

Background: PRMT5-MEP50 is an arginine methyltransferase with significant roles in development and cancer.
Results: MEP50 binds to the histone fold domain and is essential for the efficient use of SAM by PRMT5.
Conclusion: MEP50 is essential for methylation of histones H4 and H2A by PRMT5.
Significance: The mechanism of histone methylation by PRMT5-MEP50 provides novel insight into methyltransferase mechanisms and therapeutic development.
Keywords: Enzyme Kinetics, Enzyme Mechanism, Histone Methylation, Peptide Array, Protein Arginine N-methyltransferase 5 (PRMT5), WD Repeat
Abstract
The protein arginine methyltransferase PRMT5 is complexed with the WD repeat protein MEP50 (also known as Wdr77 or androgen coactivator p44) in vertebrates in a tetramer of heterodimers. MEP50 is hypothesized to be required for protein substrate recruitment to the catalytic domain of PRMT5. Here we demonstrate that the cross-dimer MEP50 is paired with its cognate PRMT5 molecule to promote histone methylation. We employed qualitative methylation assays and a novel ultrasensitive continuous assay to measure enzyme kinetics. We demonstrate that neither full-length human PRMT5 nor the Xenopus laevis PRMT5 catalytic domain has appreciable protein methyltransferase activity. We show that histones H4 and H3 bind PRMT5-MEP50 more efficiently compared with histone H2A(1–20) and H4(1–20) peptides. Histone binding is mediated through histone fold interactions as determined by competition experiments and by high density histone peptide array interaction studies. Nucleosomes are not a substrate for PRMT5-MEP50, consistent with the primary mode of interaction via the histone fold of H3-H4, obscured by DNA in the nucleosome. Mutation of a conserved arginine (Arg-42) on the MEP50 insertion loop impaired the PRMT5-MEP50 enzymatic efficiency by increasing its histone substrate Km, comparable with that of Caenorhabditis elegans PRMT5. We show that PRMT5-MEP50 prefers unmethylated substrates, consistent with a distributive model for dimethylation and suggesting discrete biological roles for mono- and dimethylarginine-modified proteins. We propose a model in which MEP50 and PRMT5 simultaneously engage the protein substrate, orienting its targeted arginine to the catalytic site.
Introduction
Protein arginine methyltransferases (PRMTs)4 methylate arginines in histones and other proteins (1). Type I enzymes monomethylate and asymmetrically dimethylate arginine (Rme1 and Rme2a) (2). PRMT5 is a Type II enzyme that catalyzes arginine monomethylation and symmetric dimethylation (Rme1 and Rme2s). The many targets of PRMT5 include ribosomal proteins, the histone chaperone nucleoplasmin, p53, and histones (2–7). We and others have shown that PRMT5 requires the WD repeat protein MEP50 (methylosome protein 50, also known as Wdr77 or androgen coreceptor p44) in vertebrates (8–12), whereas it is active independent of MEP50 in Caenorhabditis elegans. PRMT5 also partners with other cofactors (10, 12–18). The expression and activity of PRMT5-MEP50 are now clinically correlated with poor prognosis in a range of cancers, suggesting that insight into its enzymatic mechanism will be essential for highly targeted drug design potential leading to new therapeutics (19–21).
Because PRMT5 methylates multiple histone and non-histone arginines in vivo, a long-standing question is how PRMT5 selects among these many sites and multiple protein targets. Our previous work using crystallography, qualitative binding studies, and electron microscopy reconstruction suggested that substrate recruitment was primarily mediated by the MEP50 subunit. The structure of Xenopus laevis PRMT5-MEP50 that we solved (22) and the human PRMT5-MEP50 structure solved by Emtage and colleagues (23) revealed a buried and poorly accessible catalytic site. These observations are consistent with a conserved and significant stringency in substrate selection.
MEP50 is a seven-bladed WD repeat protein that is unusually acidic. We previously hypothesized that MEP50 served as a substrate presenter, similar to the initial hypothesis for the WD repeat protein Wdr5 (24) and RbAp48 (25) in binding histones H3 and H4, respectively. Our crystal structure of the XlPRMT5-MEP50·SAH complex and our electron microscopy reconstruction of the complex bound to its substrate nucleoplasmin supported this hypothesis. We and others previously showed that human PRMT5 is inactive in the absence of MEP50, consistent with a required role in binding substrate.
Quantitative enzymatic analysis of methyltransferases requires ultrasensitive techniques due to their low turnover. Previously described kinetic assays are either not commercially available, have low throughput, or provide signals too low to be of use for slow enzymes such as PRMT5. Therefore, we employ a novel ultrasensitive coupled continuous assay to measure kinetic parameters of other methyltransferases introduced previously by our colleagues (26).
Here, we test the function of MEP50 in promoting PRMT5 histone methyltransferase activity. We employ structural analysis as well as qualitative and quantitative methylation assays to measure enzymatic activity and binding affinity for histones to PRMT5-MEP50. We show that mutation within the MEP50 insertion finger impaired kinetic parameters of both histone and SAM substrates. Our results support the concept that MEP50 interacts directly with histones and the N-terminal domain of PRMT5 but may also contribute to active site remodeling in the C-terminal domain of PRMT5 for efficient methyl transfer. Our computational modeling revealed multiple modes of substrate-enzyme interaction consistent with our experimental data. These studies support the essential function of MEP50 in binding histone fold and presenting histone tail substrate to the active site of PRMT5 cross-dimer for efficient methylation.
EXPERIMENTAL PROCEDURES
Reagents
Chemicals and reagents were obtained from Sigma, Fisher, or Research Products International. [3H]Methyl- and [14C]methyl-SAM were purchased from PerkinElmer Life Sciences. SAM and SAH were purified by HPLC (Luna C18(2), Phenomenex), desalted, concentrated, and stored at −80 °C (26). Reducing agent tris(hydroxypropyl)-phosphine was from Novagen. ATP detection was achieved using the ATPLite 1Step system (PerkinElmer Life Sciences); molecular biology grade water, with additional charcoal treatment, was used for all enzymatic assays. DNase I was from New England Biolabs.
Proteins and Peptides
X. laevis PRMT5-MEP50 complex was produced and purified as described (5). XlMEP50 mutagenesis was performed by mutating Arg-42 (codon: CGC) to glutamine (codon: CAG) or glutamic acid (codon: GAG). Corresponding baculoviruses were produced in DH10Bac cells (Invitrogen), followed by co-infection of Hi5 cells with wild-type XlPRMT5. Cell lysis, nickel affinity chromatography, Superdex 200 gel filtration, and concentration were performed as before (22). Wild-type XlMEP50 and mutants (R42Q and R42E) were obtained in excess when purifying corresponding XlPRMT5-MEP50 complexes. Recombinant X. laevis core histones were expressed as His6-tagged tobacco etch virus (H2A, H3, and H4) fusion proteins, and the tag was completely removed by tobacco etch virus protease to leave a scarless histone. Recombinant nucleoplasmin from X. laevis was expressed and purified as described previously (22). The H4(1–7) and H4(1–20) peptides were from AnaSpec Inc. (catalog nos. 62754 and 62498, respectively); all other peptides were prepared by the peptide synthesis facility at Rockefeller University. Recombinant FLAG-HsPRMT5 enzyme was purchased from SinoBiologicals (catalog no. 11074-H18H; Beijing, China). Recombinant Xenopus mononucleosomes were purchased from Epicypher (catalog no. 16-0005), and octamers were produced as described (22). Enzymes for the luciferase-based assay, including the Clostridium symbiosum pyruvate phosphate dikinase (generous gift provided by Dr. Debra Dunaway-Mariano, University of New Mexico) and the adenosine phosphoribosyltransferase from Saccharomyces cerevisiae and 5′-methylthioadenosine/S-adenosyl-l-homocysteine nucleosidase from Salmonella enterica (SeMTAN) (generous gifts from Dr. Vern L. Schramm, Albert Einstein College of Medicine), were expressed and purified to homogeneity according to published protocols (27, 28).
Filter Binding Methyltransferase Assay and Fluorography
Assays were performed as described (22). Briefly, substrate protein was incubated with PRMT5-MEP50 enzyme at the indicated concentrations in 15 μl of reaction buffer (20 mm Tris, pH 8.0, 10 mm DTT, protease inhibitors, 0.5 μm [3H]methyl-SAM of specific activity 78.2 Ci mmol−1) for 20 min at 30 °C. The reaction mixture was spotted on P81 filter paper, washed with sodium carbonate buffer (0.1 m, pH 8.5), air-dried, and analyzed via scintillation counter (Wallac Winspectral 1414 LSC). Alternatively, the reaction mixture was run on an SDS-polyacrylamide gel, stained and imaged, soaked in Amplify (NAMP100, GE Healthcare), dried, and exposed to film.
Nucleosome Methyltransferase Assay
The assay was performed with 2 μg of mononucleosomes treated with 2 units of DNase I or reaction buffer alone for 1.5 h at 30 °C prior to the addition of XlPRMT5-MEP50 (300 nm final concentration) in the presence of [3H]methyl-SAM (0.3 μm final concentration; specific activity 78.2 Ci mmol−1). Similar experiments using recombinant H2A and H4 (2.5 μm) were run in parallel. After 20 min, reaction mixtures were run on an SDS-polyacrylamide gel, stained and imaged, soaked in Amplify (NAMP100, GE Healthcare), dried, and exposed to film.
FLAG Pull-down
Anti-FLAG M2 antibody-coupled agarose beads (20 μl) were incubated with equimolar amounts (50 nm) of XlMEP50 (wild type or mutants) and FLAG-tagged HsPRMT5 in 250 μl of TBS (150 mm NaCl, Tris-HCl, pH 8.0) at 4 °C for 2 h. As a negative control, XlMEP50 was incubated under the same conditions without HsPRMT5. The suspension was then transferred to a Mini-spin column (USA Scientific, Inc.) and centrifuged (30 s, 500 × g). After intense washing with TBS, the beads were boiled for 10 min in SDS loading dye for elution. The elution samples were analyzed via SDS-PAGE and Western blot with α-PRMT5 (Millipore) and α-MEP50 antibodies.
Luciferase-based Coupled Assay Buffer
The formation of SAH as a product of the methyltransferase reaction was monitored using a previous reference with the following coupling enzymes: C. symbiosum pyruvate phosphate dikinase (4.6 milliunits/well), S. cerevisiae adenosine phosphoribosyltransferase (250 microunits/well), and SeMTAN (14 microunits/well) (26). The 20× concentrated buffer B1 (1.0 m Tris acetate, pH 7.7, 20 mm phosphoenolpyruvate, 20 mm PPi, 150 mm ammonium sulfate, and 20 mm phosphoribosyl pyrophosphate; treated with charcoal and filter-sterilized) was supplemented with MgCl2 (10 mm), tris(hydroxypropyl)-phosphine (1 mm), ATPlite 1Step (note that the PerkinElmer Life Sciences substrate buffer solution supplied in this kit inhibits XlPRMT5-MEP50 and was replaced by a 50 mm Tris acetate buffer), C. symbiosum pyruvate phosphate dikinase, S. cerevisiae adenosine phosphoribosyltransferase, and SeMTAN. Then the histones/peptide substrates and XlPRMT5-MEP50 enzyme complex or CePRMT5 were added to prepare the final buffer B2. Each experiment used 40 μl of B2.
Luciferase-based Coupled Enzymatic Assays
Kinetic parameters were determined at 25 °C by monitoring luminescence using a SpectraMax L instrument configured with two photomultipliers (photo counting mode; Molecular Devices) in a 96-well half-area flat bottom plate (Corning, Inc., catalog no. 3992). Luminescence was measured in relative light units. Briefly, 40 μl of buffer B2 (containing 20–200 nm PRMT5 enzymes) was mixed with SAM substrate (10 μl of 125 μm solution, final saturating concentration of SAM at 25 μm; higher saturating concentrations are required for mutants with higher SAM Km). Under our experimental conditions, the typical Michaelis-Menten model is not suited to characterize the behavior of PRMT5 from both X. laevis and C. elegans. In vitro, these enzymes have poor activity, with turnover rates (kcat) as low as 20 h−1, and the use of high nanomolar concentration of methyltransferase is generally required. During the early stage of substrate titration (SAM or peptide substrates), enzyme and substrate concentrations have the same order of magnitude. Therefore, a significant fraction of total substrate is bound to the enzyme (i.e. enzyme-substrate complex). The Morrison kinetic model accounts for this early substrate depletion (Equation 1). Therefore, initial rates were plotted against substrate concentrations and fitted to this kinetic model to yield corresponding Km and kcat values (29),
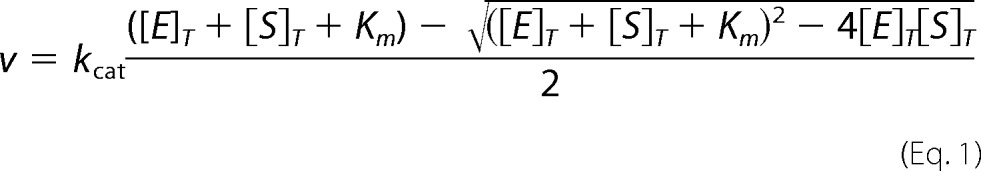 |
where [E]T and [S]T are the total concentration of enzyme and substrate, respectively.
HPLC Separation of Histones
Histones H4 (RT = 26.5 min), H2A (RT = 28.9 min), tailless H2A (RT = 32.0 min) and H3 (RT = 42.5 min) were separated onto a reversed-phase C8 column (Vydac, catalog no. 228TP101; 300 Å, 10 μm, 1.0 × 250 mm) at 65 °C using mobile phases A (water, 0.1% TFA) and B (acetonitrile, 0.1% TFA) according to the following gradient: 0–3.57 min (SAM, small peptide and salt removal; 95% A), 3.57–21 min (linear to 65% A), 21–46 min (histone separation; linear to 62% A), 46–47 min (linear 10% A), 47–57 min (higher molecular weight protein removal; constant 10% A), 57–58 min (linear 95% A), 58–78 min (equilibration; constant 95% A).
Competition Experiments between Histone H2A and Other Histones/Peptides for Binding onto XlPRMT5-MEP50 or CePRMT5
Experiments were performed in a 100-μl total volume with 50 mm MOPS, pH 7.0, 100 nm XlPRMT5-MEP50 or 100 nm CePRMT5, 20 microunits of SeMTAN, histone H2A with concentration kept at 2.0 μm, increasing amounts of histone/peptide substrates (0–12.6 μm final concentration), and 1 mm tris(hydroxypropyl)-phosphine. Reactions were initiated by adding 80 μl of the above samples to PCR tubes already loaded with 20 μl of SAM reagent (25 μm final [3H]methyl-SAM concentration; 10 μCi/reaction). After 15 min at 25 °C, TFA (3 μl, 10% (v/v)) was added to quench the reactions. Samples were kept at −80 °C before processing by HPLC. Methyl transfer onto histone H2A was then quantified by liquid scintillation counting and plotted against matching concentrations of histone/peptide competitors.
Binding Affinity of Histone H2A for XlMEP50 and XlPRMT5-MEP50
XlMEP50 was freshly prepared; a gel filtration was used as the last purification step (25 mm ADA, 100 mm NaCl, 1 mm β-mercaptoethanol, pH 6.5). Samples were prepared as described for the luciferase-based assay, with XlPRMT5-MEP50 and histone H2A concentration set at 100 nm and 2.0 μm, respectively. Exogenous XlMEP50 was added (0–20 μm), and the composition of samples was kept constant (adjusted with gel filtration buffer: 25 mm ADA, 100 mm NaCl, 1 mm β-mercaptoethanol, pH 6.5). Methyl transfer started upon the addition of SAM (25 μm final concentration). During the event, exogenous MEP50 (M) and PRMT5-MEP50 (P/M) compete for H2A (H) binding where the histone fold can bind to MEP50, forming the M*·H and P/M*·H complexes, respectively. Methyl transfer occurs when the complex P*/M*·H is formed (the histone fold is bound to the MEP50 presenter, and the histone tail is bound to the enzyme active site). The following relationships can be written: [H]T = [H]free + [P/M*·H] + [P*/M*·H] + [M*·H], the total histone concentration simplified as [H]T = [H]free + [M*·H], because [H]T ≫ [P/M]T (i.e. 2.0 μm ≫ 100 nm). Likewise, [M]T = [M]free + [M*·H] + β[M]T, the total MEP50 concentration with the factor β accounting for potential aggregation/misfolding of MEP50 occurring along the titration experiments (a value between 0 and 1, with β = 0 corresponding to no aggregation/misfolding of MEP50).
Considering the binding between exogenous MEP50 and full-length histone, the following expressions can be written,
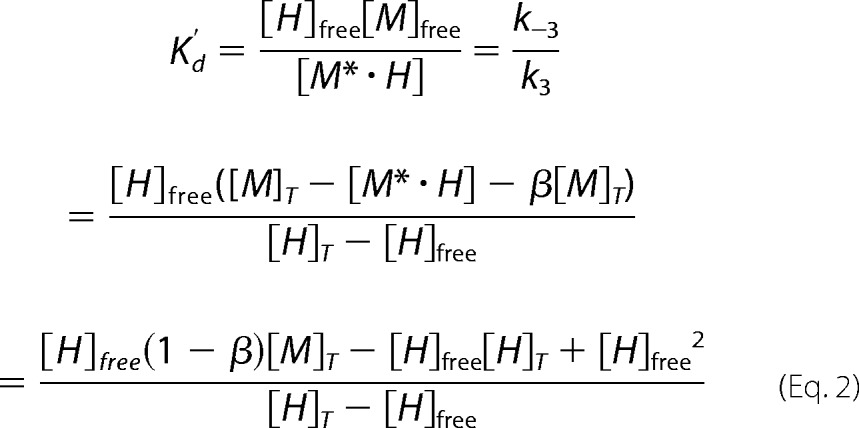 |
also equivalent to [H]free2 + ((1 − β)[M]T − [H]T + K′d[H]free − K′d[H]T = 0 and solved for the [H]free concentration,
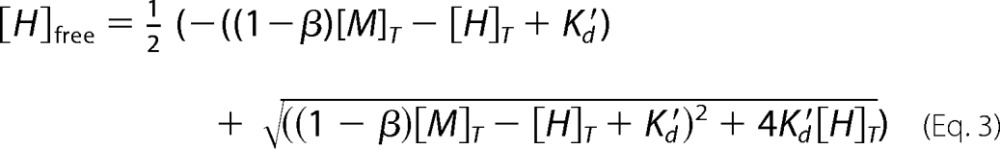 |
Considering the methyl transfer reaction, the following relationships can be written.
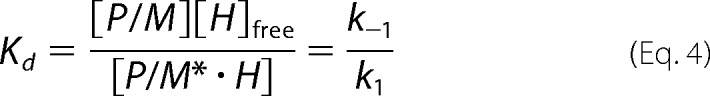 |
and by analogy,
 |
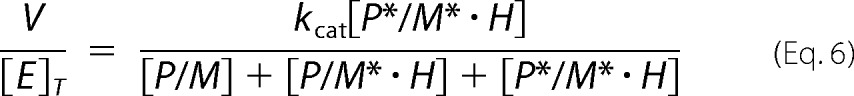 |
Equation 6 is rearranged using Km and Kd relationships leading to the expression of v/Vmax.
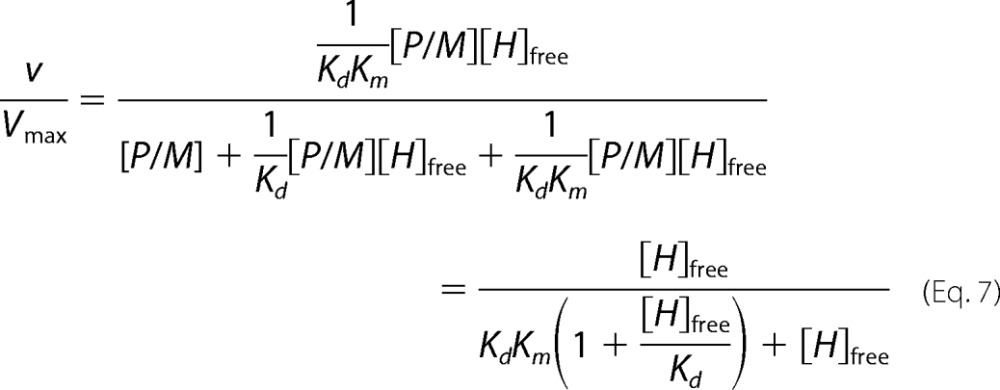 |
Finally, the solution for [H]free (Equation 3) is replaced in the v/Vmax velocity expression to obtain both Kd and K′d. Ratios between experimental initial (v) and maximal (Vmax) velocities were plotted against exogenous XlMEP50 concentrations and fitted to our binding model (Equation 7), where [M]T and [H]T are the total concentrations of MEP50 and histone H2A, respectively, [H]free is the free concentration of histone H2A at equilibrium, Km is the Michaelis constant for histone H2A when used as a substrate with XlPRMT5-MEP50, K′d is the dissociation constant of histone H2A from the complex XlMEP50·H2A, and Kd is the dissociation constant of histone H2A from XlMEP50 already complexed with XlPRMT5 (XlPRMT5-MEP50·H2A).
Peptide Array Binding Studies
A library of 20-mer peptides spanning the sequences of histones was generated with or without known modifications and in various combinations (sequences available at JPT Peptide Technologies GmbH Web site), as described previously (22). For binding studies, FLAG-HsPRMT5 and XlMEP50 were preincubated in equimolar amounts (66.5 nm) in KCl/HEPES buffer for 30 min at room temperature to form a complex. The complex was applied to individual histone code peptide microarrays for 1 h at 30 °C. For detection of binding events, the microarrays were incubated with anti-FLAG mouse monoclonal antibody (Pierce, catalog no. MA1-91878) or anti-MEP50 rabbit polyclonal antibody. Upon washing and incubation with fluorescently labeled antibody (DyLight649 anti-mouse IgG (Thermo, catalog no. 35515) or DL649 anti-rabbit IgG (Pierce, catalog no. 35565)), the microarrays were washed again and dried. Incubation with primary and secondary antibody alone was used as a control. Each microarray was scanned using GenePix Autoloader 4200AL (Molecular Devices; pixel size, 10 μm). Signal intensity was evaluated using GenepixPro software (Molecular Devices). Further evaluation and representation of results was performed using the R statistical programming system (version 2.11.1).
Structural Alignments, Binding Site Predictions, and Rigid Body Docking
All structural figures were visualized using VMD version 1.9.1 (30). Structural alignments were performed using the STAMP and Multiseq alignment tools within VMD (31, 32). Rigid body docking was performed at the ClusPro 2.0 server (33, 34).
ClusPro 2.0 Docking
Docking of H2A-H2B and H3-H4 histone dimers was done using the following attractive residues. Residue numbers are listed as Protein Data Bank chain letter-residue number in PRMT5, MEP50, and histones used for attraction in rigid body docking in ClusPro 2.0. Chain Y is the cross-dimer MEP50 from chain Q PRMT5. Chains C and D are H2A and H2B with the N-terminal tails removed, and chain A and B are H3 and H4 with the N-terminal tails removed. The residues below were assigned attractive forces in ClusPro. For H2A-H2B dimer docking: receptor attraction, Y-16, Y-19, Y-20, Y-38, Y-39, Y-40, Y-41, Y-42, Y-43, Y-44, Y-45, Y-68, Y-70, Q-318, Q-324, Q-366, Q-386, Q-396, Q-400, Q-404; ligand attraction, C-21, C-22, C-23, C-24, C-25, C-26, C-27, C-28, C-29, C-30, C-31, C-32, C-33, C-34, C-35, C-36, C-37, D-31, D-32, D-33, D-34, D-35, D-36, D-37. For H3-H4 dimer docking: receptor attraction, Y-16, Y-19, Y-20, Y-38, Y-39, Y-40, Y-41, Y-42, Y-43, Y-44, Y-45, Y-68, Y-70, Q-318, Q-324, Q-366, Q-386, Q-396, Q-400, Q-404; ligand attraction, A-61, A-62, A-63, A-64, A-65, A-66, A-67, A-68, A-69, A-70, B-24, B-25, B-26, B-27, B-28, B-29, B-30, B-31, B-32, B-33, B-34, B-35, B-36, B-37, B-38, B-39, B-40, B-85, B-86, B-87, B-88, B-89, B-90, B-91, B-92, B-93, B-94, B-95, B-96, B-97, B-98, B-99, B-100, B-101, B-102, B-103, B-104.
RESULTS
Our previous work defined two distinct relative classes of MEP50 molecules within the PRMT5-MEP50 tetramer: the directly bound MEP50 and the cross-dimer MEP50 (22). Additionally, both our solved X. laevis PRMT5-MEP50 structure and the solved human complex structure (23) demonstrated unique N- and C-terminal domains of PRMT5 simply connected by a loop. Based on its structural organization and residue conservation and our electron microscopy images of XlPRMT5-MEP50 complexed with substrate, we hypothesized that the cross-dimer MEP50 is responsible for organizing substrate for the PRMT5 catalytic domain.
Structural Arrangement of PRMT5 and MEP50
To test the conserved structural relationships between MEP50 and the distinct N- and C-terminal domains of PRMT5, we performed Cα-carbon backbone alignments between the X. laevis and human PRMT5-MEP50 structures (Protein Data Bank codes 4G56 and 4GQB, respectively) (Fig. 1A) using the STAMP alignment tool in VMD MultiSeq (31, 32). First we aligned only the entire PRMT5 molecule (as indicated by the black box in Fig. 1B and the schematic in Fig. 1A) and plotted the per-residue Cα RMSD values (Å) for PRMT5 (purple) and both MEP50 molecules (directly bound (pink) and cross-dimer (blue)) (Fig. 1B, top row). PRMT5 itself was only modestly well aligned, with many regions containing an RMSD of >2 Å. The directly bound MEP50 was induced into modest alignment by PRMT5, whereas the cross-dimer MEP50 was not aligned well at all. When we aligned the PRMT5 N-terminal domain that directly contacts MEP50, we observed tight alignment (RMSD <2 Å) for the PRMT5 domain as well as the directly bound MEP50 (Fig. 1B, second row).
FIGURE 1.

Evolutionarily conserved spatial arrangement between the PRMT5 catalytic domain and its cross-dimer paired MEP50. A, ribbon diagram of the Xenopus PRMT5 monomer (purple), its directly bound MEP50 molecule (pink), and the cross-dimer MEP50 (blue). Structural alignment windows 1, 2, and 3 are indicated on the right. Inset boxes, surface diagrams of the Xenopus (Protein Data Bank code 4G56) and human (Protein Data Bank code 4GQB) PRMT5-MEP50 structures, with the analyzed PRMT5 and MEP50 molecules colored as in A; the remainder of the structures are shown in gray. B, per-residue Cα alignments between the Xenopus and human PRMT5 windows 1, 2, and 3 as calculated with VMD MultiSeq, plotted as RMSD (Å) against amino acid position in the sequence. The PRMT5 alignment windows are boxed.
When we aligned the C-terminal domain of PRMT5 that does not directly contact either MEP50, we observed no alignment of the directly bound MEP50 but strong alignment of the cross-dimer MEP50 (RMSD <2 Å; Fig. 1B, third row). Despite the different crystal forms of the solved structures, which could possibly lead to packing artifacts, this relationship is strikingly apparent. Therefore, we concluded that a conserved relationship between the catalytic C-terminal domain of PRMT5 and the cross-dimer but non-contacting MEP50 exists. This relationship is unlikely to be allosteric due to the multiple domain boundaries and intermolecule contacts that would be required to communicate this information.
XlPRMT5 Catalytic Domain Is Inactive
We and others have previously demonstrated that PRMT5 alone has exceedingly modest histone methyltransferase activity in the absence of MEP50 (22, 23). Because we showed in Fig. 1 that a functional relationship probably exists between the catalytic domain of PRMT5 and the cross-dimer MEP50, we produced recombinant XlPRMT5 catalytic domain (PRMT5ΔN, residues 291–633) to test if the catalytic domain alone was active. This truncated protein was soluble in E. coli and sedimented primarily as monomer in analytical ultracentrifugation (Fig. 2A) (data not shown). We then measured XlPRMT5(291–633) enzymatic activity using the filter-binding methyltransferase assay on multiple known peptide and protein substrates of PRMT5-MEP50 in the absence or presence of MEP50 (Fig. 2B). XlPRMT5(291–633) did not exhibit any activity on any of these substrates, whereas intact full-length HsPRMT5 did recover activity with XlMEP50 added in trans. A 1-year exposure of a fluorogram did reveal very low levels of histone methyltransferase activity by full-length HsPRMT5 in the absence of XlMEP50, compared with robust activity of the XlPRMT5-MEP50 complex or XlMEP50 added in trans to HsPRMT5 (Fig. 2C). We concluded from these experiments that only the full-length PRMT5-MEP50 complex is able to efficiently methylate its substrates.
FIGURE 2.

Intact full-length PRMT5 complexed with MEP50 is necessary for histone methyltransferase activity. A, the catalytic C-terminal domain of XlPRMT5 (residues 291–633) is shown in purple and was expressed and purified. B, XlPRMT5(291–633) did not exhibit any activity toward histone tail peptide substrates of the intact complex (H2A(1–20), H4(1–20), H4(1–7)) or full-length histones H2A or H4. The addition of XlMEP50 to the catalytic domain did not stimulate activity, whereas the addition of XlMEP50 to full-length HsPRMT5 stimulated activity toward H2A(1–20) peptide. C, intact XlPRMT5-MEP50 complex or HsPRMT5 + XlMEP50 exhibited methyltransferase activity toward histone H4 (right two lanes). HsPRMT5 alone exhibited ultralow levels of activity toward H2A, H3, and H4, visible only after a 1-year exposure of the fluorogram (left four lanes, second panel). D, HsPRMT5 (200 nm) was preincubated with substoichiometric (1:4 and 2:4) and stoichiometric concentrations (4:4) of XlMEP50 and then assayed for methyltransferase activity against H4 peptide.
To demonstrate the strict requirement for MEP50 in promoting PRMT5 histone methyltransferase activity, we titrated XlMEP50 with constant HsPRMT5 and measured histone H2A methylation in the filter binding assay. We observed a MEP50 dose-dependent increase in activity maximal at the 4:4 stoichiometry with PRMT5, consistent with a required role for MEP50 in organizing substrate (Fig. 2D).
Histone Affinities for XlPRMT5-MEP50
Our results to this point implicate MEP50 in its essential function in promoting PRMT5 methyltransferase activity. However, in vivo, PRMT5-MEP50 has distinct protein substrate selections in different contexts. In particular, H4 Arg-3 methylation is primarily observed on chromatin, whereas H2A Arg-3 methylation is observed on soluble, cytoplasmic histones (35–37). Soluble H2A is heterodimerized with H2B but typically not complexed with H3-H4 in the cytoplasm or nucleus when in solution. Chromatin-bound histones are all found within the same nucleosome embedded in DNA.
Therefore, to determine relative substrate preferences for PRMT5-MEP50, we performed methyltransferase assays with different combinations of histone proteins (H2A and H4, both methylated by PRMT5-MEP50) and assayed the activity by fluorography. First, we titrated full-length histone H4 into reactions with constant histone H2A (Fig. 3A). Strikingly, methyl transfer onto histone H2A was readily inhibited by H4. To determine whether this significant preference for H4 is due to direct competition of the histone tail containing Arg-3, we performed a similar methyltransferase assay and titrated peptide H4(1–20) against histone H2A (Fig. 3B). This peptide substrate was a poor competitor. Because all experiments were performed under initial velocity conditions, [14C]methyl-SAM depletion was not the cause of the loss of activity when using histone/peptide competitors. This observation suggested that the competition for XlPRMT5-MEP50 may be primarily mediated through the histone fold domain of H4.
FIGURE 3.

XlMEP50 is a presenter that primarily binds histone H4 through the histone fold and exposes its N-terminal tail for methylation by XlPRMT5. A, substrate competition experiment where XlPRMT5-MEP50 activity toward histone H2A was displaced as histone H4 was titrated into the reaction (experiment performed using 50 mm MOPS, pH 7.0, 100 nm XlPRMT5-MEP50, 20 microunits of SeMTAN, 25 μm [14C]-methyl-SAM, and a H2A concentration kept constant at 2 μm while H4 was added at 0, 0.4, 0.8, 1.6, 3.2, 6.4, and 12.8 μm final concentrations). B, similar competition experiment as in A where peptide H4(1–20) was used as competing substrate (experimental conditions identical to those in A). C, quantification of methyl transfer reactions using reversed-phase HPLC. Histones H4, H2A, tailless H2A (TLH2A), and H3 were separated from each other and from radiolabeled substrate SAM using a C8 reverse HPLC column (see “Experimental Procedures”). D, substrate competition experiment where XlPRMT5-MEP50 activity toward histone H2A was displaced as histone H4, peptide H4(1–20), and histone H3 were titrated into the reaction (experiment performed using 50 mm MOPS, pH 7.0, 100 nm XlPRMT5-MEP50, 20 microunits of SeMTAN, 25 μm [3H]methyl-SAM, and a H2A concentration kept constant at 2 μm while competitors were added at 0–12.6 μm final concentrations). Transfer of [3H]methyl was quantified by liquid scintillation counting after isolation of histones. E, competition experiment similar to that in D where XlPRMT5-MEP50 was replaced by its homologous enzyme from C. elegans (experimental conditions identical to those in D). F, FLAG-HsPRMT5-XlMEP50 complex was incubated on ultrahigh density histone peptide arrays. PRMT5-MEP50 binding on the histone peptide scan data was extracted, and relative binding levels were plotted as a heat map, with no to low signal as white to light yellow, and high relative binding was plotted in red. Histone amino acid sequence numbers are represented at the top of the plots. The histone fold domain is indicated as a gray box, and the substrate residue Arg-3 (R3) on both H2A and H4 is indicated. G, relief of methyltransferase activity through the addition of MEP50. In this model used to determine the affinities of histone H2A for MEP50, P/M*·H represents the PRMT5-MEP50·histone complex, where only the histone fold is bound to the MEP50 presenter. P*/M*·H represents the PRMT5-MEP50·histone complex, where the histone fold is bound to the MEP50 presenter and the histone tail is bound to the enzyme active site. M*·H represents the complex between histone and exogenous MEP50. Each step is characterized by a constant (i.e. k1, k−1, k2, k−2, kcat, k3, and k−3). G, transferase reaction catalyzed by XlPRMT5-MEP50 was followed continuously at pH 7.7 using the luciferase-based assay with histone H2A as substrate (H2A fixed at 2 μm; see “Experimental Procedures”); XlMEP50 was added to the reactions (0–20 μm), and resulting transferase activities were recorded. Exogenous XlMEP50 competes with XlPRMT5-MEP50 for H2A binding, and methyl transfer is inhibited with increasing concentrations of XlMEP50. A dissociation constant (Kd) of H2A for both exogenous XlMEP50 (K′d H2A:MEP50) and PRMT5-associated MEP50 (Kd H2A:complex) was determined.
The response observed with fluorography during film exposure is non-linear. To overcome the drawbacks of this technique, [3H]methyl transfer was quantified by liquid scintillation counting. We performed similar competition experiments and separated histones by reversed-phase HPLC (Fig. 3C) (38). Our results confirmed the highly efficient histone H4 competition with histone H2A for Arg-3 methylation (Fig. 3D). Furthermore, histone tail peptide H4(1–20) inhibited methyl transfer toward H2A but to a lesser extent (i.e. 25% at 14 μm). With concentration levels 54 times lower than the ones required when peptide H4(1–20) was used, the histone fold domain of H4 appears to be critical for PRMT5-MEP50 complex binding and for competition with full-length histone H2A.
PRMT5 dimethylates H3 Arg-8 in vivo (39–41), but HsPRMT5-MEP50 poorly methylates H3 in vitro (42), so we tested the ability of histone H3 to displace the activity toward H2A (Fig. 3D). PRMT5-MEP50 was unable to methylate histone H3 (no incorporation of 3H-methyl at pH 7.0 when using 25 μm SAM and 100 nm enzyme). However, we observed a strong competition between histones H3 and H2A. Although both histones H4 and H3 display similar competition for PRMT5-MEP50, the highest concentrations of H3 were insufficient to achieve complete inhibition of PRMT5 methylation of histone H2A. Our results suggest the presence of specific histone fold binding regions for the XlPRMT5-MEP50 complex.
CePRMT5 Displays Affinity for the Histone Fold Domain of H4 without Assistance from a MEP50 Binding Partner
We tested the ability of CePRMT5 to bind the histone fold domain using similar competition experiments with a fixed concentration of H2A and increasing concentration of histone/peptide competitors. Although CePRMT5 does not associate with a MEP50 homologue, we did observe strong displacement of H2A by the H4 histone fold domain in comparison with the H4(1–20) peptide (Fig. 3E). Likewise, the non-substrate H3 competed efficiently against H2A for binding onto CePRMT5. These data suggest that both CePRMT5 and XlPRMT5-MEP50 enzymes may have different binding mechanisms for their protein substrates.
Histone Peptide Array Interaction Studies
To further test our hypothesis that XlPRMT5-MEP50 may bind histones H3 and H4 through their histone fold, which would explain the competition of H2A activity by these histones, we employed an extremely high density histone peptide array containing peptides covering the entire sequence of the core histones. We incubated FLAG-HsPRMT5 complexed with XlMEP50 on the array and probed with anti-FLAG and anti-MEP50 antibodies. We extracted relative binding data from these assays and plotted the signal onto the core histone sequence (Fig. 3F). Strikingly, the highest binding signals were obtained on histone fold or C-terminal peptides of histones H3 and H4. We also determined the influence of histone post-translational modifications on complex binding. In particular, pronounced loss of H4 C-terminal tail binding by HsPRMT5-XlMEP50 was observed upon phosphorylation of residue Tyr-98 (data not shown). Although this modification has not yet been observed in vivo, follow-up mass spectrometry and targeted bindings studies may specifically analyze this modification.
Furthermore, we quantified the binding affinity of histone H2A for both free XlMEP50 and the XlPRMT5-MEP50 complex, K′d and Kd, respectively. We turned to a new ultrasensitive coupled enzymatic assay to monitor PRMT5-MEP50 activity upon the addition of exogenous MEP50 under saturating concentration of SAM and fixed concentration of histone H2A (26). The assay couples conversion of SAM to SAH by PRMT5-MEP50 in a protein-dependent fashion to downstream ATP and commensurate light production by luciferase. We expect the addition of MEP50 to sequester H2A, thus preventing the histone from binding onto the PRMT5-MEP50 complex for further methylation (Fig. 3G). As a result, light production would decrease with increasing concentration of exogenous MEP50 (Equations 3 and 7). We observed an affinity (Kd) of 1.78 ± 0.06 μm between H2A and PRMT5-MEP50 (Fig. 3H), which is reminiscent of the Km measured for H2A(1–20) and histone tails (Table 1). This result supports the observations from our peptide array study with H2A residues 1–30 being responsible for most of the binding onto the PRMT5-MEP50 complex (Fig. 3F).
TABLE 1.
Kinetic parameters for XlPRMT5-MEP50, its MEP50 mutants, and CePRMT5 (boldface type)
Parameters for the SAM substrate are below the corresponding peptide substrate.
| Substrate | Sequence | Kinetic parameters |
||
|---|---|---|---|---|
| Kma | kcata | ξb | ||
| nm | h−1 | m−1 s−1 | ||
| Histone H4 | ||||
| Full-length protein | 211 ± 73 | 21 ± 2 | 2.8 × 104 | |
| H4(1–7) | H-SGRGKGG-OH | (184 ± 20) × 103 | 32 ± 2 | 48 |
| SAM | (4.7 ± 0.5) × 103 | 26.2 ± 0.6 | 1.5 × 103 | |
| H4(1–7) PRMT5-MEP50R42E | (515 ± 52) × 103 | 9.4 ± 0.3 | 5 | |
| SAM | (108 ± 31) × 103 | 10 ± 1 | 26 | |
| H4(1–21) | H-SGRGKGGKGLGKGGAKRHRKV-OH | 343 ± 42 | 36 ± 1 | 2.9 × 104 |
| H4(1–21)c HsPRMT5-MEP50 | Ac-SGRGKGGKGLGKGGAKRHRKV | 1600 ± 1000 | 10.8 ± 0.6 | 1.9 × 103 |
| H4(1–20) | H-SGRGKGGKGLGKGGAKRHRK-OH | 83 ± 8 | 37.6 ± 0.9 | 1.3 × 105 |
| SAM | (3.3 ± 0.2) × 103 | 27.4 ± 0.3 | 2.3 × 103 | |
| H4(1–20) PRMT5-MEP50R42E | (2.0 ± 0.2) × 103 | 12 ± 0.3 | 1.7 × 103 | |
| SAM | (27 ± 3) × 103 | 10.9 ± 0.4 | 112 | |
| H4(1–20) PRMT5-MEP50R42Q | 502 ± 140 | 5.4 ± 0.4 | 3.0 × 103 | |
| SAM | (5.3 ± 0.5) × 103 | 5.7 ± 0.1 | 300 | |
| H4(1–20) | Ac-SGRGKGGKGLGKGGAKRHRKK(Biot) | 139 ± 16 | 21.1 ± 0.6 | 4.2 × 104 |
| H4 K12(Ac)(1–20) | Ac-SGRGKGGKGLGK(Ac)GGAKRHRKK(Biot) | 195 ± 35 | 29 ± 1 | 3.7 × 104 |
| H4 K20(Me)2(1–20) | Ac-SGRGKGGKGLGKGGAKRHRK(Me)2K(Biot) | 124 ± 18 | 24.5 ± 0.8 | 5.5 × 104 |
| H4 K12(Ac)K20(Me)2(1–20) | Ac-SGRGKGGKGLGK(Ac)GGAKRHRK(Me)2K(Biot) | 279 ± 25 | 24.4 ± 0.7 | 2.4 × 104 |
| H4(1–20) CePRMT5 | H-SGRGKGGKGLGKGGAKRHRK-OH | (54 ± 4) × 103 | 28.6 ± 0.7 | 150 |
| SAM | (4.4 ± 0.8) × 103 | 32 ± 1 | 2.0 × 103 | |
| Histone H2A | ||||
| Hs H2A(1–20) | H-SGRGKQGGKARAKAKTRSSR-OH | 867 ± 136 | 43 ± 3 | 1.4 × 104 |
| Xl H2A.X-F(1–20) | Ac-SGRGKKVQKAASGKASRSAKA-CY-OH | (1.74 ± 0.09) × 103 | 51 ± 1 | 8.2 × 103 |
| Xl H2A.X-F R3(Me)(1–20) | Ac-SGR(Me)GKKVQKAASGKASRSAKA-CY-OH | (25 ± 2) × 103 | 40 ± 1 | 445 |
| X. laevis nucleoplasmin | ||||
| Full-length protein | (1.6 ± 0.2) × 103 | 28 ± 2 | 4.9 × 103 | |
a Kinetic parameters obtained by fitting experimental values to the Morrison kinetic model (Equation 1).
b Catalytic efficiency represented by ξ = kcat/Km.
c From published study on HsPRMT5-MEP50 (42).
Nucleosomes Are Not Substrates for PRMT5-MEP50
We previously showed that PRMT5-MEP50 is incapable of methylation of nucleosomes in vitro (22). To conclusively test this inability to methylate the typical cellular state of histones, we incubated PRMT5-MEP50 with H2A, H4, and recombinant mononucleosomes in the absence or presence of DNase I with identical buffer conditions (Fig. 4A). Mononucleosomes were not substrates of PRMT5-MEP50 compared with its significant methylation of free H2A and H4. After digestion of the DNA by DNase I, PRMT5-MEP50 did have H4 and H2A methyltransferase activity, confirming that DNA in the nucleosome directly inhibits the enzyme activity.
FIGURE 4.
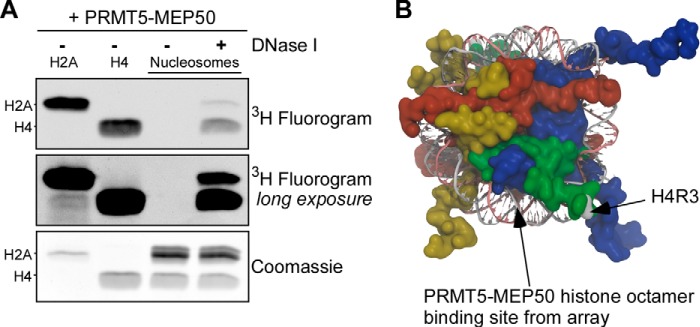
XlPRMT5-MEP50 does not methylate histones embedded in DNA in nucleosomes. A, XlPRMT5-MEP50 was incubated with H2A, H4, or recombinant mononucleosomes, in the presence or absence of DNase I. PRMT5 methylated both H2A and H4 independently (lanes 1 and 2) but did not methylate histones in mononucleosomes (lane 3). Upon DNase I treatment, histone H4 and histone H2A activity was recovered (lane 4). B, schematic representation of the nucleosome core particle (Protein Data Bank code 1KX5), with H3 shown in blue, H4 in green, H2A in red, and H2B in yellow. The targeted H4 Arg-3 is shown (gray coloring and arrow), and the region on H3-H4 corresponding to the strongest sites of interaction on the peptide array is indicated.
We used the results from our peptide array studies and mapped the domains of high binding for HsPRMT5-XlMEP50 onto the nucleosome core structure. We found that these peptides contained residues on the lateral surface of H3-H4. Binding on this surface would probably position the targeted H4 Arg-3 toward the PRMT5 catalytic site (Fig. 4B). Tight binding on this face would also orient the H2A Arg-3 away from the catalytic site, consistent with the greater activity directed toward H4 in the competition assays above. Because the highest binding regions of the octamer for HsPRMT5-XlMEP50 lie on or adjacent to the DNA superhelical path on the nucleosome, these observations may explain the absence of methyltransferase activity toward nucleosomes.
Function of the MEP50 Insertion Finger
To test our hypothesis that MEP50 orients and presents substrate to the PRMT5 catalytic domain, we inspected the structures to determine potential residues to mutate. The X. laevis and human PRMT5-MEP50 structures both contain a conserved MEP50 insertion finger that extends from the cross-dimer MEP50 and ends ∼10 Å above the catalytic domain of PRMT5 (Fig. 5A and boxed enlarged view). This insertion finger is directly on the surface that we initially hypothesized to be responsible for substrate presentation (22). X. laevis MEP50 Arg-42 is 100% conserved among vertebrate MEP50 proteins (i.e. Arg-52 in human MEP50), is at the terminus of the finger, and is ∼3.4 Å away from Glu-403 of the cross-dimer PRMT5, potentially forming a salt bridge to stabilize the finger (Fig. 5A, boxed enlarged view). We mutated this residue to glutamate or glutamine and produced PRMT5-MEP50R42E and PRMT5-MEP50R42Q protein complexes and MEP50R42E and MEP50R42Q alone (Fig. 5, B and C). The PRMT5-MEP50R42E and PRMT5-MEP50R42Q complexes were purified from insect cells in the same manner as the wild-type complex. MEP50R42E sedimented as a monomer in an analytical ultracentrifuge (data not shown). To confirm that MEP50 mutants interacted normally with PRMT5, we incubated the proteins with FLAG-tagged HsPRMT5, and both were enriched on anti-FLAG resin just like wild-type MEP50 (Fig. 5D).
FIGURE 5.

Mutations of MEP50 insertion loop residue Arg-42 affect histone methylation. A, schematic representation of the insertion loop from the cross-dimer XlMEP50 positioned adjacent to the catalytic domain of the paired PRMT5 molecular. Inset enlarged view, the only contact the cross-dimer MEP50 makes with the catalytic domain is a putative salt bridge between XlMEP50 Arg-42 and XlPRMT5 Glu-403; no other contacting residues are found. B, Coomassie-stained gel of wild-type XlPRMT5-MEP50, XlPRMT5-MEP50R42E, and XlPRMT5-MEP50R42Q complexes. C, Coomassie-stained gel of wild type and XlMEP50R42E. D, FLAG-tagged HsPRMT5 captured on anti-FLAG resin after incubation alone or with XlMEP50, XlMEP50R42E, or XlMEP50R42Q and immunoblotted for HsPRMT5 and XlMEP50, demonstrating similar interactions for the wild-type and mutated MEP50 proteins. E, filter-binding activity assays of PRMT5-MEP50, PRMT5-MEP50R42E, or PRMT5-MEP50R42Q (100 nm complex) incubated with H2A(1–20), H4(1–20), and Npm(177–196) substrate peptides or H2A and H4 full-length protein substrates. PRMT5-directed activity is represented as a percentage of wild-type activity toward the peptide/protein substrate and is the average of three independent replicates.
To determine the ability or inability of MEP50 mutants to promote PRMT5 methyltransferase activity, we first used the filter binding assay. On multiple known peptide and protein substrates, MEP50R42E only promoted low levels of PRMT5 activity (Fig. 5E). In contrast, the MEP50R42Q-containing complex, without the charge reversal that would disrupt the putative salt bridge, exhibited activity similar to that of the wild-type complex. These data support the hypothesis that cross-dimer interaction of MEP50 and the PRMT5 catalytic domain through the MEP50 insertion loop is important for methyltransferase activity.
The filter binding assays are subject to some substantial error and do not report kinetic parameters in the end point readout that we employed. Therefore, we used the luciferase-based coupled assay to determine Km and kcat of the XlPRMT5-MEP50 and mutant complexes. Our kinetic parameters for PRMT5-MEP50, the MEP50R42E, and MEP50R42Q complexes are shown in Fig. 6, A and B, and in Table 1.
FIGURE 6.

Substrate specificities for XlPRMT5-MEP50 and the impact on enzymatic efficiency upon mutation of MEP50 insertion loop residue Arg-42. Kinetic parameters for the various tested substrates (histone H4, histone peptides, and SAM) are plotted, with the kcat (h−1) on the y axis and the Km (nm; logarithmic scale) on the x axis. Highest enzymatic efficiencies are obtained with substrates found in the top left quadrant, whereas low enzymatic efficiencies are obtained with substrates found in the opposite bottom right quadrant. Arrows indicate the loss (squared values) of enzymatic efficiency upon arginine monomethylation (purple) or upon mutation of MEP50 residue Arg-42 to glutamic acid (red) and to glutamine (green). For reference, enzymatic behavior of CePRMT5 is represented in blue. A, representation of kinetic parameters for histone substrates using saturating concentration of SAM. B, representation of kinetic parameters for SAM substrate using saturating concentration of histone substrates. C, impact of XlMEP50R42Q and XlMEP50R42E on catalytic turnover (kcat; pink bars) and substrates' affinities (Km; gray bars) for both peptide and SAM substrates. The decrease of methyl transfer is represented as a percentage of wild-type kcat, whereas the loss of affinity is given as -fold increase of wild-type Km. D, histones H2A or H4 were incubated with XlPRMT5-MEP50 and SAM. Reactions were stopped at 0, 1, 5, 10, and 15 min with the addition of SDS-polyacrylamide gel loading buffer and heating to 100 °C. Reaction products were immunoblotted with monomethylarginine (R3me1)- or symmetric dimethylarginine (R3me2s)-specific antibodies.
The PRMT5-MEP50R42Q and PRMT5-MEP50R42E complexes exhibit similar loss of catalytic efficiency with H4(1–20) peptide substrate when compared with wild-type PRMT5-MEP50 (Fig. 6A, arrows with boxed change in efficiency values). Both mutations of residue Arg-42 had a similar impact on the turnover number, kcat, because mutants R42Q and R42E processed peptide substrate 4 times more slowly than the wild-type MEP50 (Fig. 6C, top pink bar graph). However, unlike the Arg to Gln mutation, the mutant R42E drastically impaired the binding of peptide substrate because its Km was 24 times higher than the wild type (Fig. 6C, top gray bar graph). These results are consistent with our hypothesis that the MEP50 insertion finger is critical for protein substrate binding.
To gain further insight into the possible interactions between MEP50 and its cross-dimer PRMT5, we determined the kinetic parameters for the SAM substrate using wild-type and MEP50 Arg-42 mutant complexes at saturating concentrations of H4(1–20) peptide substrate (Fig. 6B). Surprisingly, the Arg to Glu mutation had the highest impact on the SAM Km, with values increasing from 3.3 to 27 μm when compared with the wild-type MEP50. The Arg to Gln mutation only raised the Km for the methyl donor by a factor of 1.6 (Fig. 6C, bottom gray bar graph). To reduce potential interactions of peptide substrate with MEP50, we then measured kinetic parameters for SAM using the short peptide substrate H4(1–7) (Fig. 6B). The PRMT5-MEP50R42E complex displayed a dramatic loss of affinity for SAM with a Km 23-fold higher than that of wild-type complex (Table 1). The SAM substrate only binds to the PRMT5 active site. Therefore, our kinetic results suggest that the MEP50 insertion loop is critical for histone binding and tail orientation and is also important for configuration of catalytically efficient PRMT5.
Kinetic Parameters of XlPRMT5-MEP50 Reveal Substrate Preferences
Our complete set of histone substrate kinetic parameters is shown in Table 1 and in Fig. 6, A and B, as a semilog scatterplot of Km versus kcat (higher efficiency in the plot shown in the top left, lower efficiency in the bottom right). The enzyme exhibited slow turnover with all substrates, on the order of 10–50 h−1. The most efficient substrates were the 1–20 and 1–21 histone peptide tails from H4 and from H2A and H2A.X-F (also known as H2A.X.3), with catalytic efficiencies ranging from 2.9 × 104 to 1.3 × 105 m−1 s−1. These kinetic parameters for peptide substrates are reminiscent of results reported by Thompson and co-workers (42) using HsPRMT5-MEP50, although differences in N-terminal functionalization (amine versus acetyl) may account for subtle discrepancies (Table 1). Full-length histone H4 and nucleoplasmin were also reasonably efficient substrates. The short H4(1–7) peptide was a very poor substrate, consistent with our hypothesized role of MEP50 in organizing substrate.
Finally, we tested a monomethylated H2A.X-F R3me1 peptide in this assay to determine whether PRMT5-MEP50 is likely to be processive or distributive in its catalysis of dimethylation. The R3me1 peptide was ∼20-fold less efficiently methylated, with the majority of this effect embedded in the Km, consistent with poor substrate binding. This result is reminiscent of the previously implicated distributive mechanism of catalysis to the dimethyl state (43, 44). Additionally, we tested H2A and H4 mono- and dimethylation over time using specific antibodies. The accumulation of me2s depended on saturation of the me1 state in this assay. Although the use of antibodies is not quantitative, our results are probably incompatible with a processive methylation model and suggest a distributive model (Fig. 6D).
Prediction of Histone Binding Sites onto XlPRMT5-MEP50 Using Computational Docking
To determine potential interaction surfaces and residues on XlPRMT5-MEP50, we used the SPPIDER (45) and PredUs (46) prediction algorithms and mapped the sites identified by both (Fig. 7A). Then we docked tailless H2A-H2B dimers and H3-H4 tetramers (from Protein Data Bank entry 1KX5) with XlPRMT5-MEP50 in ClusPro 2.0 (33, 34) using experimental and predicted interaction constraints on PRMT5, MEP50, and the histones. These constraints included attractive forces on the histone regions identified on the peptide array binding studies and attractive forces on MEP50 Arg-42 and the PRMT5-MEP50 SPPIDER- and PredUs-predicted interaction sites. Sample output models for putative H2A-H2B and H3-H4 interactions are shown in Fig. 7, B and C, respectively. We conclude that our experimental and predicted binding domains are consistent with our MEP50-dependent model for histone substrate recognition.
FIGURE 7.
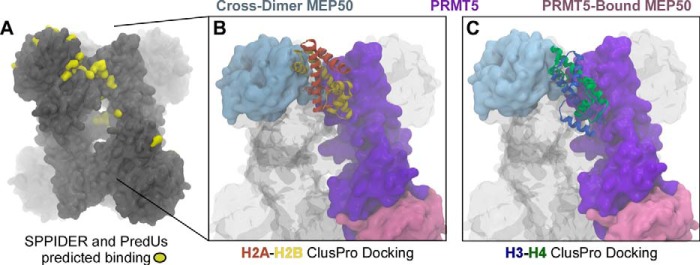
Prediction of histone binding sites onto the PRMT5-MEP50 complex. A, the predicted interacting residues on the cross-dimer pair of XlPRMT5-MEP50 was determined using the SPPIDER and PredUs algorithms and mapped onto the structure, as shown in yellow. Shown are docking of H2A-H2B dimer (orange/yellow) (B) and H3-H4 dimer (blue/green) (C) onto XlPRMT5-MEP50 (PRMT5 monomer (purple), its directly bound MEP50 molecule (pink), and the cross-dimer MEP50 (blue)), using ClusPro with attractive forces as determined by the peptide array and predictions in A.
DISCUSSION
Based on our published structure, we previously proposed the “cross-dimer” model for MEP50 presentation of substrate to PRMT5. Here we used structural analysis, biochemistry, and enzymology to test our model of MEP50-dependent histone recognition and methylation by its coordinated PRMT5. These multiple approaches provided solid support for our hypothesis that MEP50 critically enhances the histone substrate methylation and will help guide future studies to uncover specific mechanisms of recognition of other PRMT5 target proteins.
PRMT5-MEP50 Structural and Enzymatic Conservation
The structural conservation of the arrangement of MEP50 in the complex with PRMT5 gave us initial support for our model due to the clear coordination of the cross-dimer MEP50 from the catalytic domain of PRMT5. We gleaned further understanding of the role of MEP50 through structural alignment of the X. laevis PRMT5-MEP50 structure with the C. elegans PRMT5 in the absence of MEP50. We aligned the XlPRMT5 N-terminal domain, contacting XlMEP50, and the cross dimer XlPRMT5 C-terminal catalytic domain with the CePRMT5 chain and did not observe any regions of poor alignment, consistent with the absence of substantial allostery upon MEP50 binding.
Considering the relatively buried catalytic site in XlPRMT5 and its tetrameric nature, it was formally possible that MEP50 was necessary to organize substrate to overcome the entropic cost for substrate binding to PRMT5. To test this possibility, we produced the catalytic C-terminal domain of XlPRMT5 alone with the expectation that it would have robust and promiscuous activity. Strikingly, the catalytic domain did not have any appreciable histone methyltransferase activity, suggesting that the entire assembly is necessary. XlMEP50 added to the catalytic domain reaction did not stimulate activity, as expected because MEP50 only binds to the missing PRMT5 N-terminal domain. The catalytic domain alone does not dimerize due to the head-to-tail arrangement of PRMT5.
Human and X. laevis PRMT5 are highly homologous; as we previously showed, HsPRMT5 methyltransferase activity is stimulated by XlMEP50 (22). Here, we demonstrated that HsPRMT5 does have very modest histone methyltransferase activity, with a signal on the fluorogram appearing after a year of film exposure, compared with the significantly stronger activity observed after the addition of MEP50. Using our continuous luciferase-coupled assay, we did not observe any activity above background for HsPRMT5, so we are unable to assign kinetic parameters. The addition of XlMEP50 also gave a quantitative increase in methyltransferase activity when titrated into the assay, consistent with multiple active PRMT5 molecules within the tetramer.
Substantial support for the role of MEP50 in histone recognition and catalysis by PRMT5 was provided by our continuous coupled assay. In agreement with Wang et al. (43), we show that the Km for CePRMT5 methylation of H4 tail peptide is about 650 times higher than that observed for XlPRMT5-MEP50, and the catalytic efficiency for this peptide (∼150 M−1 s−1) is roughly equivalent to the loss of efficiency that we observe upon mutation of residue Arg-42 from the XlMEP50 insertion loop (∼45–75-fold). These observations that protein substrate binding, represented by substrate Km, is strongly dependent on the presence of MEP50 provide significant support for our hypothesis that MEP50 organizes substrate for PRMT5. Furthermore, the H4(1–7) short peptide would not be anticipated to be bound by MEP50, and therefore it should be poorly methylated by XlPRMT5-MEP50. Indeed, that is what we observed because this very short peptide exhibited a 184 μm Km, the highest value among all substrates assayed.
The MEP50-Histone Fold Interaction Orients the Substrate for Methylation
Competition studies between H2A and full-length histone/peptide H4 clearly highlighted the importance of histone fold domain for substrate recognition by PRMT5-MEP50. Intriguingly, the binding region for histone H2A may strongly overlap with some binding sites for histone H4, thus inhibiting methyl transfer onto H2A Arg-3. However, H3 is unable to fully abrogate the formation of H2A R3me1, so histones H2A and H3 may only share partial binding sites on the PRMT5-MEP50 complex. According to the strong competitive effects observed for both histone H3 and H4, the more physiologically relevant H3-H4 complex may be a substrate for XlPRMT5-MEP50 under specific, yet undetermined, experimental conditions.
Our histone peptide array studies are also consistent with the PRMT5-MEP50 preference for binding that we observed for full-length H3 and H4 compared with H2A. Binding studies on these ultrahigh density arrays will be generally useful for probing mechanisms of histone recognition by all histone-acting enzymes. Furthermore, our quantitative determination of binding affinity between full-length H2A and XlPRMT5-MEP50 (Kd = 1.78 ± 0.06 μm; Fig. 3H) is in good agreement with peptide array and kinetic results in which the H2A histone tail accounts for most of the binding onto the enzyme complex.
Recent publications have used kinetic and mass spectrometry analysis to model the reaction mechanism of PRMT5 and mutated PRMT1 in the generation of mono- and symmetric dimethylarginine (43, 44, 47). We independently confirmed this distributive model of progression to dimethylation through our demonstration that a synthesized monomethylated histone peptide has a 15-fold higher Km than its equivalent unmethylated peptide. We did not observe any substantive difference in the kcat between these two peptide substrates; these observations are probably incompatible with processive methylation and are therefore consistent with observations by Wang et al. (43) of a distributive mode of progression to dimethylation with CePRMT5 on monomethylated substrates. Furthermore, our highly specific R3me1 and R3me2s antibodies confirmed that the dimethylation state did not appear until saturating levels of R3me1 (Fig. 6D). Overall, these data and our prior observations of monomethylarginine in vivo point to the probably significant, but unexplored, biological role of monomethylation. Future studies will be needed to identify this biological role and potential monomethylarginine effector proteins.
PRMT5-MEP50 Exhibits Specific Histone Binding and Methyl Transfer
An enigma in the PRMT5 literature is that histone H3 and nucleosomal H4 substrates are well documented to be methylated by PRMT5 in vivo (40, 41, 48, 49), yet H3 is not a substrate in vitro. Here, we confirmed that PRMT5-MEP50 does not methylate histones in recombinant nucleosomes. The results of our histone-peptide array study and the mapping of the primary interaction domain to the histone fold of H3 and H4 are entirely consistent with the inability of PRMT5 to methylate nucleosomes and provide an example of a novel use of these arrays. Cross-talk from other post-translational modifications may explain this discrepancy, as suggested by observations showing that lysine acetylation stimulates PRMT5-MEP50 activity (50). Histones acetylated or otherwise modified in vivo may loosen histone-DNA contacts and may allow enough interaction to promote PRMT5 activity. We previously tested the enzyme complex's ability to methylate HeLa or hyperacetylated HeLa nucleosomes, but it was still unable to methylate these modified nucleosomes. Alternatively, other documented PRMT5 protein cofactors, such as COPR5 (15), Menin/Men1 (16), RioK1 (12), or ATP-dependent remodeling factors (18), may promote nucleosomal activity.
The MEP50 Insertion Finger Is Critical for the Complex's Substrate Binding and Activity
The MEP50 insertion finger directed over the cross-dimer PRMT5 catalytic domain is the WD40 repeat protein's most unique feature. We previously hypothesized that this insertion finger participates in organizing substrate for catalysis; alternatively, it may function to allosterically activate the PRMT5 catalytic domain, possibly mediated through the putative salt bridge between MEP50 Arg-42 and PRMT5 Glu-403. Our mutagenesis of MEP50 Arg-42 to glutamic acid led to a dramatic loss of catalytic efficiency for both SAM and peptide substrates with the reversal of charge (R42E). However, we observed a different behavior of the more conservative MEP50R42Q complex, with kinetic parameters for SAM substrate nearly identical to those of the wild-type XlPRMT5-MEP50. This loss of activity in the R42E mutant was primarily observed in a higher Km, with only modest kcat effects (32–40% of wild-type kcat; Fig. 6C), consistent with our hypothesis that histone/peptide substrate binding is dictated by MEP50 through intact positioning of the insertion finger. To our surprise, the Km for SAM substrate (directly bound to the C-terminal PRMT5 active site) was ∼8-fold higher upon R42E mutation, suggesting that MEP50 may indeed have some small but direct influence on catalysis, possibly mediated through the putative salt bridge to the PRMT5 catalytic domain.
RbAp46, a WD repeat protein and MEP50 analog that participates in multiple histone acetyltransferase complexes, was shown to bind histone H4 on its side face, as in our model for MEP50 binding (25). RbAp46 residues involved in H4 binding were isostructural with our predicted histone interaction domain on MEP50, providing convergent evolutionary support for our hypothesis. Furthermore, the H4 residues involved in binding RbAp46 were in the α1 helix of the histone fold (residues 24–41), consistent with the peptide array binding studies and providing support for the necessity of the histone fold interactions in the increased efficiency of full-length histone substrate methylation by PRMT5-MEP50.
We combined all of our direct observations and propose the model shown in Fig. 8. MEP50 is a presenter that 1) binds histones through their histone fold domain and 2) orients histone tail substrates toward the PRMT5 cross-dimer active site for efficient arginine methylation. We anticipate that this model will guide studies on other histone methyltransferase complexes as well as provide insight for future drug design.
FIGURE 8.
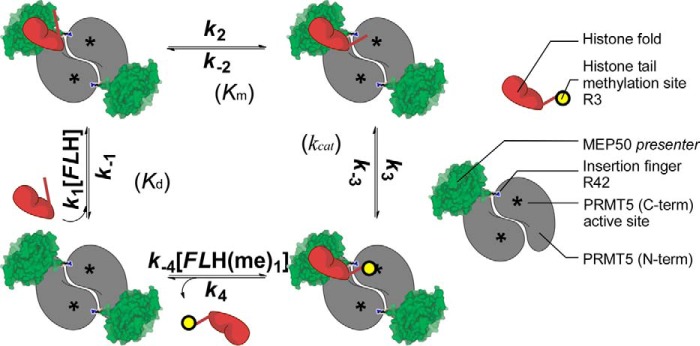
Schematic model for the sequential binding of core histone onto the MEP50 presenter and favorable orientation of the N-terminal histone tail to the PRMT5 active site for methyl transfer. XlPRMT5-MEP50 is a tetramer of heterodimers, and the cross-dimer MEP50 (green) is paired with its cognate PRMT5 molecule (gray; C-terminal active site represented by an asterisk) to promote histone methylation according to a sequential mechanism with 1) binding of the full-length histone through its histone fold (FLH in red; dissociation constant Kd), 2) favorable orientation of the histone tail toward the PRMT5 cross-dimer active site (asterisk; step characterized by a Km value), 3) methylation of Arg-3 (yellow circle with methyl group transferred from S-adenosylmethionine; step characterized by a kcat value), and 4) release of the methylated histone tail and the histone fold from the active site and the MEP50, respectively.
Acknowledgments
We thank Dr. Michael Brenowitz for assistance with analytical ultracentrifugation studies. We are grateful to the New York Structural Genomics Research Consortium and the Einstein Macromolecular Therapeutics Development Facility for baculovirus production.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM108646-01A1 (to D. S.). This work was also supported by startup funds from the Albert Einstein College of Medicine, an Alexander and Alexandrine Sinsheimer Foundation Scholar Award, and American Cancer Society Robbie Sue Mudd Kidney Cancer Research Scholar Grant 124891-RSG-13-396-01-DMC (all to D. S.). U. R. and J. S. are employees of JPT Peptide Technologies. The peptide array used in this study has been developed and manufactured by JPT. The library on this array has been extended and is marketed as “Histone Code Peptide Microarray” (His-MA_01) by JPT.
- PRMT
- protein arginine methyltransferase
- XlPRMT5
- HsPRMT5, CePRMT5, X. laevis, H. sapiens, and C. elegans PRMT5, respectively
- XlMEP50
- X. laevis MEP50
- SeMTAN
- S. enterica 5′-methylthioadenosine/S-adenosyl-l-homocysteine nucleosidase; ADA, [(carbamoylmethyl)imino]diacetic acid
- RMSD
- root mean square deviation
- SAM
- S-adenosylmethionine
- SAH
- S-adenosyl-l-homocysteine
- RT
- retention time
- R3me1 and R3me2
- mono- and dimethylated Arg-3, respectively.
REFERENCES
- 1. Di Lorenzo A., Bedford M. T. (2011) Histone arginine methylation. FEBS Lett. 585, 2024–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wolf S. S. (2009) The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol. Life Sci. 66, 2109–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wysocka J., Allis C. D., Coonrod S. (2006) Histone arginine methylation and its dynamic regulation. Front. Biosci. 11, 344–355 [DOI] [PubMed] [Google Scholar]
- 4. Lee Y. H., Stallcup M. R. (2009) Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol. Endocrinol. 23, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilczek C., Chitta R., Woo E., Shabanowitz J., Chait B. T., Hunt D. F., Shechter D. (2011) Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Biol. Chem. 286, 42221–42231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bezzi M., Teo S. X., Muller J., Mok W. C., Sahu S. K., Vardy L. A., Bonday Z. Q., Guccione E. (2013) Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 27, 1903–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scoumanne A., Zhang J., Chen X. (2009) PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 37, 4965–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friesen W. J., Wyce A., Paushkin S., Abel L., Rappsilber J., Mann M., Dreyfuss G. (2002) A novel WD repeat protein component of the methylosome binds Sm proteins. J. Biol. Chem. 277, 8243–8247 [DOI] [PubMed] [Google Scholar]
- 9. Furuno K., Masatsugu T., Sonoda M., Sasazuki T., Yamamoto K. (2006) Association of Polycomb group SUZ12 with WD-repeat protein MEP50 that binds to histone H2A selectively in vitro. Biochem. Biophys. Res. Commun. 345, 1051–1058 [DOI] [PubMed] [Google Scholar]
- 10. Le Guezennec X., Vermeulen M., Brinkman A. B., Hoeijmakers W. A., Cohen A., Lasonder E., Stunnenberg H. G. (2006) MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell Biol. 26, 843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aggarwal P., Vaites L. P., Kim J. K., Mellert H., Gurung B., Nakagawa H., Herlyn M., Hua X., Rustgi A. K., McMahon S. B., Diehl J. A. (2010) Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 18, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guderian G., Peter C., Wiesner J., Sickmann A., Schulze-Osthoff K., Fischer U., Grimmler M. (2011) RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 286, 1976–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ancelin K., Lange U. C., Hajkova P., Schneider R., Bannister A. J., Kouzarides T., Surani M. A. (2006) Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat. Cell Biol. 8, 623–630 [DOI] [PubMed] [Google Scholar]
- 14. Friesen W. J., Paushkin S., Wyce A., Massenet S., Pesiridis G. S., Van Duyne G., Rappsilber J., Mann M., Dreyfuss G. (2001) The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol. Cell Biol. 21, 8289–8300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lacroix M., El Messaoudi S., Rodier G., Le Cam A., Sardet C., Fabbrizio E. (2008) The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep. 9, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gurung B., Feng Z., Iwamoto D. V., Thiel A., Jin G., Fan C. M., Ng J. M., Curran T., Hua X. (2013) Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res. 73, 2650–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karkhanis V., Hu Y.-J., Baiocchi R. A., Imbalzano A. N., Sif S. (2011) Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 36, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pal S., Vishwanath S. N., Erdjument-Bromage H., Tempst P., Sif S. (2004) Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell Biol. 24, 9630–9645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan F., Alinari L., Lustberg M. E., Martin L. K., Cordero-Nieves H. M., Banasavadi-Siddegowda Y., Virk S., Barnholtz-Sloan J., Bell E. H., Wojton J., Jacob N. K., Chakravarti A., Nowicki M. O., Wu X., Lapalombella R., Datta J., Yu B., Gordon K., Haseley A., Patton J. T., Smith P. L., Ryu J., Zhang X., Mo X., Marcucci G., Nuovo G., Kwon C. H., Byrd J. C., Chiocca E. A., Li C., Sif S., Jacob S., Lawler S., Kaur B., Baiocchi R. A. (2014) Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 74, 1752–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bao X., Zhao S., Liu T., Liu Y., Liu Y., Yang X. (2013) Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J. Histochem. Cytochem. 61, 206–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gu Z., Gao S., Zhang F., Wang Z., Ma W., Davis R. E., Wang Z. (2012) Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem. J. 446, 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ho M.-C., Wilczek C., Bonanno J. B., Xing L., Seznec J., Matsui T., Carter L. G., Onikubo T., Kumar P. R., Chan M. K., Brenowitz M., Cheng R. H., Reimer U., Almo S. C., Shechter D. (2013) Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One 8, e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Antonysamy S., Bonday Z., Campbell R. M., Doyle B., Druzina Z., Gheyi T., Han B., Jungheim L. N., Qian Y., Rauch C., Russell M., Sauder J. M., Wasserman S. R., Weichert K., Willard F. S., Zhang A., Emtage S. (2012) Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U.S.A. 109, 17960–17965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruthenburg A. J., Wang W., Graybosch D. M., Li H., Allis C. D., Patel D. J., Verdine G. L. (2006) Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol. 13, 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murzina N. V., Pei X. Y., Zhang W., Sparkes M., Vicente-Garcia J., Pratap J. V., McLaughlin S. H., Ben-Shahar T. R., Verreault A., Luisi B. F., Laue E. D. (2008) Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure 16, 1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hemeon I., Gutierrez J. A., Ho M. C., Schramm V. L. (2011) Characterizing DNA methyltransferases with an ultrasensitive luciferase-linked continuous assay. Anal. Chem. 83, 4996–5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sturm M. B., Schramm V. L. (2009) Detecting ricin: sensitive luminescent assay for ricin A-chain ribosome depurination kinetics. Anal. Chem. 81, 2847–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H. C., Ciskanik L., Dunaway-Mariano D., von der Saal W., Villafranca J. J. (1988) Investigations of the partial reactions catalyzed by pyruvate phosphate dikinase. Biochemistry 27, 625–633 [DOI] [PubMed] [Google Scholar]
- 29. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 30. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 31. Russell R. B., Barton G. J. (1992) Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins 14, 309–323 [DOI] [PubMed] [Google Scholar]
- 32. Roberts E., Eargle J., Wright D., Luthey-Schulten Z. (2006) MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics 7, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Comeau S. R., Gatchell D. W., Vajda S., Camacho C. J. (2004) ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 32, W96–W99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Comeau S. R., Gatchell D. W., Vajda S., Camacho C. J. (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20, 45–50 [DOI] [PubMed] [Google Scholar]
- 35. Shechter D., Nicklay J. J., Chitta R. K., Shabanowitz J., Hunt D. F., Allis C. D. (2009) Analysis of histones in Xenopus laevis. I. A distinct index of enriched variants and modifications exists in each cell type and is remodeled during developmental transitions. J. Biol. Chem. 284, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nicklay J. J., Shechter D., Chitta R. K., Garcia B. A., Shabanowitz J., Allis C. D., Hunt D. F. (2009) Analysis of histones in Xenopus laevis. II. Mass spectrometry reveals an index of cell type-specific modifications on H3 and H4. J. Biol. Chem. 284, 1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tee W. W., Pardo M., Theunissen T. W., Yu L., Choudhary J. S., Hajkova P., Surani M. A. (2010) Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 24, 2772–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shechter D., Dormann H. L., Allis C. D., Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 39. Fabbrizio E., El Messaoudi S., Polanowska J., Paul C., Cook J. R., Lee J. H., Negre V., Rousset M., Pestka S., Le Cam A., Sardet C. (2002) Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3, 641–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Majumder S., Alinari L., Roy S., Miller T., Datta J., Sif S., Baiocchi R., Jacob S. T. (2010) Methylation of histone H3 and H4 by PRMT5 regulates ribosomal RNA gene transcription. J. Cell Biochem. 109, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang L., Pal S., Sif S. (2008) Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell Biol. 28, 6262–6277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang M., Fuhrmann J., Thompson P. R. (2014) Protein arginine methyltransferase 5 catalyzes substrate dimethylation in a distributive fashion. Biochemistry 53, 7884–7892 [DOI] [PubMed] [Google Scholar]
- 43. Wang M., Xu R. M., Thompson P. R. (2013) Substrate specificity, processivity, and kinetic mechanism of protein arginine methyltransferase 5. Biochemistry 52, 5430–5440 [DOI] [PubMed] [Google Scholar]
- 44. Gui S., Gathiaka S., Li J., Qu J., Acevedo O., Hevel J. M. (2014) A remodeled protein arginine methyltransferase 1 (PRMT1) generates symmetric dimethylarginine. J. Biol. Chem. 289, 9320–9327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Porollo A., Meller J. (2007) Prediction-based fingerprints of protein-protein interactions. Proteins 66, 630–645 [DOI] [PubMed] [Google Scholar]
- 46. Zhang Q. C., Deng L., Fisher M., Guan J., Honig B., Petrey D. (2011) PredUs: a web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 39, W283–W287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang W.-L., Anderson L. C., Nicklay J. J., Chen H., Gamble M. J., Shabanowitz J., Hunt D. F., Shechter D. (2014) Phosphorylation and arginine methylation mark histone H2A prior to deposition during Xenopus laevis development. Epigenetics Chromatin 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao Q., Rank G., Tan Y. T., Li H., Moritz R. L., Simpson R. J., Cerruti L., Curtis D. J., Patel D. J., Allis C. D., Cunningham J. M., Jane S. M. (2009) PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 16, 304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pal S., Baiocchi R. A., Byrd J. C., Grever M. R., Jacob S. T., Sif S. (2007) Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 26, 3558–3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feng Y., Wang J., Asher S., Hoang L., Guardiani C., Ivanov I., Zheng Y. G. (2011) Histone H4 acetylation differentially modulates arginine methylation by an in cis mechanism. J. Biol. Chem. 286, 20323–20334 [DOI] [PMC free article] [PubMed] [Google Scholar]