ± 0.01 | −8.1 ± 0.2 | −0.016 ± 0.002 | −8.1 ± 0.1 | 0.10 ± 0.01 | −9.1 ± 0.1 | 44 ± 3 | −8.2 ± 0.2 | 75 ± 5 |
| CCL5 | −8.2 ± 0.1 | −8.4 ± 0.1 | −9.1 ± 0.5 | −0.09 ± 0.02 | −9.2 ± 0.1 | −0.10 ± 0.01 | −9.0 ± 0.3 | −0.020 ± 0.001 | −8.1 ± 0.2 | 0.10 ± 0.01 | −10.3 ± 0.3 | 34 ± 2 | −8.0 ± 0.2 | 91 ± 3 |
| CCL8 | −8.2 ± 0.1 | −8.4 ± 0.1 | −8.2 ± 0.1 | −0.09 ± 0.01 | −8.1 ± 0.3 | −0.06 ± 0.01 | −8.1 ± 0.4 | −0.015 ± 0.003 | −7.0 ± 0.2 | 0.08 ± 0.01 | −9.2 ± 0.2 | 39 ± 3 | −7.4 ± 0.1 | 89 ± 3 |
| CCL13 | −7.2 ± 0.1 | −7.4 ± 0.1 | ND | ND | −7.6 ± 0.1 | −0.05 ± 0.01 | ND | ND | ND | ND | ND | ND | ND | ND |
FIGURE 3.

Panel of G proteins activated by CCR5. Real time measurement of BRET signal in HEK293T cells coexpressing CCR5 and G protein biosensors (black bars) or G protein biosensors only (open bars) and stimulated for 1 min with 100 nm of the chemokines CCL3, CCL4, CCL5, CCL7, CCL8, and CCL13 or 1 μm of the small molecules TAK779 (T) and Maraviroc (M) is shown. The results are expressed as the difference in BRET signal measured in the presence and absence of stimulation. The data represent the means ± S.E. of at least six independent experiments. Statistical significance between cells expressing or not CCR5. (***, p < 0.001; **, p < 0.01; *, p < 0.1) and between chemokines (###, p < 0.001; ##, p < 0.01; #, p < 0.1) was assessed using Tukey's test.
FIGURE 4.
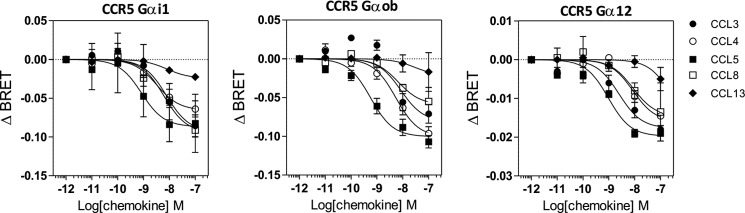
Activation of Gi1, Gob, and G12 by CCR5. Real time measurement of BRET signal in HEK293T cells coexpressing CCR5 and G protein biosensors and stimulated with increasing concentration of chemokines is shown. The results are expressed as the differences in BRET signal measured in the presence and absence of chemokines. The data represent the means ± S.E. of three independent experiments.
FIGURE 5.
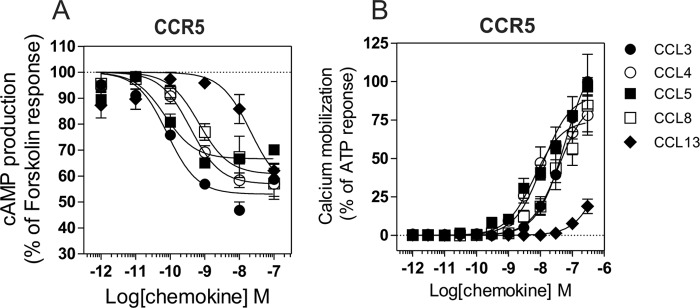
Inhibition of cAMP and calcium mobilization triggered by CCR5. A, measurement of BRET signal in HEK293T cells coexpressing the cAMP biosensor and CCR5 and stimulated sequentially by chemokines and forskolin. The results were normalized for the basal signal in absence of stimulation (0%), and the maximal response was obtained with forskolin only (100%). The data represent the means ± S.E. of three independent experiments. B, calcium mobilization was measured in CHO-K1 cells using the aequorin-based functional assay. Cells expressing CCR5 were stimulated with increasing concentrations of chemokines, and luminescence was recorded for 30 s. The results were normalized for the basal luminescence in absence of agonist (0%), and the maximal response was obtained with 25 μm ATP (100%). The data represent the means ± S.E. of three independent experiments.
Activation of G Protein Subtypes by CCR2
Because CCL7, CCL8, and CCL13 also bind to the closely related receptor CCR2, we next tested which G protein subtypes were activated by these chemokines when bound to CCR2. We showed that these chemokines competed with radiolabeled CCL2 for CCR2 binding with calculated pIC50 values compatible with previous estimates (Fig. 2 and Table 3) (20). CCL7, CCL8, and CCL13 (at 100 nm) activated Gαi and Gαo proteins at similar potencies to CCL2, which was used as a positive control (Figs. 6 and 7 and Table 3). However, CCL8 and CCL13 were less efficient agonists than CCL2 and CCL7 in most assays. In contrast to its effect on CCR5, CCL7 efficiently activated Gαi/o proteins through CCR2, in agreement with its known role as a CCR2 agonist. Interestingly, CCL2 and CCL7 also activated Gα12 with potencies similar to those of Gαi and Gαo. In contrast, CCL13 poorly activated Gα12 in comparison with Gαi and Gαo, whereas CCL8 was almost silent in this assay. The relative efficacy of these four CCR2 agonists therefore differs depending on the specific G protein and the activation readout. Furthermore, because our results suggest that CCL8 and CCL13 bind to CCR2 less efficiently than CCL2 and CCL7, the relative potency of various chemokines in the various G protein assays does not strictly follow their binding affinity for CCR2. This is therefore suggestive of signaling bias. Similar to CCR5, none of the Gαq, Gα11, Gαs, or Gα13 proteins was activated upon chemokine binding to CCR2 (Fig. 6 and data not shown). Finally, we showed that all chemokines inhibited cAMP accumulation with relative potencies compatible with binding data (Fig. 8A). In contrast, CCL2 induced significantly more calcium influx than CCL7, despite their similar binding affinities. Likewise, CCL13 induced significantly higher calcium influx than CCL8, despite a lower binding affinity. The calculated calcium mobilization efficacies were similar for the various chemokines, with the exception of CCL8, for which no values could be determined (Fig. 8B and Table 3).
TABLE 3.
Binding and signaling parameters of CCR2
Binding and functional parameters were measured on HEK293 or CHO-K1 cells expressing CCR2. The pIC50, pEC50, and Emax values were obtained from experiments as displayed in Figs. 2, 7, 8, and 9. The values represent the means ± S.E. of at least three independent experiments. ND, not determined.
| Ligands | [125I]CCL2 |
Gαi1 |
Gαob |
Gα12 |
β-Arr2 |
cAMP |
Ca2+ |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pIC50 | pKi | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | |
| CCL2 | −9.6 ± 0.1 | −10.0 ± 0.1 | −8.7 ± 0.2 | −0.13 ± 0.01 | −8.0 ± 0.1 | −0.15 ± 0.01 | −8.4 ± 0.4 | −0.042 ± 0.004 | −8.5 ± 0.1 | 0.14 ± 0.01 | −9.1 ± 0.3 | 31 ± 4 | −8.2 ± 0.1 | 86 ± 5 |
| CCL7 | −9.2 ± 0.1 | −9.6 ± 0.1 | −8.4 ± 0.2 | −0.10 ± 0.01 | −8.2 ± 0.1 | −0.12 ± 0.01 | −8.2 ± 0.2 | −0.030 ± 0.003 | −7.7 ± 0.1 | 0.12 ± 0.01 | −9.3 ± 0.2 | 29 ± 4 | −7.5 ± 0.2 | 86 ± 8 |
| CCL8 | −8.5 ± 0.1 | −8.9 ± 0.1 | −8.5 ± 0.2 | −0.06 ± 0.01 | −8.8 ± 0.4 | −0.05 ± 0.01 | ND | ND | −7.7 ± 0.1 | 0.03 ± 0.01 | −8.4 ± 0.2 | 31 ± 4 | ND | ND |
| CCL13 | −7.9 ± 0.1 | −8.3 ± 0.1 | −8.5 ± 0.4 | −0.06 ± 0.01 | −7.9 ± 0.2 | −0.08 ± 0.01 | ND | ND | −7.0 ± 0.1 | 0.14 ± 0.01 | ND | ND | ND | ND |
FIGURE 6.

Panel of G proteins activated by CCR2. Real time measurement of BRET signal in HEK293T cells coexpressing CCR2 and G protein biosensors (black bars) or G protein biosensors only (open bars) and stimulated for 1 min with 100 nm of the chemokines CCL2, CCL7, CCL8, and CCL13 or 1 μm of the small molecules TAK779 (T) and Maraviroc (M) is shown. The results are expressed as the difference in BRET signal measured in the presence and absence of stimulation. The data represent the means ± S.E. of at least six independent experiments. Statistical significance between cells expressing or not CCR2 (***, p < 0.001; **, p < 0.01; *, p < 0.1) and between chemokines (###, p < 0.001; ##, p < 0.01; #, p < 0.1) was assessed using Tukey's test.
FIGURE 7.
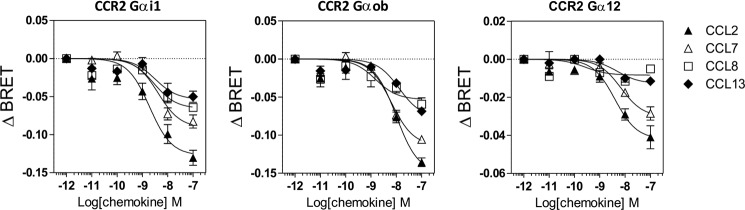
Activation of Gi1, Gob, and G12 by CCR2. Real time measurement of BRET signal in HEK293T cells coexpressing CCR2 and G protein biosensors and stimulated with increasing concentrations of chemokines is shown. The results are expressed as the differences in BRET signal measured in the presence and absence of chemokines. The data represent the means ± S.E. of three independent experiments.
FIGURE 8.
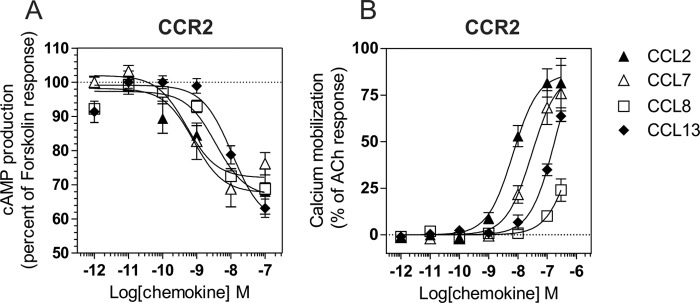
Inhibition of cAMP and calcium mobilization triggered by CCR2. A, measurement of BRET signal in HEK293T cells coexpressing the cAMP biosensor and CCR2 and stimulated sequentially by chemokines and forskolin. The results were normalized for the basal signal in absence of stimulation (0%), and the maximal response was obtained with forskolin only (100%). The data represent the means ± S.E. of three independent experiments. B, calcium mobilization was measured in HEK293 cells using the aequorin-based functional assay. Cells expressing CCR2 were stimulated with increasing concentrations of chemokines and luminescence was recorded for 30 s. The results were normalized for the basal luminescence in absence of agonist (0%), and the maximal response was obtained with 50 μm acetylcholine (100%). The data represent the means ± S.E. of three independent experiments.
Arrestin Recruitment by CCR5 and CCR2
We finally investigated differences in β-arrestin-2 recruitment to CCR2 and CCR5 in response to the various chemokines. Stimulation by chemokines induced a progressive increase in the energy transfer between β-arrestin-2-Rluc and CCR5-Venus, indicating recruitment of the arrestin to CCR5 (Fig. 9). CCL5 and CCL8 recruited β-arrestin-2 with the fastest kinetics of the chemokines tested (Tau values: CCL3, 5.0 min; CCL4, 6.5 min; CCL5, 3.5 min; and CCL8, 3.5 min). The BRETmax values reached for the various chemokines were reflective of the respective pEC50 values combined with the lack of saturation for some chemokines at 100 nm. Very low β-arrestin-2 recruitment to CCR5 was detected upon CCL13 stimulation, consistent with the weak activation detected in other readouts for this chemokine. Stimulating CCR5 with increasing concentrations of chemokines showed that all tested chemokines activated β-arrestin-2 recruitment with potencies comparable with those estimated for G protein activation (Fig. 9 and Table 2). No differences in arrestin recruitment were observed for CCL5 and CCL3/CCL4 in this assay, suggesting the absence of strong signaling bias between G protein- and arrestin-dependent pathways for CCR5 agonists. We also investigated the recruitment of β-arrestin-2 by CCR2 and showed that the various chemokines triggered the recruitment of β-arrestin-2-Rluc to CCR2-Venus, but with variable kinetics and BRETmax values (Fig. 9 and Table 3). In this assay, CCL2 was more potent than CCL7 despite similar affinities, and CCL13 efficacy was substantially higher than that of CCL8, which is similar to our results for calcium mobilization. These data are in agreement with a previous report (20) and support a model with bias in the activation of signaling pathways by CCR2 agonists.
FIGURE 9.
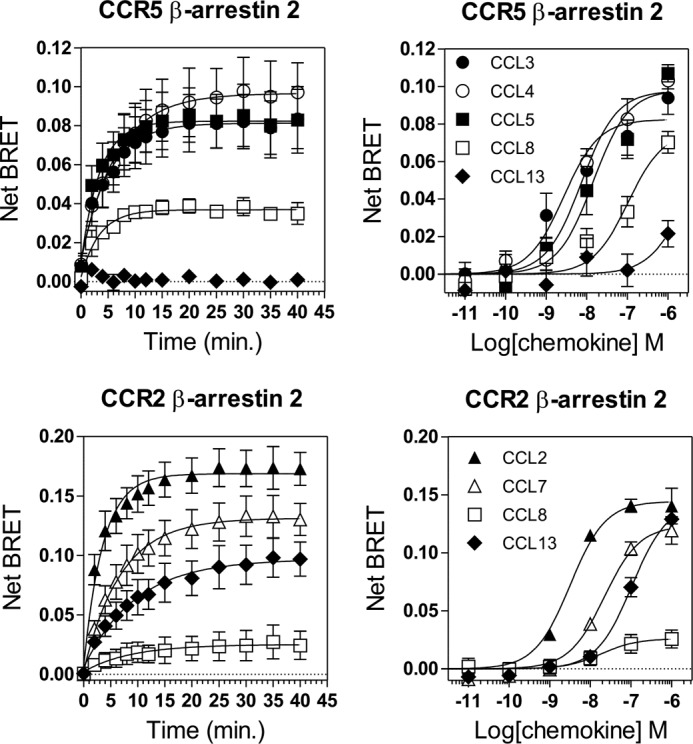
Recruitment of β-arrestin 2 by CCR5 and CCR2. A–D, real time measurement of BRET signal in HEK293T cells expressing either β-arrestin2-RLuc8 and CCR5-Venus (A and B) or β-arrestin2-RLuc8 and CCR2-Venus (C and D) and stimulated with chemokines. For kinetics, BRET signals were measured after addition of 100 nm chemokines. For dose-response curves, BRET was recorded 30 min after stimulation with various concentrations of chemokines. The results are expressed as net BRET, corresponding to the difference in BRET signal between cells expressing arrestin plus receptor and cells expressing arrestin only. The data represent the means ± S.E. of three independent experiments.
Assessment of Chemokines Bias
We performed a quantitative analysis of chemokines bias based on functional parameters from Tables 2 and 3 as previously described for other chemokine receptors (15). Bias factors between pathways were determined for most chemokines with the exception of the cases in which signaling parameters could not be determined. Most of the calculated bias values ranged between 0 and 1, suggesting no or weak bias, and none of these biases was statistically significant (Fig. 10 and 11 and Tables 4 and 5). Only few bias factors raised above 1 with statistical significance. At CCR5, CCL5 and CCL8 showed some level of bias for Gαi1 and Gαob relative to arrestin, the bias of CCL5 between Gαob and arrestin being the only one to reach statistical significance. Similarly, CCL8 showed at CCR2 a statistically significant bias for Gαob activation relative to arrestin. These results suggest the existence of some bias between G protein and arrestin activation. At CCR5, significant bias could also be determined for some chemokines (CCL4 and CCL8) between calcium and arrestin or cAMP. However, we have to keep in mind that calcium mobilization with CCR5 was performed in another cell type, which may influence the estimation of bias. It is also of note that CCL8 showed at CCR5 significant bias for Gαi1 and Gαob relative to cAMP. This result may appear somehow paradoxical because inhibition of cAMP constitutes a representative readout of Gαi/o protein activation. Nevertheless, it may not be relevant to compare cAMP inhibition and a single G protein or to calculate bias between assays displaying different levels of amplification.
FIGURE 10.

Chemokine bias factors at CCR5. Bias factors between different pathways were calculated for each chemokine, using CCL3 as the reference chemokine. Bias factors that are significantly different (Tukey's test) from the reference chemokine CCL3 are in bold and colored according to the preferred pathway. The data represent the means ± S.E. from Table 4. ND, not determined.
FIGURE 11.

Chemokine bias factors at CCR2. Bias factor between different pathways were calculated for each chemokine, using CCL2 as the reference chemokine. Bias factors that are significantly different (Tukey's test) from the reference are in bold and colored according to the preferred pathway. The data represent the means ± S.E. from Table 4. ND, not determined.
TABLE 4.
Bias factors at CCR5
The bias factors were calculated as described under “Experimental Procedures.” The agonist CCL3 was arbitrarily defined as the reference with a bias factor of 0. The values represent the means ± S.E. of at least three independent experiments. ND, not determined.
| Bias (β) | Gαi1:Gαob | Gαi1:Gα12 | Gαi1:Arr | Gαi1:cAMP | Gαi1:Ca | Gαob:G12 | Gαob:Arr | Gαob:cAMP | Gαob:Ca | Gα12:Arr | Gα12:cAMP | Gα12:Ca | Arr:cAMP | Arr:Ca | cAMP:Ca |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCL3 | 0.00 ± 0.76 | 0.00 ± 0.67 | 0.00 ± 0.60 | 0.00 ± 0.58 | 0.00 ± 0.56 | 0.00 ± 0.68 | 0.00 ± 0.62 | 0.00 ± 0.60 | 0.00 ± 0.58 | 0.00 ± 0.50 | 0.00 ± 0.47 | 0.00 ± 0.45 | 0.00 ± 0.37 | 0.00 ± 0.34 | 0.00 ± 0.30 |
| CCL4 | −0.06 ± 0.61 | 0.80 ± 0.57 | 0.59 ± 0.50 | 0.93 ± 0.50 | −0.54 ± 0.48 | 0.86 ± 0.55 | 0.65 ± 0.48 | 0.99 ± 0.47 | −0.48 ± 0.46 | −0.21 ± 0.43 | 0.13 ± 0.42 | −1.34 ± 0.41 | 0.34 ± 0.32 | −1.13 ± 0.30 | −1.47 ± 0.29a |
| CCL5 | −0.33 ± 0.74 | 0.47 ± 0.70 | 1.58 ± 0.68 | 1.01 ± 0.66 | 0.34 ± 0.63 | 0.80 ± 0.53 | 1.91 ± 0.50a | 1.35 ± 0.48 | 0.67 ± 0.44 | 1.11 ± 0.43 | 0.55 ± 0.40 | −0.13 ± 0.36 | −0.56 ± 0.37 | −1.24 ± 0.32 | −0.68 ± 0.28 |
| CCL8 | 0.13 ± 0.64 | 0.72 ± 0.65 | 1.72 ± 0.50 | 2.59 ± 0.49a | 0.03 ± 0.44 | 0.59 ± 0.71 | 1.59 ± 0.58 | 2.46 ± 0.56a | −0.10 ± 0.53 | 0.99 ± 0.60 | 1.87 ± 0.58 | −0.70 ± 0.55 | 0.88 ± 0.41 | −1.69 ± 0.36a | −2.56 ± 0.33a |
| CCL13 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
a Bias factors with statistically significant difference with the CCL3 reference (p < 0.05).
TABLE 5.
Bias factors at CCR2
The bias factors were calculated as described under “Experimental Procedures.” The agonist CCL2 was arbitrarily defined as the reference with a bias factor of 0. The values represent the means ± S.E. of at least three independent experiments. ND, not determined.
| Bias (β) | Gαi1Gαob | Gαi1:Gα12 | Gαi1:Arr | Gαi1:cAMP | Gαi1:Ca | Gαob:Gα12 | Gαob:Arr | Gαob:cAMP | Gαob:Ca | Gα12:Arr | Gα12:cAMP | Gα12:Ca | Arr:cAMP | Arr:Ca | cAMP:Ca |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCL2 | 0.00 ± 0.46 | 0.00 ± 0.49 | 0.00 ± 0.43 | 0.00 ± 0.62 | 0.00 ± 0.44 | 0.00 ± 0.32 | 0.00 ± 0.52 | 0.00 ± 0.49 | 0.00 ± 0.24 | 0.00 ± 0.27 | 0.00 ± 0.52 | 0.00 ± 0.30 | 0.00 ± 0.46 | 0.00 ± 0.17 | 0.00 ± 0.64 |
| CCL7 | −0.72 ± 0.58 | −0.26 ± 0.61 | 0.27 ± 0.56 | −0.83 ± 0.68 | ND | 0.46 ± 0.32 | 0.98 ± 0.48 | −0.11 ± 0.44 | 0.72 ± 0.26 | 0.52 ± 0.29 | −0.57 ± 0.48 | 0.26 ± 0.33 | −1.10 ± 0.41 | −0.26 ± 0.22 | 0.83 ± 0.56 |
| CCL8 | −1.09 ± 0.83 | ND | 0.80 ± 0.73 | −0.07 ± 0.81 | ND | ND | 1.89 ± 0.96a | 1.02 ± 0.53 | ND | ND | ND | ND | −0.87 ± 0.37 | ND | ND |
| CCL13 | −0.86 ± 0.60 | ND | 0.26 ± 0.54 | ND | ND | ND | 1.12 ± 0.57 | ND | ND | ND | ND | ND | ND | ND | ND |
a Bias factors with statistically significant differences with the CCL2 reference (p < 0.05).
DISCUSSION
Over the past decade, biased signaling has emerged as a novel and challenging concept in the GPCR field. This concept relies on the hypothesis that receptors can oscillate among multiple conformational states, each of which is able to activate its own set of signaling pathways (7, 21). Selective activation of signaling pathways by specific GPCR ligands has been demonstrated for several receptors, including angiotensin AT1R, dopamine D2R, serotonin 5-HT2CR, and the pituitary adenylate cyclase-activating peptide receptors (22–24). Differential effects of chemokines on their common receptor was reported in terms of signaling cascade activation, receptor phosphorylation, or internalization (14, 14, 25–29). So far, one of the prototypical examples of signaling bias in GPCRs is the chemokine receptor CCR7. It was originally shown that both natural ligands of CCR7, CCL19 and CCL21, equally activate G protein-dependent signaling, whereas only CCL19 is able to promote efficient β-arrestin recruitment and MAPK phosphorylation (11–13). The precise molecular mechanisms underlying this signaling bias is not known, although the involvement of CCR7 phosphorylation by selective GRKs has been suggested (11). Although discrepancies regarding the extent of arrestin activation and MAPK phosphorylation exist between studies, they all supported that CCL19 and CCL21 lead to similar G protein-dependent signaling (11, 12). In the present study, we confirmed that CCL21 binding induces β-arrestin recruitment with a lower potency than CCL19, but in contrast to previous reports, we also showed that CCL21 is less potent at activating G proteins, as measured by BRET biosensors directly monitoring the conformational changes associated to Gαi or Gαo activation. We also confirmed that the lower Gαi/o activation following stimulation by CCL21 is associated with a reduced cAMP accumulation, in perfect agreement with the role of Gαi/o proteins in inhibiting cAMP generation. Similarly, CCL21 activates less efficiently calcium mobilization and ERK1/2 phosphorylation, indicating that several signals downstream of G proteins and arrestin behave similarly. Collectively, our data suggest that CCL21 interacts with CCR7 as efficiently as CCL19 but is less potent in activating G proteins and recruiting β-arrestin, thus challenging the notion of bias claimed between these two pathways. The exact reason for the discrepancy between our results and those of Kohout et al. (12) is not known for sure but may be linked to the various expression systems used in the original study, whereas we performed all our assays in HEK293T cells. In line with this hypothesis, we found that calcium mobilization and Erk1/2 phosphorylation can be activated by CCL21 as efficiently as CCL19 in CHO cells expressing CCR7 (not shown). It should thus be kept in mind that the nature of the cells used to monitor receptor signaling can impact some assays. BRET biosensors could thus constitute an interesting alternative tool to investigate early signaling events at the level of G protein activation because they measure conformational changes of the ligand-receptor-G protein complex.
We also investigated the putative signaling selectivity at two other chemokine receptors, CCR2 and CCR5, and compared the data generated using the G protein biosensors with those obtained with β-arrestin recruitment, cAMP accumulation, and calcium mobilization assays. We first identified the set of G protein subtypes activated by CCR2 and CCR5 and showed that both receptors activate proteins of the Gαi, Gαo, and Gα12 subfamilies. Not surprisingly, an antagonist chemokine was silent in such assays, whereas small molecule inverse agonists produced changes in some BRET signals compatible with an inhibition of the constitutive activity of these receptors. These results confirm that G protein biosensors constitute valuable tools to discriminate properties of receptor ligands. All agonist chemokines activated G proteins with potencies that generally correlate with the potencies calculated for β-arrestin recruitment and their binding affinities for the receptors. Gαi1 activation and arrestin recruitment have previously been reported to be correlated for CCR2 (20). There were, however, some changes in the relative efficiency of the chemokines according to the assay used. CCL5 was, for example, more potent on CCR5 than CCL3 and CCL4 in some G protein assays, whereas these three chemokines displayed similar binding parameters and did not differ significantly in other assays. CCL8 also displayed a lower relative potency on CCR5 in the β-arrestin-2 recruitment assay. Quantitative analysis revealed that CCL5 and CCL8 display some selectivity for G proteins activation over β-arrestin-2 recruitment, even if only one bias value reached statistical significance. Likewise, CCL7 and CCL8 displayed lower relative potencies on CCR2 in the calcium mobilization and β-arrestin recruitment assays, whereas they behaved similarly to CCL2 and CCL13 in other assays. CCL8 also activated Gαi1 and Gαi2 much more efficiently than Gα12 through CCR2. Quantitative analysis showed that CCL8 displayed significant bias for Gαob activation relative to arrestin, but no other significant bias could be determined. One explanation could be that most of our concentration-response data showed moderate variations, yielding bias values ranging between 0 and 1. Mathematical calculation of bias factors is therefore useful to quantify to some extent the selectivity of a ligand for one pathway relative to another. However, it remains to be determined precisely whether those values reflect distinct physiological responses.
Collectively, these data suggest that different chemokines acting on CCR5 or CCR2 may trigger overlapping but distinct sets of G protein subtypes, providing some selectivity in downstream signaling cascades. Finally, our data also revealed that CCR5 and CCR2 activate Gα12, whereas CCR7 does not at all. Our results also revealed that CCR5/2 activate Gα12 but not Gα13, which belongs to the same family. This activation profile is clearly distinct from other receptors that activate both Gα12 and Gα13, such as the thromboxane receptor (TPaR) (17), illustrating the utility of biosensors in discriminating the activation of G protein subtypes that cannot be otherwise measured by “classical” readouts.
In summary, we showed here that G protein BRET biosensors enable the pragmatic analysis of G protein subtype activation by chemokine receptors upon interaction with their various ligands. We also showed that the chemokines tested in this study activated G proteins with potencies that generally match those detected for β-arrestin recruitment and other assays, but with subtle changes in the rank order according to assay. Thus, the behavior of chemokines contrast to the situation encountered in other studies with some synthetic small molecules that show agonism in one assay but antagonism in another (30, 31). Collectively, our results suggest the existence of a moderate signaling bias between chemokines acting on the same receptor. Although variations in G proteins or arrestins activation were detected, the structural basis underlying biased signaling remains to be identified precisely. It also appears that the bias remains relatively subtle for natural ligands such as chemokines, whereas more overt bias has been described for synthetic small molecule agonists.
This work was supported by the Fonds de la Recherche Scientifique Médicale of Belgium, the Actions de Recherche Concertées, and the Interuniversity Attraction Poles Programme (P7–14), Belgian State, Belgian Science Policy.
- GPCR
- G protein-coupled receptor
- BRET
- bioluminescence resonance energy transfer.
REFERENCES
- 1. Lagerström M. C., Schiöth H. B. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug. Discov. 7, 339–357 [DOI] [PubMed] [Google Scholar]
- 2. Strange P. G. (2008) Signaling mechanisms of GPCR ligands. Curr. Opin. Drug. Discov. Devel. 11, 196–202 [PubMed] [Google Scholar]
- 3. Gether U. (2000) Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 21, 90–113 [DOI] [PubMed] [Google Scholar]
- 4. Marrari Y., Crouthamel M., Irannejad R., Wedegaertner P. B. (2007) Assembly and trafficking of heterotrimeric G proteins. Biochemistry 46, 7665–7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rajagopal S., Rajagopal K., Lefkowitz R. J. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug. Discov. 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whalen E. J., Rajagopal S., Lefkowitz R. J. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 17, 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kenakin T. (2011) Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 336, 296–302 [DOI] [PubMed] [Google Scholar]
- 8. Shenoy S. K., Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fuxe K., Kenakin T. (2010) The changing world of G protein-coupled receptors. J. Recept. Signal. Transduct. Res. 30, 271. [DOI] [PubMed] [Google Scholar]
- 10. Bonecchi R., Galliera E., Borroni E. M., Corsi M. M., Locati M., Mantovani A. (2009) Chemokines and chemokine receptors: an overview. Front. Biosci. (Landmark Ed) 14, 540–551 [DOI] [PubMed] [Google Scholar]
- 11. Zidar D. A., Violin J. D., Whalen E. J., Lefkowitz R. J. (2009) Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. U.S.A. 106, 9649–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kohout T. A., Nicholas S. L., Perry S. J., Reinhart G., Junger S., Struthers R. S. (2004) Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J. Biol. Chem. 279, 23214–23222 [DOI] [PubMed] [Google Scholar]
- 13. Byers M. A., Calloway P. A., Shannon L., Cunningham H. D., Smith S., Li F., Fassold B. C., Vines C. M. (2008) Arrestin 3 mediates endocytosis of CCR7 following ligation of CCL19 but not CCL21. J. Immunol. 181, 4723–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rajagopal S., Bassoni D. L., Campbell J. J., Gerard N. P., Gerard C., Wehrman T. S. (2013) Biased agonism as a mechanism for differential signaling by chemokine receptors. J. Biol. Chem. 288, 35039–35048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. El-Asmar L., Springael J. Y., Ballet S., Andrieu E. U., Vassart G., Parmentier M. (2005) Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol. Pharmacol. 67, 460–469 [DOI] [PubMed] [Google Scholar]
- 16. Galés C., Van Durm J. J., Schaak S., Pontier S., Percherancier Y., Audet M., Paris H., Bouvier M. (2006) Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786 [DOI] [PubMed] [Google Scholar]
- 17. Saulière A., Bellot M., Paris H., Denis C., Finana F., Hansen J. T., Altié M. F., Seguelas M. H., Pathak A., Hansen J. L., Sénard J. M., Galés C. (2012) Deciphering biased-agonism complexity reveals a new active AT(1) receptor entity. Nat. Chem. Biol. 8, 622–630 [DOI] [PubMed] [Google Scholar]
- 18. Barak L. S., Salahpour A., Zhang X., Masri B., Sotnikova T. D., Ramsey A. J., Violin J. D., Lefkowitz R. J., Caron M. G., Gainetdinov R. R. (2008) Pharmacological characterization of membrane-expressed human trace amine-associated receptor 1 (TAAR1) by a bioluminescence resonance energy transfer cAMP biosensor. Mol. Pharmacol. 74, 585–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blanpain C., Migeotte I., Lee B., Vakili J., Doranz B. J., Govaerts C., Vassart G., Doms R. W., Parmentier M. (1999) CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood 94, 1899–1905 [PubMed] [Google Scholar]
- 20. Berchiche Y. A., Gravel S., Pelletier M. E., St-Onge G., Heveker N. (2011) Different effects of the different natural CC chemokine receptor 2b ligands on β-arrestin recruitment, Gαi signaling, and receptor internalization. Mol. Pharmacol. 79, 488–498 [DOI] [PubMed] [Google Scholar]
- 21. Kenakin T. P. (2012) Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br. J. Pharmacol. 165, 1659–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mottola D. M., Kilts J. D., Lewis M. M., Connery H. S., Walker Q. D., Jones S. R., Booth R. G., Hyslop D. K., Piercey M., Wightman R. M., Lawler C. P., Nichols D. E., Mailman R. B. (2002) Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J. Pharmacol. Exp. Ther. 301, 1166–1178 [DOI] [PubMed] [Google Scholar]
- 23. Berg K. A., Maayani S., Goldfarb J., Clarke W. P. (1998) Pleiotropic behavior of 5-HT2A and 5-HT2C receptor agonists. Ann. N.Y. Acad. Sci. 861, 104–110 [DOI] [PubMed] [Google Scholar]
- 24. Spengler D., Waeber C., Pantaloni C., Holsboer F., Bockaert J., Seeburg P. H., Journot L. (1993) Differential signal transduction by five splice variants of the PACAP receptor. Nature 365, 170–175 [DOI] [PubMed] [Google Scholar]
- 25. Oppermann M., Mack M., Proudfoot A. E., Olbrich H. (1999) Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J. Biol. Chem. 274, 8875–8885 [DOI] [PubMed] [Google Scholar]
- 26. Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mack M., Luckow B., Nelson P. J., Cihak J., Simmons G., Clapham P. R., Signoret N., Marsh M., Stangassinger M., Borlat F., Wells T. N., Schlöndorff D., Proudfoot A. E. (1998) Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J. Exp. Med. 187, 1215–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kouroumalis A., Nibbs R. J., Aptel H., Wright K. L., Kolios G., Ward S. G. (2005) The chemokines CXCL9, CXCL10, and CXCL11 differentially stimulate Gαi-independent signaling and actin responses in human intestinal myofibroblasts. J. Immunol. 175, 5403–5411 [DOI] [PubMed] [Google Scholar]
- 29. Tian Y., New D. C., Yung L. Y., Allen R. A., Slocombe P. M., Twomey B. M., Lee M. M., Wong Y. H. (2004) Differential chemokine activation of CC chemokine receptor 1-regulated pathways: ligand selective activation of Gα14-coupled pathways. Eur. J. Immunol. 34, 785–795 [DOI] [PubMed] [Google Scholar]
- 30. Saita Y., Kodama E., Orita M., Kondo M., Miyazaki T., Sudo K., Kajiwara K., Matsuoka M., Shimizu Y. (2006) Structural basis for the interaction of CCR5 with a small molecule, functionally selective CCR5 agonist. J. Immunol. 177, 3116–3122 [DOI] [PubMed] [Google Scholar]
- 31. Ferain T., Hoveyda H., Ooms F., Schols D., Bernard J., Fraser G. (2011) Agonist-induced internalization of CC chemokine receptor 5 as a mechanism to inhibit HIV replication. J. Pharmacol. Exp. Ther. 337, 655–662 [DOI] [PubMed] [Google Scholar]