

Background: Ligation of cancer cell surface GRP78 by activated α2-macroglobulin (α2M*) promotes proliferation and blocks apoptosis.
Results: α2M* treatment of prostate cancer cells enhances the Warburg effect and up-regulates lipid metabolism in an insulin-like manner.
Conclusion: α2M* exerts insulin-like effects on prostate cancer cells.
Significance: Targeting cell surface GRP78-dependent cancer cell regulation, such as by antibodies, offers a unique potential therapeutic approach.
Keywords: Glycolysis, Lipogenesis, Prostate Cancer, Signal Transduction, Warburg Effect, Aerobic Glycolysis, α-2-Macroglobulin and Metabolic Regulation, Antibody Directed Against the Carboxyl-terminal Domain of GRP78, Cell Surface GRP78
Abstract
Ligation of cell surface GRP78 by activated α2-macroglobulin (α2M*) promotes cell proliferation and suppresses apoptosis. α2M*-treated human prostate cancer cells exhibit a 2–3-fold increase in glucose uptake and lactate secretion, an effect similar to insulin treatment. In both α2M* and insulin-treated cells, the mRNA levels of SREBP1-c, SREBP2, fatty-acid synthase, acetyl-CoA carboxylase, ATP citrate lyase, and Glut-1 were significantly increased together with their protein levels, except for SREBP2. Pretreatment of cells with α2M* antagonist antibody directed against the carboxyl-terminal domain of GRP78 blocks these α2M*-mediated effects, and silencing GRP78 expression by RNAi inhibits up-regulation of ATP citrate lyase and fatty-acid synthase. α2M* induces a 2–3-fold increase in lipogenesis as determined by 6-[14C]glucose or 1-[14C]acetate incorporation into free cholesterol, cholesterol esters, triglycerides, free fatty acids, and phosphatidylcholine, which is blocked by inhibitors of fatty-acid synthase, PI 3-kinase, mTORC, or an antibody against the carboxyl-terminal domain of GRP78. We also assessed the incorporation of [14CH3]choline into phosphatidylcholine and observed similar effects. Lipogenesis is significantly affected by pretreatment of prostate cancer cells with fatostatin A, which blocks sterol regulatory element-binding protein proteolytic cleavage and activation. This study demonstrates that α2M* functions as a growth factor, leading to proliferation of prostate cancer cells by promoting insulin-like responses. An antibody against the carboxyl-terminal domain of GRP78 may have important applications in prostate cancer therapy.
Introduction
Prostate cancer, the most commonly diagnosed malignancy of men, may acquire androgen independence, a poor prognostic indicator associated with a more metastatic phenotype (1–3). Growth factors play a role in the progression of these cancers by ligating cell surface-binding sites to induce receptor autophosphorylation at specific tyrosine residues. This results in the assembly of multiprotein complexes, which activate signal transduction mechanisms, promoting tumor cell proliferation (4–9). Activation of PI 3-kinase results in its recruitment to the inner surface of the plasma membrane, where it generates 3′-phosphorylated phosphoinositides, which regulate downstream signal transduction by activating Akt, mTORC1, and mTORC2 (4–12).
α2-Macroglobulin (α2M)2 is a broad spectrum proteinase inhibitor synthesized by hepatocytes and macrophages (13–17); however, tumors and their associated stroma also produce this protein (18). Prostate cancer cells secrete prostate-specific antigen, a chymotrypsin-like serine proteinase, metalloproteinases, and other proteinases, which activate α2M in the tumor microenvironment (18). When α2M reacts with a proteinase, the “bait region” in each subunit is cleaved, and consequently, β-cysteinyl-γ-glutamyl thiol esters in all four of its subunits rupture, followed by a large conformational change in the molecule (13). This exposes a receptor recognition site in the carboxyl-terminal domain of each subunit (19). Small nucleophiles, such as methylamine or ammonia, can directly rupture the thiol esters, triggering a similar conformational change exposing these sites (13). Two receptors bind activated forms of α2M (α2M*), the LDL receptor-related protein (LRP) and cell surface-associated GRP78 (glucose-regulated protein of Mr ∼78,000) (20–23). Although GRP78 is primarily a resident endoplasmic reticulum chaperone (24), it appears on the cell surface of many types of malignant cells, including human prostate cancer (20–23), where it functions as a very high affinity α2M* signaling receptor linked to pro-proliferative, pro-migratory, and anti-apoptotic signaling cascades (6–9, 22, 23, 25–39). Patients with many forms of cancer, including prostate, may develop auto-antibodies that bind to the amino-terminal domain of GRP78 close to or at the α2M*-binding site (40–42). These antibodies are receptor agonists in contrast to antibodies directed against the carboxyl-terminal domain of GRP78, which are receptor antagonists and promote cell death and apoptosis by impairing PI 3-kinase/PDK/Akt/mTORC1/mTORC2 signaling (18, 23, 25, 29, 36, 38–45).
Cancers undergo metabolic reprogramming, most importantly effecting energy metabolism (46–54). Cancer cells show an increased uptake of glucose that is decoupled from pyruvate oxidation via aerobic glycolysis, resulting in lactic acid accumulation, known as the Warburg effect (55). Cancer cells also demonstrate alterations of fatty acid, triglyceride, and cholesterol metabolism (56). Irrespective of the concentration of extracellular lipids, cancer cells synthesize and accumulate fatty acids de novo in part because of the elevated expression of fatty-acid synthase, a key metabolic enzyme catalyzing the synthesis of long chain fatty acids (46–54, 57–64). Furthermore, fatty acid oxidation is a dominant pathway for energy generation in many tumors (65). PI 3-kinase/Akt/mTORC signaling stimulates fatty acid synthesis by activating ATP citrate lyase, and it stimulates lipogenic gene expression via activation and nuclear localization of the transcription factor SREBP1 (sterol regulatory element-binding protein) (64, 66–71). Inhibition of ATP citrate lyase induces growth arrest and apoptosis in prostate cancer cells (72). Cholesterol accumulation also occurs in prostate cancer, and dysregulation of its biosynthetic pathway is associated with malignant transformation (59, 73–75). Cholesterol is an important component of biological membranes because it modulates the fluidity of lipid bilayers and forms lipid rafts that coordinate the activation of certain signal transduction pathways (59, 73–75). The intracellular pool of cholesterol esters is a storage form of cholesterol that prevents its toxic effects (76). The accumulation of cholesterol esters is induced by the loss of PTEN, up-regulation of the PI 3-kinase/Akt/mTORC pathway, and activation of SREBP. Whereas SREBP1 mainly regulates fatty acid, phospholipid, and triacylglyceride biosynthesis, SREBP2 regulates cholesterol biosynthesis (77). SREBPs traffic to the Golgi apparatus where they are processed by two proteinases to liberate a soluble fraction that translocates to the nucleus. Here, SREBPs activate transcription by binding to sequences in the promoters of target genes. Insulin-mediated stimulation of SREBP1-c processing and SREBP1-c mRNA induction requires PI 3-kinase/Akt/mTORC1 signaling, and either rapamycin or PI 3-kinase inhibitors block its activation (47, 49, 59, 64, 68, 69, 78, 79).
Glucose-derived carbons are channeled into fatty acids, which are incorporated into glycerolipids (46–59, 80, 81). Fatty-acid synthase inhibition decreases tumor growth by suppressing the synthesis of phosphatidylcholine and other phospholipids necessary for membrane biogenesis, lipid raft formation, and the production of proactive lipids (80, 81). The hydrolysis of phosphatidylcholine mediates mitogenic signal transduction events in cells, and the products of its metabolism, such as diacylglycerol and arachidonic acid metabolites, are second messengers essential for mitogenic activity. Previous studies demonstrate that α2M* up-regulates the synthesis and activity of cPLA2, phospholipase D, and COX-2 (82–84).
We previously reported that binding of α2M* to GRP78 on the surface of various tumor cells, including prostate cancer, induces proliferation and survival by activating PI 3-kinase/Akt/mTORC signaling. In this study, we determined whether α2M* enhances the Warburg effect in prostate cancer cells causing proliferation. We report here that α2M* up-regulates aerobic glycolysis in prostate cancer cells as determined by increased glucose uptake, increased lactate secretion, and up-regulation of Glut-1 in the presence of oxygen. The synthesis of fatty acids, cholesterol, triglycerides, and phosphatidylcholine with corresponding increases in the expression of SREBP1-c, SREBP2, ATP citrate lyase, and acetyl-CoA carboxylase is observed. Treatment with an antibody directed against the carboxyl-terminal domain of GRP78 (anti-CTD) inhibits α2M*-induced cell proliferation and lipogenesis as determined by studies with 1-[14C]acetate, 6-[14C]glucose, and [14CH3]choline. A similar effect was seen with inhibitors of PI 3-kinase, Akt-1, mTORC1, mTORC2, fatty-acid synthase, and SREBP activation with either α2M* or insulin treatment, the latter was used as a positive control. The effects of ligating cell surface GRP78 with α2M* are strikingly similar to the effects induced by insulin.
EXPERIMENTAL PROCEDURES
Materials
Culture media were purchased from Invitrogen. α2M* was prepared as described previously (32). Antibodies against fatty-acid synthase, acetyl-CoA carboxylase, and SREBP1-c were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody against the carboxyl-terminal domain of GRP78 (anti-CTD) was purchased from Aventa Biopharmaceutical Corp. (San Diego). Antibodies against β-actin were from Sigma. 1-[14C]Acetate (specific activity of 45–60 mCi/mmol), 6-[14C]glucose (specific activity of 50–62 mCi/mmol), 1-[14C]deoxyglucose (specific activity of 45–60 mCi/mol), [3H]leucine (specific activity of 115 mCi/mol), [3H]thymidine (specific activity of 71.5 mCi/mmol), and [14CH3]choline (specific activity of 50–62 mCi/mmol) were from PerkinElmer Life Sciences. SilicagelH-coated glass plates were from Anal Tech (Newark, DE), Torin1, KU0037694, fatostatin A, and C-75 were from Tocris (Bristol, UK). Orlistat and lipid standards used in TLC were from Sigma. MKK2206 was from Selleckchem.com. Rapamycin and LY294002 were from Biomol (Plymouth, PA). All other chemicals were of analytical grade and purchased locally.
Cancer Cell Lines
We used two prostate cancer cell lines that express GRP78 on their cell surface, 1-LN and DU-145 (32). 1-LN cells originally were the kind gift of Dr. Phillip Walther, Department of Surgery, Duke University Medical Center. They now reside in our laboratory and are available on request. The DU-145 cell line was obtained from ATCC (Manassas, VA). These cells were grown as needed in multiwell plates (300 × 103 cell/well) in RPMI 1640 medium containing 10% fetal bovine serum, 2 mm glutamine, 12.5 units of penicillin/ml, 6.5 μg/ml streptomycin, and 10 nm insulin (RPMI-S) at 37 °C in a humidified CO2 (5%) incubator. At ∼90% confluency, the medium was aspirated, and the monolayers were washed with ice-cold Hanks' balanced salt solution containing 10 mm HEPES (pH 7.4) and 3.5 mm NaHCO3 (HHBSS) before use in the studies described below. Although the focus of this and previous studies has been androgen-independent tumors, in some experiments we employed hormone-dependent LnCap prostate cancer cells that, like 1-LN and DU-145 cells but unlike PC-3 cells, express GRP78 on their cell surface. LnCap cells were obtained from ATCC and grown as above.
Protein Synthesis in Prostate Cancer Cells Stimulated with α2M* or Insulin and the Effect of PI 3-Kinase/Akt/mTORC and Fatty-acid Synthase Inhibitors
1-LN and DU-145 cells grown in 48-well plates were preincubated with the Akt1 inhibitor MKK-2206 (8 nm/60 min), fatty-acid synthase inhibitors C-75 (20 μg/ml/2 h), orlistat (10 μm/2 h), and mTORC1 and mTORC2 inhibitors KU0036794 (1 μm/1 h) followed by the addition of α2M* (100 pm) or insulin (200 nm) to the respective wells. [3H]Leucine (2 μCi/ml) was added to the cells and incubated overnight. The reactions were terminated by aspirating the medium, and the cells were washed twice with ice-cold 5% TCA, followed by three washes with ice-cold PBS. The cells were lysed in a volume of NaOH (40 °C/2 h). Protein was estimated in an aliquot (85), and the lysates were counted in a liquid scintillation counter (32).
[3H]Thymidine Incorporation in Prostate Cancer Cells Stimulated with α2M* or Insulin and the Effect of PI 3-Kinase/Akt/mTORC or Fatty-acid Synthase Inhibitors
1-LN and DU-145 cells in 48-well plates were grown and processed as described above except that [3H]thymidine (2 μCi/ml) was added to the cells. The reactions were stopped by aspirating the medium, and the cells were washed twice with ice-cold 5% TCA, followed by three washes with ice-cold PBS. The cells were lysed in a volume of 1 m NaOH, the protein estimated in an aliquot (85), and samples were counted in a liquid scintillation counter. The effects of inhibitors on protein and DNA synthesis were evaluated by incubating prostate cancers with these inhibitors alone, and incorporation of [3H]leucine or [3H]thymidine into protein and DNA was determined as above. Inhibitor effects on prostate cancer cell proliferation was also evaluated by counting the cells in a hemocytometer at “0” time and after 16 h of incubation.
Glucose Uptake by Prostate Cancer Cells Stimulated with α2M* or Insulin
1-LN and DU-145 cells were grown in 6-well plates as described above in serum-free medium. The serum-free medium was aspirated, and DMEM containing 25 mm glucose was added followed by the addition of buffer, α2M*, insulin, or anti-CTD prior to the addition of α2M*. A final concentration of 1 μCi/ml [14C]deoxyglucose was then added. The cells were incubated as described above for 0 and 60 min. The reactions were terminated by aspirating the medium. The cells were washed with ice-cold PBS, and the cells were lysed in lysis buffer containing 50 mm Tris·HCl (pH 7.5), 120 mm NaCl, 1% Nonidet P-40, 2.5 mm sodium fluoride, 1 mm sodium phosphate, 0.1 mm sodium orthovanadate, 1 mm PMSF, 1 mm benzamidine, and leupeptin (20 μg/ml) (32). The samples were counted in a scintillation counter.
Lactate Secretion in 1-LN and DU-145 Cells Stimulated with α2M* or Insulin
Lactate secretion (3 × 106 cells/well) was determined in 6-well plates according to the manufacturer's instructions using a kit (Enzo Life Sciences, Farmingdale, NY). The cells incubated overnight in DMEM were stimulated with buffer, α2M*, insulin, or anti-CTD prior to α2M*. Lactate secretion was measured in aliquots of the medium removed at 0 and 60 min of incubation.
Real Time PCR of SREBP1, SREBP2, Fatty-acid Synthase, ATP Citrate Lyase, Acetyl-CoA Carboxylase, and Glut-1 in Prostate Cancer Cells Stimulated with α2M* or Insulin
1-LN and DU-145 prostate cancer cells were grown in 6-well plates and washed twice with HHBSS. The cells were exposed to either buffer, α2M*, or insulin and incubated as described above. The reactions were terminated by aspirating the medium. Total cellular RNA was extracted using the RNeasy minikit (Qiagen, Valencia, CA) according to the manufacturer's instructions and quantified by absorbance at 260 nm. Total RNA (500 ng) was pre-digested with DNase I (Invitrogen) for 15 min and used for DNA synthesis with RNase H reverse transcriptase (Invitrogen) and random primers according to the manufacturer's instructions. Real time formation of product was monitored in a Stratagene Mx3005P system (Agilent Technologies Inc., Santa Clara, CA) using SYBR Green I detection under the following conditions: 10 min at 95 °C for 1 cycle, 15 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C for 40 cycles. The sequences of primers were as follows: fatty-acid synthase, forward 5′-TCG TGG GCT ACA GCA TGGT-3′ and reverse 5′-CCC TCT GAA GTC GAA GAA GAA-3′; acetyl-CoA carboxylase, forward 5′-CTG TAG AAA CCC GGA CAG TAG AAC-3′ and reverse 5′-GGT CAG CAT ACA TCT CCA TGT G-3′; ATP citrate lyase, forward 5′-TGC TCG ATT ATG CAC TGG AAGT-3′ and reverse 5′-ATG AAC CCC CAT ACT CCT TCC CAG-3′; SREBP1, forward 5′-GGA CTT CGA GCA AGA GAT GG-3′ and reverse 5′-ATG TGG CAG CAG GAC GTG GAG AC-3′; SREBP2, forward 5′-CCT GCC CCT CTC CTT CCT CT-3′ and reverse 5′-TGC CCT GCC ACC TAT CCT CT-3′; Glut-1, forward 5′-AAC TCT TCA GCC AGG GTC CAC-3′ and reverse 5′-CAC AGT GAA GAT GAT GAA GAC-3′; human β actin, forward 5′-GGA CTT CGA GCA AGA GAT GG-3′ and reverse 5′- AGC ACT GTG TTG GCG TAC AG-3′.
MxPro QPCR software (Stratagene Agilent Technologies, Santa Cruz, CA) was used to select a threshold level of fluorescence in the linear log phase of the PCR product accumulation. The expression of target genes was normalized to the expression of β-actin to determine the fold change over buffer-treated cells.
Effect of Silencing GRP78 Gene Expression in 1-LN and DU-145 Cells on α2M*-induced Fatty-acid Synthase and ATP Citrate Lyase Expression
To determine the requirement of cell surface GRP78 for α2M*-induced up-regulation of lipogenesis, we silenced the expression of GRP78 by RNAi. In our earlier publications, we used two GRP78-targeting RNA sequences for gene expression silencing and found identical effects on GRP78 expression and downstream signaling (29). We also found that transfection of cells with GRP78 dsRNA down-regulates both the total cellular GRP78 pool and cell surface-localized GRP78 by ∼60–65% (29). Therefore, in this study, we used only one GRP78-targeting RNA sequence to silence the expression of GRP78. For the GRP78 dsRNA, sense (5′-AAA ACA GCA AUU AGU AAA GTT-3′) and antisense (5′-CUU UAC UAA UUG CUG UAU UTT-3′) oligonucleotides were prepared and annealed by Ambion (Austin, TX). Transfection of 1-LN and DU-145 cells with GRP78 dsRNA was performed as described previously (29). Down-regulation of GRP78 upon transfection with GRP78 dsRNA was evaluated by Western blotting. Briefly, 1-LN and DU-145 cell monolayers (1.5 × 106 cells/well in 6-well plates) were incubated as described above and transfected with 100 nm annealed GRP78 dsRNA, and control cells were transfected with Lipofectamine as described previously (29). Forty eight hours after transfection, the control cells were stimulated with either buffer or α2M*. Cells transfected with scrambled dsRNA (100 nm) and treated with α2M* were used as the control. The reactions were terminated by aspirating the medium, and the cells were lysed in lysis buffer on ice for 15 min. The lysates were then transferred to tubes and centrifuged, and the protein concentration in the supernatant was determined. Equal amounts of lysate protein were electrophoresed, transferred to a membrane, and immunoblotted with anti-fatty-acid synthase or ATP citrate lyase antibodies. The protein bands were detected and quantified by enhanced chemifluorescence and phosphorimaging. The membrane was reprobed for the protein loading control, actin. Consistent with previous studies, the GRP78 protein was reduced 60–70%.
1-[14C]Acetate Incorporation into Lipids of Prostate Cancer Cells Stimulated with α2M* or Insulin and the Effect of Pretreatment with PI 3-Kinase/Akt/mTORC and Fatty-acid Synthase Inhibitors
1-LN and DU-145 cells (3 × 106 cells/well) incubated overnight in 6-well plates were washed with RPMI-S medium, and RPMI-S medium was added prior to the addition of 1-[14C]acetate (1 μCi/well). The cells were incubated for 4 h as described above. The reactions were terminated by aspirating the medium, and the cells were washed three times with ice-cold PBS, and RPMI-S medium was added to each well. The cells were pretreated with either LY294002, rapamycin, torin, KU0036795, C-75, or anti-CTD and then stimulated with buffer, α2M*, or insulin. The reactions were terminated by aspirating the medium, and the cells were washed three times with ice-cold buffer and lysed in lysis buffer for 15 min over ice. The cell lysates were transferred into Eppendorf tubes and centrifuged at 4 °C for 5 min at 1000 rpm; the supernatant was transferred to new Eppendorf tubes, and their protein content was determined. Equal amounts of lysate proteins (250 μg) were used for lipid extraction. The lipids were extracted in screw cap glass tubes by adding chloroform/methanol/water (2:1:0.5 v/v/v) to the lysates. The mixture was vortexed and incubated at 4 °C overnight (86). The tubes were centrifuged at 1000 rpm for 5 min at 4 °C, and the aqueous phase was removed. The organic phase was washed with 1 ml of water by centrifugation as described above. The aqueous phase was removed, and the organic phase was dried under N2. The lipid residues were dissolved in chloroform, and an aliquot was counted in a liquid scintillation counter. The effect of inhibitors alone on lipogenesis from 1-[14C]acetate in 1-LN and DU-145 cells was evaluated by incubating these cells with the inhibitors under identical conditions.
1-[14C]Acetate Incorporation into Nonsaponifiable (Cholesterol) and Saponifiable (Fatty Acid) Fractions of Prostate Cancer Cells Treated with α2M* or Insulin and the Effect of PI 3-Kinase/Akt/mTORC and Fatty-acid Synthase Inhibitors
1-[14C]Acetate (1 μCi/ml/4 h)-labeled 1-LN and DU-145 cells were pretreated with inhibitors of PI 3-kinase/Akt/mTORC and fatty-acid synthase and stimulated with α2M* or insulin as described above. Lysate protein, 250 μg, was used for lipid extraction as above. The lipid extracts in glass tubes were evaporated to dryness under N2. The dried lipid extracts were saponified by refluxing with 30% alcoholic KOH for 30 min at 70 °C in a water bath. The saponification was terminated by adding water, and the nonsaponifiable lipid was extracted three times with petroleum ether (bp. 40–60 °C). The combined lipid extracts were washed with water. The lipid extracts were evaporated to dryness under N2, and the dried lipid residue was dissolved in chloroform, and an aliquot of the lipid extract was counted in a liquid scintillation counter. The alkali digest remaining after extraction of the nonsaponifiable fraction was acidified to pH 2 with 6 n H2SO4, and the fatty acids were extracted three times with petroleum ether (bp 40–60 °C). The combined extracts were washed with water. The lipid extracts were evaporated to dryness under N2, and the dried lipid residue was dissolved in chloroform, and an aliquot was counted in a liquid scintillation counter.
In a separate set of studies, the lipid extracts were evaporated to dryness under N2, and the lipid residues were dissolved chloroform and subjected to TLC fractionation on Silica Gel H-coated plates, preheated at 110 °C for 1 h. An aliquot of the respective lipid extracts and the lipid standards, cholesterol ester, tripalmitin, palmitic acid, cholesterol, and phosphatidylcholine were applied to the TLC plates. The plates were developed to half the distance in CHCl3/CH3OH/H2O (60:30:5 v/v/v). The plates were removed, air-dried, and again developed to half-distance in the same solvent system. The plates were air-dried and developed to the full distance in hexane/ether/acetic acid (80:20:1.5 v/v/v/). The plates were dried and exposed to I2 in a closed glass chamber in a chemical hood until the lipid spots appeared. The lipid spots on the plates corresponding to the lipid standards, cholesterol ester (Rf value 0.83), tripalmitin (Rf value 0.79), palmitic acid (Rf value 0.75), cholesterol (Rf value 0.57), and phosphatidylcholine (Rf value 0.24) were scraped into vials and counted in a liquid scintillation counter.
Lipogenesis from 6-[14C]Glucose in Prostate Cancer Cells Stimulated with α2M* or Insulin and the Effect of PI 3-Kinase/Akt/mTORC and Fatty-acid Synthase Inhibitors
Lipogenesis from 6-[14C]glucose in prostate cancer cells was determined as described above except that instead of 1-[14C]acetate, we used 6-[14C]glucose (1 μCi/ml 4 h) to label the 1-LN and DU-145 cells. Pretreatment of 6-[14C]glucose-labeled cells with PI 3-kinase/Akt/mTORC and fatty-acid synthase inhibitors, stimulation of cells with α2M* or insulin, and lipid extraction were performed as for the 1-[14C]acetate-labeled cells. An aliquot of lipid extract was used for liquid scintillation counting.
Determination of SREBP1 Cleavage in Prostate Cancer Cells Stimulated with α2M or Insulin by Western Blotting
1-LN and DU-145 cells were incubated overnight in RPMI-S media in 6-well plates and treated with α2M* (100 pm/25 min) or insulin (200 nm/20 min), alone or in the presence of fatostatin A (20 μm/2 h). The reactions were terminated by aspirating the medium and washed three times with ice-cold PBS. The monolayers were lysed in lysis buffer containing 50 mm Tris·HCl (pH 7.5), 120 mm NaCl, 1% nonidet-P-40, 2.5 mm sodium fluoride, 1 mm sodium phosphate, 0.1 mm sodium orthovanadate, 1 mm PMSF, 1 mm benzamidine, leupeptin 20 μg/ml) and protease-inhibitor tablets (1 tablet/10 ml) (Roche Applied Science) over ice for 15 min. The lysates were transferred to Eppendorf tubes, centrifuged at 1000 rpm/5 min/4 °C; supernatants were removed into new tubes and their protein contents determined. Equal amounts of lysate proteins were electrophoresed on 10% gels, and protein bands were transferred to PVDF membranes that were immunoblotted with anti-SREBP1 antibody. SREBP1 cleavage was determined by the generation of cleaved SREBP1, and the molecular mass of ∼74 kDa from was SREBP1 (molecular mass ∼151 kDa).
Effect of Incubation Time on Expression of SREBP1-c, ATP Citrate Lysate, and Fatty-acid Synthase in Prostate Cancer Cells Stimulated with α2M*
1-LN and DU-145 cells were stimulated with buffer or α2M* and incubated for 0, 10, 20, 40, and 60 min as described above. The reactions were terminated by aspirating the medium, and the cells were lysed in lysis buffer for 15 min on ice. The cell lysates were transferred to Eppendorf tubes and briefly centrifuged. Then the supernatants were transferred to new tubes and their protein contents determined. A volume of 4× sample buffer was added to equal amounts of protein lysates, contents were mixed and boiled for 5 min, electrophoresed, and then transferred to PVDF membranes. The respective membranes were immunoblotted with antibodies against SREBP1-c, ATP-citrate lyase, or fatty-acid synthase. The bands were visualized and quantified with a PhosphorImager. The membranes were reprobed for actin as the protein loading control. The specificity of the antibodies used in this study was determined by treating the cells with nonimmune antibodies of the appropriate species and isotype, and the cell lysates were processed as described above. Under the experimental conditions, no reactivity of these antibodies was observed.
Effect of Inhibiting the Proteolytic Cleavage of SREBPs by Fatostatin A on Cell Proliferation in Prostate Cancer Cells Stimulated with α2M* or Insulin
1-LN, DU-145, and LnCap cells in 48-well plates were grown as described above. Fatostatin A (20 μm/2 h) or anti-CTD was added to the wells, and the cells were incubated at 37 °C in a humidified incubator (5% CO2). At certain time points, the cells were stimulated with buffer, α2M*, or insulin, followed immediately by the addition of [3H]leucine (2 μCi/ml) or [3H]thymidine (2 μCi/ml) and incubated overnight. The incorporation of [3H]leucine into the cell proteins and of [3H]thymidine into DNA was measured as described above.
Effect of Fatostatin A on α2M* or Insulin-stimulated Lipogenesis from 1-[14C]Acetate in Prostate Cancer Cells
1-LN, DU-145, and LnCap cells in 6-well plates were prelabeled with 1-[14C]acetate (1 μCi/ml/4 h) and washed twice with ice-cold PBS, followed by the addition of RPMI-S medium to each well. Fatostatin A or anti-CTD was added to the wells and incubated. Then buffer, α2M*, or insulin was added, and the cells were incubated. The reactions were terminated by aspirating the medium, and the cells were washed with PBS and lysed in lysis buffer on ice for 15 min. The incorporation of 1-[14C]acetate into cellular lipids was measured as described above.
Effect of Fatostatin A Treatment on Phosphatidylcholine Synthesis in Prostate Cancer Cells Stimulated with α2M* or Insulin
1-LN and DU-145 cells in 6-well plates were labeled with [14CH3]choline (4 μCi/ml/4 h) (87). The reactions were terminated by aspirating the medium, and the cells were washed twice with cold PBS, and RPMI-S medium was added to each well. In separate experiments, fatostatin A or anti-CTD was added to the wells. After incubation, the cells were stimulated with either buffer, α2M*, or insulin and incubated as described above. The reactions were terminated by aspirating the medium, and the cells were lysed in lysis buffer on ice for 15 min. Lipids from the cell lysates were extracted as described above. The incorporation of [14CH3]choline into phosphatidylcholine was measured by TLC as described above.
Statistical Analysis
The statistical significance of the data was determined by Student's t test. The values significantly different at the 5% level are denoted by an asterisk.
RESULTS
Fatty-acid Synthase or PI 3-Kinase/Akt/mTORC Signaling Pathway Inhibitors Suppress α2M* or Insulin-induced Protein and DNA Synthesis
We first assessed whether lipogenesis is involved in α2M*-induced cell proliferation by pretreating 1-LN and DU-145 cells with specific inhibitors of Akt1, mTORC1, mTORC2, or fatty-acid synthase, because these regulate cell proliferation by modulating SREBP processing and fatty-acid synthase activity. We used MKK-2206, a specific inhibitor of Akt1, KU0063794, an inhibitor of mTORC1 and mTORC2, C-75, which inactivates the β-ketoacyl reductase and thiol esterase activities of fatty-acid synthase, and orlistat, which inactivates fatty-acid synthase by forming a covalent adduct with the thiol esterase domain of the enzyme. Stimulation of 1-LN and DU-145 cells with α2M* caused an ∼1.5–2-fold increase in protein and DNA synthesis, which was similar to insulin up-regulated protein (Fig. 1, A and B) and DNA (Fig. 1, C and D) synthesis. The data are corroborated by cell growth studies that show similar results to these findings (Fig. 1, E and F). These inhibitors at the concentration used did not induce cell death as determined by incorporation of [3H]leucine into protein and [3H]thymidine into DNA as compared with buffer-treated cells (Fig. 1, G and H). Down-regulation of α2M* and insulin-dependent protein and DNA synthesis in these cells by inhibitors demonstrates that Akt1 and mTORC signaling and fatty-acid synthase up-regulation are necessary for macromolecular synthesis.
FIGURE 1.

Inhibition of α2M* (black bars) and insulin (white bars)-induced protein synthesis in 1-LN (A) and DU-145 (B) prostate cancer cells is shown. DNA synthesis for 1-LN (C) and DU-145 (D) cells, respectively, is shown. Cell growth studies are shown in E and F, respectively. The bars in A–D are as follows: 1, buffer; 2, α2M* or insulin; 3, MKK-2206 then α2M* or insulin; 4, KU0063794 then α2M* or insulin; 5, C-75 then α2M* or insulin; and 6, orlistat then α2M* or insulin. Protein synthesis data are the mean ± S.E. from three experiments. The effects of inhibitors on cell growth of 1-LN (E) and DU-145 (F) cells stimulated with α2M* (black bars) or insulin (white bars) are shown. The bars at 16 h in E and F are as above. The effects of inhibitors alone on protein synthesis (G) and DNA synthesis (H) in 1-LN (black bars) and DU-145 (white bars) prostate cancer cells are shown. The bars in both G and H are as follows: 1, buffer; 2, MKK-2206; 3, KU0063794; 4, C-75; and 5, orlistat. The values in G and H are the mean ± S.E. from three experiments. Values significantly different at 5% level from buffer or inhibitor-treated cells are marked with an asterisk.
α2M* Up-regulates the Expression of SREBP1-c, Fatty-acid Synthase, and ATP Citrate Lyase in 1-LN and DU-145 Prostate Cancer Cells
Because inhibition of fatty-acid synthase suppresses protein and DNA synthesis in 1-LN and DU-145 cells stimulated with α2M* or insulin, we next evaluated SREBP1-c, ATP citrate lyase, and fatty-acid synthase expression in these cells by Western blotting (Fig. 2, A and B). Stimulation of 1-LN and DU-145 cells with α2M* caused a 1.5–2.5-fold up-regulation of SREBP1, SREBP2, fatty-acid synthase, and ATP citrate lyase in 1-LN (Fig. 2A) and DU-145 cells (Fig. 2B). We do not understand the decline in SREBP1 and ATP citrate lyase expression in α2M*-stimulated 1-LN and DU-145 cells after 15 min of incubation. Most likely it is due to their rapid turnover and/or precursor-product relationship in a downstream cascade causing activation of lipogenic responses. We also measured the transcriptional up-regulation of SREBP1-c, SREBP2, fatty-acid synthase, acetyl-CoA carboxylase, and ATP citrate lyase by assaying their mRNA levels by real time PCR. Treatment of 1-LN and DU-145 cells with α2M* and insulin increased the mRNA expression relative to β-actin by ∼1.5–2.5-fold compared with buffer-treated cells (Fig. 2, C and D). Although α2M* and insulin caused a 1.5–2.5-fold increase in the mRNA levels, the mRNA expression varied among the genes studied. The expression level of SREBP1-c protein was very low compared with the other gene products (Fig. 2, C and D). These results show that similar to insulin, α2M* up-regulates lipogenic pathways in prostate cancer cells.
FIGURE 2.

Effect of time of incubation of 1-LN (A) and DU-145 cells (B) with α2M* (100 pm) on the expression of SREBP1 (○), ATP citrate lyase (black bars), and fatty-acid synthase (▩) is shown. The values are expressed in arbitrary units ( × 102) and are the mean ± S.E. from three to four independent experiments. The abbreviations used in the figure are as follows: ATP-CYL, ATP citrate lyase; FAS, fatty-acid synthase; ACC, CoA carboxylase; and SREBP, sterol regulatory binding protein 1. Real time mRNA PCR of SREBP1, SREBP2, ATP citrate lyase, acetyl-CoA carboxylase, and fatty-acid synthase in 1-LN and DU-145 cells stimulated with α2M or insulin (C). The mRNA levels from three to four experiments are expressed as fold change over the mRNA levels of buffer-treated cells. The mRNA levels are expressed relative to β-actin. Values significantly different at 5% level from buffer-treated cells are marked with an asterisk.
Elevated Glucose Uptake in Prostate Cancer Cells Treated with α2M*
Prostate carcinoma cells show increased expression of Glut-1 and increased glucose uptake and utilization to support androgen-independent proliferation (53, 88). Therefore, we measured glucose uptake in 1-LN and DU-145 prostate cells stimulated with α2M* or insulin by incubating cells with 1-[14C]deoxyglucose (Fig. 3, A and B). At 60 min of incubation, α2M* and insulin-stimulated cells showed a 2–3-fold increase in glucose uptake compared with buffer-treated cells (Fig. 3, A and B). Pretreatment of 1-LN and DU-145 cells with anti-CTD significantly reduced α2M*-mediated glucose uptake. Glut-1 mRNA levels in both 1-LN and DU-145 cells treated with α2M* or insulin increased 1.5–4-fold compared with buffer-treated cells (Fig. 3C). Expression of Glut-1 was more pronounced in 1-LN cells compared with DU-145 cells stimulated with α2M* or insulin (Fig. 3C).
FIGURE 3.

1-[14C]Deoxyglucose uptake by 1-LN (A) and DU-145 (B) cells is shown. 1-[14C]Deoxyglucose uptake in cells treated with buffer (black bars), α2M* (light gray bars); insulin (dark gray bars), or anti-CTD antibody then α2M* for 60 min (white bars) is shown. 1-[14C]Deoxyglucose uptake is expressed as cpm/mg/supernatant and is the mean ± S.E. from four experiments. Values significantly different at the 5% level at the 60-min period from buffer and anti-CTD are indicated by an asterisk. C, Glut-mRNA levels determined by real time PCR in 1-LN (black bars) and DU-145 (white bars) cells treated with α2M* or insulin. Glut-1 mRNA levels are expressed as fold change over mRNA levels in buffer-treated cells and are significantly different (asterisk) at 5% levels from buffer-treated cells and are denoted by an asterisk. D, lactate secretion in1-LN (black bars) and DU-145 (white bars) cells treated with the following: 1, buffer; 2, α2M*; 3, insulin; or 4, anti-CTD then α2M*. Lactate secretion is expressed as micromolars/60 min and is the mean ± S.E. from three experiments. Values significantly different at the 5% level from buffer and anti-CTD-treated cells are indicated by an asterisk.
Increased Lactate Secretion in Prostate Cancer Cells Stimulated with α2M* or Insulin
We next determined the secretion of lactate in prostate cells stimulated with α2M* or insulin (Fig. 3D) (89–91). α2M* or insulin-stimulated cells showed an ∼2-fold increase in lactate secretion compared with buffer-treated cells. Importantly, anti-CTD blocked both the increased glucose uptake (Fig. 3, A and B) and lactate secretion (Fig. 3D) in 1-LN and DU-145 prostate cancer cells stimulated with α2M*. Previous studies of head and neck tumors have shown a correlation of high lactate levels with an increased incidence of metastasis (91). We hypothesize a similar behavior in prostate cancer.
Lipogenesis from 1-[14C]Acetate Is Up-regulated in Prostate Cancer Cells Stimulated with α2M* or Insulin
Next, we studied the activities of lipogenic enzymes in prostate cancer cells via the incorporation of the labeled precursors 1-[14C]acetate and 6-[14C]glucose into the total cellular lipids, cholesterol, fatty acids, and phosphatidylcholine. In both 1-LN and DU-145 cells, α2M* and insulin up-regulated 1-[14C]acetate incorporation into total lipids by 2–3-fold compared with buffer-treated cells (Figs. 3B and 4A). Pretreatment of cells with PI 3-kinase, mTORC1, mTORC2, or fatty-acid synthase inhibitors nearly abolished α2M* and insulin-induced lipogenesis. Importantly, anti-CTD also abrogated α2M*-induced lipogenesis in these cells. We also evaluated whether inhibitors used in this study caused reduced lipogenesis due to initiation of cell death by incubating cancer cells with inhibitors alone and processed cells for lipid extraction and counting for 1-[14C]acetate incorporation. The incorporation was comparable with that observed in buffer-treated cells showing thereby that inhibitors did not suppress 1-[14C]acetate incorporation into lipid by inducing cell death (data not shown).
FIGURE 4.

Induction of lipogenesis by α2M* or insulin in prostate cancer cells. A, 1-LN cells, inhibition of α2M* (black bars) or insulin (white bars)-induced lipogenesis from 1-[14C]acetate in 1-LN prostate cancer cells by fatty-acid synthase inhibitors. The bars in the figure are as follows: 1, buffer; 2, α2M* or insulin; 3, LY294002 (20 μm/25 min) and then α2M* or insulin; 4, rapamycin (100 nm/20 min) and then α2M or insulin; 5, torin and then α2M* or insulin; 6, C-75 and then α2M or insulin; 7, KU0063794 and then α2M* or insulin; and 8, anti-CTD and then α2M or insulin. B, inhibition of α2M* (black bars) or insulin (white bars)-induced lipogenesis from 1-[14C]acetate in DU-145 cells by the fatty-acid synthase inhibitors in DU-145 cells. The bars in the figure are same as in A. The values are the mean ± S.E. from three experiments in both cases. Values significantly different at the 5% level in A and B from buffer and inhibitor-treated cells are denoted by an asterisk. C, inhibition of α2M* (black bars) or insulin (white bars)-induced lipogenesis from 6-[14C]glucose in 1-LN prostate cancer cells pretreated with fatty-acid synthase inhibitors. The bars in the figure are as follows: 1, buffer; 2, α2M* or insulin; 3, LY294002 and then α2M* or insulin; 4, rapamycin and then α2M* or insulin; 5, torin and then α2M* or insulin; 6, C-75 and then α2M or insulin; 7, KU0063794 and then α2M* or insulin; 8, MKK2206 and then α2M* or insulin; 9, anti-CTD and then α2M*. D, suppression of α2M* (black bars) or insulin (white bars)-induced lipogenesis from 6-[14C]glucose in DU-145 cells pretreated with fatty-acid synthase inhibitors as in C. The results are the mean ± S.E. from three experiments. Values significantly different at the 5% levels in from buffer-treated and inhibitor-treated cells are denoted by an asterisk. Treatment of 1-LN or DU-145 cells with inhibitors alone did not effect 1-[14C]acetate or 6-[14C]glucose measurements (data not shown).
Lipogenesis Is Up-regulated in Prostate Cancer Cells Stimulated with α2M* or Insulin
We next studied the incorporation of 6-[14C]glucose into the cellular lipids of 1-LN and DU-145 cells stimulated with α2M* or insulin (Fig. 4, C and D). We also examined the modulation of 6-[14C]glucose incorporation into cellular lipids using inhibitors of PI 3-kinase, Akt, mTORC1, mTORC2, fatty-acid synthase, or anti-CTD. Similar to lipogenesis from 1-[14C]acetate, lipogenesis from 6-[14C]glucose was up-regulated 2–3-fold in these cells. Pretreatment of cells prior to stimulation with α2M* or insulin with PI 3-kinase/Akt/mTORC1, mTORC2, or fatty-acid synthase inhibitors abrogated α2M* or insulin-induced increased lipogenesis from 6-[14C]glucose, consistent with the role of PI 3-kinase/Akt/mTORC1 and mTORC2 signaling and fatty-acid synthase activity in lipogenic responses in these cells. Anti-CTD also showed a similar effect on α2M* effects.
α2M* and Insulin Increase the Incorporation of 1-[14C]Acetate into Nonsaponifiable and Saponifiable Lipids of 1-LN and DU-145 Prostate Cancer Cells
To access 1-[14C]acetate conversion into fatty acids and cholesterol, we saponified cellular lipids and separated the nonsaponifiable fraction, which includes cholesterol, and the saponifiable fraction, which includes fatty acids from glycerides and phospholipids. To understand the role of PI 3-kinase/Akt/mTORC signaling in the synthesis of these lipids, we pretreated prostate cancer cells with specific inhibitors of these signaling pathways prior to stimulation with α2M* or insulin. Each caused a severalfold increase in 1-[14C]acetate incorporation into both nonsaponifiable (Fig. 5, A and B) and saponifiable (Fig. 5, C and D) fractions, indicating increased cholesterol and fatty acid synthesis in 1-LN and DU-145 cells.
FIGURE 5.

α2M (black bars) and insulin (white bars) up-regulate the incorporation of 1-[14C]acetate into a nonsaponifiable lipid fraction (cholesterogenesis) in 1-LN cells (A) and in DU-145 cells (B). The bars in A and B are as follows: 1, buffer; 2, α2M* and then α2M* or insulin; 3, LY294002 and then α2M* or insulin; 4, rapamycin and then α2M or insulin; 5, torin and then α2M* or insulin; 6, C-75 and then α2M* or insulin; 7, KU0063794 and then α2M* or insulin; 8, anti-CTD and then α2M*. The incorporation of 1-[14C]acetate into nonsaponifiable fractions (cholesterogenesis) in both A and B is expressed as cpm/mg cell protein and is the mean ± S.E. from three experiments. Values significantly different at the 5% level from buffer and inhibitor-treated cells in both A and B are denoted by an asterisk. Up-regulation of 1-[14C]acetate incorporation into saponifiable fractions (fatty acids) by α2M* (black bars) or insulin (white bars) in 1-LN cells (C) and DU-145 (D) cells. The bars in C and D are as follows: 1, buffer; 2, α2M* or insulin; 3, LY294002 and then α2M* or insulin; 4, rapamycin and then α2M* or insulin; 5, torin and then α2M* or insulin; 6, C-75 and then α2M* or insulin; 7, KU0063794 and then α2M* or insulin; 8, anti-CTD and then α2M*. The incorporation of 1-[14C]acetate into saponifiable fraction (fatty acids) in C and D is expressed as cpm/mg cell protein and is the mean ± S.E. from three experiments. Values significantly different from buffer and inhibitor-treated cells in both C and D are denoted by an asterisk.
Thin Layer Chromatography of 1-[14C]Acetate-labeled Lipids of Prostate Cancer Cells Stimulated with α2M* or Insulin
In the next series of experiments, we separated 1-[14C]acetate-labeled lipids into cholesterol, esterified cholesterol, triglycerides, free fatty acids, and phosphatidylcholine by thin layer chromatography to study the synthesis of individual lipids. A representative thin layer chromatogram is shown in Fig. 6A. Cholesterol was the major lipid in both 1-LN and DU-145 prostate cancer cells representing ∼75–80% of the total lipids, followed by phosphatidylcholine, which formed ∼15% of total cellular lipids (Fig. 6A). The synthesis of esterified cholesterol from 1-[14C]acetate-labeled cholesterol and fatty acyl-CoA was increased 2–2.5-fold in both 1-LN (Fig. 6, B and C) and DU-145 cells (Fig. 6, D and E) stimulated with α2M* or insulin. Pretreatment of cells before stimulation with α2M* or insulin combined with inhibitors of PI 3-kinase/Akt/mTORC and fatty-acid synthase significantly decreased the esterification of cholesterol (Fig. 6). Similarly, the ligation of GRP78 with anti-CTD reduced cholesterol esterification (Fig. 6).
FIGURE 6.

Thin layer chromatography of 1-[lsqb]14C]acetate-labeled lipid extracts of 1-LN and DU-145 cells treated with α2M* or insulin. A, representative thin layer chromatogram of cell lipids of 1-LN cells. A similar pattern was obtained with DU-145 cells (data not shown). B, fatty-acid synthase inhibitors inhibit α2M-induced increased incorporation of 1-[14C]acetate into esterified cholesterol (black) triglycerides (red), fatty acids (green), cholesterol (blue), and phosphatidylcholine (white) fractions of 1-LN cells. The bars are in the figure are as follows: 1, buffer; 2, α2M*; 3, LY294002 and then α2M*; 4, rapamycin and then α2M*; 5, torin and then α2M*; 6, C-75 and then α2M*; 7, KU0063794 and then α2M*; 8, anti-CTD and then α2M*. C, inhibition of insulin-induced increase 1-[14C]acetate incorporation into cellular lipids fraction of 1-LN cells by prior treatment of cells with fatty-acid synthase inhibitors. Bars in the figure are as in B except treatment with anti-CTD or KU0063794, which was not performed. Fatty-acid synthase inhibitors suppress α2M* (D) and insulin-induced increase (E) of [14C]acetate incorporation into cellular lipids of DU-145 cells. The bars in figure in D are the same as in B, and the bars in figure in E are the same as in C. The incorporation of 1-[14C]acetate into various lipid fractions in B–E is expressed as cpm/mg protein and is mean ± S.E. from three experiments. Values significantly different at 5% levels from buffer and inhibitor-treated cells in B–E are denoted by an asterisk.
Stimulation of 1-LN (Fig. 6, B and C) and DU-145 prostate cancer cells (Fig. 6, D and E) also caused an increased synthesis of triglycerides incorporating fatty acids synthesized from 1-[14C]acetate (Fig. 6). However, the magnitude of synthesis varied in these cells stimulated with α2M* or insulin. Synthesis of triglycerides and free fatty acids from 1-[14C]acetate in 1-LN and DU-145 cells was significantly suppressed by inhibition of PI 3-kinase/Akt/mTORC signaling or fatty-acid synthase, as well as by anti-CTD treatment prior to α2M* addition (Fig. 6, B and C, and D and E).
Stimulation of 1-LN (Fig. 6, B and C) and DU-145 (Fig. 6, D and E) prostate cancer cells with α2M* or insulin up-regulated cholesterol synthesis from 1-[14C]acetate by about 2–3-fold compared with buffer-treated cells. This increased cholesterol synthesis causes the accumulation of cholesterol in prostate cancer cells under our experimental conditions (Fig. 6A). Cholesterol synthesis is regulated by PI 3-kinase/Akt/mTORC signaling and was demonstrated by inhibiting these pathways. Inhibition of fatty-acid synthase by C-75 nearly abrogated cholesterol synthesis. Down-regulation of GRP78 by anti-CTD also inhibited cholesterol synthesis.
We next measured the synthesis of phosphatidylcholine in prostate cancer cells stimulated with α2M* or insulin. We quantified the levels of 1-[14C]acetate-labeled fatty acids incorporated into phosphatidylcholine by thin layer chromatography (Fig. 6B). Similar to insulin, α2M*-treated prostate cancer cells up-regulated phosphatidylcholine synthesis 2–3-fold (Fig. 6). Pretreatment of prostate cancer cells with inhibitors of PI 3-kinase/Akt/mTORC or fatty-acid synthase significantly reduced α2M*/insulin-induced increased phosphatidylcholine synthesis from 1-[14C]acetate-labeled fatty acids.
Inhibition of SREBP1 Proteolytic Cleavage by Fatostatin A Suppresses Cell Proliferation, Lipogenesis, and Phosphatidylcholine Biosynthesis in Prostate Cancer Cells Stimulated with α2M* or Insulin
Fatostatin A inhibits proteolytic cleavage of SREBP1 and thus inhibits downstream signaling cascades (91, 92). Stimulation of prostate cancer cells with α2M*, similar to insulin, up-regulated protein synthesis 2–3-fold in 1-LN, DU-145, and LnCap cells compared with buffer-treated controls, although pretreatment of cells with fatostatin A nearly abolished these effects (Fig. 7, A and C). α2M* and insulin also up-regulated [3H]thymidine uptake, indicating increased DNA synthesis 2–3-fold, although pretreatment with fatostatin A nearly abrogated these effects (Fig. 7, B and D). The effects of fatostatin treatment on SREBP1 cleavage is shown in Fig. 7, E and F. Fatostatin A inhibits proteolytic cleavage of SREBP1 and thus inhibits signaling cascades. We determined the effect of fatostatin A on SREBP1 cleavage by Western blotting in 1-LN and DU-145 cells. Cells stimulated with α2M* or insulin demonstrated significant cleavage of SREBP1 (molecular mass ∼151 kDa) to mature SREBP1 (molecular mass ∼74 kDa).
FIGURE 7.

Inhibition of proteolytic cleavage of SREBP1 by fatostatin A on α2M*-dependent metabolic regulation. Fatostatin abrogates α2M* or insulin-induced protein and DNA synthesis in1-LN (black bars) and DU-145 (white bars) cells (A and B). The bars are as follows: 1, buffer; 2, α2M*; 3, insulin; 4, fatostatin A (20 μm/2 h) and then α2M*; 5, fatostatin A and then insulin; and 6, fatostatin. Similar results were obtained with LnCAP cells (C and D). The bars are as above and the mean ± S.E. from three experiments. Values significantly different at the 5% levels from buffer and inhibitor-treated cells are denoted by an asterisk. A representative immunoblot of three experiments showing cleaved SREBP1 (kDa ∼151) into mature cleaved SREBP1 (kDa ∼74) in α2M* or insulin-treated cells and its inhibition by fatostatin A (E and F). The lanes in immunoblots are as follows: lane 1, α2M*; lane 2, insulin; lane 3, fatostatin A then α2M*; lane 4, fatostatin A then insulin; or lane 5, buffer.
Because of the critical regulation of lipogenesis by the transcription factor, SREBP, we next determined the total cellular lipogenesis from 1-[14C]acetate in 1-LN and DU-145 prostate cancer cells pretreated with fatostatin A (Fig. 8, A and B). Fatostatin A significantly reduced 1-[14C]acetate incorporation into total lipids of prostate cancer cells stimulated with α2M* or insulin (Fig. 8, A and B). Pretreatment of 1-LN and DU-145 and LnCap prostate cancer cells with PI 3-kinase/Akt/mTORC and fatty-acid synthase inhibitors and fatostatin A inhibitor of SREBP cleavage significantly suppressed the α2M* or insulin-induced increase in total lipogenesis from 1-[14C]acetate (Fig. 8, A–C). PI 3-kinase/Akt/mTORC is upstream of SREBPs, and fatty-acid synthase, a target gene of SREBP1-c, is downstream; therefore, it is expected that impairment of SREBP1 activation and hence activation of target genes is significantly inhibited. These results are consistent with genetic manipulations of SREBPs and RNAi silencing of SREBPs, which impair lipogenesis and cell proliferation (46–54).
FIGURE 8.

Fatostatin A suppresses α2M* or insulin-induced increased lipogenesis from 1-[14C]acetate in 1-LN (A) and DU-145 (B) and LnCap (C) prostate cancer cells. The bars in figure in A and B are as follows: 1, buffer; 2, α2M*; 3, insulin; 4, fatostatin A and then α2M*; 5, fatostatin A and then insulin; and 6, anti-CTD and then α2M*. The bars in figure in C are as follows: 1, buffer; 2, α2M*; 3, insulin; 4, torin and then α2M* or insulin; 5, C-75 and then α2M* or insulin; 6, fatostatin A and then α2M* or insulin; 7, fatostatin A. 1-[14C]Acetate incorporation in A–C is expressed as cpm/mg cell protein and is mean ± S.E. from three experiments. Values significantly different at 5% levels from buffer and inhibitor-treated cells in A–C are denoted by an asterisk. Inhibition of SREBP1 cleavage by fatostatin A suppresses α2M* or insulin-induced increased de novo phosphatidylcholine synthesis from [14CH3]choline in 1-LN (D) and DU-145 (E). The bars in both D and E are as follows: 1, buffer; 2, α2M*; 3, insulin; 4, fatostatin A and then α2M*; 5, fatostatin A and then insulin; 6, anti-CTD and then α2M*. The synthesis of phosphatidylcholine in both panels is expressed as cpm/mg cell protein and is the mean ± S.E. from three experiments. Values significantly different at 5% levels from buffer and fatostatin A-treated cells in both panels are denoted by an asterisk.
Next, using thin layer chromatography, we showed that both α2M* and insulin increased phosphatidylcholine synthesis 2–3-fold from 1-[14C]acetate-labeled acyl-CoAs in 1-LN and DU-145 prostate cancer cells, which was greatly suppressed by inhibitors of PI 3-kinase/Akt/mTOR and fatty-acid synthase (Fig. 6). SREBP is involved in de novo phosphatidylcholine synthesis (88, 93, 94). To further understand the regulation of de novo phosphatidylcholine by SREBP, we pretreated [14CH3]choline-labeled 1-LN and DU-145 prostate cancer cells with fatostatin A prior to stimulation with α2M* or insulin, extracted lipids, fractionated phosphatidylcholine by thin layer chromatography, and counted the band in a scintillation counter. Both α2M* and insulin up-regulated phosphatidylcholine synthesis 2-fold in both 1-LN (Fig. 8E) and DU-145 cells (Fig. 8F). Inhibition of proteolytic cleavage of SREBP by fatostatin A significantly inhibited α2M* and insulin-induced de novo phosphatidylcholine synthesis in both 1-LN and DU-145 cells (Fig. 8, E and F).
Down-regulation of GRP78 Suppresses the Expression of the SREBP Target Genes ATP Citrate Lyase and Fatty-acid Synthase in 1-LN and DU-145 Prostate Cancer Cells Stimulated with α2M*
To further assess the role of the cell surface GRP78 receptor in α2M*-induced lipogenesis, we silenced the expression of GRP78 using RNAi and quantified the expression of two key enzymes, ATP-citrate lyase and fatty-acid synthase, in 1-LN and DU-145 prostate cancer cells stimulated with α2M*. Silencing the expression of GRP78 significantly reduced α2M* up-regulation of ATP citrate lyase and fatty-acid synthase (Fig. 9, A and B) in 1-LN and DU-145 cells compared with buffer-treated cells.
FIGURE 9.

Down-regulation of cell surface GRP78 by RNAi suppresses α2M*-induced expression of ATP citrate lyase (black bars) and fatty-acid synthase (white bars) in 1-LN (A) and DU-145 (B) prostate cancer cells. The panels in both immunoblots are as follows: 1, Lipofectamine + α2M*; 2, GRP78 dsRNA (100 nm/48 h)/Lipofectamine and then α2M*; and 3, scrambled dsRNA (100 nm/48 h)/Lipofectamine and then α2M*. The changes in expression of target proteins are in arbitrary units and are the mean ± S.E. from four experiments. Values significantly different at the 5% level for GRP78 dsRNA-transfected cells compared with the controls are denoted by an asterisk. A representative immunoblot of fatty-acid synthase and ATP citrate lyase in 1-LN (A) and DU-145 (B) cells from four experiments is shown below the respective bar diagrams.
DISCUSSION
Rapidly proliferating tumor cells exhibit increased glucose uptake and increased secretion of lactate in the presence of abundant oxygen, known as the Warburg effect, which is a hallmark of aggressive tumors (55). The yield of ATP by anaerobic glucose consumption is low, two molecules of ATP/molecule of glucose compared with the 38 ATP molecules/glucose obtained by the complete oxidation of glucose (55). The high glycolytic flux, however, allows the amount of cellular ATP produced from glycolysis to exceed that produced from oxidative phosphorylation (46–64, 78). Activation of the PI 3-kinase/Akt/mTORC pathways in tumor cells enhances the expression of genes and up-regulates enzyme activities causing increased glycolysis and lactate production (46–64, 78).
α2M* binds to LRP and cell surface GRP78 with very distinct binding characteristics and cellular responses (18, 20–23, 25–39). The Kd value for α2M* binding to GRP78 is 50–100 pm compared with ∼5 nm for LRP, whereas the receptor number of GRP78 is ∼5000/cell compared with ∼200,000/cell for LRP (87–89). At ligand concentrations near the Kd value of the ligand, binding of α2M* to GRP78 but not LRP activates phospholipase β/γ, generating an increase in [Ca2+]i, inositol 1,4,5-trisphosphate, and diacylglycerol (18, 20–23, 25–39). GRP78 is expressed on rheumatoid, but not normal, synovial fibroblasts (32), 1-LN and DU-145, but not PC-3 prostate cancer cells, and highly activated but not resident peritoneal macrophages (6–8, 18, 22, 32–39, 45–47, 95, 96). In all these cases, the effects correlate with the presence of GRP78 on the cell surface. α2M* binding to cell surface GRP78 up-regulates protein and DNA synthesis, which is blocked by inhibitors of the PI 3-kinase/Akt/mTORC signaling pathway. In contrast to amino-terminal domain-dependent pro-proliferative signaling, down-regulation of GRP78 by RNAi or ligation by anti-CTD induces apoptosis by up-regulating p53 levels and caspase activation (38, 39). Because of the similarities observed between signaling events elicited in various cancer cell types by growth factors resulting in the Warburg effect, we ascertained whether α2M*-stimulated prostate cancer cells also exhibit aerobic glycolysis and associated metabolic alterations. The primary focus of these studies is on androgen-independent human cancer cells as in most of our previous studies. These are the most aggressive cancers with the highest metastatic potential. Indeed, it is these patients who are most likely to develop autoantibodies to the amino-terminal domains, which are receptor agonists (18, 40, 41). Nevertheless, in this study we have performed studies of the androgen-dependent LnCAP human prostate cancer line with similar results. We focused on glucose uptake, lactate secretion, lipogenesis, and fatty-acid synthase-dependent activities. The salient findings of this study are as follows. 1) Inhibition of PI 3-kinase/Akt/mTORC or fatty-acid synthase activation significantly suppresses both protein synthesis as measured by [3H]leucine incorporation and DNA synthesis as measured by [3H]thymidine incorporation in 1-LN and DU-145 prostate cancer cells treated with either α2M* or insulin. 2) α2M*-treated prostate cancer cells show an ∼2-fold transient increase in SREBP1 and ATP citrate lyase after a 5–10-min incubation, although there is approximately a 2–3-fold sustained increase in fatty-acid synthase after 25 min of incubation. 3) mRNA levels of SREBP1-c, SREBP2, fatty-acid synthase, ATP citrate lyase, and acetyl-CoA carboxylase are up-regulated 1.5–2.5-fold compared with buffer-treated cells for both 1-LN and DU-145 prostate cancer cells treated with α2M* or insulin. 4) 1-[14C]Deoxyglucose uptake and lactate secretion in 1-LN and DU-145 cells show a 2–2.5-fold increase in α2M* or insulin-treated cells, which is attenuated by anti-CTD in α2M*-treated cells. 5) mRNA levels of Glut-1 in 1-LN and DU-145 cells treated with α2M* or insulin show a 1.5–3-fold increase compared with buffer-treated cells. 6) Stimulation of 1-LN and DU-145 prostate cancer cells with α2M* or insulin shows a 2–2.5-fold increase in lipogenesis as measured by the incorporation of 1-[14C]acetate. Pretreatment of cells with inhibitors of PI 3-kinase/Akt/mTORC or fatty-acid synthase significantly suppresses α2M* or insulin-induced lipogenesis in these cells, and anti-CTD has a similar effect on α2M*-treated cells. 7) α2M* or insulin-induced lipogenesis from 6-[14C]glucose in 1-LN and DU-145 is also inhibited by pretreatment of cells with inhibitors of PI 3-kinase/Akt/mTORC, and fatty-acid synthase, and anti-CTD has a similar effect on α2M*-dependent effects. 8) α2M* and insulin induce a severalfold increase in cholesterol and fatty acid synthesis as measured by the incorporation of 1-[14C]acetate into the nonsaponifiable fraction and saponifiable fraction, respectively, which is suppressed by pretreatment of cells with inhibitors of PI 3-kinase/Akt/mTORC or fatty-acid synthase, as well as anti-CTD-treated α2M* exposed cells. 9) Thin layer chromatographic fractionation of 1-[14C]acetate-labeled cell lipids of 1-LN and DU-145 cells stimulated with α2M* or insulin shows a significant increase in the synthesis of cholesterol esters, triglycerides, free fatty acids, cholesterol, and phosphatidylcholine. Pretreatment of labeled cells with inhibitors of PI 3-kinase/Akt/mTORC or fatty-acid synthase significantly suppressed α2M* or insulin-induced synthesis of cholesterol esters, triglycerides, free fatty acids, cholesterol, and phosphatidylcholine, and anti-CTD suppresses α2M*-dependent effects. 10) Inhibition of proteolytic cleavage of SREBP by fatostatin A significantly suppresses α2M* or insulin-induced protein and DNA synthesis, lipogenesis from 1-[14C]acetate, and de novo phosphatidylcholine synthesis from [14CH3]choline. 11) Down-regulation of cell surface GRP78 by RNAi significantly suppresses the expression of fatty-acid synthase and ATP citrate lyase in 1-LN and DU-145 cells compared with buffer-treated controls. These data demonstrate that α2M*, similar to insulin, functions as a growth factor, inducing the Warburg effect and altering lipid metabolism in 1-LN and DU-145 prostate cancer cells. These effects promote prostate cancer cell proliferation, which is blocked by both inhibitors of PI 3-kinase/Akt/mTORC or fatty-acid synthase and silencing or ligation of cell surface GRP78 by RNAi or by anti-CTD, respectively.
Based on our data, we postulate the following mechanism of α2M*-induced metabolic changes in cancer cells. Binding of α2M* to cell surface GRP78 activates PI 3-kinase/Akt/mTORC signaling, which enhances aerobic glycolysis as evidenced by increased glucose uptake and lactate secretion in these cells. PI 3-kinase/Akt/mTORC signaling activates SREBPs and initiates cell proliferation as evidenced by increased lipogenesis, protein, and DNA synthesis. Increased protein expression of fatty-acid synthase, ATP citrate lyase, and SREBP1-c and increased mRNA levels of SREBP1-c, SREBP2, ATP citrate lyase, acetyl-CoA carboxylase, and fatty-acid synthase correlates with these data. This increased metabolic activity of prostate cancer cells is suppressed by pretreatment of cells with inhibitors of PI 3-kinase/Akt/mTORC and fatty-acid synthase. These effects are mediated by SREBP, which is supported by the observation that inhibition of SREBP proteolytic cleavage by fatostatin A significantly suppresses α2M*-induced protein synthesis, DNA synthesis, and lipogenesis. The ligation of cell surface GRP78 by α2M* is a key player in inducing metabolic alterations in prostate cancer cells because down-regulating GRP78 by RNAi significantly suppresses the activation of two lipogenic enzymes, acetyl-CoA carboxylase and fatty-acid synthase. The key role of GRP78 is also evident in experiments in which we ligated GRP78 with antibody directed against its (CTD. This results in significantly decreased protein and DNA synthesis accompanied by reduced glucose uptake, lactate secretion, lipogenesis, and phosphatidylcholine synthesis. This down-regulation of GRP78 functions to inhibit the Warburg effect and may be a potential target for cancer therapeutic development. A schematic representation of α2M* and cell surface GRP78 in inducing aerobic glycolysis in prostate cancer cells is shown in Fig. 10.
FIGURE 10.
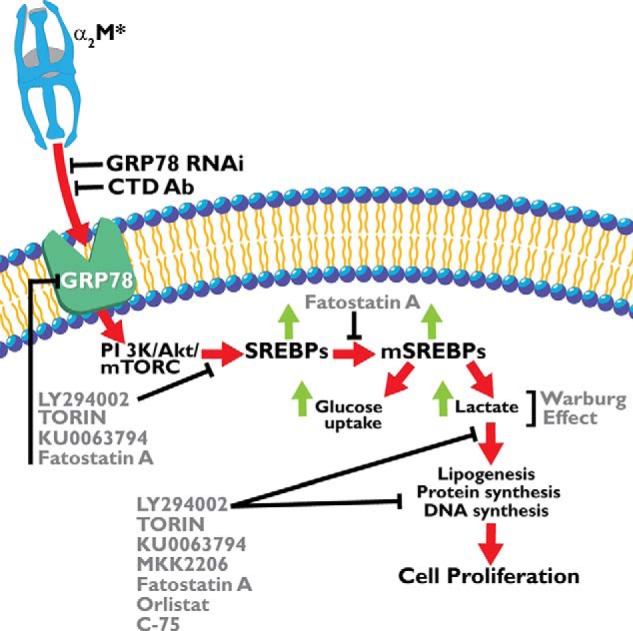
Schematic representation of cellular events involved in α2M*-induced aerobic glycolysis in prostate cancer cells. The abbreviations are as in the cleaved or active form, except for mSREBPS1, which indicates the mature form.
In conclusion, the metabolic effects on prostate cancer cells treated with α2M* were highly consistent with insulin. The α2M/complement gene family evolved at least 600 million years ago (16). In horseshoe crabs, α2M functions as both a proteinase inhibitor and complement protein, but through gene duplication and divergent evolution, these functions became discrete to either α2M or complement (16). The insulin gene evolved 500 million years ago as a regulator of cellular metabolism (97). Because of the similarities on cellular responses elicited upon binding of α2M* and insulin to their receptors, we propose that although α2M evolved as a proteinase inhibitor, during later evolutionary phases, it acquired functions as a growth factor. In this context, GRP78 on the cell surface may be viewed as a proto-oncogene (98).
Footnotes
- α2M
- α2-macroglobulin
- α2M*
- activated α2-macroglobulin
- SREBP
- sterol regulatory element-binding protein
- PI
- phosphatidylinositol
- CTD
- carboxyl-terminal domain
- LRP
- LDL receptor-related protein
- mTOR
- mammalian target of rapamycin
- mTORC
- mTOR complex.
REFERENCES
- 1. Jemal A., Thomas A., Murray T., Thun M. (2002) Cancer statistics. CA Cancer J. Clin. 52, 23–47 [DOI] [PubMed] [Google Scholar]
- 2. Abate-Shen C., Shen M. M. (2000) Molecular genetics of prostate cancer. Genes Dev. 14, 2410–2434 [DOI] [PubMed] [Google Scholar]
- 3. Montano X., Djamgoz M. B. (2004) Epidermal growth factor, neurotrophins and the metastatic cascade in prostate cancer. FEBS Lett. 571, 1–8 [DOI] [PubMed] [Google Scholar]
- 4. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 5. Manning B. D., Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Misra U. K., Pizzo S. V. (2012) Upregulation of mTORC2 activation by the selective agonist of EPAC, 8-CPT-2Me-cAMP, in prostate cancer cells: assembly of a multiprotein signaling complex. J. Cell Biochem. 113, 1488–1500 [DOI] [PubMed] [Google Scholar]
- 7. Misra U. K., Pizzo S. V. (2005) Coordinate regulation of forskolin-induced cellular proliferation in macrophages by protein kinase A/cAMP-response element-binding protein (CREB) and Epac1-Rap1 signaling: effects of silencing CREB gene expression on Akt activation. J. Biol. Chem. 280, 38276–38289 [DOI] [PubMed] [Google Scholar]
- 8. Misra U. K., Pizzo S. V. (2014) Activated α2-macroglobulin binding to cell surface GRP78 induces T-loop phosphorylation of Akt1 by PDK1 in association with Raptor. PLoS One 9, e88373. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9. Misra U. K., Pizzo S. V. (2009) Epac1-induced cellular proliferation in prostate cancer cells is mediated by B-Raf/ERK and mTOR signaling cascades. J. Cell Biochem. 108, 998–1011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Bayascas J. R. (2010) PDK1: the major transducer of PI 3-kinase actions. Curr. Top. Microbiol. Immunol. 346, 9–29 [DOI] [PubMed] [Google Scholar]
- 11. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 12. Misra U. K., Pizzo S. V. (2013) Raptor association with PDK1 is necessary for T-loop phosphorylation of Akt in prostate cancer cells stimulated with 8-CPT-2-Me-cAMP. J. Biochem. Pharmacol. Res. 1, 219–227 [Google Scholar]
- 13. Hart J. P., Pizzo S. V. (2006) in Principles and Clinical Practice (Coleman R. W., Clowes A. W., Goldhaber S. Z., Marder B. J., George J. N., eds) 5th Ed., pp. 395–407, Lippincott, Williams and Wilkins, Baltimore [Google Scholar]
- 14. Feldman S. R., Pizzo S. V. (1984) Comparison of the binding of chicken α2-macroglobulin and ovomacroglobulin to the mammalian α2-macroglobulin receptor. Arch. Biochem. Biophys. 235, 267–275 [DOI] [PubMed] [Google Scholar]
- 15. Feldman S. R., Pizzo S. V. (1986) Purification and characterization of a “half-molecule” α2-macroglobulin from the southern grass frog: absence of binding to the mammalian α2-macroglobulin receptor. Biochemistry 25, 721–727 [DOI] [PubMed] [Google Scholar]
- 16. Enghild J. J., Thøgersen I. B., Salvesen G., Fey G. H., Figler N. L., Gonias S. L., Pizzo S. V. (1990) α2-Macroglobulin from Limulus polyphemus exhibits proteinase inhibitory activity and participates in a hemolytic system. Biochemistry 29, 10070–10080 [DOI] [PubMed] [Google Scholar]
- 17. Thøgersen I. B., Salvesen G., Brucato F. H., Pizzo S. V., Enghild J. J. (1992) Purification and characterization of an α2-macroglobulin proteinase inhibitor from the mollusc Octopus vulgaris. Biochem. J. 285, 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Misra U. K., Payne S., Pizzo S. V. (2011) Ligation of prostate cancer cell surface GRP78 activates a proproliferative and antiapoptotic feedback loop: a role for secreted prostate-specific antigen. J. Biol. Chem. 286, 1248–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feldman S. R., Gonias S. L., Pizzo S. V. (1985) Model of α2-macroglobulin structure and function. Proc. Natl. Acad. Sci. U.S.A. 82, 5700–5704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Misra U. K., Chu C. T., Rubenstein D. S., Gawdi G., Pizzo S. V. (1993) Receptor-recognized α2-macroglobulin-methylamine elevates intracellular calcium, inositol phosphates and cyclic AMP in murine peritoneal macrophages. Biochem. J. 290, 885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Misra U. K., Chu C. T., Gawdi G., Pizzo S. V. (1994) Evidence for a second α2-macroglobulin receptor. J. Biol. Chem. 269, 12541–12547 [PubMed] [Google Scholar]
- 22. Misra U. K., Gonzalez-Gronow M., Gawdi G., Hart J. P., Johnson C. E., Pizzo S. V. (2002) The role of Grp 78 in α2-macroglobulin-induced signal transduction. Evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP 78-mediated signal transduction. J. Biol. Chem. 277, 42082–42087 [DOI] [PubMed] [Google Scholar]
- 23. Misra U. K., Gonzalez-Gronow M., Gawdi G., Wang F., Pizzo S. V. (2004) A novel receptor function for the heat shock protein Grp78: silencing of Grp78 gene expression attenuates α2M*-induced signalling. Cell. Signal. 16, 929–938 [DOI] [PubMed] [Google Scholar]
- 24. Ma Y., Hendershot L. M. (2004) The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer 4, 966–977 [DOI] [PubMed] [Google Scholar]
- 25. Misra U. K., Deedwania R., Pizzo S. V. (2006) Activation and cross-talk between Akt, NF-κB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J. Biol. Chem. 281, 13694–13707 [DOI] [PubMed] [Google Scholar]
- 26. Gonzalez-Gronow M., Pizzo S. V., Misra U. K. (2013) Cellular Trafficking of Cell Stress Proteins in Health and Diseases (Henderson B., Pockley A. G., eds), pp. 229–242, Springer Science Business Media, Dordrecht, The Netherlands [Google Scholar]
- 27. Misra U. K., Gawdi G., Gonzalez-Gronow M., Pizzo S. V. (1999) Coordinate regulation of the α2-macroglobulin signaling receptor and the low density lipoprotein receptor-related protein/α2-macroglobulin receptor by insulin. J. Biol. Chem. 274, 25785–25791 [DOI] [PubMed] [Google Scholar]
- 28. Asplin I. R., Misra U. K., Gawdi G., Gonzalez-Gronow M., Pizzo S. V. (2000) Selective upregulated expression of the α2-macroglobulin signaling receptor in highly metastatic 1-LN prostate carcinoma cells. Arch. Biochem. Biophys. 383, 135–141 [DOI] [PubMed] [Google Scholar]
- 29. Misra U. K., Deedwania R., Pizzo S. V. (2005) Binding of activated α2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J. Biol. Chem. 280, 26278–26286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Misra U. K., Pizzo S. V. (1998) Binding of receptor-recognized forms of α2-macroglobulin to the α2-macroglobulin signaling receptor activates phosphatidylinositol 3-kinase. J. Biol. Chem. 273, 13399–13402 [DOI] [PubMed] [Google Scholar]
- 31. Misra U. K., Akabani G., Pizzo S. V. (2002) The role of cAMP-dependent signaling in receptor-recognized forms of α2-macroglobulin-induced cellular proliferation. J. Biol. Chem. 277, 36509–36520 [DOI] [PubMed] [Google Scholar]
- 32. Misra U. K., Gonzalez-Gronow M., Gawdi G., Pizzo S. V. (1997) Up-regulation of the α2-macroglobulin signaling receptor in rheumatoid synovial fibroblasts. J. Biol. Chem. 272, 497–502 [DOI] [PubMed] [Google Scholar]
- 33. Misra U. K., Pizzo S. V. (1998) Ligation of the α2M signalling receptor elevates the levels of p21Ras-GTP in macrophages. Cell. Signal. 10, 441–445 [DOI] [PubMed] [Google Scholar]
- 34. Misra U. K., Pizzo S. V. (1998) Ligation of the α2M signaling receptor with receptor-recognized forms of α2-macroglobulin initiates protein and DNA synthesis in macrophages. The effect of intracellular calcium. Biochim. Biophys. Acta 1401, 121–128 [DOI] [PubMed] [Google Scholar]
- 35. Misra U. K., Pizzo S. V. (2004) Potentiation of signal transduction mitogenesis and cellular proliferation upon binding of receptor-recognized forms of α2-macroglobulin to 1-LN prostate cancer cells. Cell. Signal. 16, 487–496 [DOI] [PubMed] [Google Scholar]
- 36. Misra U. K., Pizzo S. V. (2012) Receptor-recognized α2-macroglobulin binds to cell surface-associated GRP78 and activates mTORC1 and mTORC2 signaling in prostate cancer cells. PLoS One 7, e51735. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37. Misra U. K., Pizzo S. V. (2007) Upregulation of AKT1 protein expression in forskolin-stimulated macrophage: evidence from ChIP analysis that CREB binds to and activates the AKT1 promoter. J. Cell. Biochem. 100, 1022–1033 [DOI] [PubMed] [Google Scholar]
- 38. Misra U. K., Pizzo S. V. (2010) Ligation of cell surface GRP78 with antibody directed against the COOH-terminal domain of GRP78 suppresses Ras/MAPK and PI 3-kinase/AKT signaling while promoting caspase activation in human prostate cancer cells. Cancer Biol. Ther. 9, 2 142–152 [DOI] [PubMed] [Google Scholar]
- 39. Misra U. K., Mowery Y., Kaczowka S., Pizzo S. V. (2009) Ligation of cancer cell surface GRP78 with antibodies directed against its COOH-terminal domain up-regulates p53 activity and promotes apoptosis. Mol. Cancer Ther. 8, 1350–1362 [DOI] [PubMed] [Google Scholar]
- 40. Gonzalez-Gronow M., Cuchacovich M., Llanos C., Urzua C., Gawdi G., Pizzo S. V. (2006) Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum. Cancer Res. 66, 11424–11431 [DOI] [PubMed] [Google Scholar]
- 41. Mintz P. J., Kim J., Do K. A., Wang X., Zinner R. G., Cristofanilli M., Arap M. A., Hong W. K., Troncoso P., Logothetis C. J., Pasqualini R., Arap W. (2003) Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotechnol. 21, 57–63 [DOI] [PubMed] [Google Scholar]
- 42. Arap M. A., Lahdenranta J., Mintz P. J., Hajitou A., Sarkis A. S., Arap W., Pasqualini R. (2004) Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell 6, 275–284 [DOI] [PubMed] [Google Scholar]
- 43. de Ridder G. G., Gonzalez-Gronow M., Ray R., Pizzo S. V. (2011) Autoantibodies against cell surface GRP78 promote tumor growth in a murine model of melanoma. Melanoma Res. 21, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Ridder G. G., Ray R., Pizzo S. V. (2012) A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Res. 22, 225–235 [DOI] [PubMed] [Google Scholar]
- 45. Kanoh Y., Ohtani N., Mashiko T., Ohtani S., Nishikawa T., Egawa S., Baba S., Ohtani H. (2001) Levels of α2 macroglobulin can predict bone metastases in prostate cancer. Anticancer Res. 21, 551–556 [PubMed] [Google Scholar]
- 46. DeBerardinis R. J., Lum J. J., Hatzivassiliou G., Thompson C. B. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 [DOI] [PubMed] [Google Scholar]
- 47. Cantor J. R., Sabatini D. M. (2012) Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 882–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Menendez J. A., Lupu R. (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 [DOI] [PubMed] [Google Scholar]
- 49. Ros S., Santos C. R., Moco S., Baenke F., Kelly G., Howell M., Zamboni N., Schulze, (2012) Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2, 328–343 [DOI] [PubMed] [Google Scholar]
- 50. Flavin R., Zadra G., Loda M. (2011) Metabolic alterations and targeted therapies in prostate cancer. J. Pathol. 223, 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baenke F., Peck B., Miess H., Schulze A. (2013) Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis. Model Mech. 6, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lunt S. Y., Vander Heiden M. G. (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464 [DOI] [PubMed] [Google Scholar]
- 53. Gatenby R. A., Gillies R. J. (2004) Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899 [DOI] [PubMed] [Google Scholar]
- 54. Vander Heiden M. G. (2011) Targeting cancer metabolism: a therapeutic window opens. Nat. Rev. Drug Discov. 10, 671–684 [DOI] [PubMed] [Google Scholar]
- 55. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 56. Medes G., Thomas A., Weinhouse S. (1953) Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 13, 27–29 [PubMed] [Google Scholar]
- 57. Rossi S., Graner E., Febbo P., Weinstein L., Bhattacharya N., Onody T., Bubley G., Balk S., Loda M. (2003) Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol. Cancer Res. 1, 707–715 [PubMed] [Google Scholar]
- 58. Van de Sande T., Roskams T., Lerut E., Joniau S., Van Poppel H., Verhoeven G., Swinnen J. V. (2005) High-level expression of fatty-acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. J. Pathol. 206, 214–219 [DOI] [PubMed] [Google Scholar]
- 59. Santos C. R., Schulze A. (2012) Lipid metabolism in cancer. FEBS J. 279, 2610–2623 [DOI] [PubMed] [Google Scholar]
- 60. Yoshii Y., Furukawa T., Oyama N., Hasegawa Y., Kiyono Y., Nishii R., Waki A., Tsuji A. B., Sogawa C., Wakizaka H., Fukumura T., Yoshii H., Fujibayashi Y., Lewis J. S., Saga T. (2013) Fatty acid synthase is a key target in multiple essential tumor functions of prostate cancer: uptake of radiolabeled acetate as a predictor of the targeted therapy outcome. PloS One 8, e64570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Van de Sande T., De Schrijver E., Heyns W., Verhoeven G., Swinnen J. V. (2002) Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in the overexpression of fatty-acid synthase in LNCaP prostate cancer cells. Cancer Res. 62, 642–646 [PubMed] [Google Scholar]
- 62. Hatzivassiliou G., Zhao F., Bauer D. E., Andreadis C., Shaw A. N., Dhanak D., Hingorani S. R., Tuveson D. A., Thompson C. B. (2005) ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8, 311–321 [DOI] [PubMed] [Google Scholar]
- 63. Griffiths B., Lewis C. A., Bensaad K., Ros S., Zhang Q., Ferber E. C., Konisti S., Peck B., Miess H., East P., Wakelam M., Harris A. L., Schulze A. (2013) Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Porstmann T., Griffiths B., Chung V.-L., Delpuech O., Griffiths J. R., Downward J., Schulze A. (2005) PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24, 6465–6481 [DOI] [PubMed] [Google Scholar]
- 65. Liu Y. (2006) Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 9, 230–234 [DOI] [PubMed] [Google Scholar]
- 66. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goldstein J. L., DeBose-Boyd R. A., Brown M. S. (2006) Protein sensors for membrane sterols. Cell 124, 35–46 [DOI] [PubMed] [Google Scholar]
- 68. Shao W., Espenshade P. J. (2012) Expanding roles for SREBP in metabolism. Cell Metab. 16, 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yellaturu C. R., Deng X., Park E. A., Raghow R., Elam M. B. (2009) Insulin enhances the biogenesis of nuclear sterol regulatory element-binding protein (SREBP)-1c by posttranscriptional down-regulation of Insig-2A and its dissociation from SREBP cleavage-activating protein (SCAP).SREBP-1c complex. J. Biol. Chem. 284, 31726–31734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Owen J. L., Zhang Y., Bae S.-H., Farooqi M. S., Liang G., Hammer R. E., Goldstein J. L., Brown M. S. (2012) Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. U.S.A. 109, 16184–16189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bauer D. E., Hatzivassiliou G., Zhao F., Andreadis C., Thompson C. B. (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 [DOI] [PubMed] [Google Scholar]
- 72. Beckers A., Organe S., Timmermans L., Scheys K., Peeters A., Brusselmans K., Verhoeven G., Swinnen J. V. (2007) Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 67, 8180–8187 [DOI] [PubMed] [Google Scholar]
- 73. Pelton K., Freeman M. R., Solomon K. R. (2012) Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 12, 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krycer J. R., Brown A. J. (2013) Cholesterol accumulation in prostate cancer: a classic observation from a modern perspective. Biochim. Biophys. Acta 1835, 219–229 [DOI] [PubMed] [Google Scholar]
- 75. Lingwood D., Simons K. (2010) Lipid rafts as a membrane-organizing principle. Science 327, 46–50 [DOI] [PubMed] [Google Scholar]
- 76. Yue S., Li J., Lee S.-Y., Lee H.-J., Shao T., Song B., Cheng L., Masterson T. A., Liu X., Ratliff T. L., Cheng J.-X. (2014) Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 19, 393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brown M. S., Goldstein J. L. (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 [DOI] [PubMed] [Google Scholar]
- 78. Porstmann T., Santos C. R., Griffiths B., Cully M., Wu M., Leevers S., Griffiths J. R., Chung Y.-L., Schulze A. (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Peterson T. R., Sengupta S. S., Harris T. E., Carmack A. E., Kang S. A., Balderas E., Guertin D. A., Madden K. L., Carpenter A. E., Finck B. N., Sabatini D. M. (2011) mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ridgway N. D. (2013) The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 48, 20–38 [DOI] [PubMed] [Google Scholar]
- 81. Glunde K., Bhujwalla Z. M., Ronen S. M. (2011) Choline metabolism in malignant transformation. Nat. Rev. Cancer 11, 835–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Misra U. K., Pizzo S. V. (2002) Regulation of cytosolic phospholipase A2 activity in macrophages stimulated with receptor-recognized forms of alpha 2-macroglobulin: role in mitogenesis and cell proliferation. J. Biol. Chem. 277, 4069–4078 [DOI] [PubMed] [Google Scholar]
- 83. Misra U. K., Pizzo S. V. (1999) Upregulation of macrophage plasma membrane and nuclear phospholipase D activity on ligation of the alpha2-macroglobulin signaling receptor: involvement of heterotrimeric and monomeric G proteins. Arch. Biochem. Biophys. 363, 68–80 [DOI] [PubMed] [Google Scholar]
- 84. Misra U. K., Pizzo S. V. (2001) Induction of cyclooxygenase-2 synthesis by ligation of the macrophage alpha(2)-macroglobulin signalling receptor. Cell. Signal. 13, 801–808 [DOI] [PubMed] [Google Scholar]
- 85. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 86. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 87. Kennedy E. P., Weiss S. B. (1956) The function of cytidine coenzymes in the biosynthesis of phospholipids. J. Biol. Chem. 222, 193–214 [PubMed] [Google Scholar]
- 88. Vander Heiden M. G., Cantley L. C., Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kato Y., Ozawa S., Miyamoto C., Maehata Y., Suzuki A., Maeda T., Baba Y. (2013) Acidic extracellular microenvironment and cancer. Cancer Cell Int. 13, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Walenta S., Salameh A., Lyng H., Evensen J. F., Mitze M., Rofstad E. K., Mueller-Klieser W. (1997) Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am. J. Pathol. 150, 409–415 [PMC free article] [PubMed] [Google Scholar]
- 91. Kamisuki S., Mao Q., Abu-Elheiga L., Gu Z., Kugimiya A., Kwon Y., Shinohara T., Kawazoe Y., Sato S., Asakura K., Choo H. Y., Sakai J., Wakil S. J., Uesugi M. (2009) A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 16, 882–892 [DOI] [PubMed] [Google Scholar]
- 92. Li X., Chen Y. T., Hu P., Huang W. C. (2014) Fatostatin displays high anti-tumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 13, 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. van der Sanden M. H., Houweling M., van Golde L. M., Vaandrager A. B. (2003) Inhibition of phosphatidylcholine synthesis induces expression of the endoplasmic reticulum stress and apoptosis-related protein CCAAT/enhancer-binding protein-homologous protein (CHOP/GADD153). Biochem. J. 369, 643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lagace T. A., Storey M. K., Ridgway N. D. (2000) Regulation of phosphatidylcholine metabolism in Chinese hamster ovary cells by the sterol regulatory element-binding protein (SREBP)/SREBP cleavage-activating protein pathway. J. Biol. Chem. 275, 14367–14374 [DOI] [PubMed] [Google Scholar]
- 95. Bhattacharjee G., Misra U. K., Gawdi G., Cianciolo G., Pizzo S. V. (2001) Inducible expression of the alpha2-macroglobulin signaling receptor in response to antigenic stimulation: a study of second messenger generation. J. Cell. Biochem. 82, 260–270 [DOI] [PubMed] [Google Scholar]
- 96. Misra U. K., Payne S., Pizzo S. V. (2013) The monomeric receptor binding domain of tetrameric α2-macroglobulin binds to cell surface GRP78 triggering equivalent activation of signaling cascades. Biochemistry 52, 4014–4025 [DOI] [PubMed] [Google Scholar]
- 97. Steiner D. F., Chan S. J., Welsh J. M., Kwok S. C. (1985) Structure and evolution of the insulin gene. Annu. Rev. Genet. 19, 463–484 [DOI] [PubMed] [Google Scholar]
- 98. Adamson E. D. (1987) Oncogenes in development. Development 99, 449–471 [DOI] [PubMed] [Google Scholar]