

Background: Nucleoside transport is an important therapeutic target against cancer.
Results: Cell-based studies show that a metal-containing nucleoside displays anti-cancer effects and diagnostic properties against adherent cancers.
Conclusion: A metal-containing nucleoside functions as a biophotonic substrate for a nucleoside transporter.
Significance: A novel nucleoside with combined therapeutic and diagnostic activities provides a tool to treat cancer.
Keywords: Cancer Therapy, Molecular Imaging, Molecular Pharmacology, Nucleoside/Nucleotide Analogue, Transport
Abstract
Nucleoside transport is an essential process that helps maintain the hyperproliferative state of most cancer cells. As such, it represents an important target for developing diagnostic and therapeutic agents that can effectively detect and treat cancer, respectively. This report describes the development of a metal-containing nucleoside designated Ir(III)-PPY nucleoside that displays both therapeutic and diagnostic properties against the human epidermal carcinoma cell line KB3-1. The cytotoxic effects of Ir(III)-PPY nucleoside are both time- and dose-dependent. Flow cytometry analyses validate that the nucleoside analog causes apoptosis by blocking cell cycle progression at G2/M. Fluorescent microscopy studies show rapid accumulation in the cytoplasm within 4 h. However, more significant accumulation is observed in the nucleus and mitochondria after 24 h. This localization is consistent with the ability of the metal-containing nucleoside to influence cell cycle progression at G2/M. Mitochondrial depletion is also observed after longer incubations (Δt ∼48 h), and this effect may produce additional cytotoxic effects. siRNA knockdown experiments demonstrate that the nucleoside transporter, hENT1, plays a key role in the cellular entry of Ir(III)-PPY nucleoside. Collectively, these data provide evidence for the development of a metal-containing nucleoside that functions as a combined therapeutic and diagnostic agent against cancer.
Introduction
One hallmark of cancer is its hyperproliferative nature, which is characterized by uncontrollable DNA synthesis. This feature provides an important focal point for therapeutic intervention. Indeed, ∼20% of all anti-cancer drugs are classified as anti-metabolites that affect DNA synthesis (1). Widely used anti-cancer agents include the nucleoside analogs gemcitabine and fludarabine, which target enzymes, such as ribonucleotide reductase and DNA polymerases, that are essential for DNA replication (2). The basis for their pharmacological effects rely heavily on modifications to the (deoxy)ribose moiety (Fig. 1A) (3). In these cases, sugar modifications allow the corresponding deoxynucleoside triphosphates to function as non-obligate chain terminators of DNA synthesis, which accounts for much of their anti-cancer activity (4–7).
FIGURE 1.

A, chemical structures of natural nucleosides (adenosine and cytosine), conventional anti-cancer nucleosides (fludarabine and gemcitabine), and the metal-containing nucleoside, Ir(III)-PPY nucleoside. B, two-and three-dimensional models for adenosine and Ir(III)-PPY nucleoside, which provide a more accurate comparison of shape and size of the metal-containing nucleoside analog versus a natural nucleoside substrate. Models were developed using Spartan version 4.0 software.
The therapeutic activity of many nucleoside analogs is often limited by their cellular uptake and subsequent metabolism to the corresponding nucleoside triphosphate (8–11). In fact, the hydrophilic nature of most nucleoside analogs requires an active transport system to catalyze efficient cellular uptake. Indeed, the cellular levels of nucleoside transporters can be used as predictive factors for patient responses to gemcitabine against pancreatic (12) and lung (13) cancer. However, there are several technological problems associated with easily identifying which transporter(s) is responsible for their uptake. Much of this challenge arises from the existence of two distinct families of nucleoside transporters. These include equilibrative nucleoside transporters (ENTs)2 and concentrative nucleoside transporters (CNTs). An additional level of complexity is the number of isoforms in each family. For example, humans possess four different ENT isoforms (designated hENT1–hENT4) and three distinct CNT isoforms (designated hCNT1–hCNT3). Each hENT isoform catalyzes the bidirectional transport of nucleosides following a concentration gradient and displays distinct transport activities for pyrimidine and purine (deoxy) nucleosides (14–16). In contrast, hCNTs catalyze the transport of (deoxy)nucleosides against a gradient by coupling nucleoside movement with sodium or proton co-transport (17–20). hCNT1 and hCNT2 translocate pyrimidine and purine (deoxy) nucleoside, respectively, via a sodium-dependent mechanism. hCNT3 shows broad substrate specificity and possesses the unique ability to translocate nucleosides in both sodium- and proton-coupled manners (17–20). Whereas both classes of nucleoside transporters are promiscuous in the ability to transport pyrimidine and purine nucleosides, most rely exclusively on the presence of a ribose or deoxyribose moiety for substrate recognition (14–20).
Because nucleoside transporters play key roles in the uptake of anti-cancer nucleoside analogs, an important goal is to develop chemical entities that can accurately and easily measure their activities at the cellular and organismal level. Most contemporary approaches use isotopically labeled nucleosides to quantify cellular uptake. This reliance has several logistical problems, such as special requirements for synthesis (21) and the use of discontinuous time-based assays (22) to monitor the influx and/or efflux of a nucleoside. Finally, the use of radiolabeled nucleosides has obvious limitations in measuring nucleoside transport activity and tissue distribution in humans.
To combat these deficiencies, we recently developed a metal-containing nucleoside analog, designated Ir(III)-PPY nucleoside, which contains iridium embedded within a bis-cyclometalated scaffold attached to a deoxyriboside (Fig. 1A) (23). This cyclometalated complex of iridium(III) was chosen due to its high stability in aqueous environments present in a living cell (24). In addition, iridium complexes are high yielding phosphorescent compounds that can be rationally tuned to emit visible light across a range of wavelengths (25). At face value, the size of the iridium complex attached to the deoxyribose moiety appears to be very large and would prohibit its utilization as a substrate for any nucleoside transporter. However, modeling studies provided in Fig. 1B demonstrate that the three-dimensional structure of Ir(III)-PPY nucleoside is compact and spherical, possessing an overall volume (596.3 A3) that is only ∼2.5-fold larger than deoxyadenosine (228.5 A3). Based on these features, the goal here is to further establish that Ir(III)-PPY nucleoside functions as a bona fide substrate for a nucleoside transporter. Here we provide further biochemical evidence that this novel metal-containing nucleoside indeed enters cells and displays both therapeutic and diagnostic activity against cancer cells. Cell-based studies demonstrate that Ir(III)-PPY nucleoside produces cytotoxic effects against an adherent cancer cell line, KB3-1. In addition, the metal-containing nucleoside rapidly enters cells primarily through the activity of a specific nucleoside transporter, hENT1. Co-localization and cell fractionation studies demonstrate that Ir(III)-PPY nucleoside accumulates in the nucleus and mitochondria of cancer cells in a time- and dose-dependent manner. The localization of Ir(III)-PPY nucleoside in these organelles coincides with their ability to produce anti-cancer effects by affecting DNA synthesis and the stability of mitochondria.
EXPERIMENTAL PROCEDURES
Materials
All chemical reagents were purchased from Sigma-Aldrich. KB3-1 and KB-V1 cells were a generous gift from Dr. Michael Gottesman (NCI, National Institutes of Health, Bethesda, MD). Human dermal microvascular endothelial cells and dermal fibroblast cells were obtained from ATCC (Manassas, VA). Phosphate-buffered saline (PBS), antibiotic and antifungal agents, amphotericin, propidium iodide, PrestoBlue, DAPI, Alexa Fluor 588, and an apoptosis assay kit containing Alexa Fluor 488-labeled annexin V that is used for cell culture studies were from Invitrogen. l-Mimosine was purchased from MP BioMedicals, LLC. Ir(III)-PPY nucleoside was synthesized and purified as described previously (23).
Cell Culture
All cells were cultured at 37 °C in humidified air and 5% CO2. The epidermal carcinoma cell line KB3-1 was maintained in Dulbecco's modified Eagle's medium (Mediatech, Manassas, VA) with 10% fetal bovine serum (USA Scientific), 100 units/ml penicillin (Invitrogen), 100 μg/ml streptomycin, and 250 μg/ml gentamycin. The doubling time was ∼24 h. KB-V1 cells were grown and maintained under the same conditions with the addition of 100 nm vinblastine. Human dermal microvascular endothelial cells were cultivated using vascular cell basal medium supplemented with an endothelial cell growth kit containing VEGF (ATCC). Dermal fibroblast cells were maintained in Dulbecco's modified Eagle's medium (Mediatech) with 10% fetal bovine serum (USA Scientific), 100 units/ml penicillin (Invitrogen), 100 μg/ml streptomycin, and 250 μg/ml gentamycin.
Cell Viability Assays
The cytotoxicity of Ir(III)-PPY nucleoside was quantified using a PrestoBlue assay according to the manufacturer's protocols (Invitrogen). Cells were plated at a density of 20,000 cells/well in 96-well plates and then incubated for 24 h after plating to ensure adhesion. Ir(III)-PPY nucleoside was added to wells in a dose-dependent manner (1–100 μm) and treated for variable time periods (24–72 h). In all cases, the final concentration of the co-solvent, DMSO, was maintained at 0.1%. After the desired end point, the original medium was replaced with 90 μl of fresh medium and 10 μl of PrestoBlue reagent. The fluorescence of each sample was measured using a microplate reader (excitation/emission = 560/590 nm). Raw data were normalized to 100% viability (0.1% DMSO control), and IC50 values were obtained through a fit of the data to Equation 1,
Assessment of Cell Death via Apoptosis Versus Necrosis
Cells were treated with 0.1% DMSO (vehicle) and Ir(III)-PPY nucleoside (10 or 50 μm) for variable time periods (24–72 h). Cells were harvested by centrifugation, washed in PBS, and resuspended in 100 μl of binding buffer containing 5 μm annexin V-Alexa Fluor 488 conjugate. Cells were treated with 1 μg/ml propidium iodide (PI) and incubated at room temperature for 15 min, followed by flow cytometry analysis. In dual parameter studies, cells treated with PI were also treated with an additional 400 μl of 1× annexin-binding buffer. In either case, cells were analyzed using band pass filters with wavelengths of 525/40 nm and 620/30 nm with a flow cytometer (Beckman Coulter XL).
For experiments interrogating nuclear morphology, KB3-1 cells were grown overnight in 12-well cell culture dishes using an initial density of 100,000 cells/ml. Cells were then treated with variable concentrations of Ir(III)-PPY nucleoside for time periods ranging from 4 to 24 h. Images were obtained using an EVOSfl Advanced microscope using DAPI stain (excitation/emission = 360/447 nm). Detection of Ir(III)-PPY nucleoside was achieved using excitation and emission wavelengths of 312 and 510 nm, respectively.
Cell cycle analysis using PI staining was performed by treating KB3-1 cells (200,000 cells/ml) with variable concentrations of Ir(III)-PPY nucleoside in the presence or absence of NBMPR for time periods varying from 1 to 2 days. After the appropriate time, cells were treated with 0.25% trypsin-EDTA and harvested by centrifugation at 3,000 rpm for 10 min. The supernatant was removed, and the cells were washed with 1× PBS. The cells were then fixed with methanol and stored at −20 °C overnight. Cell pellets were dislodged using a 0.5-ml solution of 10 μg/ml PI and 2 mg/ml of RNase A in saponin-based permeabilization and wash buffer and then incubated at room temperature for 15 min prior to flow cytometry analysis. Experiments using l-mimosine to induce cell cycle arrest were performed using similar approaches. In this case, KB3-1 cells were first pretreated for 24 h with 400 μm l-mimosine, a compound that causes cell cycle arrest between late S phase and early G1 phase.
Cellular Uptake of Ir(III)-PPY Nucleoside
Uptake of Ir(III)-PPY nucleoside into KB3-1 cells was visualized by microscope imaging. Cells were plated on 35-mm dish glass bottom microwell dishes and preincubated in the absence or presence of NBMPR. After 24 h, cells were co-cultivated with Ir(III)-PPY nucleoside for 1–2 days. At the appropriate times, cells were washed twice with PBS and exposed to fresh medium in the presence of 1 μg/ml DAPI, Alexa Fluor 568 phalloidin (1:2,000), and/or 10 nm MitoPT-TMRE for up to 15 min. After this time, cells were washed twice with 1× PBS, fixed with 4% paraformaldehyde, and washed twice more with 1× PBS. Images were obtained using an EVOSfl Advanced microscope.
Quantification of Ir(III)-PPY nucleoside uptake into KB3-1 cells was performed using a microplate assay as described previously (23). Briefly, cells were plated overnight onto 12-well plates at a density of 200,000 cells/well. The cells were then preincubated with DMSO, NBMPR, or dipyridamole for 24 h prior to treatment with variable concentrations of Ir(III)-PPY nucleoside for 1 or 2 days. Cells were treated with 0.25% trypsin-EDTA and harvested by centrifugation. The supernatant was removed and then washed twice with 1× PBS. Cells were lysed with 0.1% Triton X-100 in 1× PBS and then measured using a fluorescence plate reader (excitation, 340 nm; emission, 480 nm).
siRNA Knockdown of hENT1
KB3-1 cells were cultured in 6-well culture plates to 60–70% confluence in growth medium without antibiotics. After washing with PBS, cells were treated with Lipofectamine 2000 (Invitrogen) containing targeting hENT1 siRNA (s4694, Ambion, Foster City, CA) at a final concentration of 125 pmol in Opti-MEM (Invitrogen). The sense and antisense siRNA oligonucleotides used to generate siRNA sequence specifically targeting hENT1 (NM_001078174) are as follows: 5′-AUCGUGCUCAUUAAUUCATT-3′ (sense) and 5′-UGAAUUAAUGAGCACGAUCTT-3′ (antisense). Cells were incubated at 37 °C in a humidified CO2 incubator for 6 h. After changing growth medium, cells were treated with variable concentrations of Ir(III)-PPY nucleoside. At 24 h post-treatment, cells were harvested, and lysates were separated using SDS-PAGE. Western blotting of antibodies specific for hENT1 (rabbit polyclonal IgG, Santa Cruz Biotechnology) and β-actin (mouse monoclonal IgG) were detected using HRP-conjugated anti-rabbit and HRP-conjugated anti-mouse antibody (Santa Cruz Biotechnology, Inc.). Western blots were developed using the chemiluminescent peroxidase substrate for detection (Lumi-light Western blotting substrate, Roche Applied Science).
Isolation of Cytosolic, Mitochondrial, and Nuclear Fractions
Sample preparation was performed using the Abcam cell fractionation kit (Abcam, Inc., Cambridge, MA) and protocols established by the manufacturer. Briefly, KB3-1 cells were harvested and washed in buffer A (supplied by manufacturer). Cell density was measured via manual counting using trypan blue staining and a hematocytometer. 50 μl of buffer B (supplied by the manufacturer) was added to the cell suspension, and the solution was incubated for 7 min on a shaker with gentle agitation at room temperature. Samples were then centrifuged at 300 × g for 3 min at room temperature. The resulting supernatant containing the cytosolic fraction was transferred to a fresh tube for subsequent quantitative analyses. Enrichment of mitochondria was similarly performed by resuspending the aforementioned cell pellet in 50 μl of buffer C (supplied by the manufacturer). This solution was incubated for 10 min on a shaker with gentle agitation at room temperature and then centrifuged at 300 × g for 3 min at room temperature. The supernatant containing the mitochondrial fraction was transferred to a fresh tube for subsequent analyses. Enrichment of the nuclear fraction was accomplished by resuspending the cell pellet in 50 μl of buffer D (supplied by the manufacturer). The solution was again incubated for 10 min on a shaker with gentle agitation at room temperature and then centrifuged at 300 × g for 3 min at room temperature. The supernatant contains the nuclear fractions. Quantification of Ir(III)-PPY nucleoside present in each fraction (cytoplasm, mitochondria, and nucleus) was measured using a fluorescent plate reader (Spectramax M4, Molecular Devices) at the optimal excitation (340 nm) and emission (480 nm) wavelengths as described previously (23).
RESULTS
Anti-cancer Effects of Ir(III)-PPY Nucleoside
The ability of Ir(III)-PPY nucleoside to produce cytostatic and/or cytotoxic effects was tested against two isogenic human epidermal carcinoma cell lines, KB3-1 (multidrug resistance-negative) and KB-V1 (multidrug resistance-positive). Both types of cells were treated with increasing concentrations (0–100 μm) of Ir(III)-PPY nucleoside for time intervals of up to 3 days. At 24-h intervals, cell viability and proliferation were measured using the PrestoBlue assay kit. Cell viability is expressed as the optical density ratio of cells treated with Ir(III)-PPY nucleoside compared with cells treated with 0.1% DMSO. The cytotoxic effects of Ir(III)-PPY nucleoside against KB3-1 cells are both time- and dose-dependent (Fig. 2A). The time dependence is evident as the IC50 value of Ir(III)-PPY nucleoside decreases from 56 μm at 24 h to 30 μm after 48 h, and this value further decreases to 15 μm after 72 h post-treatment (Table 1). Ir(III)-PPY nucleoside displays significantly weaker antiproliferative effects against the multidrug resistance-positive cell line, KB-V1 (Fig. 2B). The reduced efficacy against KB-V1 cells suggests that Ir(III)-PPY nucleoside first enters the cell through active transport and is then exported out of the cell by P-glycoprotein activity (26).
FIGURE 2.

A, dose- and time-dependent effects of Ir(III)-PPY nucleoside against the human epidermal carcinoma cell line, KB3-1. 0.1% DMSO was used as the vehicle, whereas Ir(III)-PPY nucleoside concentration was varied from 1 to 100 μm. An IC50 value of 30 ± 5 μm was obtained for the Ir(III)-PPY nucleoside after 48 h of treatment. B, dose- and time-dependent effects of Ir(III)-PPY nucleoside against the multidrug resistance-positive carcinoma cell line, KB-V1. C, structural comparison of Ir(III)-PPY nucleoside with Ir(III)PPY3, the metal complex lacking the deoxyribose moiety. Ir(III)-PPY nucleoside displays cytotoxic effects against KB3-1 cells that are time- and dose-dependent. In contrast, Ir(III)PPY3 does not produce cytostatic or cytotoxic effects even at high concentrations (50 μm) for extended time periods (3 days of treatment). The various treatments are shown as follows: 0.1% DMSO (white bars), 10 μm Ir(III)PPY3 (black bars), 10 μm Ir(III)-PPY nucleoside (blue bars), 50 μm Ir(III)PPY3 (black hashed bars), and 50 μm Ir(III)-PPY nucleoside (blue hashed bars). Error bars, S.E.
TABLE 1.
Summary of IC50 values for Ir(III)-PPY nucleoside in the KB3-1 and KB-V1 cells
Each value represents the mean ± S.D. for three independent experiments.
| Time | KB3-1 | KB-V1 |
|---|---|---|
| h | μm | μm |
| 24 | 56 ± 3 | >400 |
| 48 | 30 ± 5 | >100 |
| 72 | 15.2 ± 1.3 | ∼100 |
We next confirmed that nucleoside transport activity is required for the anti-cancer effects of Ir(III)-PPY nucleoside by measuring the ability of a cyclometalated iridium complex lacking a deoxyribose moiety, designated Ir(III)PPY3, to affect cellular proliferation. Fig. 2C compares the time- and dose-dependent effects of Ir(III)PPY3 versus Ir(III)-PPY nucleoside. These data clearly show that Ir(III)PPY3 does not produce any cytostatic or cytotoxic effects at concentrations as high as 50 μm over a 3-day time interval. Thus, the presence of the deoxyribose moiety is essential for the pharmacological effects of Ir(III)-PPY nucleoside and suggests that uptake is dependent upon the activity of a nucleoside transporter.
Non-invasion Detection of Ir(III)-PPY Nucleoside Uptake via Fluorescence Microscopy
We previously used fluorescence microscopy to measure Ir(III)-PPY nucleoside uptake into KB3-1 cells (23). In addition, we demonstrated that only high concentrations of deoxyadenosine (200 μm) could block Ir(III)-PPY nucleoside uptake (23). These data suggest that one or more hENT family members participates in its uptake. To verify that uptake is dependent on the presence of a deoxyribose moiety, fluorescence microscopy was used to compare the cellular uptake of Ir(III)PPY3 versus Ir(III)-PPY nucleoside. Images provided in Fig. 3A clearly show that Ir(III)PPY3 does not enter the cells, whereas treatment with an identical concentration of Ir(III)-PPY nucleoside results in significant intracellular accumulation under the same time frame tested (Δt = 2 days). These imaging data, coupled with the inability of Ir(III)PPY3 to produce anti-cancer effects, verify that uptake of the cyclometalated iridium complex depends on the presence of a deoxyribose moiety and confirms that at least one nucleoside transporter is involved in the uptake of Ir(III)-PPY nucleoside.
FIGURE 3.
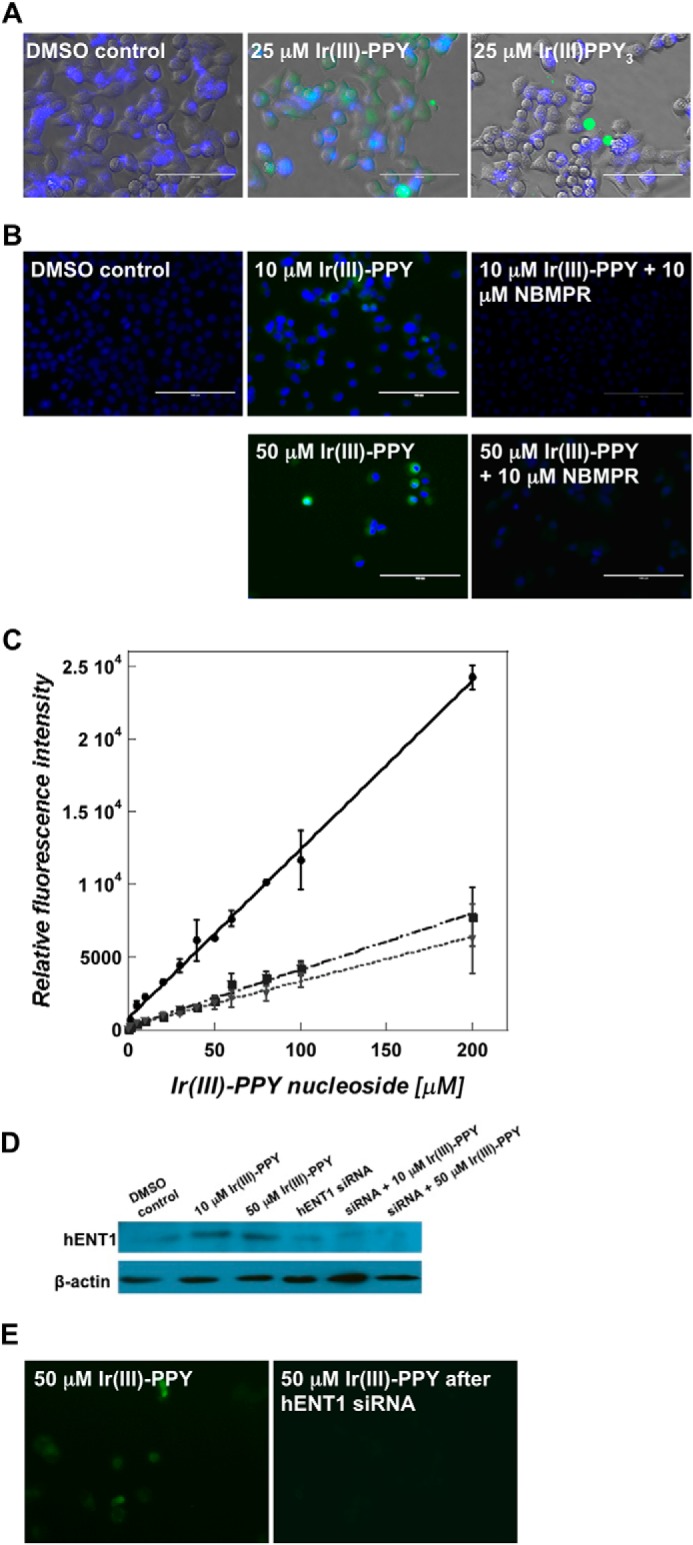
A, fluorescence microscope images of KB3-1 cells treated with 0.1% DMSO, 25 μm Ir(III)PPY3, or 25 μm Ir(III)-PPY nucleoside (Δt = 24 h post-treatment). Ir(III)-PPY nucleoside shows green fluorescence, whereas nuclei were stained with DAPI (blue) (magnification, ×40). B, fluorescence microscope images of 10 or 50 μm Ir(III)-PPY nucleoside uptake in the absence or presence of NBMPR in KB3-1 cells (Δt = 24 h post-treatment). Ir(III)-PPY nucleoside shows green fluorescence, whereas nuclei were stained with DAPI (blue) (magnification, ×40). C, the accumulation of Ir(III)-PPY nucleoside in KB3-1 cells was measured in the absence or presence of nucleoside transport inhibitors. Whole cell fluorescence intensities of Ir(III)-PPY nucleoside were measured using a fluorescence plate reader assay (excitation/emission = 312/510). The various treatments are shown as follows: Ir(III)-PPY nucleoside alone (solid line), Ir(III)-PPY nucleoside in the presence of 10 μm NBMPR (dashed and dotted line), and Ir(III)-PPY nucleoside in the presence of 1 μm dipyridamole (dotted line). D, Western blot analysis of cells treated with and without siRNA targeting hENT1. Cells treated with siRNA show reduced levels of the nucleoside transporter. E, cells treated with siRNA targeting hENT1 (right panel) show reduced uptake of Ir(III)-PPY nucleoside compared with cells treated with scrambled siRNA (left panel) (magnification, ×40). Error bars, S.E.
Based on this finding, we interrogated the involvement of hENT family members by testing the ability of the hENT inhibitor, NBMPR, to block Ir(III)-PPY nucleoside uptake. Microscopy images provided in Fig. 3B show that cells pretreated with 10 μm NBMPR have lower fluorescence intensities after exposure to 50 μm Ir(III)-PPY nucleoside compared with cells not treated with the inhibitor. This result is consistent with a role of hENT1 in the transport of Ir(III)-PPY nucleoside.
Ir(III)-PPY nucleoside uptake into KB3-1 cells was further quantified using a fluorescent plate reader assay (23). In this analysis, cells were pretreated with 1 μm dipyridamole (an inhibitor of hENT1 and hENT2) or 10 μm NBMPR for 24 h and then co-cultivated with variable concentrations of Ir(III)-PPY nucleoside for an additional 48 h. Fig. 3C shows a dose-dependent increase in fluorescence signal in cells treated with Ir(III)-PPY nucleoside alone. However, pretreatment with either dipyridamole or NBMPR causes a 4-fold decrease in Ir(III)-PPY nucleoside uptake. This result is consistent with cell viability experiments showing that pretreatment with NBMPR reduces the cytotoxic effects of Ir(III)-PPY nucleoside and suggests that hENT1 is primarily responsible for Ir(III)-PPY nucleoside transport. We note, however, that uptake at high concentrations of Ir(III)-PPY nucleoside (>50 μm) is not completely blocked by either inhibitor. Thus, although active transport mediated by hENT1 predominates, the metal-containing nucleoside can also enter cells via passive diffusion at high concentrations. Cellular entry by active and passive transport is not unusual because this phenomenon has been documented with many conventional nucleoside analogs (27, 28). For example, the primary route for the uptake of ara-C at low concentrations (∼1 μm) is via hENT1 (27, 28). However, at higher concentrations that exceed 50 μm, passive diffusion becomes the primary route for uptake because simple inward diffusion rates exceed those of hENT1 activity (29).
Regardless, to further demonstrate that hENT1 catalyzes Ir(III)-PPY nucleoside transport, we performed a series of siRNA knockdown experiments targeting this nucleoside transporter. Western blot analyses provided in Fig. 3D demonstrate that siRNA treatment specifically targeting hENT1 leads to a reduction in the level of this nucleoside transporter. Fluorescence microscopy studies on DMSO and siRNA-treated cells validate that reducing hENT1 expression causes a concomitant decrease in Ir(III)-PPY nucleoside uptake (Fig. 3E). Collectively, the results obtained using pharmacological inhibitors against hENT1 and siRNA techniques to reduce hENT1 levels suggest that the activity of this nucleoside transporter is essential for Ir(III)-PPY nucleoside uptake. However, we note that these results do not completely eliminate roles of other hENT or hCNT family members.
Ir(III)-PPY Nucleoside Causes Cell Death by a Unique Mechanism
The mechanism by which Ir(III)-PPY nucleoside causes cell death was evaluated using dual parameter flow cytometry to measure PI uptake coupled with Alexa Fluor 488 annexin V conjugate staining. In this analysis, live cells are negative for either fluorophore and are easily distinguished from cells that are early apoptotic (annexin V-positive and PI-negative), late apoptotic (PI- and annexin V-positive), or necrotic (PI-positive and annexin V-negative). Data provided in Fig. 4A demonstrate that treatment with 10 and 50 μm Ir(III)-PPY nucleoside, respectively, causes significant increases in early and late stage apoptosis. In addition, Ir(III)-PPY nucleoside causes apoptosis in a dose- and time-dependent manner, consistent with experiments measuring cellular viability (see above).
FIGURE 4.

A, dual parameter flow cytometry of KB3-1 cells treated with 0.1% DMSO 10 μm Ir(III)-PPY nucleoside or 50 μm Ir(III)-PPY nucleoside. Treatment with 50 μm Ir(III)-PPY nucleoside for 48 h produces a robust apoptotic effect. B, DAPI staining demonstrates cell cycle-specific toxicity by Ir(III)-PPY nucleoside in a dose- and time-dependent manner. C, cell cycle analysis of KB3-1 cells treated with Ir(III)-PPY nucleoside. Treatment with 50 μm Ir(III)-PPY nucleoside for 48 h causes a significant perturbation in cell cycle progression because cells are unable to complete mitosis. The anti-cancer effects of Ir(III)-PPY nucleoside can be blocked by pretreating cells with the hENT inhibitor, NBMPR. D, KB3-1 cells were synchronized at the G1 phase of the cell cycle by treating with 400 μm l-mimosine for 24 h. After removal of medium containing l-mimosine, fresh medium was added containing 0.1% DMSO, 10 μm Ir(III)-PPY nucleoside, or 50 μm Ir(III)-PPY nucleoside. The 0 h control represents treatment with l-mimosine alone.
DAPI staining was next used to investigate morphological changes associated with exposure to Ir(III)-PPY nucleoside (30). Fig. 4B shows that increasing concentrations of Ir(III)-PPY nucleoside causes a concomitant decrease in the number of KB3-1 cells. These cell killing effects are time-dependent and recapitulate results obtained using dual parameter flow cytometry (see above). Furthermore, these images show that Ir(III)-PPY nucleoside causes chromatin condensation in treated cells (highlighted by white arrows). Rather than appearing round, as expected for a classical apoptotic pathway, the nuclei appear elongated and reminiscent of cells undergoing mitosis (31). These data suggest that Ir(III)-PPY nucleoside causes cell death by affecting cell cycle progression, specifically at G2/M.
This hypothesis was tested using flow cytometry and PI staining to measure the effects of Ir(III)-PPY nucleoside on cell cycle progression. KB3-1 cells treated with 0.1% DMSO (negative control) show a normal cell cycle distribution for asynchronous diploid cells (Fig. 4C, left). In this case, most cells exist at G1 (53.1 ± 3.3%) and S phase (32.5 ± 2.3%) with a smaller population at G2/M (14.4 ± 1.2%). Treatment with 50 μm Ir(III)-PPY nucleoside (Fig. 4C, center) significantly perturbs cell cycle progression. However, these effects could not be accurately determined using a standard diploid model. Instead, cell cycle distributions were defined using a diploid-tetraploid model representing a heterogeneous population of cells containing two and four sets of chromosomes, respectively (32). Using this model, the diploid cell population shows significant alterations in cell cycle progression, as reflected in a decrease in G1 (19.9 ± 7.1%) coupled with increases at S phase (43.2 ± 7.8%) and G2/M (36.9 ± 2.9%). Tetraploid cells also show significant perturbations in cell cycle progression, as reflected in decreases at G1 (34.2 ± 1.0%) and S phase (20.5 ± 6.6%) coupled with an increase at G2/M (45.3 ± 5.7%). Taken together, these data demonstrate that Ir(III)-PPY nucleoside causes aneuploidy by affecting cell cycle progression in diploid cells. Finally, the effects of Ir(III)-PPY nucleoside on cell cycle progression are blocked by pretreating cells with 10 μm NBMPR (Fig. 3C, right). In this case, only diploid cells are detected, and cell populations are essentially identical compared with DMSO treatment (G1 = 54.0 ± 3.6%, S phase = 34.3 ± 2.9%, and G2/M = 11.7 ± 1.3%). This ability of NBMPR to inhibit the pharmacological effects of Ir(III)-PPY nucleoside provides additional evidence for hENT1 in transporting the metal-containing nucleoside. Table 2 summarizes cell populations in KB3-1 cells treated with 0.1% DMSO, Ir(III)-PPY nucleoside alone, and Ir(III)-PPY nucleoside with NBMPR.
TABLE 2.
Summary of the effects of Ir(III)-PPY nucleoside on cell cycle progression
| Condition | G1 | S | G2/M |
|---|---|---|---|
| % | % | % | |
| DMSO | 53.1 ± 3.3 | 32.5 ± 2.3 | 14.4 ± 1.2 |
| 50 μm Ir(III)-PPY nucleoside (D)a | 19.9 ± 7.1 | 43.2 ± 7.8 | 36.9 ± 2.9 |
| 50 μm Ir(III)-PPY nucleoside (T)b | 34.2 ± 1.0 | 20.5 ± 6.6 | 45.3 ± 5.7 |
| 50 μm Ir(III)-PPY nucleoside + 10 μm NBMPR | 54.0 ± 3.6 | 34.3 ± 2.9 | 11.7 ± 1.3 |
a Diploid cells.
b Tetraploid cells.
The ability of Ir(III)-PPY nucleoside to block cell cycle progression at G2/M is unusual because most nucleoside analogs affect either G1 or S phase (33–35). Analogs such as gemcitabine and fludarabine cause these effects by inhibiting nucleotide biosynthesis and/or DNA polymerase activity. In addition, inhibiting nucleoside transporter activity does not typically block cell cycle progression at G2/M. For example, Sakumura et al. (36) showed that dilazep, an adenosine uptake inhibitor, blocks cells at both G1 and S phase. Thus, the unique effects of Ir(III)-PPY nucleoside on cell cycle progression prompted us to further investigate its activity against KB3-1 cells synchronized at G1 by treatment with l-mimosine (37). Cells treated with 400 μm l-mimosine for 24 h accumulate at G1 (Fig. 4D). After this time period, the medium was replaced with fresh medium containing 0.1% DMSO (control) or Ir(III)-PPY nucleoside (10 or 50 μm), and the cells were treated for an additional 48 h. KB3-1 cells treated with DMSO or 10 μm Ir(III)-PPY nucleoside re-establish a normal cell cycle pattern after 48 h (Fig. 4D). However, treatment with 50 μm Ir(III)-PPY nucleoside causes accumulation at G2/M. Collectively, these results indicate that the metal-containing nucleoside induces cell cycle arrest, most likely by inhibiting mitosis.
There are several mechanisms that could account for this effect on mitosis. One possibility raised by a reviewer is that Ir(III)-PPY nucleoside disrupts cell cycle progression by intercalating into DNA (38). Preliminary results using gel electrophoresis techniques indicate that Ir(III)-PPY nucleoside does not bind to plasmid DNA, even when used at high concentrations of 50 μm nucleoside.3 Although Ir(III)-PPY nucleoside does not appear to bind nonspecifically to plasmid DNA, it is possible that the metal-containing nucleoside binds to quadruplex DNA that can form at telomeric DNA. Another possibility is that Ir(III)-PPY nucleoside inhibits the activity of kinases associated with cell cycle progression. In fact, we reported on the ability of gold-containing indolyl-compounds to inhibit several important kinases involved in cell cycle progression, including AKT2, Aurora kinase A, and Aurora kinase B with micromolar potencies (39). We are currently pursuing efforts to determine whether the ability of Ir(III)-PPY nucleoside to perturb cell cycle progression results from interactions with specific sequences of DNA, kinase inhibition, or other mechanisms.
Ir(III)-PPY Nucleoside Is a Cancer-selective Theranostic Agent
The potential safety of Ir(III)-PPY nucleoside as an anti-cancer agent was examined by quantifying its cytostatic and cytotoxic effects against two non-cancerous human cell lines, human dermal microvascular endothelial cells and dermal fibroblast cells. Experiments were performed as described earlier in which each cell line was treated with increasing concentrations (0–100 μm) of Ir(III)-PPY nucleoside, and cell viability was then measured at 24-h intervals. Representative data provided in Fig. 5A show that Ir(III)-PPY nucleoside produces negligible cytostatic or cytotoxic effects against either cell line, even at high concentrations (100 μm). This result contrasts data obtained using KB3-1 cells in which 100 μm Ir(III)-PPY nucleoside produces significant anti-cancer effects within 24 h.
FIGURE 5.
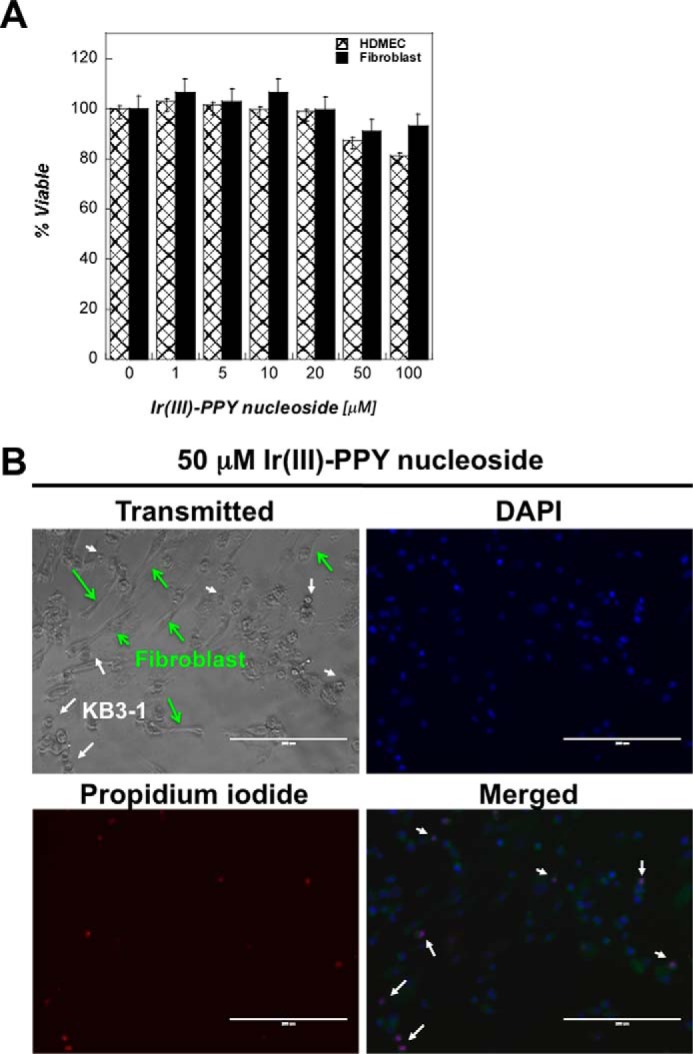
A, comparison of the anti-cancer effects of Ir(III)-PPY nucleoside measured against two non-cancerous cell lines (human dermal microvascular endothelial cells (HDMEC) and dermal fibroblast cells) versus the cancerous cell line KB3-1. All three cell lines were treated with variable concentrations of Ir(III)-PPY nucleoside (1–100 μm) for 24 h. DMSO was used as the vehicle control. B, DAPI and propidium iodide staining demonstrates that treatment with 50 μm Ir(III)-PPY nucleoside produces apoptotic effects against KB3-1 cells (cancer) but not dermal fibroblast cells (non-cancerous) after 24 h of exposure. Green arrows, position of fibroblasts; white arrows, position of KB3-1 cells (magnification, ×20). Error bars, S.E.
The selectivity of Ir(III)-PPY nucleoside was further demonstrated by simultaneously treating co-cultivated cancerous (KB3-1) and non-cancerous (dermal fibroblast) cells. Both cell lines were grown together at equal densities and then treated with 50 μm Ir(III)-PPY nucleoside. After 24 h, cells were analyzed by standard light microscopy techniques to differentiate KB3-1 cells (small circular cells) from dermal fibroblasts (large elongated cells). Fig. 5B shows that there are fewer KB3-1 cells compared with dermal fibroblasts. In addition, cell staining with DAPI and PI as markers for apoptosis demonstrated that Ir(III)-PPY nucleoside induces apoptosis in KB3-1 cells but not in fibroblasts (Fig. 5B). These results demonstrate that Ir(III)-PPY nucleoside is more selective toward killing cancer cells than non-cancerous cells.
Subcellular Localization of Ir(III)-PPY Nucleoside
To further examine the mechanism by which Ir(III)-PPY nucleoside causes cell death, we used high field microscopy techniques to determine the cellular localization of Ir(III)-PPY nucleoside. Imaging experiments were performed, treating KB3-1 cells with Ir(III)-PPY nucleoside for time periods of 4–48 h. At various times, cells were then co-stained to identify various cellular organelles, including the nucleus, mitochondria, and cytoskeleton. Fig. 6A shows the results of KB3-1 cells treated with 10 or 50 μm Ir(III)-PPY nucleoside for 48 h and then stained with DAPI or MitoPT-TMRE to identify the nucleus and mitochondria, respectively. Cells treated with 10 μm Ir(III)-PPY nucleoside show moderate levels of green fluorescence, indicative of uptake of the metal-containing nucleoside. The merged image of green fluorescence (Ir(III)-PPY nucleoside) coupled with DAPI staining shows significant accumulation of the metal-containing nucleoside in the nucleus. However, localization of Ir(III)-PPY nucleoside is also detected within the mitochondria of KB3-1 cells.
FIGURE 6.

A, measuring the subcellular localization of Ir(III)-PPY nucleoside by fluorescence microscopy. KB3-1 cells were treated with DMSO, 10 μm Ir(III)-PPY nucleoside, or 50 μm Ir(III)-PPY nucleoside for 48 h. Ir(III)-PPY nucleoside shows green fluorescence. Nuclei were stained with DAPI (blue). Mitochondria were stained with MitoPT (red). Merged images show that Ir(III)-PPY nucleoside primarily accumulates in the cytoplasm and nucleus at low concentrations (10 μm). However, an increase in mitochondrial accumulation is observed at higher concentrations (50 μm). Images were obtained using an EVOSfl Advanced microscope (magnification, ×40). B, higher magnification images (magnification, ×60) show that Ir(III)-PPY nucleoside accumulates in the nucleus and mitochondria of KB3-1 cells in a time- and dose-dependent manner. Ir(III)-PPY nucleoside shows green fluorescence. Nuclei were stained with DAPI (blue). Mitochondria were stained with MitoPT (red) (magnification, ×60).
Several more pronounced effects are observed in cells treated with 50 μm Ir(III)-PPY nucleoside. First, fewer cells are detected, and this reduction is consistent with the dose-dependent cytotoxicity of Ir(III)-PPY nucleoside (vide supra). Second, the remaining cells show high levels of green fluorescence, indicative of an increase in the cellular accumulation of the nucleoside analog. Furthermore, merged images show significant co-localization of Ir(III)-PPY nucleoside in the nucleus of KB3-1 after 48 h of exposure (Fig. 6B). This localization within the nucleus is consistent with the ability of Ir(III)-PPY nucleoside to alter cell cycle progression. A surprising feature, however, is that treatment with 50 μm Ir(III)-PPY nucleoside reduces the number of mitochondria, a phenomenon consistent with mitophagy (40). It is currently unclear how Ir(III)-PPY nucleoside causes mitophagy. However, a recent report from Liu et al. (41) showed that iridium complexes containing C∧N-chelated and pyridine ligands display anti-cancer activity by producing reactive oxygen species, which subsequently leads to a loss of mitochondrial membrane potential. Although we have yet to examine this potential mechanism, the report by Liu et al. (41) highlights how iridium complexes can exert distinct biological effects on cancer cells.
Fractionation studies were also performed to quantify the amount of Ir(III)-PPY nucleoside in the cytosol, nucleus, and mitochondria of treated KB3-1 cells. Representative data provided in Fig. 7A plot the normalized intensity of fluorescence in nuclear, mitochondrial, and cytosolic fractions in cells treated with Ir(III)-PPY nucleoside for 24 h. At low Ir(III)-PPY nucleoside concentrations (10 μm), there are small but significant amounts of the nucleoside present in all three fractions. However, the majority of nucleoside exists in the nuclear fraction compared with either the cytosol of mitochondrial fractions. Enhanced nuclear accumulation of Ir(III)-PPY nucleoside occurs in a dose-dependent manner because increasing the concentration to 50 μm leads to much higher amounts in the nucleus compared with the cytosolic and mitochondrial fractions. This distribution correlates well with the results of microscopy studies (Fig. 6B) and validate the use of imaging to visualize the intracellular localization of the metal-containing nucleoside.
FIGURE 7.
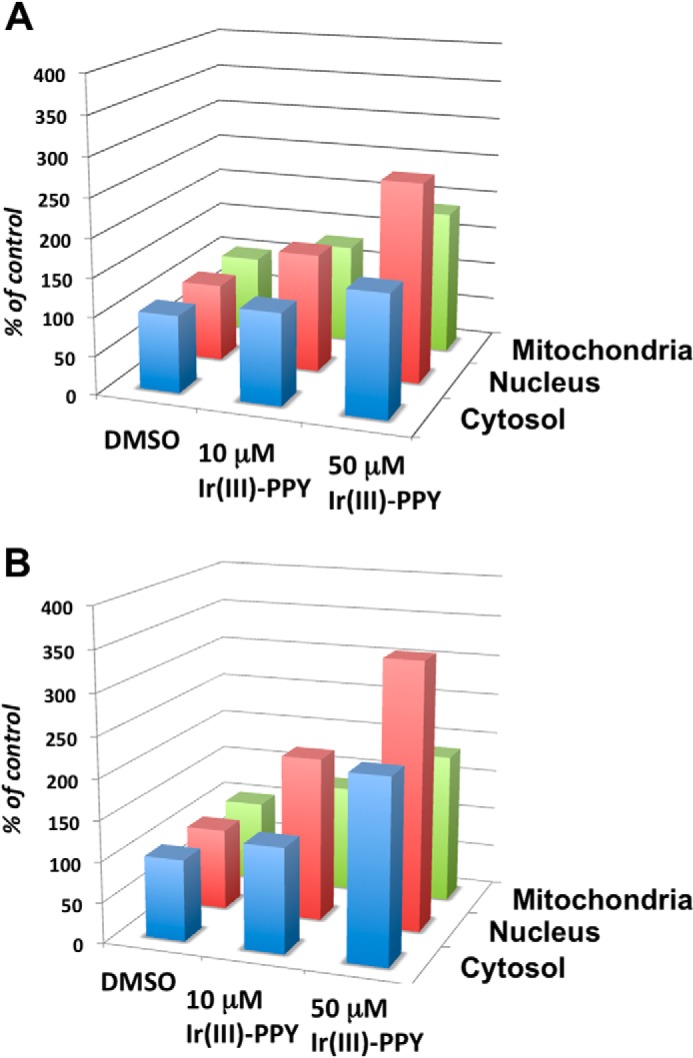
Results of fractionation studies to quantify the amount of Ir(III)-PPY nucleoside in the cytosol, nucleus, and mitochondria of treated KB3-1 cells. Cells were treated with 0.1% DMSO, 10 μm Ir(III)-PPY nucleoside, and 50 μm Ir(III)-PPY nucleoside. After 24 h (A) or 48 h (B) post-treatment, cells were harvested, and fractions corresponding to the cytosol, mitochondria, and nucleus were isolated. The relative amount of Ir(III)-PPY nucleoside measured using its fluorescence properties as described under “Experimental Procedures.” Error bars are omitted for clarity. In all cases, the S.E. associated with each value is less than 10%.
Fig. 7B shows that the subcellular localization of Ir(III)-PPY nucleoside is time-dependent. This is evident because the amount of metal-containing nucleoside in the nucleus nearly doubles from 24 to 48 h of exposure. Similar time-dependent increases in nucleoside accumulation are observed in the cytosolic and mitochondrial fractions. Again, the time dependence of enhanced nuclear localization is consistent with the microscopy analyses provided in Fig. 6B. Collectively, these data indicate that Ir(III)-PPY nucleoside can easily enter the nucleus of a cell, presumably by transport through the nuclear pore complex. Most likely, Ir(III)-PPY nucleoside enters the nucleus via passage through nuclear pore complexes that contain a central hole ∼50 nm in diameter. Because Ir(III)-PPY nucleoside is significantly smaller (∼1 nm), it probably passes through the nuclear pore complex without assistance from other cellular components.
DISCUSSION
Nucleoside transporters play essential roles in the maintenance of normal cellular homeostasis. These proteins are also important in pathological conditions, such as cancer. In particular, nucleoside transporters, such as hENT1, help to mediate the efficient uptake of pyrimidine and purine nucleoside analogs that function as anti-cancer agents. This later aspect highlights the important role of nucleoside transporters in optimizing the therapeutic response to chemotherapy. Indeed, the absence of certain nucleoside transporters can produce drug resistance (12), whereas overexpression can increase drug selectivity and improve sensitivity (13). hENT1 is particularly important because its levels are a positive predictor for response to gemcitabine in patients with advanced pancreatic cancer (12) and metastatic lung disease (13). As such, quantifying nucleoside transporter activity can predict therapeutic responses to nucleoside analogs. Unfortunately, the ability to obtain this information conveniently and rapidly is difficult because it requires the use of invasive techniques such as histological, genomic, and proteomic analyses of patient samples. Furthermore, quantifying protein and mRNA levels provides limited information because they cannot directly measure the cellular activity of a specific nucleoside transporter. Based on data provided here, we propose that Ir(III)-PPY nucleoside represents a significant advance in this area because it can directly, efficiently, and conveniently monitor the activity a specific nucleoside transporter via non-invasive techniques. This is supported by fluorescence microscopy experiments that prove that Ir(III)-PPY nucleoside rapidly enters cells within 4 h. Quantitative fluorescence measurements further demonstrate that NBMPR blocks the uptake of Ir(III)-PPY nucleoside by greater than 4-fold. Genetic approaches using siRNA knockdown further validated that reducing hENT1 levels causes a concomitant decrease in the uptake of Ir(III)-PPY nucleoside. Collectively, these results indicate that hENT1 plays a primary role in transporting the metal-containing nucleoside into cells. This is important because the ability of Ir(III)-PPY nucleoside to non-invasively quantify the cellular activity of hENT1 could identify patients that would respond favorably to currently used anti-cancer nucleosides, such as gemcitabine (42, 43).
Other fluorescent nucleoside analogs have been developed as surrogates to analyze nucleoside uptake inside living cells. For example, Zhang et al. (44) described confocal microscopy studies using two fluorescent analogs, designated FuPmR and dFuPmR, to study nucleoside influx and subsequent intracellular distribution. Although both analogs are transported into cells, they function as highly promiscuous substrates for several human nucleoside transporters (hENT1, hENT2, hCNT1, hCNT2, and hCNT3) ectopically expressed in Saccharomyces cerevisiae or Xenopus oocytes (44). Our data show that Ir(III)-PPY nucleoside is unique because it specifically monitors the cellular activity of hENT1. In addition, the rapid uptake of Ir(III)-PPY nucleoside (∼4 h) is advantageous for “real-time” monitoring of hENT1 activity.
Another distinctive feature is the ability of Ir(III)-PPY nucleoside to produce cytotoxic effects in both time- and dose-dependent manners. Although flow cytometry analyses show that cancer cells treated with Ir(III)-PPY nucleoside undergo apoptosis, the mechanism underlying this effect is very complex. Our data show that Ir(III)-PPY nucleoside arrests cell cycle progression at G2/M, probably by blocking mitosis. Furthermore, microscopy and fractionation studies show that Ir(III)-PPY nucleoside accumulates in the nucleus within 24 h, and this phenomenon is consistent with the ability of the nucleoside to affect cell cycle progression. The nuclear accumulation of Ir(III)-PPY nucleoside contrasts the activity of other iridium-containing compounds that produce anti-cancer effects (45, 46). Indeed, these compounds distribute diffusely in the cytoplasm rather than localizing within specific organelles. Other metal-containing complexes, including those containing triphenylphosphine moieties, tend to accumulate in mitochondria (47). This pattern of subcellular localization probably occurs because these positively charged compounds can interact with the negatively charged mitochondrial membrane. Our microscopy and cell fractionation data also show that Ir(III)-PPY nucleoside accumulates in the mitochondria in a time- and dose-dependent manner. This accumulation may occur through electrostatic interactions with the mitochondrial membrane as described above. However, Ir(III)-PPY nucleoside could also be transported into this organelle through recognition of the deoxyribose moiety. This is reasonable because nucleoside transporter activity is strongly dependent upon the presence of a 3′-OH group on the sugar of a nucleoside (48–50). Because optimal activity of the mitochondria and nucleus rely heavily on nucleoside metabolism, it is likely that these organelles interact with Ir(III)-PPY nucleoside using similar transport mechanisms.
The uptake of Ir(III)-PPY nucleoside into mitochondria might also explain why high levels of Ir(III)-PPY nucleoside induce mitophagy, a metabolic process that mediates the selective elimination of damaged mitochondria (40). One proposed role of mitophagy is to remove damaged or dysfunctional mitochondria. This process serves an important role because mitochondria are essential for many diverse biological functions, including ATP biosynthesis and apoptosis (51–53). Ir(III)-PPY nucleoside may trigger mitophagy by inducing metabolic stress caused by perturbing normal nucleoside metabolism or by producing reactive oxygen species, as described by Liu et al. (41), using other iridium complexes. Current efforts are under way to examine these potential mechanisms.
In conclusion, this report describes the application of a unique biophotonic probe that can monitor the activity of a specific nucleoside transporter, hENT1. Although the data presented here are consistent with the involvement of hENT1 in the uptake of Ir(III)-PPY nucleoside, we have not completely eliminated the role of other hENT or hCNT family members. As such, we are currently exploring their potential roles using siRNA methods as reported here for hENT1. Regardless, a key innovation of this work is the use of non-invasive methods to directly visualize uptake of the metal-containing nucleoside and to correlate its anti-cancer activity with cellular uptake and localization. We envision that this unique nucleoside analog can be employed in a variety of cell-based studies to comprehensively understand the complex biology of nucleoside metabolism and thus represents a new technological platform to overcome existing deficiencies with currently used nucleoside analogs.
This research was supported by National Science Foundation Grant CBET-1066107 (to A. J. B. and T. G.).
A. J. Berdis, unpublished data.
- ENT
- equilibrative nucleoside transporter
- CNT
- concentrative nucleoside transporter
- PI
- propidium iodide
- NBMPR
- nitrobenzylthioinosine.
REFERENCES
- 1. Parker W. B. (2009) Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 109, 2880–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crawford S. (2013) Is it time for a new paradigm for systemic cancer treatment? Lessons from a century of cancer chemotherapy. Front. Pharmacol. 4, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berdis A. J. (2008) DNA polymerases as therapeutic targets. Biochemistry 47, 8253–8260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tiwari K. N., Cappellacci L., Montgomery J. A., Secrist J. A., 3rd (2003) Synthesis and anti-cancer activity of some novel 5-azacytosine nucleosides. Nucleosides Nucleotides Nucleic Acids 22, 2161–2170 [DOI] [PubMed] [Google Scholar]
- 5. Wang M., Liu Y., Liu S., Zheng D. (2004) 8-Chloro-adenosine sensitizes a human hepatoma cell line to TRAIL-induced apoptosis by caspase-dependent and -independent pathways. Oncol. Rep. 12, 193–199 [PubMed] [Google Scholar]
- 6. Curbo S., Johansson M., Balzarini J., Lewis L. D., Karlsson A. (2009) Acute cytotoxicity of arabinofuranosyl nucleoside analogs is not dependent on mitochondrial DNA. Exp. Cell Res. 315, 2539–2543 [DOI] [PubMed] [Google Scholar]
- 7. Su C. H., Chu W. C., Lan K. H., Li C. P., Chao Y., Lin H. C., Lee S. D., Tsai Y. C., Lee W. P. (2012) Gemcitabine causes telomere attrition by stabilizing TRF2. Eur. J. Cancer. 48, 3465–3474 [DOI] [PubMed] [Google Scholar]
- 8. Mackey J. R., Yao S. Y., Smith K. M., Karpinski E., Baldwin S. A., Cass C. E., Young J. D. (1999) Gemcitabine transport in Xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J. Natl. Cancer. Inst. 91, 1876–1881 [DOI] [PubMed] [Google Scholar]
- 9. Baldwin S. A., Mackey J. R., Cass C. E., Young J. D. (1999) Nucleoside transporters: molecular biology and implications for therapeutic development. Mol. Med. Today 5, 216–224 [DOI] [PubMed] [Google Scholar]
- 10. Zhang J., Visser F., King K. M., Baldwin S. A., Young J. D., Cass C. E. (2007) The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 26, 85–110 [DOI] [PubMed] [Google Scholar]
- 11. López-Guerra M., Trigueros-Motos L., Molina-Arcas M., Villamor N., Casado F. J., Montserrat E., Campo E., Colomer D., Pastor-Anglada M. (2008) Identification of TIGAR in the equilibrative nucleoside transporter 2-mediated response to fludarabine in chronic lymphocytic leukemia cells. Haematologica 93, 1843–1851 [DOI] [PubMed] [Google Scholar]
- 12. Spratlin J., Sangha R., Glubrecht D., Dabbagh L., Young J. D., Dumontet C., Cass C., Lai R., Mackey J. R. (2004) The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin. Cancer Res. 10, 6956–6961 [DOI] [PubMed] [Google Scholar]
- 13. Sève P., Dumontet C. (2005) Chemoresistance in non-small cell lung cancer. Curr. Med. Chem. Anticancer Agents 5, 73–88 [DOI] [PubMed] [Google Scholar]
- 14. Molina-Arcas M., Trigueros-Motos L., Casado F. J., Pastor-Anglada M. (2008) Physiological and pharmacological roles of nucleoside transporter proteins. Nucleosides Nucleotides Nucleic Acids 27, 769–778 [DOI] [PubMed] [Google Scholar]
- 15. Löffler M., Morote-Garcia J. C., Eltzschig S. A., Coe I. R., Eltzschig H. K. (2007) Physiological roles of vascular nucleoside transporters. Arterioscler. Thromb. Vasc. Biol. 27, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 16. Young J. D., Yao S. Y., Sun L., Cass C. E., Baldwin S. A. (2008) Human equilitractive nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38, 995–1021 [DOI] [PubMed] [Google Scholar]
- 17. Pastor-Anglada M., Cano-Soldado P., Errasti-Murugarren E., Casado F. J. (2008) SLC28 genes and concentrative nucleoside transporter (CNT) proteins. Xenobiotica 38, 972–994 [DOI] [PubMed] [Google Scholar]
- 18. Ritzel M. W., Yao S. Y., Ng A. M., Mackey J. R., Cass C. E., Young J. D. (1998) Molecular cloning, functional expression and chromosomal localization of a cDNA encoding a human Na+/nucleoside cotransporter (hCNT2) selective for purine nucleosides and uridine. Mol. Membr. Biol. 15, 203–211 [DOI] [PubMed] [Google Scholar]
- 19. Ritzel M. W., Ng A. M., Yao S. Y., Graham K., Loewen S. K., Smith K. M., Hyde R. J., Karpinski E., Cass C. E., Baldwin S. A., Young J. D. (2001) Recent molecular advances in studies of the concentrative Na+-dependent nucleoside transporter (CNT) family: identification and characterization of novel human and mouse proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). Mol. Membr. Biol. 18, 65–72 [DOI] [PubMed] [Google Scholar]
- 20. Slugoski M. D., Ng A. M., Yao S. Y., Smith K. M., Lin C. C., Zhang J., Karpinski E., Cass C. E., Baldwin S. A., Young J. D. (2008) A proton-mediated conformational shift identifies a mobile pore-lining cysteine residue (Cys-561) in human concentrative nucleoside transporter 3. J. Biol. Chem. 283, 8496–8507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wiebe L. I., Knaus E. E., Morin K. W. (1999) Radiolabelled pyrimidine nucleosides to monitor the expression of HSV-1 thymidine kinase in gene therapy. Nucleosides Nucleotides 18, 1065–1066 [DOI] [PubMed] [Google Scholar]
- 22. Thampy K. G., Barnes E. M., Jr. (1983) Adenosine transport by primary cultures of neurons from chick embryo brain. J. Neurochem. 40, 874–879 [DOI] [PubMed] [Google Scholar]
- 23. Maity A., Choi J.-S., Teets T. S., Deligonul N., Berdis A. J., Gray T. G. (2013) Cyclometalated iridium(III) complexes with deoxyribose substituents. Chemistry 19, 15924–15932 [DOI] [PubMed] [Google Scholar]
- 24. Patra M., Gasser G. (2012) Organometallic compounds: an opportunity for chemical biology? Chembiochem 13, 1232–1252 [DOI] [PubMed] [Google Scholar]
- 25. Shavaleev N. M., Monti F., Costa R. D., Scopelliti R., Bolink H. J., Ortí E., Accorsi G., Armaroli N., Baranoff E., Grätzel M., Nazeeruddin M. K. (2012) Bright blue phosphorescence from cationic bis-cyclometalated iridium(III) isocyanide complexes. Inorg. Chem. 51, 2263–2271 [DOI] [PubMed] [Google Scholar]
- 26. Hopper-Borge E., Xu X., Shen T., Shi Z., Chen Z. S., Kruh G. D. (2009) Human multidrug resistance protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Res. 69, 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weinstein H. J., Griffin T. W., Feeney J., Cohen H. J., Propper R. D., Sallan S. E. (1982) Pharmacokinetics of continuous intravenous and subcutaneous infusions of cytosine arabinoside. Blood 59, 1351–1353 [PubMed] [Google Scholar]
- 28. Ho D. H., Frei E., 3rd (1971) Clinical pharmacology of 1-β-d-arabinofuranosyl cytosine. Clin. Pharmacol. Ther. 12, 944–954 [DOI] [PubMed] [Google Scholar]
- 29. Capizzi R. L., Yang J. L., Cheng E., Bjornsson T., Sahasrabudhe D., Tan R. S., Cheng Y. C. (1983) Alteration of the pharmacokinetics of high-dose ara-C by its metabolite, high ara-U in patients with acute leukemia. J. Clin. Oncol. 1, 763–771 [DOI] [PubMed] [Google Scholar]
- 30. Hunter A. L., Choy J. C., Granville D. J. (2005) Detection of apoptosis in cardiovascular diseases. Methods Mol. Med. 112, 277–289 [DOI] [PubMed] [Google Scholar]
- 31. Vagnarelli P. (2012) Mitotic chromosome condensation in vertebrates. Exp. Cell Res. 318, 1435–1441 [DOI] [PubMed] [Google Scholar]
- 32. Fukunaga M. (2001) Flow cytometric and clinicopathologic study of complete hydatidiform moles with special reference to the significance of cytometric aneuploidy. Gynecol. Oncol. 81, 67–70 [DOI] [PubMed] [Google Scholar]
- 33. Shi Z., Azuma A., Sampath D., Li Y. X., Huang P., Plunkett W. (2001) S-phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res. 61, 1065–1072 [PubMed] [Google Scholar]
- 34. Sigmond J., Peters G. J. (2005) Pyrimidine and purine analogues, effects on cell cycle regulation and the role of cell cycle inhibitors to enhance their cytotoxicity. Nucleosides Nucleotides Nucleic Acids 24, 1997–2022 [DOI] [PubMed] [Google Scholar]
- 35. Wang Q., Liu X., Wang Q., Zhang Y., Jiang J., Guo X., Fan Q., Zheng L., Yu X., Wang N., Pan Z., Song C., Qi W., Chang J. (2011) FNC, a novel nucleoside analogue inhibits cell proliferation and tumor growth in a variety of human cancer cells. Biochem. Pharmacol. 81, 848–855 [DOI] [PubMed] [Google Scholar]
- 36. Sakumura T., Fujii Z., Umemoto S., Murakami T., Kawata Y., Fujii K., Minami M., Sasaki K., Matsuzaki M. (2000) Dilazep, a nucleoside transporter inhibitor, modulates cell cycle progression and DNA synthesis in rat mesangial cells in vitro. Cell Prolif. 33, 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krude T. (1999) Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp. Cell Res. 247, 148–159 [DOI] [PubMed] [Google Scholar]
- 38. Wheate N. J., Brodie C. R., Collins J. G., Kemp S., Aldrich-Wright J. R. (2007) DNA intercalators in cancer therapy: organic and inorganic drugs and their spectroscopic tools of analysis. Mini Rev. Med. Chem. 7, 627–648 [DOI] [PubMed] [Google Scholar]
- 39. Craig S., Gao L., Lee I., Gray T., Berdis A. J. (2102) Gold-containing indoles as anticancer agents that potentiate the cytotoxic effects of ionizing radiation. J. Med. Chem. 55, 2437–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ding W. X., Yin X. M. (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Z., Romero-Canelón I., Habtemariam A., Clarkson G. J., Sadler P. J. (2014) Potent half-sandwich iridium(III) anticancer complexes containing C∧N-chelated and pyridine ligands. Organometallics 33, 5324–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alexander R. L., Greene B. T., Torti S. V., Kucera G. L. (2005) A novel phospholipid gemcitabine conjugate is able to bypass three drug-resistance mechanisms. Cancer Chemother. Pharmacol. 56, 15–21 [DOI] [PubMed] [Google Scholar]
- 43. Zhu H., Liu Z., Tang L., Liu J., Zhou M., Xie F., Wang Z., Wang Y., Shen S., Hu L., Yu L. (2012) Reversal of P-gp and MRP1-mediated multidrug resistance by H6, a gypenoside aglycon from Gynostemma pentaphyllum, in vincristine-resistant human oral cancer (KB/VCR) cells. Eur. J. Pharmacol. 696, 43–53 [DOI] [PubMed] [Google Scholar]
- 44. Zhang J., Sun X., Smith K. M., Visser F., Carpenter P., Barron G., Peng Y., Robins M. J., Baldwin S. A., Young J. D., Cass C. E. (2006) Studies of nucleoside transporters using novel autofluorescent nucleoside probes. Biochemistry 45, 1087–1098 [DOI] [PubMed] [Google Scholar]
- 45. Yu M., Zhao Q., Shi L., Li F., Zhou Z., Yang H., Yi T., Huang C. (2008) Cationic iridium(III) complexes for phosphorescence staining in the cytoplasm of living cells. Chem. Commun. (Camb.) 14, 2115–2117 [DOI] [PubMed] [Google Scholar]
- 46. Fernández-Moreira V., Thorp-Greenwood F. L., Coogan M. P. (2010) Application of d6 transition metal complexes in fluorescence cell imaging. Chem. Commun. (Camb.) 46, 186–202 [DOI] [PubMed] [Google Scholar]
- 47. Shabaik Y.H., Millard M., Neamati N. (2013) Mechanistic evaluation of a novel small molecule targeting mitochondria in pancreatic cancer cells. PLoS One 8, e54346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang C., Swaan P. W., Ngo L. Y., Lum P. Y., Patil S. D., Unadkat J. D. (2004) Molecular requirements of the human nucleoside transporters hCNT1, hCNT2, and hENT1. Mol. Pharmacol. 65, 558–570 [DOI] [PubMed] [Google Scholar]
- 49. Vickers M. F., Kumar R., Visser F., Zhang J., Charania J., Raborn R.T., Baldwin S.A., Young J. D., Cass C. E. (2002) Comparison of the interaction of uridine, cytidine, and other pyrimidine nucleoside analogues with recombinant human equilibrative nucleoside transporter 2 (hENT2) produced in Saccharomyces cerevisiae. Biochem. Cell Biol. 80, 639–644 [DOI] [PubMed] [Google Scholar]
- 50. Griffith D. A., Jarvis S. M. (1996) Nucleoside and nucleobase transport systems of mammalian cells. Biochim. Biophys. Acta 1286, 153–181 [DOI] [PubMed] [Google Scholar]
- 51. Bereiter-Hahn J., Jendrach M. (2010) Mitochondrial dynamics. Int. Rev. Cell Mol. Biol. 284, 1–65 [DOI] [PubMed] [Google Scholar]
- 52. Hedskog L., Zhang S., Ankarcrona M. (2012) Strategic role for mitochondria in Alzheimer's disease and cancer. Antioxid. Redox Signal. 16, 1476–1491 [DOI] [PubMed] [Google Scholar]
- 53. Oberst A., Bender C., Green D. R. (2008) Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 15, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]