

Background: The role of protein-tyrosine phosphatases (PTP) in ERBB2 signaling is undefined.
Results: Phosphatidic acid (PA) activated PTPD2; inhibition of PA production or PTPD2 expression attenuated ERBB2-mediated morphological changes in mammary epithelial cells.
Conclusion: The PLD2-PTPD2 axis is required for ERBB2 signaling.
Significance: PA-regulated PTPD2 activity is a novel, positive element of ERBB2 signaling, which may offer a new therapeutic strategy in breast cancer.
Keywords: Breast Cancer, Cell Signaling, Lipid Signaling, Phosphatidic Acid, Phospholipase D, Phosphotyrosine Signaling, Receptor-tyrosine Kinase, Signal Transduction, ERBB2, Protein-tyrosine Phosphatase
Abstract
We used a loss-of-function screen to investigate the role of classical protein-tyrosine phosphatases (PTPs) in three-dimensional mammary epithelial cell morphogenesis and ERBB2 signaling. The study revealed a novel role for PTPD2 as a positive regulator of ERBB2 signaling. Suppression of PTPD2 attenuated the ERBB2-induced multiacinar phenotype in three-dimensional cultures specifically by inhibiting ERBB2-mediated loss of polarity and lumen filling. In contrast, overexpression of PTPD2 enhanced the ERBB2 phenotype. We also found that a lipid second messenger, phosphatidic acid, bound PTPD2 in vitro and enhanced its catalytic activity. Small molecule inhibitors of phospholipase D (PLD), an enzyme that produces phosphatidic acid in cells, also attenuated the ERBB2 phenotype. Exogenously added phosphatidic acid rescued the PLD-inhibition phenotype, but only when PTPD2 was present. These findings illustrate a novel pathway involving PTPD2 and the lipid second messenger phosphatidic acid that promotes ERBB2 function.
Introduction
Reversibility of protein-tyrosine phosphorylation is critical for the dynamic regulation of signal transduction pathways and is maintained by the complementary activity of two families of enzymes, protein-tyrosine kinases (PTKs)2 and protein-tyrosine phosphatases (PTPs). Disturbance of the balance between their functions results in aberrant phosphorylation patterns and has been implicated in the etiology of several human diseases, including cancer. Considerable progress has been made in understanding the function of PTKs in cancer. Therapeutic initiatives aimed at modulating tyrosine phosphorylation have traditionally focused on targeting PTKs (1, 2); however, tumor-derived mutations have also been described in several PTP genes and many of these enzymes have been implicated as products of oncogenes or tumor suppressors in human cancer (3, 4). Yet the functional role played by individual PTPs in cell transformation is still not well understood.
The receptor tyrosine kinase ERBB2 is amplified or overexpressed in ∼25% of breast cancer patients and plays a causal role in mammary carcinogenesis (5, 6). ERBB2 overexpression is an adverse prognostic feature in early stages of the disease and correlates with aggressive tumor development (6, 7). The anti-ERBB2 drug, Herceptin (trastuzumab), is widely employed for the treatment of metastatic ERBB2-positive breast cancer, yet ∼70% of patients are resistant to the drug or develop resistance over the course of therapy (8, 9). Consequently, a deeper understanding of the regulation of ERBB2 signaling is of critical importance in the search for alternative or complementary therapeutic strategies.
Considering that ERBB2 initiates a signal transduction pathway that propagates via tyrosine phosphorylation, it is intuitive that PTPs would play a crucial role in regulation. There are several studies that implicate specific PTPs as negative regulators of signaling pathways initiated by ERBB2. For example, loss-of-function analyses of PTPs in mammary epithelial cells have revealed a negative role for PTPRR, PTPRG, PTPN23 (10), and RPTPα (11) in ERBB2-dependent cell migration. In contrast, some PTPs have also been shown to act as positive regulators of ERBB2-driven carcinogenesis. Inhibition of PTP1B in mouse models of ERBB2-driven breast cancer delayed the onset of mammary tumors and reduced the incidence of metastasis (12, 13). SHP2 is required for ERK activation in ERBB2-mediated carcinogenesis (14). Members of the PTP family have also been implicated in the sensitivity of breast cancer cells to therapy. For example, the activation of PTEN underlies the activity of Herceptin, and PTEN deficiency has been found to correlate with Herceptin resistance (15). These findings suggest that PTPs function as specific regulators of ERBB2 signal transduction pathways and highlight the importance of investigating possible roles for other members of the PTP family in regulating the pathophysiology of ERBB2-driven breast cancers.
Here we report an RNAi-mediated loss-of-function screen to study the role of PTPs in mammary epithelial morphogenesis. We used MCF10A cells expressing an inducible form of ERBB2 to investigate the effect of suppressing individual PTPs on mammary acinus architecture in three-dimensional organotypic cultures. We identified a novel role for PTPD2 as a positive regulator of ERBB2 signaling; suppression of PTPD2 attenuated the ERBB2-induced phenotype in three-dimensional cultures, whereas overexpression of PTPD2 enhanced it. In addition, we report that the catalytic activity of PTPD2 was up-regulated by phosphatidic acid (PA) in vitro. Consistent with an important role for PA in ERBB2 signaling in cells, we show that phospholipase D2 (PLD2) an enzyme that produces PA in cells, was itself required for ERBB2 signaling in a manner that was dependent on PTPD2. We propose a novel regulatory pathway involving PLD2-mediated production of phosphatidic acid and the regulation of PTPD2 function as being critical for optimal ERBB2 signaling.
EXPERIMENTAL PROCEDURES
Materials
Growth factor reduced MatrigelTM was from BD Biosciences, AP1510 was from ARIAD Pharmaceuticals, anti-PTPD2 antibody was from R&D Systems, anti-Ki67 antibody was from Zymed Laboratories Inc., anti-GM130 antibody was from BD Transduction Labs, anti-phosphotyrosine antibody 4G10 was from Millipore, HA antibody was from Covance, phospho-Akt, total Akt, phospho-Erk1/2, total Erk1/2, and cleaved caspase-3 antibodies were from Cell Signaling Technologies, poly-HEMA, YAP1 antibody, and 5-fluro-2-indolyl des-chlorohalopemide (FIPI) were from Sigma, lipid strips were from Echelon Biosciences, l-α-dipalmitoyl-[glycerol-14C(U)]phosphatidic acid was from PerkinElmer Life Sciences, unlabeled l-α-phosphatidic acid and phosphatidylserine were from Avanti Polar Lipids, DiFMUP was from Molecular Probes, PLD1 inhibitor VUO359595 and PLD2 inhibitor VUO364739 were a kind gift from Alex Brown, Vanderbilt University, and SU6656 was from Calbiochem.
Constructs
PTPD2 catalytic domain (886–1187) in pNIC28Bsa4 vector was a gift from Stefan Knapp (University of Oxford). PTP1B(1–321) is described in Ref. 16. YAP1 shRNA constructs in the pLKO1 vector were obtained from Addgene; plasmids 42540 and 42541 (17). Full-length FLAG-tagged PTPD2 construct was kindly provided by Yeesim Khew-Goodall (Centre for Cancer Biology, SA Pathology, Adelaide/University of Adelaide). To stably overexpress PTPD2 in 10A.B2 cells, the cDNA was subcloned into the retroviral pBabe vector.
Cell Culture and Generation of Stable Cell Lines
The 10A.B2 cell line was cultured as described in Ref. 18. For stable suppression of PTPD2, we expressed pMLP retroviral vector using the targeting sequences GACGGTGTGGCATTTACAA (shRNA#1) and GCCACAAGATATCAGTATT (shRNA#2) as described in Ref. 10 and selected stable cell lines using puromycin (2 μg/ml).
Three-dimensional Morphogenesis Assay
Morphogenesis assays were performed in MatrigelTM as described in Ref. 18. Each experiment was conducted at least twice in duplicate. Beginning day 4, the overlay medium was replenished with 1 μm AP1510 (to stimulate chimeric ERBB2) or vehicle control. The morphology of the acini was followed by phase microscopy until day 16. For PLD inhibition, FIPI or PLD isoform-specific inhibitors were added to overlay medium at concentrations of 1 and 2.5 μm, respectively. For SRC inhibition, SU6656 was added to overlay medium at a concentration of 5 μm (10).
Microscopy and Quantitation of Acini
Phase images from at least 32 random fields per condition were taken using a Zeiss Axiovert 200M microscope. The surface area of each acinus at day 16 was measured using Axiovision 4.4 software (Zeiss). Structures having the appearance of multiple partially, or fully filled acini within the same basement membrane and beyond a predetermined size cutoff (set individually for each experiment) were designated “multiacinar structures.” 150 acini were counted per condition and the number of multiacinar structures formed was calculated as a percentage of the total number of structures imaged. The average of two wells was used to calculate the fold-difference as compared to shLuc hairpin-expressing control. Two-way analysis of variance (ANOVA) was used to determine the statistical significance of the data collected.
Preparation of Lysates and Immunoblotting
To harvest total cellular lysates from 10A.B2 cell lines, a fully confluent plate of cells was washed with 5 ml of 1× phosphate-buffered saline (PBS) and extracted in 500 μl of RIPA buffer (50 mm Tris-HCl, 150 mm NaCl, 1% (v/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, pH 8.0, containing 2 mm sodium orthovanadate and Roche protease inhibitor mixture). Cells were lysed on a rotating wheel at 4 °C for 30 min. Thereafter, the lysate was cleared by centrifugation at 12,000 × g for 10 min. The supernatant was quantitated using Bradford reagent. Equal amounts of protein were resolved by SDS-PAGE and detected by immunoblotting. To check the activation status of effectors downstream of ERBB2, confluent 10A.B2 cells were washed twice with 1× PBS and starved in DMEM/F-12 medium (without any additives) for 14–16 h. Cells were then stimulated in DMEM/F-12 containing 1 μm AP1510 for the desired duration and lysed for immunoblotting.
Immunofluorescence
For immunofluorescence visualization of Ki67, GM130, and caspase-3, acini were fixed on days 16, 18 and 8, respectively, as described in Ref. 18. Microscopy was performed on Zeiss Axiovert 200M microscope using AxioVison 4.4 and ApoTome imaging system. Acini with 3 or more Ki67-positive cells were designated “Ki67-positive acini.” Data are represented as percentage of Ki67-positive acini out of a total of 50 acini counted per condition.
Detachment Culture
Tissue culture plates were coated with 12 mg/ml of poly-HEMA and incubated at 37 °C until dry. 10A.B2 cell lines were plated in complete growth medium with or without 1 μm AP1510 in suspension at a density of 200,000 cells/ml for 48 h. Thereafter, cells were collected and washed in 1× PBS before lysis for immunoblotting.
Purification of Recombinant PTP1B and His-tagged PTPD2 Catalytic Domains
PTP1B(1–321) and His6-tagged PTPD2 catalytic domain (Stefan Knapp lab) were introduced into the bacterial strain BL21-RIL for recombinant protein production. Briefly, 5 ml of overnight bacterial culture was added to 500 ml of LB medium and incubated until A600 reached 0.6. Cultures were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and allowed to grow for 4 h at 18 °C. Cells were harvested and lysed in 25 ml of Lysis buffer (50 mm HEPES, 100 mm NaCl, 1% (v/v) Triton X-100, 0.5% (v/v) DMSO, pH 7.0) containing protease inhibitors by sonication. The sonicated lysate was cleared by centrifugation at 45,000 × g for 1 h at 4 °C. The supernatant was filtered through a 0.45-μm filter and loaded onto a Ni2+-NTA column. The column was washed with 50 mm HEPES, 250 mm NaCl, 1 mm DTT, and 50 mm imidazole (pH 7.0). Bound His6-tagged protein was eluted using 300 mm imidazole.
Lipid Binding Assays
[14C]DPPA was resuspended in 20 mm imidazole, 1 mm EDTA, 1 mm DTT (pH 7.0), and vesicles were prepared by sonication until the solution became clear. Recombinant catalytic domain of PTPD2 or PTP1B bound to Ni2+-NTA beads was incubated for 30 min at room temperature with various concentrations of [14C]DPPA. The PTP-[14C]DPPA complex was separated from unbound [14C]DPPA by centrifugation, and bound radioactivity was measured by liquid scintillation counting. To assess the extent of any quenching during liquid scintillation counting, the highest concentration of [14C]DPPA was incubated with varying amounts of Ni2+-NTA-agarose beads, and radioactivity in the absence and presence of the beads was compared. No apparent change in the radioactivity of [14C]DPPA was observed even when Ni2+-NTA-agarose beads were included at 20-fold excess over the amount of beads used in the binding assays.
Phosphatase Activity Assays
For phosphatase assays, varying concentrations of DiFMUP (0–500 μm) was added to assay buffer (50 mm HEPES, 100 mm NaCl, 0.01% (v/v) Tween, 0.1% (v/v) DMSO, 2 mm DTT, 2 mm EDTA, pH 6.5) containing 0.1 μm purified PTPD2 in a final volume of 100 μl. The fluorescence emitted at 450 nm was monitored continuously for 20 min using a Gemini XPS fluorescence plate reader. For assays using radiolabeled substrate, reduced carboxamidomethylated and maleylated lysozyme was phosphorylated on tyrosine to a stoichiometry of 0.8 mol of 32P/mol of protein using recombinant GST-FER kinase and [γ-32P]ATP, and activity was measured as described previously (19, 20).
RESULTS
Loss-of-function Screen of Classical PTPs to Identify Regulators of Mammary Epithelial Morphogenesis
To investigate the roles of classical PTPs in mammary epithelial cells, we employed a loss-of-function screen combined with a three-dimensional organotypic culture model system. We used 10A.B2 cells, MCF10A cells that ectopically express a chimeric form of ERBB2, which can be selectively activated using a small molecule dimerizer, AP1510 (21). We expressed a library of shRNAs (10) against classical PTPs in 10A.B2 cells to study systematically the effect of loss of individual PTPs, either alone, or in combination with ERBB2 activation, on the architecture of mammary acini-like structures formed in three-dimensional culture in Matrigel.
To make the screen more manageable, we tested shRNAs in pools. We chose 4 shRNAs per PTP and grouped them into 2 pools of 2 hairpins each. In the absence of AP1510 stimulation, we found that the shRNA pools against 3 PTPs (PTPRK, PTP-BAS, and PTPRU) disrupted acinus morphology, resulting in the formation of partially filled, disorganized structures (Fig. 1A). In the context of AP1510-induced ERBB2 signaling, we found that shRNA pools against 8 PTPs had distinct effects on acinus architecture. The multiacinar phenotype characteristic of ERBB2 activation was enhanced by shRNA pools against PTPRU, PTPRM, PTPRZ1, and PTPRO, suggesting a negative effect of these PTPs on signaling. In contrast, shRNA pools against PTPRS, PTPD2, PTPTYP, and PTPHe attenuated the ERBB2 phenotype, consistent with a positive role for these PTPs (Fig. 1B). The results suggest distinct and specific functional roles played by different PTPs in signal transduction pathways that regulate mammary acinus architecture.
FIGURE 1.

Loss-of-function screen of classical PTPs to identify regulators of mammary epithelial morphogenesis. The effect of shRNA pools against the indicated PTPs on 10A.B2 acinus morphology was assessed by three-dimensional culture in Matrigel in the absence (A) or presence (B) of AP1510. 10A.B2 cells were infected with pools of short hairpins (designated A or B) or control short hairpin targeting firefly luciferase (shLuc). Data are represented as percentage of acini that formed multiacinar structures, mean ± S.D. (A) or percent change in multiacinar structures formed relative to AP1510-stimulated shLuc controls (B).
We were interested in PTPs that had not been characterized extensively and acted as positive regulators of ERBB2 signaling as they may represent a novel avenue for therapeutic intervention in ERBB2-positive tumors. Both shRNA pools against PTPD2 showed a significant decrease in the AP1510-induced multiacinar phenotype (Fig. 1B), suggesting that the phenotype was produced by at least two distinct short hairpins. Consequently, we focused our efforts on PTPD2.
PTPD2 Plays a Positive Role in ERBB2 Signaling
We deconvoluted the pools of short hairpins against PTPD2 into individual shRNAs and selected two that efficiently suppressed expression, as verified by immunoblotting (Fig. 2A). Then, we tested whether these individual shRNAs recapitulated the phenotype observed with the pools used in the screen. Activation of ERBB2 induced the formation of multiacinar structures in control acini; however, the abundance and size of these structures was significantly decreased in cells expressing the PTPD2 shRNAs (Fig. 2B).
FIGURE 2.

Suppression of PTPD2 inhibited the AP1510-induced phenotype in three-dimensional cultures. a, immunoblot showing the PTPD2 protein level in 10A.B2 cells expressing control or individual shRNAs targeting PTPD2. The expression of β-actin was used as the loading control. b, representative phase-contrast images of acinar structures, expressing the indicated shRNAs and treated with AP1510 or vehicle control for 12 days (left). For quantitation of the multiacinar phenotype, data are expressed as percentage of acini that form multiacinar structures ± S.E. (***, p < 0.0005) (right, top). Size distribution of multiacinar structures are expressed as area in μm2 (right, bottom). c, representative images of acinar structures stimulated with AP1510 or vehicle and immunostained for proliferation marker Ki67 (red). Nuclei were costained with DAPI in blue (left). Quantitation of Ki67-positive acini are indicated (right). d, representative images of cross-sections of acinar structures stimulated with AP1510 or vehicle control and immunostained for cis-Golgi marker GM130 (red). Nuclei were costained with DAPI in blue. Inset shows the representative AP1510-stimulated structures with arrows indicating mislocalization of GM130 to lateral or basal surfaces. Scale bar represents 100 μm.
Activation of ERBB2 in mammary acini induces the formation of multiacinar structures by causing hyperproliferation, lumen filling, and disruption of apico-basal polarity (22). We tested which of these events was affected by loss of PTPD2. ERBB2-induced changes in proliferation were measured in acini expressing control or PTPD2 shRNA by staining with an antibody against Ki67 (a marker of proliferating cells). Interestingly, ERBB2-induced proliferation in shPTPD2 acini was similar to that of control acini (Fig. 2C).
The outermost cells in mammary acini undergo apico-basal polarization as a result of which GM130, a cis-Golgi marker, is localized to the side of the cells facing the prospective lumen (23). Activation of ERBB2 disrupts this axis of polarity, as a result of which GM130 is mislocalized. Accordingly, in AP1510-stimulated control acini, we observed that GM130 was localized to the basolateral surface of the cells (Fig. 2D, white arrowheads). However, in PTPD2 shRNA-expressing cells, GM130 was localized to the apical surface despite ERBB2 activation with AP1510 (Fig. 2D).
Activation of ERBB2 also causes lumen filling by overriding apoptotic signals in ECM-detached lumenal cells (22, 23). Activation of chimeric ERBB2 in control 10A.B2 cells resulted in the formation of acini with filled lumens, whereas it failed to cause lumen filling in structures derived from cells expressing PTPD2 shRNA (shPTPD2 cells) (Fig. 2D). We observed that compared with control acini, shPTPD2 acini showed increased staining for caspase-3 (a marker of apoptotic cells) in the context of ERBB2 activation (Fig. 3). Together, these data suggest that PTPD2 was required specifically for ERBB2-mediated cell survival and disruption of apico-basal polarity, but not for ERBB2-mediated hyperproliferation.
FIGURE 3.

Suppression of PTPD2 enhanced apoptosis in AP1510-induced three-dimensional cultures. Representative images of cross-sections of acinar structures stimulated with AP1510 or vehicle control and immunostained for the apoptosis marker, cleaved caspase-3 (red). Nuclei were costained with DAPI in blue.
To validate further the role of PTPD2 in ERBB2 signaling, we tested the effect of PTPD2 overexpression (Fig. 4A) in 10A.B2 cells. We observed that when stimulated with AP1510, cells that overexpressed PTPD2 showed an enhanced multiacinar phenotype as compared with controls (Fig. 4B).
FIGURE 4.

Overexpression of PTPD2 enhanced the AP1510-induced phenotype. A, immunoblot showing the PTPD2 protein level in 10A.B2 cells expressing vector control or PTPD2 cDNA. The expression of β-actin was used as the loading control. B, representative phase-contrast images of acini expressing empty vector or PTPD2 cDNA and treated with AP1510 or vehicle control for 12 days (left). For quantitation of the multiacinar phenotype, data are expressed as percentage of acini that form multiacinar structures, mean ± S.E. (**, p < 0.005) (right). Scale bar represents 100 μm.
To investigate whether there were alterations in the copy number or mRNA expression level of the PTPN14 gene, which encodes PTPD2, in human breast cancer, we used cBioPortal to analyze 463 tumor samples from the TCGA data set (24, 25). The results indicated that PTPD2 was altered in 14% of the human tumor samples analyzed, a majority of the alterations being amplification or mRNA up-regulation events. This was comparable with PTP1B, which is established to act as a positive regulator of ERBB2 signaling in breast cancer (12, 13). Together, these results are consistent with a pro-oncogenic role for PTPD2 in human breast cancer.
Effect of PTPD2 Suppression on ERBB2 Signaling
Following dimerization, ERBB2 undergoes autophosphorylation, creating phosphotyrosine sites that recruit and activate signaling complexes. This results in the activation of effector pathways, chiefly, RAS/MAPK and PI3K/AKT (26). We evaluated the effects of PTPD2 suppression on the activation of these pathways following ERBB2 stimulation. Under conditions of acute activation of chimeric ERBB2 (≤60 min), suppression of PTPD2 did not have a significant effect on the overall tyrosine phosphorylation of ERBB2 in cells cultured in 2D. However, we observed a clear inhibition of Akt signaling in PTPD2 knockdown cells as compared with control (Fig. 5A). Any effect of PTPD2 knockdown on ERK signaling was less pronounced.
FIGURE 5.
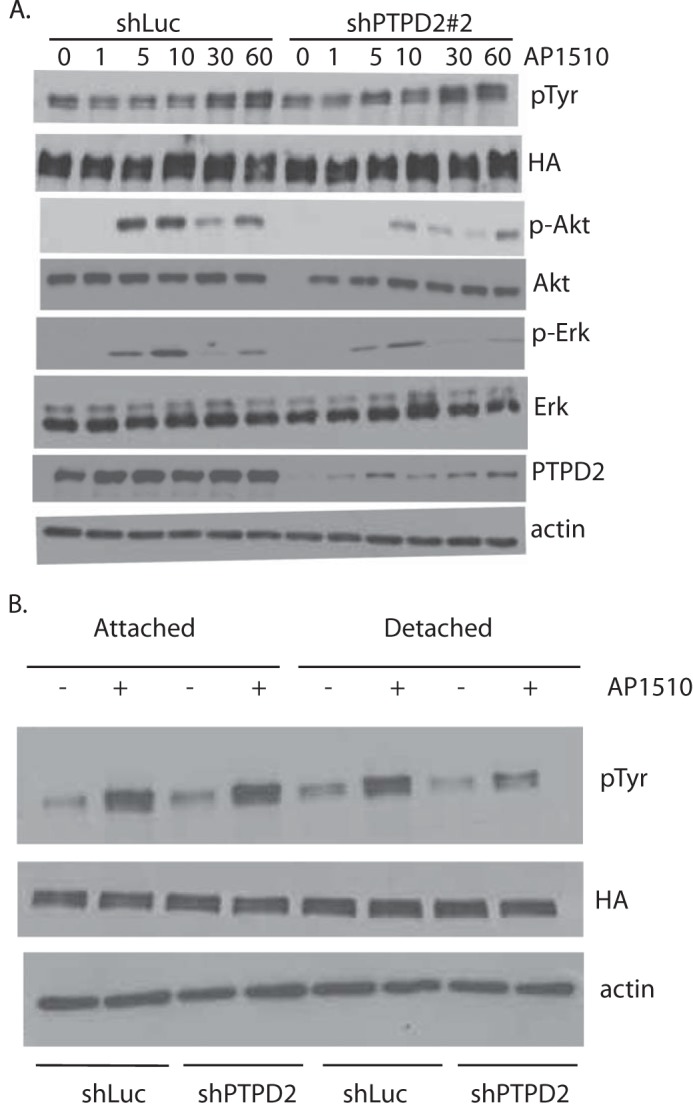
Suppression of PTPD2 attenuated ERBB2 effector pathways. A, time course of phosphorylation-induced activation of the indicated mediators of ERBB2 signaling was measured by immunoblotting. 10A.B2 cells that expressed either control shLuc or shPTPD2 hairpins were serum starved overnight and subjected to ERBB2 activation with AP1510 for the indicated time (min) before cell lysis. β-Actin was used as the loading control. B, 10A.B2 cells expressing control shLuc or shPTPD2 hairpins were plated under ECM-attached or ECM-detached conditions for 48 h, with or without AP1510. Phosphorylation status of ERBB2 was measured by immunoblotting with pan-phosphotyrosine (pTyr) antibody 4G10 and total ERBB2 was assessed using antibody to the HA epitope tag.
In light of our observation that loss of PTPD2 prevents ERBB2-mediated survival of cells in the lumen, we also tested the effect of PTPD2 loss on ERBB2 signaling in cells cultured in suspension (27). Although we observed no apparent difference in AP1510-induced ERBB2 phosphorylation between control cells and PTPD2-knockdown cells cultured under attached conditions, loss of PTPD2 in suspension culture resulted in a decrease in AP1510-induced ERBB2 phosphorylation as compared with shLuc controls (Fig. 5B). This requirement for PTPD2 for optimal ERBB2 activation in ECM-detached (suspension culture) cells is consistent with a role of the phosphatase as a positive regulator of signaling.
We tested whether PTPD2 exerted its positive effects by acting on known substrates or components of the ERBB2 signaling pathway. Some reports implicate PTPD2 in the regulation of the transcription factor, YAP1 (28). We tested whether, under conditions of ERBB2 activation, PTPD2 regulated YAP1 tyrosine phosphorylation, thereby affecting its function as a transcription factor. However, we did not observe tyrosine phosphorylation of YAP1 in 10A.B2 cells, either under basal conditions or following AP1510 stimulation. Furthermore, we found that suppression of YAP1 with 2 different functional shRNAs did not affect the multiacinar phenotype of ERBB2 activation (Fig. 6, a and b), suggesting that YAP1 is not a critical substrate of PTPD2 in ERBB2 signaling. SRC family kinases are also known to be critical downstream regulators of ERBB2 signaling (29, 30). We tested whether PTPD2 cooperated with ERBB2 signaling by modulating the activation of SRC. We observed that whereas a small molecule inhibitor of SRC, SU6656, inhibited the multiacinar phenotype in control 10A.B2 cells, it did not affect the more aggressive multiacinar phenotype of PTPD2-overexpressing cells (Fig. 7). Suppression of PTPD2 also did not affect SRC phosphorylation status. This suggests that PTPD2 does not act through SRC in this context.
FIGURE 6.
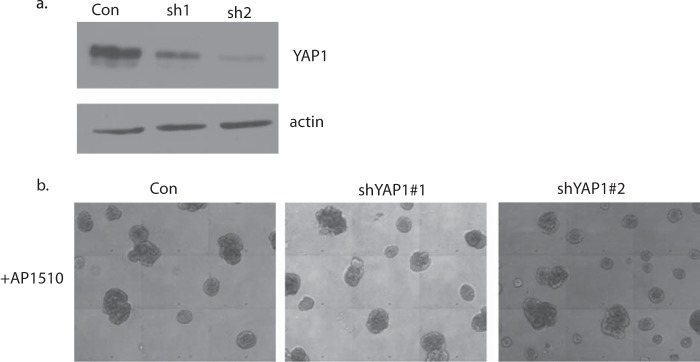
Suppression of YAP1 did not affect the AP1510-induced multiacinar phenotype in three-dimensional cultures. a, immunoblot showing the YAP1 protein level in 10A.B2 cells expressing control shLuc or shRNAs targeting YAP1. The expression of β-actin was used as the loading control. b, representative phase-contrast images of acinar structures expressing the indicated shRNAs and treated with AP1510 for 12 days. Scale bar represents 100 μm.
FIGURE 7.

Effect of SRC inhibitor SU6656 on the AP1510-induced multiacinar phenotype with and without PTPD2 overexpression. Representative phase-contrast images of AP1510-stimulated acinar structures expressing vector control or PTPD2 and treated with vehicle (DMSO) or SRC inhibitor SU6656 (5 μm). Scale bar represents 100 μm.
Phosphatidic Acid Bound to PTPD2 in Vitro and Enhanced Its Catalytic Activity
According to published reports, the activity of PTPD2 toward phosphotyrosine substrates is less robust than that of other classical PTPs (31). This has been attributed to the presence of an isoleucine in place of the consensus tyrosine residue that defines the depth of the catalytic cleft and stabilizes the interaction with the incoming phosphotyrosine substrate. It has been suggested that this deviation in the primary sequence might influence the substrate preference of PTPD2 (31, 32). Dual specificity phosphatases also lack this critical tyrosine, and present a more open, shallow active site cleft. We did not observe dephosphorylation of phosphoserine and phosphothreonine-containing peptides by the recombinant catalytic domain of PTPD2. Furthermore, some dual specificity phosphatases, including the lipid phosphatase PTEN, are also known to recognize phospholipids as substrates; however, when tested against a panel of phospholipids, we observed that both wild type and substrate-trapping mutant forms of PTPD2 specifically bound to phosphatidic acid (Fig. 8A), indicating that it is not a substrate but rather a binding partner of PTPD2.
FIGURE 8.

Phosphatidic acid bound PTPD2 in vitro and augmented its catalytic activity. A, protein-lipid overlay to measure binding of the catalytic domain of PTPD2 to a lipid array. Strips containing the indicated phospholipids were probed with 0.5 μg/ml of bacterially expressed, His-tagged catalytic domain of wild type (left) or substrate-trapping mutant form (right) of PTPD2. Bound protein was detected by immunoblotting with anti-His antibody. B, recombinant PTPD2 catalytic domain (●) or PTP1B (■) was incubated with various concentrations of [14C]DPPA, and the PTP-[14C]DPPA complex was separated from unbound lipid by centrifugation, and bound radioactivity was measured by scintillation counting. C, [14C]DPPA binding to full-length PTPD2 performed as in B. D, recombinant PTPD2 catalytic domain was left untreated (○) or incubated with phosphatidic acid for 30 min (●), and the phosphatase activity was measured using DiFMUP as substrate. E, effect of phosphatidylserine on PTPD2 catalytic activity was assessed as in D. F, PTPD2 (left) or PTP1B (right) were left untreated (open symbols) or incubated with [14C]DPPA (closed symbols) for 30 min, and the phosphatase activity was measured using 32P-reduced carboxamidomethylated and maleylated lysozyme (RCML) as substrate.
This lipid-protein interaction was investigated further by performing binding assays with liposomes of [14C]DPPA. We found that recombinant PTPD2 bound strongly to the radiolabeled lipid. Recombinant PTP1B(1–321), on the other hand, bound poorly in comparison, consistent with specificity in the binding of PTPD2 to DPPA (Fig. 8B). Full-length PTPD2, immunoprecipitated from cells, also efficiently bound to [14C]DPPA (Fig. 8C).
We tested the functional significance of PA binding to PTPD2 function. When recombinant PTPD2 was treated with PA, we observed an increase in the activity of the enzyme toward DiFMUP, an artificial substrate (Fig. 8D). This suggested that phosphatidic acid might act as a positive regulator of the catalytic activity of PTPD2. This effect was not seen when PTPD2 was treated with phosphatidylserine (Fig. 8E).
We also tested whether PA had a similar effect on the catalytic activity of full-length PTPD2 toward a protein substrate. We found that PA binding resulted in an ∼3-fold increase in the catalytic activity of full-length PTPD2 (immunoprecipitated from cells) toward phosphorylated reduced carboxamidomethylated and maleylated lysozyme (RCML) (Fig. 8F, left) without altering substrate affinity. This indicates that PA binding to PTPD2 increased substrate turnover. Full-length PTP1B, derived from cells, was not activated by PA and served as a negative control in this experiment (Fig. 8F, right). Taken together, these data suggest that PA binds to PTPD2 in vitro and augments its catalytic activity.
Small Molecule Inhibitors of PLD Suppressed the Multiacinar Phenotype by Inhibiting ERBB2-mediated Lumen Filling
We hypothesized that phosphatidic acid-mediated regulation of PTPD2 was crucial for the function of the phosphatase in cells as a positive regulator of ERBB2 signaling. Therefore, we tested whether manipulation of the levels of phosphatidic acid in cells would attenuate the multiacinar phenotype induced by ERBB2 activation in three-dimensional cultures. FIPI is a well characterized small molecule that has been used widely to inhibit PLD, an enzyme that generates phosphatidic acid from phosphatidylcholine (33). In the absence of signaling from chimeric ERBB2, FIPI had no effect on the morphogenesis of 10A.B2 cells. However, FIPI treatment inhibited the AP1510-induced multiacinar phenotype of 10A.B2 cells in three-dimensional culture (Fig. 9A, compare first and second panel). FIPI treatment reduced the number and size of multiacinar structures formed upon AP1510 stimulation. This suggests that the function of PLD enzyme and, by extension, the levels of phosphatidic acid in the cell, was important for ERBB2 signaling.
FIGURE 9.

Small molecule inhibitors of PLD suppressed the multiacinar phenotype. A, representative phase-contrast images of 10A.B2 acinar structures treated with vehicle (DMSO), dual PLD inhibitor (FIPI), PLD1-specific inhibitor (VUO359595), or PLD2-specific inhibitor (VUO364739), with or without AP1510 stimulation. B, for quantitation of the multiacinar phenotype, data are expressed as percentage of acini that formed multiacinar structures, mean ± S.E. (***, p < 0.0005) (left). Size distribution of multiacinar structures expressed as area in μm2 (right). C, AP1510-stimulated acinar structures treated with DMSO or PLD2-specific inhibitor were immunostained for proliferation marker Ki67 (red). Nuclei were costained with DAPI in blue. D, cross-sectional view of AP1510-stimulated acini treated as in C with DAPI-stained nuclei in blue. White arrows indicate acini with clear lumen. Scale bar represents 100 μm.
Two isoforms of mammalian PLD have been identified, PLD1 and PLD2, which differ in basal activity, localization, and regulatory functions (34). To investigate the relative importance of each isoform in the regulation of ERBB2 signaling, we treated 10A.B2 acini with previously reported isoform-selective inhibitors (35, 36). We found that a PLD1-specific inhibitor (VUO359595) did not have a significant effect on the multiacinar phenotype. However, the PLD2-specific inhibitor VUO364739 suppressed the AP1510-induced multiacinar phenotype of ERBB2 as compared with vehicle-treated control (Fig. 9A, compare first panel with third and fourth panels). Treatment with the PLD1-specific inhibitor resulted in a small decrease in the size distribution of multiacinar structures formed as compared with control, whereas the PLD2-specific inhibitor caused a dramatic reduction in the size distribution of the multiacinar structures resulting from AP1510 stimulation (Fig. 9B). This result implicates the PLD2 isoform as a positive regulator of ERBB2 signaling.
We observed that RNAi-mediated suppression of PTPD2 attenuated the formation of multiacinar structures by inhibiting specifically ERBB2-mediated lumen filling and loss of polarity, but not ERBB2-induced hyperproliferation (Fig. 2, c and d). We tested whether inhibition of PLD2 also affected the formation of multiacinar structures in the same manner. The results of PLD inhibition with the PLD2 isoform-specific inhibitor, VUO364739, were strikingly similar to those observed with loss of PTPD2 in the context of ERBB2 activation. There was no difference in ERBB2-induced proliferation in acini treated with the PLD2 inhibitor as compared with DMSO-treated controls (Fig. 9C). However, ERBB2 activation inhibited lumen filling in structures treated with the PLD2 inhibitor (Fig. 9D). The small molecule inhibitor of PLD2 essentially phenocopied shRNA-mediated suppression of PTPD2 in the context of ERBB2 signaling, suggesting that PLD2 and PTPD2 might be acting in the same effector pathway that regulates ERBB2-mediated lumen filling and loss of polarity.
Suppression of PTPD2 Inhibited Phosphatidic Acid-mediated Rescue of the PLD Inhibition Phenotype
It has been shown previously that exogenously added phosphatidic acid is incorporated into cell membranes and can elicit cellular responses (37). We tested whether rescue of the PLD inhibition phenotype by exogenously added PA was dependent on PTPD2. In FIPI-treated control acini, we observed that the addition of phosphatidic acid to the three-dimensional overlay medium rescued the effect of the PLD inhibitor. In shPTPD2 acini, however, phosphatidic acid did not rescue the FIPI treatment phenotype, suggesting that PTPD2 was required for signaling downstream of the lipid (Fig. 10, A and B). This result suggests that PTPD2 is required for the functional role played by PLD and, by extension, phosphatidic acid in ERBB2 signaling.
FIGURE 10.

Suppression of PTPD2-inhibited phosphatidic acid-mediated rescue of the PLD inhibition phenotype. A, phase-contrast images of AP1510-stimulated acinar structures expressing the indicated shRNAs, and treated with vehicle (DMSO), dual PLD inhibitor, FIPI, or FIPI and phosphatidic acid. Insets show representative acini. B, for quantitation of the multiacinar phenotype, data are expressed as percentage of acini that form multiacinar structures, mean ± S.E. shLuc (control) and shPTPD2 acini are represented by white and black bars, respectively. Scale bar represents 100 μm.
DISCUSSION
In this study, we have identified PTPD2 as a novel, positive regulator of the ERBB2 signaling pathway. We observed that suppression of PTPD2 attenuated the phenotype characteristic of ERBB2 activation in three-dimensional cultures, suggesting that this enzyme is critical for the ERBB2 signaling pathway to function optimally. Importantly, we observed that PTPD2 was required specifically for ERBB2-mediated lumen filling and loss of polarity, but not for hyperproliferation. This is different from PTP1B, which also acts as a positive regulator of ERBB2 signaling, but is required for ERBB2-mediated proliferation as well as cell survival (38). This evidence highlights the specificity of PTPs in the regulation of signaling pathways downstream of ERBB2.
We report that PTPD2 specifically bound to PA from among a panel of phosphorylated lipids. Lipid species have been shown to function as critical second messengers in signal transduction pathways, both by recruiting signaling molecules to cellular membranes (39) and modulating enzyme catalytic activity (40). PA, for example, has been shown to modulate the activity of several enzymes involved in signal transduction including isoforms of cAMP-specific phosphodiesterases (41) and the protein kinase Raf-1 (42). Of particular relevance to our findings, it has been reported that PA can bind to and augment the catalytic activity of the phosphotyrosine-specific PTP, SHP-1 (43). Consistent with these observations, we found that PA binding to PTPD2 resulted in enhancement of the phosphatase activity of PTPD2 toward phosphorylated RCML (reduced carboxamidomethylated and maleylated lysozyme) substrate.
We found that inhibition of PLD (the enzyme that produces phosphatidic acid from phosphatidylcholine) also suppressed the multiacinar phenotype of activated ERBB2 in three-dimensional cultures, suggesting a positive role for this enzyme in the ERBB2 signaling pathway. It has been reported that PLD2 forms a complex with the EGF receptor and undergoes tyrosine phosphorylation following stimulation with EGF (44). Although we did not observe tyrosine phosphorylation of PLD2 upon acute stimulation of chimeric ERBB2, it is possible that there exist other mechanisms of PLD2 activation in the context of ERBB2 signaling. Consistent with a pro-oncogenic role for this enzyme, a high level of PLD activity has been observed in breast cancers (45, 46). We also observed that exogenously added phosphatidic acid can rescue the PLD inhibition phenotype, but only when PTPD2 was present. This result supports a central role for PTPD2 downstream of PLD2 activity in ERBB2 signaling. We speculate that enhancement of the catalytic function of PTPD2 by PLD2-generated phosphatidic acid is important for the dephosphorylation of critical downstream mediators of ERBB2 signaling.
Consistent with a positive signaling function, overexpression of PTPD2 in epithelial Madin-Darby canine kidney cells has been shown to cause increased cell motility and changes in gene expression observed in epithelial cancer cell lines that have undergone epithelial-mesenchymal transition, a process associated with cancer progression (47). There is an interesting difference in reports of the functional role of PTPD2 in breast cancer. In studies carried out in parental MCF10A cells, which are dependent on EGF signaling, it has been reported that suppression of PTPD2 results in the disruption of MCF10A acinus morphology, suggesting that it may have a tumor suppressor function (48, 49). In our study, we have reported that suppression of PTPD2 rescues the multiacinar phenotype caused by ERBB2 activation, suggesting a pro-oncogenic role for PTPD2 in the context of ERBB2 activation in 10A.B2 cells. The observation that PTPD2 has functionally distinct roles under these two conditions underscores the fact that some PTPs may function negatively or positively depending on the signaling context. For example, PTP1B is known to play a pro-oncogenic role in the context of ERBB2-driven mammary carcinogenesis (12, 13). However, in ovarian cancer cells, PTP1B inhibits pro-survival signals induced by insulin-like growth factor-1 (50). SHP2 plays a well characterized oncogenic role as an activator of RAS signaling in a variety of tumors (3). However, deficiency of PTPN11, the gene that encodes SHP2, has also been shown to underlie excessive proliferation in cartilage tumors, consistent with a tumor suppressor function in this context (51).
There are precedents in literature to suggest that proliferation might be regulated independently of cell survival and apico-basal polarity downstream of ERBB2. A mutant form of the polarity protein Par6, which is unable to bind to atypical protein kinase C, reverses the multiacinar and cell survival phenotypes characteristic of ERBB2, without affecting hyperproliferation (52). Another study provides evidence that proliferation and polarity are regulated independently, downstream of a β4 integrin-ERBB2 complex during ERBB2-driven carcinogenesis (53). It remains to be tested whether PTPD2 functions in ERBB2 signaling by regulating one or more of these effector pathways or an as yet uncharacterized pathway. Our observations suggest that PTPD2 is at least required for ERBB2 to override apoptotic signals. One possibility is that ERBB2 could be signaling to anti-apoptotic pathways via PTPD2. However, it has also been suggested that polarity proteins themselves can directly regulate cell death pathways (52), raising the possibility that PTPD2 could be regulating these pathways indirectly by acting on polarity proteins.
The challenges underlying the use of existing anti-ERBB2 therapies necessitate further investigation of the mechanisms that regulate this signaling pathway to identify new therapeutic targets. Our findings illustrate a novel signaling axis comprising PLD2 and PTPD2 that acts positively to regulate ERBB2 signaling. It is important to gain a mechanistic understanding of signaling events downstream of these molecules and to validate our findings in vivo using disease-relevant animal models. Together, these studies could provide a basis for prognostic and therapeutic interventions targeting PTPD2 in ERBB2-positive breast cancers.
Acknowledgments
We thank Michael Frohman (Stony Brook University) for helpful discussions. We also thank Alex Brown (Vanderbilt University), Yeesim Khew-Goodall (Centre for Cancer Biology, SA Pathology, Adelaide/University of Adelaide), and Stefan Knapp for their generous gift of PLD isoform-specific inhibitors, full-length PTPD2 cDNA construct, and His6-tagged PTPD2 catalytic domain (886–1187), respectively.
This work was supported, in whole or in part, by National Institutes of Health Grant CA53840 and Cold Spring Harbor Laboratory Cancer Center Support Grant CA45508 (to N. K. T.).
- PTK
- protein-tyrosine kinase
- PTP
- protein-tyrosine phosphatase
- PLD
- phospholipase D
- PA
- phosphatidic acid
- PTEN
- phosphatase and tensin homolog
- YAP1
- Yes-associated protein 1
- DPPA
- dipalmitoyl phosphatidic acid
- DiFMUP
- 6,8-difluoro-4-methylumbelliferyl phosphate
- FIPI
- 5-fluro-2-indolyl des-chlorohalopemide
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Sliwkowski M. X., Mellman I. (2013) Antibody therapeutics in cancer. Science 341, 1192–1198 [DOI] [PubMed] [Google Scholar]
- 2. Hunter T. (2009) Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol. 21, 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Julien S. G., Dubé N., Hardy S., Tremblay M. L. (2011) Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 11, 35–49 [DOI] [PubMed] [Google Scholar]
- 4. Ostman A., Hellberg C., Böhmer F. D. (2006) Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 6, 307–320 [DOI] [PubMed] [Google Scholar]
- 5. King C. R., Kraus M. H., Aaronson S. A. (1985) Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science 229, 974–976 [DOI] [PubMed] [Google Scholar]
- 6. Slamon D. J., Clark G. M., Wong S. G., Levin W. J., Ullrich A., McGuire W. L. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 [DOI] [PubMed] [Google Scholar]
- 7. Yu D., Hung M. C. (2000) Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene 19, 6115–6121 [DOI] [PubMed] [Google Scholar]
- 8. Cobleigh M. A., Vogel C. L., Tripathy D., Robert N. J., Scholl S., Fehrenbacher L., Wolter J. M., Paton V., Shak S., Lieberman G., Slamon D. J. (1999) Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 17, 2639–2648 [DOI] [PubMed] [Google Scholar]
- 9. Vogel C. L., Cobleigh M. A., Tripathy D., Gutheil J. C., Harris L. N., Fehrenbacher L., Slamon D. J., Murphy M., Novotny W. F., Burchmore M., Shak S., Stewart S. J., Press M. (2002) Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 20, 719–726 [DOI] [PubMed] [Google Scholar]
- 10. Lin G., Aranda V., Muthuswamy S. K., Tonks N. K. (2011) Identification of PTPN23 as a novel regulator of cell invasion in mammary epithelial cells from a loss-of-function screen of the “PTP-ome”. Genes Dev. 25, 1412–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boivin B., Chaudhary F., Dickinson B. C., Haque A., Pero S. C., Chang C. J., Tonks N. K. (2013) Receptor protein-tyrosine phosphatase α regulates focal adhesion kinase phosphorylation and ErbB2 oncoprotein-mediated mammary epithelial cell motility. J. Biol. Chem. 288, 36926–36935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bentires-Alj M., Neel B. G. (2007) Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 67, 2420–2424 [DOI] [PubMed] [Google Scholar]
- 13. Julien S. G., Dubé N., Read M., Penney J., Paquet M., Han Y., Kennedy B. P., Muller W. J., Tremblay M. L. (2007) Protein-tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 39, 338–346 [DOI] [PubMed] [Google Scholar]
- 14. Bentires-Alj M., Gil S. G., Chan R., Wang Z. C., Wang Y., Imanaka N., Harris L. N., Richardson A., Neel B. G., Gu H. (2006) A role for the scaffolding adapter GAB2 in breast cancer. Nat. Med. 12, 114–121 [DOI] [PubMed] [Google Scholar]
- 15. Nagata Y., Lan K. H., Zhou X., Tan M., Esteva F. J., Sahin A. A., Klos K. S., Li P., Monia B. P., Nguyen N. T., Hortobagyi G. N., Hung M. C., Yu D. (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127 [DOI] [PubMed] [Google Scholar]
- 16. Krishnan N., Koveal D., Miller D. H., Xue B., Akshinthala S. D., Kragelj J., Jensen M. R., Gauss C. M., Page R., Blackledge M., Muthuswamy S. K., Peti W., Tonks N. K. (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rosenbluh J., Nijhawan D., Cox A. G., Li X., Neal J. T., Schafer E. J., Zack T. I., Wang X., Tsherniak A., Schinzel A. C., Shao D. D., Schumacher S. E., Weir B. A., Vazquez F., Cowley G. S., Root D. E., Mesirov J. P., Beroukhim R., Kuo C. J., Goessling W., Hahn W. C. (2012) β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Debnath J., Muthuswamy S. K., Brugge J. S. (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268 [DOI] [PubMed] [Google Scholar]
- 19. Meng T. C., Hsu S. F., Tonks N. K. (2005) Development of a modified in-gel assay to identify protein-tyrosine phosphatases that are oxidized and inactivated in vivo. Methods 35, 28–36 [DOI] [PubMed] [Google Scholar]
- 20. Tonks N. K., Diltz C. D., Fischer E. H. (1988) Purification of the major protein-tyrosine phosphatases of human placenta. J. Biol. Chem. 263, 6722–6730 [PubMed] [Google Scholar]
- 21. Muthuswamy S. K., Gilman M., Brugge J. S. (1999) Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol. Cell. Biol. 19, 6845–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Muthuswamy S. K., Li D., Lelievre S., Bissell M. J., Brugge J. S. (2001) ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Debnath J., Mills K. R., Collins N. L., Reginato M. J., Muthuswamy S. K., Brugge J. S. (2002) The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111, 29–40 [DOI] [PubMed] [Google Scholar]
- 24. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 27. Grassian A. R., Coloff J. L., Brugge J. S. (2011) Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harbor Symp. Quant. Biol. 76, 313–324 [DOI] [PubMed] [Google Scholar]
- 28. Huang J. M., Nagatomo I., Suzuki E., Mizuno T., Kumagai T., Berezov A., Zhang H., Karlan B., Greene M. I., Wang Q. (2013) YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene 32, 2220–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Belsches-Jablonski A. P., Biscardi J. S., Peavy D. R., Tice D. A., Romney D. A., Parsons S. J. (2001) Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene 20, 1465–1475 [DOI] [PubMed] [Google Scholar]
- 30. Muthuswamy S. K., Siegel P. M., Dankort D. L., Webster M. A., Muller W. J. (1994) Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol. Cell. Biol. 14, 735–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barr A. J., Ugochukwu E., Lee W. H., King O. N., Filippakopoulos P., Alfano I., Savitsky P., Burgess-Brown N. A., Müller S., Knapp S. (2009) Large-scale structural analysis of the classical human protein-tyrosine phosphatome. Cell 136, 352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barr A. J., Debreczeni J. E., Eswaran J., Knapp S. (2006) Crystal structure of human protein-tyrosine phosphatase 14 (PTPN14) at 1.65-Å resolution. Proteins 63, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 33. Su W., Yeku O., Olepu S., Genna A., Park J. S., Ren H., Du G., Gelb M. H., Morris A. J., Frohman M. A. (2009) 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jenkins G. M., Frohman M. A. (2005) Phospholipase D: a lipid centric review. Cell. Mol. Life Sci. 62, 2305–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lavieri R., Scott S. A., Lewis J. A., Selvy P. E., Armstrong M. D., Alex Brown H., Lindsley C. W. (2009) Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors: Part II. identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Bioorg. Med. Chem. Lett. 19, 2240–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lavieri R. R., Scott S. A., Selvy P. E., Kim K., Jadhav S., Morrison R. D., Daniels J. S., Brown H. A., Lindsley C. W. (2010) Design, synthesis, and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamides: discovery of an isoform-selective small molecule phospholipase D2 inhibitor. J. Med. Chem. 53, 6706–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishikimi A., Fukuhara H., Su W., Hongu T., Takasuga S., Mihara H., Cao Q., Sanematsu F., Kanai M., Hasegawa H., Tanaka Y., Shibasaki M., Kanaho Y., Sasaki T., Frohman M. A., Fukui Y. (2009) Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science 324, 384–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arias-Romero L. E., Saha S., Villamar-Cruz O., Yip S. C., Ethier S. P., Zhang Z. Y., Chernoff J. (2009) Activation of Src by protein-tyrosine phosphatase 1B Is required for ErbB2 transformation of human breast epithelial cells. Cancer Res. 69, 4582–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao R., Fu X., Li Q., Krantz S. B., Zhao Z. J. (2003) Specific interaction of protein-tyrosine phosphatase-MEG2 with phosphatidylserine. J. Biol. Chem. 278, 22609–22614 [DOI] [PubMed] [Google Scholar]
- 40. Liscovitch M., Cantley L. C. (1994) Lipid second messengers. Cell 77, 329-334 [DOI] [PubMed] [Google Scholar]
- 41. Némoz G., Sette C., Conti M. (1997) Selective activation of rolipram-sensitive, cAMP-specific phosphodiesterase isoforms by phosphatidic acid. Mol. Pharmacol. 51, 242–249 [DOI] [PubMed] [Google Scholar]
- 42. Ghosh S., Bell R. M. (1997) Regulation of Raf-1 kinase by interaction with the lipid second messenger, phosphatidic acid. Biochem. Soc. Trans. 25, 561–565 [DOI] [PubMed] [Google Scholar]
- 43. Frank C., Keilhack H., Opitz F., Zschörnig O., Böhmer F. D. (1999) Binding of phosphatidic acid to the protein-tyrosine phosphatase SHP-1 as a basis for activity modulation. Biochemistry 38, 11993–12002 [DOI] [PubMed] [Google Scholar]
- 44. Slaaby R., Jensen T., Hansen H. S., Frohman M. A., Seedorf K. (1998) PLD2 complexes with the EGF receptor and undergoes tyrosine phosphorylation at a single site upon agonist stimulation. J. Biol. Chem. 273, 33722–33727 [DOI] [PubMed] [Google Scholar]
- 45. Fiucci G., Czarny M., Lavie Y., Zhao D., Berse B., Blusztajn J. K., Liscovitch M. (2000) Changes in phospholipase D isoform activity and expression in multidrug-resistant human cancer cells. Int. J. Cancer 85, 882–888 [DOI] [PubMed] [Google Scholar]
- 46. Welsh C. J., Yeh G. C., Phang J. M. (1994) Increased phospholipase D activity in multidrug resistant breast cancer cells. Biochem. Biophys. Res. Commun. 202, 211–217 [DOI] [PubMed] [Google Scholar]
- 47. Wyatt L., Wadham C., Crocker L. A., Lardelli M., Khew-Goodall Y. (2007) The protein-tyrosine phosphatase Pez regulates TGFβ, epithelial-mesenchymal transition, and organ development. J. Cell Biol. 178, 1223–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu X., Yang N., Figel S. A., Wilson K. E., Morrison C. D., Gelman I. H., Zhang J. (2013) PTPN14 interacts with and negatively regulates the oncogenic function of YAP. Oncogene 32, 1266–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang W., Huang J., Wang X., Yuan J., Li X., Feng L., Park J. I., Chen J. (2012) PTPN14 is required for the density-dependent control of YAP1. Genes Dev. 26, 1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fan G., Lin G., Lucito R., Tonks N. K. (2013) Protein-tyrosine phosphatase 1B antagonized signaling by insulin-like growth factor-1 receptor and kinase BRK/PTK6 in ovarian cancer cells. J. Biol. Chem. 288, 24923–24934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang W., Wang J., Moore D. C., Liang H., Dooner M., Wu Q., Terek R., Chen Q., Ehrlich M. G., Quesenberry P. J., Neel B. G. (2013) Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aranda V., Haire T., Nolan M. E., Calarco J. P., Rosenberg A. Z., Fawcett J. P., Pawson T., Muthuswamy S. K. (2006) Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat. Cell Biol. 8, 1235–1245 [DOI] [PubMed] [Google Scholar]
- 53. Guo W., Pylayeva Y., Pepe A., Yoshioka T., Muller W. J., Inghirami G., Giancotti F. G. (2006) β4 Integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 126, 489–502 [DOI] [PubMed] [Google Scholar]