

Background: Structures of GltPh have been solved in the absence of a lipid bilayer.
Results: Transport rates of GltPh are influenced by lipid bilayer composition.
Conclusion: Transmembrane orientation and lipid-protein interactions influence transport in GltPh.
Significance: This provides a specific example of how interactions between lipid headgroups and membrane-bound proteins can influence function.
Keywords: Amino Acid Transport, Glutamate, Lipid Bilayer, Membrane Protein, Membrane Transporter Reconstitution, EAAT, GltPh, Cation-π
Abstract
The aspartate transporter from Pyrococcus horikoshii (GltPh) is a model for the structure of the SLC1 family of amino acid transporters. Crystal structures of GltPh provide insight into mechanisms of ion coupling and substrate transport; however, structures have been solved in the absence of a lipid bilayer so they provide limited information regarding interactions that occur between the protein and lipids of the membrane. Here, we investigated the effect of the lipid environment on aspartate transport by reconstituting GltPh into liposomes of defined lipid composition where the primary lipid is phosphatidylethanolamine (PE) or its methyl derivatives. We showed that the rate of aspartate transport and the transmembrane orientation of GltPh were influenced by the primary lipid in the liposomes. In PE liposomes, we observed the highest transport rate and showed that 85% of the transporters were orientated right-side out, whereas in trimethyl PE liposomes, 50% of transporters were right-side out, and we observed a 4-fold reduction in transport rate. Differences in orientation can only partially explain the lipid composition effect on transport rate. Crystal structures of GltPh revealed a tyrosine residue (Tyr-33) that we propose interacts with lipid headgroups during the transport cycle. Based on site-directed mutagenesis, we propose that a cation-π interaction between Tyr-33 and the lipid headgroups can influence conformational flexibility of the trimerization domain and thus the rate of transport. These results provide a specific example of how interactions between membrane lipids and membrane-bound proteins can influence function and highlight the importance of the role of the membrane in transporter function.
Introduction
Transport proteins undergo a series of conformational changes within the membrane to drive the movement of substrate across the cell membrane. Historically, the cell membrane was thought to serve solely as an impermeable barrier delineating the extracellular and cytosolic compartments. However, we now know that the cell membrane is a mutable and dynamic organelle that is made up of a complex repertoire of lipid species (1), and it has been demonstrated that membrane lipids may modulate the function of proteins within the membrane. Some proteins require the presence of particular lipids in the membrane to act as cofactors for protein function (2), whereas some lipid species stabilize otherwise energetically unfavorable conformations of a protein (3). Additionally, the function of membrane-bound proteins may be influenced by more diffuse, nonspecific features of the membrane such as lateral pressure or hydrophobic thickness (4).
Glutamate is the predominant excitatory neurotransmitter in the central nervous system and is involved in a variety of neurological functions including memory and cognition. The excitatory amino acid transporters act to terminate glutamate-mediated excitatory neurotransmission (5). The structure of the Na+-dependent aspartate transporter from Pyrococcus horikoshii (GltPh)3 has been solved in various conformations (6–9). This archaeal homologue represents a structural paradigm for members of the SLC1 transporter family, which includes the human excitatory amino acid transporters and neutral amino acid transporters (5). GltPh is a trimer with each protomer composed of eight transmembrane helices and two re-entrant hairpin loops (HP1 and HP2) that form part of the substrate binding site (7). Each monomer is made up of two domains (see Fig. 1A): a rigid trimerization domain, which forms the interprotomer contacts and defines the interface with the lipid bilayer, and a dynamic transport domain, which alternates the exposure of the substrate and ion binding sites to the cytoplasmic and extracellular milieu (7, 8).
FIGURE 1.
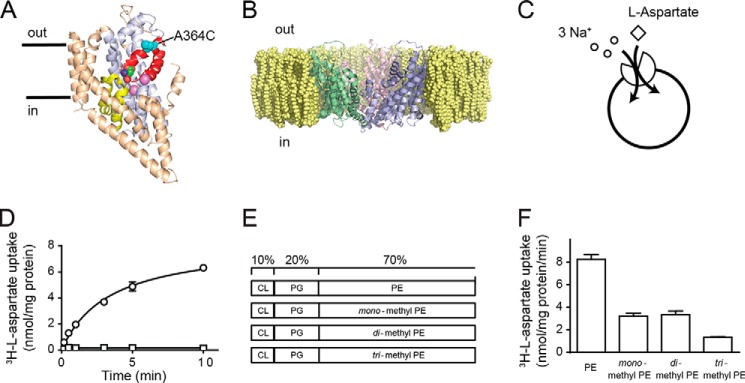
Rates of aspartate transport by GltPh are influenced by the lipid bilayer composition. A, structure of a single protomer of GltPh in the plane of the membrane with bound l-aspartate and Na+ (purple spheres). The transport domain is in light blue, HP1 is yellow, HP2 is red, and the trimerization domain is in wheat. Residue Ala-364 is represented as cyan spheres. B, crystal structure of GltPh (Protein Data Bank code 2NWX) viewed from the plane of the membrane in a simulated PE lipid bilayer. The figure was made using the program PyMOL (38). C, transporter function was studied by reconstituting purified protein into liposomes. A schematic representation of GltPh transport stoichiometry is shown. D, transport was measured in the presence of an inwardly directed Na+ gradient (open circles). Internal liposome buffer contained 100 mm KCl, 20 mm HEPES-Tris pH, 7.5. External buffer contained 100 mm NaCl, 20 mm HEPES-Tris, pH 7.5, 1 μm valinomycin, 100 nm l-[3H]aspartate. No transport was observed in the absence of an inwardly directed Na+ gradient (open squares). E, schematic representation of liposome lipid compositions, which comprise 10% CL, 20% PG, and 70% PE or its monomethyl, dimethyl, or trimethyl derivatives. F, initial rates of transport for wild-type GltPh reconstituted in liposomes as shown in E. Data represent the mean of experiments performed in triplicate, and error bars indicate S.E.
X-ray crystallography reveals atomic resolution structures; however, the conditions amenable for traditional x-ray crystallography do not reflect the dynamic and heterogeneous environment of a lipid bilayer. There can be over 1000 different species of lipids in a cell membrane due to the inherent changeability of headgroup chemistry, number and length of acyl chains, and degree of saturation (1). Fig. 1B shows the crystal structure of GltPh in a simulated phosphatidylethanolamine (PE) bilayer. Although this is a simplified view of what GltPh may look like in a lipid bilayer, it highlights the potential interactions between GltPh and the lipids of the bilayer, which are not accounted for in the current x-ray crystal structures. Structural and functional analyses of GltPh have improved our understanding of the mechanism of ion binding, anion permeation, and substrate transport in the SLC1 transporter family (6, 7, 10, 13), but little is known about the influence of lipid bilayer composition on the function of GltPh. In this study, we investigated whether the function of GltPh is influenced by the lipid environment by reconstituting purified protein into liposomes of varying lipid headgroup chemistry and measuring the transport of radiolabeled aspartate. We showed that GltPh was sensitive to the lipid environment and propose a role for lipids in influencing the transmembrane orientation of the transporter. We also identified a tyrosine residue (Tyr-33) in transmembrane domain 1 (TM1) that was exposed to the lipid bilayer and influenced the rate of transport. These results provide a specific example of how interactions between membrane lipids and membrane-bound proteins can influence function.
MATERIALS AND METHODS
Protein Purification
All mutations were made using the Q5 site-directed mutagenesis kit (New England Biolabs), and cysteine substitutions were introduced into a cysteine-less GltPh mutant in which the single native cysteine had been mutated to serine (C321S); this mutant is fully active (11, 12). GltPh was grown and purified as described previously (13). Briefly, membranes containing His-GltPh were isolated and solubilized with n-dodecyl β-d-maltopyranoside (Anatrace), and protein was purified using nickel-nitrilotriacetic acid resin (Qiagen). The histidine tag was subsequently removed by digestion with thrombin (10 units/mg of protein), and the protein further purified on a size exclusion column where the detergent was exchanged to n-decyl β-d-maltopyranoside (Anatrace).
Reconstitution
Pure lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Liposome compositions used were as follows: PE liposomes, 70% dioleoyl phosphatidylethanolamine, 20% phosphatidylglycerol (PG), 10% cardiolipin (CL); monomethyl PE liposomes, 70% N-methyl phosphatidylethanolamine, 20% PG, 10% CL; dimethyl PE liposomes, 70% N,N-dimethyl phosphatidylethanolamine, 20% PG, 10% CL; trimethyl PE liposomes, 70% dioleoyl phosphatidylcholine, 20% PG, 10% CL.
Pure protein was reconstituted into liposomes as described previously (13). Lipids were mixed, dried under nitrogen, and resuspended in internal buffer (100 mm KCl, 20 mm HEPES-Tris, pH 7.5) by sonication using a cylindrical sonicator (Laboratory Supplies Co.). The lipid suspension was frozen in liquid nitrogen and thawed at least six times. Liposomes were formed by extrusion through 400-nm polycarbonate membranes (Avanti Polar Lipids) and treated with Triton X-100 at a ratio determined by swelling of vesicles measured by light scattering of vesicles (absorbance at 540 nm) (13, 14). Protein was added at 2.5 μg of protein/mg of lipid. The protein/lipid mixture was left at room temperature for 30 min before detergent was removed using SM2 Biobeads (Bio-Rad). The proteoliposome mixture was incubated with gentle agitation with four consecutive batches of Biobeads (120 mg/ml). Liposomes were concentrated by centrifugation at 150,000 × g for 30 min, resuspended at 100 mg of lipid/ml, and either used immediately or flash frozen in liquid nitrogen and stored at −80 °C.
Transport Assays
Transport function of GltPh was measured by l-[3H]aspartate radiolabel uptake (13). Unless stated otherwise, liposomes (100 mg/ml) were diluted 133-fold into uptake buffer (100 mm NaCl, 20 mm HEPES-Tris, pH 7.5, 1 μm valinomycin, 100 nm l-[3H]aspartate) at 30 °C. At each time point, an aliquot was removed and diluted 10-fold into ice-cold quench buffer (100 mm LiCl, 20 mm HEPES-Tris, pH 7.5) followed by immediate filtration over nitrocellulose filters (0.22-μm pore size, Millipore). The filters were washed with quench buffer under vacuum; filters were combined with scintillation fluid and assayed for radioactivity using a Trilux β counter (PerkinElmer Life Sciences).
The Na+ dependence of l-[3H]aspartate transport was determined by varying the extraliposomal Na+ concentration from 0.3 to 100 mm. Choline chloride was used to balance osmolarity. The K0.5 of aspartate transport was measured by varying the extraliposomal l-[3H]aspartate concentration from 3 to 1000 nm, maintaining NaCl concentrations at 100 mm. The influence of lipid bilayer composition on the transport-active limb of the GltPh transport cycle was explored by loading liposomes with 100 mm NaCl, 20 mm HEPES-Tris, pH 7.5, 100 μm l-aspartate and diluting liposomes into uptake buffer as described previously (11). Initial rates of transport represent the amount of l-[3H]aspartate transport over the linear portion of the time course.
Thiol modification was carried out by treating liposomes with 1 mm 2-(trimethylammonium)ethyl methanethiosulfonate (MTSET) for 5 min at room temperature. This isolated the inside out orientation of GltPh. MTSET is membrane-impermeable, and inhibition is complete under these conditions. Higher concentrations of MTSET or longer reaction times did not result in more than a 50% reduction in transport (see Fig. 2B). The reducing agents dithiothreitol (DTT; 1 mm) and tris(2-carboxyethyl) phosphine (TCEP; 20 mm) were used to reduce GltPh thiol modification. To isolate the right-side out (RSO) orientation of GltPh, liposomes containing 1 mm MTSET were diluted into uptake buffer containing 20 mm TCEP, which is able to rescue the RSO orientation. The uptake was performed as soon as the liposomes were in the TCEP-containing buffer. The rate of transport was ∼50% of that of untreated liposomes, which demonstrates that the reduction of A364C by excess TCEP is immediate. The residual amount of MTSET was very low (15 μm, which was ∼6-fold lower than the concentration of MTSET that gave 10% inhibition (100 μm; see Fig. 2B).
FIGURE 2.
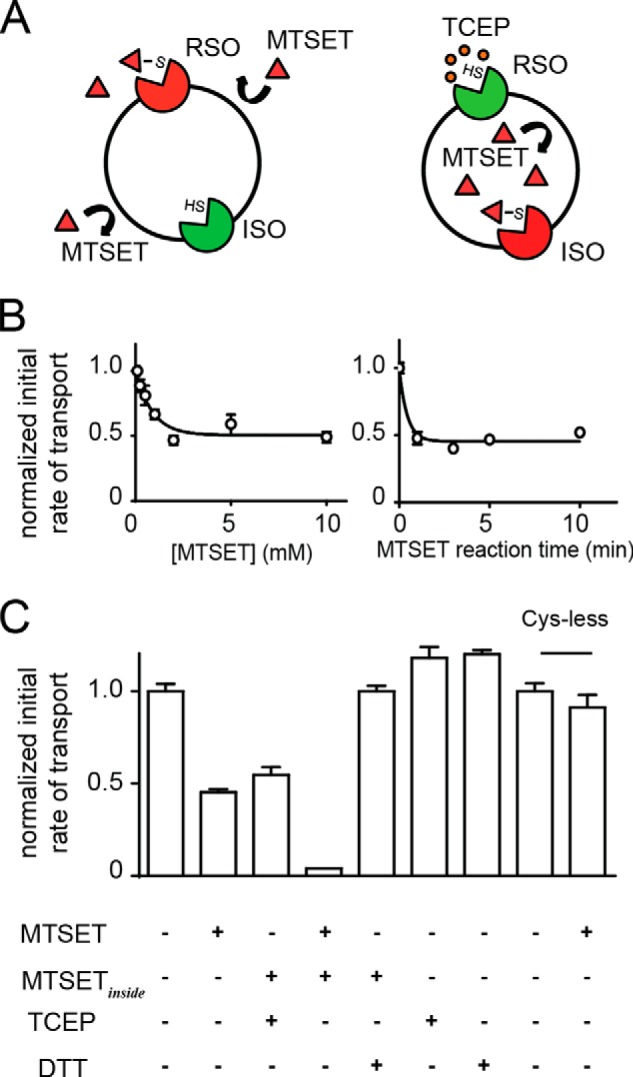
Thiol modification strategies to observe sided properties of GltPh. A, A364C was only accessible from one side of the membrane. Following treatment of liposomes with 1 mm MTSET (red triangles) A364C is modified resulting in inhibition of the RSO transporters while the ISO transporters are still functional as A364C is unmodified (represented by SH). The strategy for modifying ISO transporters involved loading liposomes with MTSET (MTSETinside) (100 mm KCl, 20 mm HEPES-Tris, 1 mm MTSET) followed by performing uptake in buffer containing TCEP (orange circles) (100 mm NaCl, 20 mm HEPES-Tris, pH 7.5, 1 μm valinomycin, 20 mm TCEP, 100 nm l-[3H]aspartate) to selectively rescue RSO transporters. B, concentration-response data for the effect of increasing incubation concentrations of MTSET on A364C in liposomes. Data were fit to an inhibitor concentration-response curve for display purposes. 1 mm MTSET was incubated with liposomes containing A364C for the indicated times, and then uptake was performed in buffer containing 100 mm NaCl, 20 mm HEPES-Tris, pH 7.5, 1 μm valinomycin, 100 nm l-[3H]aspartate. Transport rates were normalized to untreated liposomes. C, initial rates of transport of A364C were reduced by ∼50% with MTSET treatment or when MTSET-loaded liposomes (MTSETinside) were treated with TCEP. Full inhibition of both transporter populations was achieved by treating MTSET-loaded liposomes (MTSETinside) with MTSET. Inhibition by both transporter populations was fully reversible following treatment with DTT. DTT and TCEP had no effect on untreated A364C in liposomes. Application of MTSET was shown to have no effect on Cys-less GltPh reconstituted in liposomes. Data represent the mean of experiments performed in triplicate, and error bars indicate S.E.
Biochemical Evaluation of Transmembrane Orientation
Liposomes were reconstituted with His-tagged GltPh (20 μg of protein/mg of lipid) and subjected to treatment with thrombin (10 units/mg of protein). Proteolysis was terminated by the addition of 10 mm EDTA and 1 mm AEBSF. Liposomes were centrifuged at 150,000 × g for 20 min, and pellets were resuspended in 12% SDS loading buffer and loaded on a 10–20% SDS-polyacrylamide gel.
Amido Black Quantification
A modified Amido Black assay (15), which is used to quantify microgram quantities of protein in the presence of milligram quantities of lipids, which can often interfere with more traditional visual colorimetric assays, was conducted. Liposome samples were dissolved with SDS, and protein was precipitated with trichloroacetic acid (Sigma) followed by vortexing. Samples were filtered under vacuum, and mixed cellulose ester membranes were stained with Amido Black stain. Protein spots were excised and soaked in elution solution, and the absorbance of the eluate was measured at 530 nm.
Data Analysis
Analysis of kinetic data was conducted using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). All values presented are the mean ± S.E. of experiments performed in triplicate. One-way analysis of variance was performed with a Dunnett's post hoc test. The p values <0.05 were interpreted as statistically significant and are indicated in figures as * for p < 0.05, ** for p < 0.01, and *** for p < 0.001.
RESULTS
The Lipid Bilayer Influences Aspartate Transport Rates by GltPh
The aim of this study was to investigate the role of the lipid environment in GltPh function by varying the lipid composition of the liposomes in which GltPh is reconstituted. Aspartate transport via GltPh is coupled to the co-transport of three Na+ ions, and in the presence of an inwardly directed Na+ gradient, GltPh will transport aspartate into liposomes (Fig. 1, C and D). A common lipid condition used when studying GltPh is a 3:1 mixture of Escherichia coli polar lipid extract to trimethyl PE. Trimethyl PE is commonly referred to as 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; however, we have chosen to use the N-methyl nomenclature here. The E. coli polar lipid extract is composed of ∼10% CL, a bulky mitochondrial membrane lipid; ∼20% PG; and ∼70% PE (10, 12, 13, 16).
Purified GltPh protein was reconstituted in liposomes in which the primary phospholipid species was PE or its monomethyl, dimethyl, or trimethyl derivative while maintaining a constant amount of CL (10%) and PG (20%) (Fig. 1E). Similar lipid compositions have been used in previous studies to investigate the influence of lipid bilayer composition on two multidrug transporters, LmrP (17) and HorA (18). When GltPh was reconstituted in liposomes consisting primarily of PE, the initial rate of aspartate transport was 8.25 ± 0.45 nmol/mg of protein/min. Interestingly, when the primary lipid species was mono- or dimethyl PE, the rates were reduced by ∼50% to 3.21 ± 0.26 and 3.34 ± 0.33 nmol/mg of protein/min, respectively. Furthermore, when the primary lipid species was trimethyl PE, the rates of transport were reduced by a further 25% to 1.33 ± 0.06 nmol/mg of protein/min (Fig. 1F). All rates were normalized to the amount of protein per lipid that was determined by a modified Amido Black assay (15) as there was some variability in the reconstitution efficiency in the different lipid compositions.
Kinetic Analysis of Transport by GltPh
We sought to determine whether our observed influence of the lipid bilayer composition was due to changes in the apparent affinity of Na+ and/or aspartate for GltPh. In the presence of saturating Na+ concentrations, we determined that the half-maximal rates of aspartate transport were not significantly different comparing the mono-, di-, or trimethyl lipid compositions with PE (Table 1). The Na+ dependence of transport was also determined by measuring the rate of transport with increasing concentrations of Na+ in the presence of saturating l-[3H]aspartate, and similarly, no difference was found between the different lipid compositions (Table 1). These results indicate that the differences in initial rates of l-[3H]aspartate transport via GltPh in the different lipid compositions are not due to differences in the apparent affinity of l-aspartate or Na+.
TABLE 1.
The effect of lipid bilayer composition on kinetics of transport by GltPh
Shown are the initial rate of l-[3H]aspartate transport as a function of [l-[3H]aspartate] in the outside buffer in the presence of saturating Na+ concentrations and the initial rate of l-[3H]aspartate transport as a function of [Na+] in the outside buffer in the presence of saturating l-[3H]aspartate concentrations. Kinetic parameters for GltPh reconstituted in varying lipid compositions were derived by fitting the l-aspartate concentration-response data to the Michaelis-Menten equation and fitting the Na+ concentration-response data to the Hill equation. The values are averaged from experiments performed in triplicate, and errors indicate S.E. No significant difference was observed for K0.5 values.
| 3:1 E. coli polar lipid extract:trimethyl PE | PE | Monomethyl PE | Dimethyl PE | Trimethyl PE | |
|---|---|---|---|---|---|
| Aspartate K0.5 (nm) | 84.7 ± 8.6 | 57.9 ± 8.8 | 62.8 ± 5.9 | 67.5 ± 10.6 | 82.4 ± 14.6 |
| Aspartate Vmax (nmol mg−1 min−1) | 10.9 ± 0.3 | 11.1 ± 0.4 | 6.1 ± 0.1 | 3.4 ± 0.1 | 1.6 ± 0.1 |
| Na+ K0.5 (mm) | 2.6 ± 0.2 | 3.2 ± 0.5 | 3.2 ± 0.6 | 3.4 ± 0.5 | 3.0 ± 0.2 |
Transmembrane Orientation of GltPh in Liposomes
When GltPh is reconstituted into the 3:1 E. coli polar lipid extract:trimethyl PE lipid composition, the transmembrane orientation distribution is ∼50% in the RSO and ∼50% in the inside out (ISO) orientations (13). To investigate whether transporter orientation in the different lipid compositions was responsible for the variations in rates of transport observed, we established a method for determining the orientation of transporters in liposomes using a cysteine-substituted GltPh construct and a thiol modification assay similar to a method used by Tsai et al. (19) for the arginine/agmatine antiporter AdiC.
When studying excitatory amino acid transporter 1 in Xenopus laevis oocytes in which the transporters are presumed to be correctly oriented, modification of a cysteine residue introduced into HP2 (V452C) with the membrane-impermeable methanesulfonate reagent MTSET inhibits l-[3H]glutamate transport by >90% (20). This near-complete inhibition of transport suggests that V452C is accessible to the extracellular environment, and modification with MTSET prevents excitatory amino acid transporter 1 from transporting glutamate. In GltPh, the equivalent residue in HP2 is Ala-364 (Fig. 1A). The A364C mutation was introduced into a cysteine-less GltPh background, purified to homogeneity, and reconstituted into the control lipid mixture (3:1 E. coli polar lipids:trimethyl PE). Fig. 2A illustrates the strategy for selectively isolating the ISO (left) and RSO (right) orientations of GltPh. An MTSET concentration-response and modification time course was performed to determine the conditions required to completely modify A364C (Fig. 2B). When A364C-containing liposomes were treated with 1 mm MTSET for 5 min, rates of transport were reduced by ∼50%, which indicates that 50% of the transporter population (RSO) had been silenced (Fig. 2C). The reciprocal modification of the ISO transporters was performed by incorporation of 1 mm MTSET on the inside of the liposome followed by application of the membrane-impermeable reducing agent TCEP, which selectively rescues the RSO transporters, to the outside of the liposomes. These results demonstrate that the RSO and ISO populations were each contributing ∼50% of the transport of untreated liposomes (Fig. 2C). When 1 mm MTSET was present on both sides of the liposome, transport was reduced to background levels, suggesting that both RSO and ISO transporters had been silenced by MTSET modification. Function could be rescued to pre-MTSET modification levels by treatment with DTT, which is a membrane-permeable reducing agent. Finally, we showed that transport by Cys-less GltPh was not affected by application of MTSET (Fig. 2C).
To confirm that the ∼50% reduction in transport represents ∼50% of the protein oriented in RSO or ISO orientation, we reconstituted His-tagged GltPh (GltPh-His) into the 3:1 control condition and treated these liposomes with thrombin. Samples were separated on a 10–20% acrylamide gel, and two bands that correspond to GltPh-His and cleaved GltPh were observed. We performed a time course of thrombin digestion. After 1 min, two bands appeared, the reaction was stable up to 6 h, and the bands were of equal density (Fig. 3A). This demonstrates that thrombin can only access around half of the protein to cleave the His tag and correlates with our uptake data where MTSET treatment reduced transport rates by ∼50%. Taken together, these results demonstrate that A364C is only accessible from one side of the membrane, and through the use of cysteine-reactive methanesulfonate agents and reducing agents, we can selectively modify each population of transporters (RSO or ISO).
FIGURE 3.
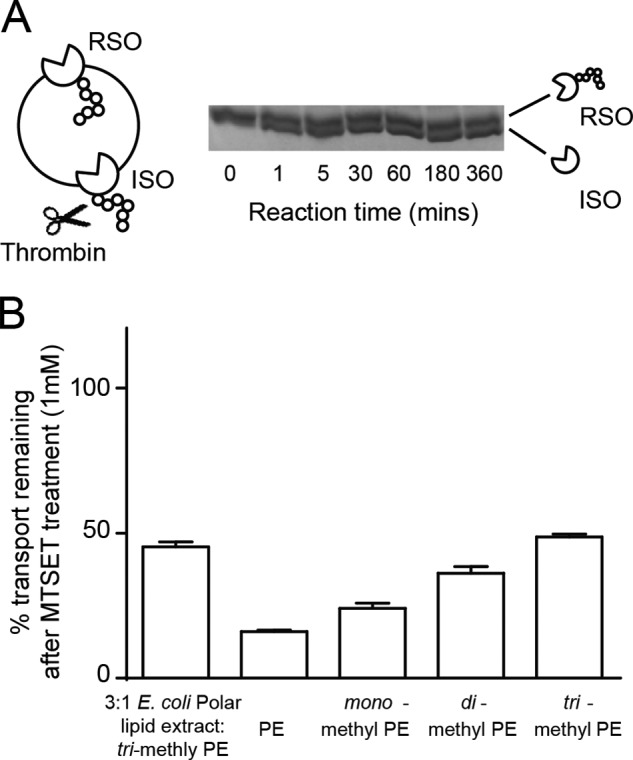
Inhibition of RSO transporters reveals the distribution of transmembrane orientation. A, strategy for thrombin cleavage of His-tagged GltPh reconstituted in 3:1 E. coli polar lipid extract:trimethyl PE. The hexahistidine tag is susceptible to thrombin cleavage (scissors). Thrombin (10 units/mg of protein) was added to liposomes containing His-GltPh (20 μg/mg of lipid). At the indicated time points, the digest was stopped with 1 mm AEBSF and 10 mm EDTA. Samples were run on a 10–20% SDS-polyacrylamide gel. B, percentage of transport remaining after 1 mm MTSET (in 100 mm KCl, 20 mm HEPES-Tris, pH 7.5) incubation of A364C reconstituted in liposomes. Data were normalized to transport of untreated liposomes in each lipid composition. Data represent the mean of experiments performed in triplicate, and error bars indicate S.E.
This thiol modification strategy allowed us to probe the sidedness of the transporters to determine the kinetic parameters of the RSO and ISO populations as well as understand the effect that lipid bilayer composition has on the transmembrane orientation of GltPh. We measured half-maximal rates of aspartate transport in the presence of 100 mm Na+ and found that GltPh has an aspartate K0.5 of 39.3 ± 6.7 and 140.7 ± 26.2 nm for the RSO and ISO orientations, respectively (Table 2). This observation supports the role of GltPh as a concentrative aspartate transporter that has a higher affinity for aspartate when the binding site is facing the external side of the cell. We also determined that the Na+ dependence of transport was similar for both RSO and ISO orientations with Na+ K0.5 values of 2.2 ± 0.2 and 2.1 ± 0.3 mm, respectively (Table 2).
TABLE 2.
Orientation of substrate binding site determines aspartate K0.5
Treatment of A364C-GltPh in liposomes with thiol-modifying agents allows determination of sided kinetics of transport. Shown are the initial rate of l-[3H]aspartate transport as a function of [l-[3H]aspartate] in the outside buffer in the presence of saturating Na+ concentrations and the initial rate of l-[3H]aspartate transport as a function of [Na+] in the outside buffer in the presence of saturating l-[3H]aspartate concentrations. Kinetic parameters for GltPh were derived by fitting the l-aspartate concentration-response data to the Michaelis-Menten equation and fitting the Na+ concentration-response data to the Hill equation. The values are averaged from experiments performed in triplicate, and errors indicate S.E.
| ISO | RSO | |
|---|---|---|
| Aspartate K0.5 (nm) | 140.7 ± 26.2 | 39.3 ± 6.7 |
| Na+ K0.5 (mm) | 2.1 ± 0.3 | 2.2 ± 0.2 |
We then used this strategy to selectively inhibit the RSO transporters to investigate the effect of the different lipid compositions on the orientation of GltPh in the liposomes. When A364C was reconstituted into PE liposomes, MTSET application reduced the initial rates of transport by 84.0 ± 0.5% (Fig. 3B), indicating that ∼84% of the transporters were in the RSO orientation. When reconstituted in monomethyl and dimethyl PE liposomes, rates were reduced by 76.0 ± 1.9 and 64.0 ± 2.3%, respectively. Finally, l-[3H]aspartate transport in trimethyl PE liposomes was reduced by 51 ± 1% following treatment with MTSET (Fig. 3B). These results demonstrate that when GltPh was reconstituted in lipids in which the headgroups are unmethylated (such as PE) the transporters are more correctly oriented. In addition, the outwardly directed substrate binding site of GltPh had a 3.5-fold higher affinity for aspartate than the inwardly directed binding site (Table 2). In combination, these results only account for an ∼18% difference in the rate of transport we observed and cannot fully account for the ∼75% difference in transport rate observed between PE- and trimethyl PE liposomes. Therefore, other factors to explain these differences must exist.
Lipid Bilayer Composition Influences Substrate-bound Isomerization of GltPh
The transport cycle of GltPh begins as an empty transporter with binding sites for Na+ and aspartate that are outward facing (Fig. 4A). Once Na+ and aspartate have bound (step i), the loaded transporter isomerizes from outward facing to inward facing (step ii). The strong electrochemical gradient for Na+ prompts unbinding of the coupled ions and aspartate (step iii) to leave an empty transporter with binding sites for Na+ and aspartate facing inward that then relocates (step iv) back to the outward facing state. As we have shown that there was no appreciable difference in the apparent affinity (K0.5) for aspartate and Na+ in the various lipid compositions (Table 1), we hypothesized that isomerization of the substrate-loaded (step ii) or substrate-free (step iv) transporter was influenced by lipid bilayer composition. To isolate the substrate-loaded limb of the transport cycle (step ii), counterflow conditions were imposed in which liposomes loaded with 100 mm NaCl and 100 μm l-aspartate were diluted into a buffer containing equimolar Na+ and trace amounts of l-[3H]aspartate. Under these conditions, external l-[3H]aspartate was exchanged for internal unlabeled l-aspartate, providing us with a measure of the substrate-loaded isomerization limb of the GltPh transport cycle (step ii) (Fig. 4A, inset).
FIGURE 4.
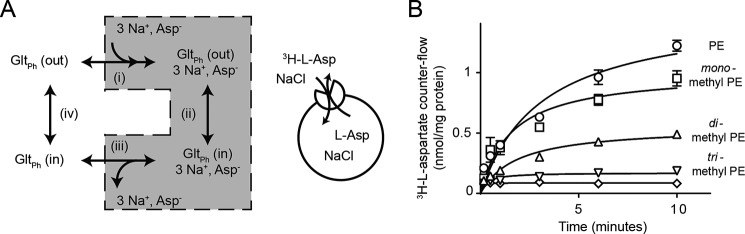
Lipid bilayer composition influences substrate-loaded isomerization of GltPh. A, schematic of GltPh transport cycle where the empty transporter (GltPh (out)) is in the outward facing state and may bind Na+ and aspartate (step i) to form the substrate-loaded complex (GltPh, 3 Na+, Asp−). This complex can isomerize from the outward facing state to the inward facing state (step ii). The substrate and co-transported ions dissociate (step iii), leaving an empty transporter in the inward facing state (GltPh (in)) to relocate to the outward facing state (step iv). Inset, schematic of experimental conditions for counterflow experiments where Na+- and aspartate-loaded liposomes were used to isolate the transport-active limb of the transport cycle (shaded). B, time course of l-[3H]aspartate accumulation in liposomes. Counterflow assays were performed with internal buffer containing 100 mm NaCl, 20 mm HEPES-Tris pH 7.5, 100 μm unlabeled l-aspartate and an external buffer containing 100 mm NaCl, 20 mm HEPES-Tris, 100 nm l-[3H]aspartate. Symbols represent PE liposomes (open circles), monomethyl PE liposomes (open squares), dimethyl PE liposomes (open triangles), and trimethyl PE liposomes (inverted open triangles). Background is shown as open diamonds. Data represent the mean of experiments performed in triplicate, and error bars indicate S.E.
Under counterflow conditions, the highest rates of exchange were observed in PE liposomes at a rate of 128.8 ± 9.5 nmol/mg of protein/min. Monomethyl and dimethyl PE liposomes had rates of transport of 111.3 ± 5.1 and 69.3 ± 5.2 nmol/mg of protein/min, respectively. Finally, the lowest rate of 19.4 ± 2.1 nmol/mg of protein/min was observed in trimethyl PE liposomes (Fig. 4B). This trend is strikingly similar to the initial rates of transport that we observed in the complete transport cycle in the various lipid compositions. The exchange conditions isolate the binding/unbinding of Na+ and aspartate as well as the isomerization of the substrate-bound transporter. As GltPh orientation can only account for 18% of the difference in rate observed between PE and trimethyl PE liposomes, we propose that the lipid composition also influences the ability of the substrate-loaded transporter to move through the membrane.
Rationale for Site-specific Interactions
The structure of the intermediate outward facing state (Protein Data Bank code 3V8G) of GltPh proposes that although the trimerization domain is primarily rigid there are small movements into the surrounding lipid bilayer (9). The ∼7-Å flexing movement of TM1 (Fig. 5A) in particular is believed to accommodate the rotation of the transport domain as it completes its isomerization between the outward facing state and inward facing state. We asked whether a residue on TM1 could affect the favorability of this movement into the surrounding lipid bilayer, for example through direct protein-lipid interactions or steric bulk, and could account for the sensitivity of GltPh to the lipid environment. In GltPh, the upper portion of TM1 contains a tyrosine (Tyr-33) residue that may be involved in site-specific interactions (Fig. 5A) consistent with x-ray crystallographic structures of bovine PE-binding protein and murine PE-binding protein bound to zwitterionic lipids (21, 22).
FIGURE 5.

Functional properties of tyrosine 33 mutants. A, superimposition of the trimerization domain of the outward facing (green; Protein Data Bank code 2NWX), intermediate outward facing (pink; Protein Data Bank code 3V8G), and inward facing (blue; Protein Data Bank code 3KBC) crystal structures. Structures were aligned to TM4. The transport domain was omitted for clarity. Tyr-33 is shown as spheres. The figure was made using the program PyMOL (38). B, size exclusion column profile for wild-type GltPh (black), Y33F (red), Y33S (orange), Y33W (yellow), Y35F (green), and Y35S (blue). Inset, SDS-PAGE of purified wild-type GltPh (lane 2), Y33F (lane 3), Y33S (lane 4), Y33W (lane 5), Y35F (lane 6), and Y35S (lane 7). Lane 1 contains ladder. C, -fold change in l-[3H]aspartate transport rates for wild-type and mutant GltPh reconstituted in PE liposomes. D, -fold change in l-[3H]aspartate transport rates for wild-type and mutant GltPh reconstituted in trimethyl PE liposomes. Data represent the mean of experiments performed in triplicate, and error bars indicate S.E. with p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) compared with WT in each lipid species.
The involvement of Tyr-33 in a lipid-protein interaction was investigated through mutagenesis. The phenol side chain of tyrosine imparts both an aromatic nature and hydrogen-bonding capacity. To dissect these properties, Y33F was generated to remove the hydrogen-bonding capacity of the phenol hydroxyl, and Y33S was generated to maintain hydrogen-bonding capacity of the side chain but remove the aromaticity and side chain bulk. Mutant transporters were purified to homogeneity and shown to be functional when reconstituted into control lipid conditions with no significant change in the K0.5 for aspartate compared with wild-type GltPh (Fig. 5B and Table 3). The mutant transporters were then reconstituted into liposomes composed of PE and trimethyl PE, and rates of transport were examined.
TABLE 3.
The effect of mutagenesis of Tyr-33 on kinetics of transport by GltPh
Shown are kinetic parameters for mutant GltPh reconstituted in liposomes composed of a 3:1 mixture of E. coli polar lipid extract:trimethyl PE. Data were derived by fitting the l-aspartate concentration-response data to the Michaelis-Menten equation. The values are averaged from experiments performed in triplicate, and errors indicate S.E. No significant difference was observed for K0.5 values.
| WT | Y33F | Y33S | Y33W | Y35F | Y35S | |
|---|---|---|---|---|---|---|
| Aspartate K0.5 (nm) | 84.7 ± 8.6 | 86.3 ± 13.0 | 116.0 ± 19.8 | 61.0 ± 9.0 | 57.5 ± 9.5 | 57.7 ± 10.6 |
When reconstituted into PE liposomes, Y33F showed initial rates of transport comparable with wild-type GltPh (WT), whereas the rate of transport of Y33S was 1.9 ± 0.1-fold higher than that of WT (Fig. 5C). These effects were even more marked when Y33S was reconstituted into trimethyl PE liposomes, which displayed a 3.3 ± 0.1-fold increase in initial rate (Fig. 5D). These results are consistent with a cation-π interaction occurring between Tyr-33 and the zwitterionic lipid headgroups. By maintaining this π interaction in the Y33F mutant, we observed rates comparable with WT. When we removed the potential of cation-π interactions entirely via Y33S, rates increased significantly. Y33W was also generated but was found to function ∼0.64-fold less efficiently in PE liposomes and 0.81-fold less efficiently in trimethyl PE liposomes (Fig. 5, C and D). Tryptophan residues at the edges of transmembrane domains typically serve to anchor transmembrane helices to the lipid bilayer (23), and the Y33W mutation may reduce the ability of the transporter to undergo important conformational changes within the lipid bilayer. To address the site specificity of this putative cation-π interaction, we created similar mutations at a nearby tyrosine residue (Tyr-35). Although changes were observed when Y35 was mutated to Phe and Ser, the nature of the residue at position 35 was not affected by lipid composition to the same extent as the residue at position Tyr-33 (Fig. 5, C and D).
DISCUSSION
In this study, we investigated the effect of the lipid environment on the function of the aspartate transporter GltPh, a prokaryotic homologue of the SLC1 family of membrane transporters. We demonstrated a functional effect of the headgroup chemistry of the lipids in which GltPh was reconstituted that was due to a combination of transmembrane orientation and constraining cation-π interactions between the lipid headgroups and a tyrosine residue in TM1.
A common lipid composition used when studying reconstituted membrane proteins is a 3:1 mixture of E. coli polar lipid extract to trimethyl PE. In this system, GltPh is orientated ∼50% RSO and 50% ISO (13). We have developed a novel thiol modification system using a single cysteine mutant (GltPh A364C) and methanesulfonate reagents that allows us to study the sided kinetics of GltPh. Using this method, we showed that in liposomes in which the predominant lipid is PE the ratio of RSO to ISO changed to 85% RSO and 15% ISO. This suggests that the nature of lipid headgroups is an important factor in determining transporter orientation that presumably is due to specific lipid headgroup-protein interactions. The effect of lipid composition orienting membrane proteins has also been observed with proteorhodopsin in lipid bilayers (24). This method allowed us to determine the K0.5 for aspartate and Na+ for each face of the transporter. Although we saw no difference in the affinity of Na+ for either side of the transporter, we observed that the affinity for aspartate was ∼3.5-fold higher for the RSO compared with ISO transporters. This is in contrast to work conducted by Reyes et al. (25) who observed similar affinities for aspartate for both sides of the transporter when measuring binding to detergent-solubilized protein. In contrast, our transport assays represent the ensemble of binding, unbinding, and substrate translocation (K0.5) of GltPh reconstituted into a lipid bilayer and are in agreement with studies on the human and salamander glutamate transporters also showing a higher affinity for glutamate for the extracellular facing binding site (26–28). The difference in K0.5 for the RSO compared with ISO transporters that we observed can partially explain the different rates observed in the PE liposomes compared with the trimethyl PE liposomes, but other factors are required to fully explain the observed effects (Fig. 6A).
FIGURE 6.
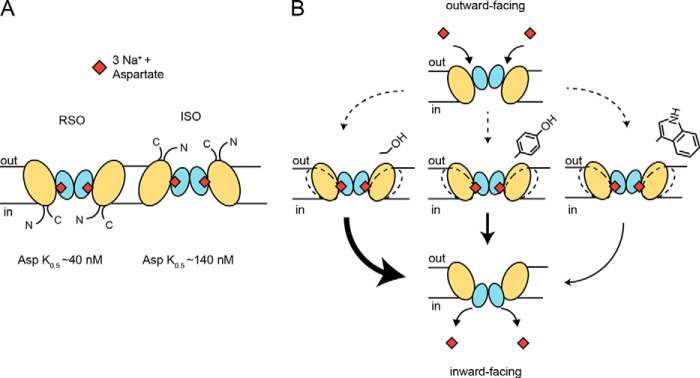
Schematic of transporter modulation by lipid bilayer composition. A, GltPh reconstituted in liposomes orients as either the RSO or ISO transmembrane orientation, and these two populations have different K0.5 values for aspartate transport. The transport domain is represented in blue, and the trimerization domain is colored wheat. B, movements of the trimerization domain into the surrounding lipid bilayer are made more or less favorable (arrow thickness) depending on the amino acid residue present at position 33. Movement of the trimerization domain into the surrounding lipid bilayer is required for transport.
The outward occluded and inward occluded crystal structures of GltPh suggest that the transport domain undergoes a large movement (∼18 Å) relative to the stable trimerization domain during transport (8). However, subtle movements of the trimerization domain have been observed, and it was proposed that these movements are required to accommodate the large movement of the transport domain (9). We propose that the likelihood of residues engaging a cation-π interaction varies with the different lipid headgroups, which in turn affect the mobility of TM1 and thus the rate of translocation. When we consider the chemistry of these lipid headgroup moieties, continuing along the series from trimethyl PE to PE, the positive charge of the headgroup becomes more labile and is less able to engage a cation-π interaction. Removing this putative cation-π interaction by altering the lipids into which GltPh is reconstituted allows the requisite movement of TM1 and thus translocation to be more dynamic.
Our results also demonstrate lipid-specific effects that can be augmented by the nature of a residue at the extracellular edge of TM1 that is predicted to face into the lipid membrane. When the Y33S mutant was reconstituted in the strong cation-π partner lipid trimethyl PE, it displayed an approximately 3-fold increase in initial rates compared with WT reconstituted in the same lipid. We suggest that this is because the constraining cation-π interaction is now absent due to the serine mutation. Conversely, the Y33W mutant reduced transport rates compared with wild type, which may be explained by the side chain forming stronger cation-π interactions, anchoring TM1 of GltPh to the membrane, slowing important structural movements required for the isomerization of the substrate-loaded transporter. These results further support our proposal that movements of TM1 into the surrounding lipid bilayer are determined by both the lipid bilayer composition and the chemical nature of residues on TM1 (Fig. 6B). Although our results provide specific examples of how the lipid bilayer can influence transport rates in GltPh, they do not preclude the influence of diffuse, nonspecific features of the bilayer such as intrinsic curvature or steric bulk on transport rates.
Other studies investigating the dynamics of GltPh using a variety of methods have also observed an effect of lipids in determining transport properties. Electron paramagnetic resonance studies by Hänelt et al. (29) have shown a pronounced role of the lipid bilayer to favor intermediate conformations (like that of the intermediate outward facing state), and single molecule fluorescence resonance energy transfer studies suggest that the intermediate conformations of GltPh are rate-determining (30, 31). Finally, a molecular dynamics study using GltPh found that inclusion of constraints imposed by the lipid bilayer was required to permit the conformational changes necessary to allow alternating access of the transporter (32). Our data reveal an effect of the lipid bilayer on isomerization of the Na+ and substrate-loaded transporter (Fig. 4) and further support the growing evidence for a specific role of the lipid bilayer in GltPh function.
The influence of the lipid bilayer composition appears to be a feature shared across a variety of membrane-bound proteins (33–37). Hakizimana et al. (17) and Gustot et al. (18) have observed an influence of lipid bilayer composition on the multidrug transporters LmrP and HorA, respectively. A conserved aspartate residue in LmrP is believed to participate in a hydrogen bond with the lipid headgroups of PE, monomethyl PE, and dimethyl PE. However, transport is abrogated when LmrP is reconstituted in trimethyl PE because this requisite hydrogen bonding is no longer possible (17). In HorA, the tilt angle of the α helices vary by ∼10° when PE was compared with trimethyl PE, resulting in an uncoupling of ATP hydrolysis and substrate transport (18).
In addition to influences of transmembrane orientation, we have identified a site-specific, lipid-specific interaction between GltPh and the lipid bilayer that we propose controls the substrate-loaded isomerization step. GltPh cannot progress through the transport cycle without entering intermediate conformations, and lipid bilayer composition, reported through Tyr-33, influences the favorability of GltPh to enter these rate-determining intermediate conformations. Our results offer a putative mechanism for GltPh to be coupled to the chemical composition of the membrane.
Acknowledgment
We thank members of the Transporter Biology Group at the University of Sydney for helpful discussions.
Note Added in Proof
The following review of the effects of the lipid environment on membrane protein topology and function has been added to those cited in the version of this article that was published on February 20, 2015 as a Paper in Press: Bogdanov, M., Dowhan, W., and Vitrac, H. (2014) Lipids and topological rules governing membrane protein assembly. Biochim. Biophys. Acta 1843, 1475–1488.
This work was supported in part by National Health and Medical Research Council of Australia Project Grant APP1048784.
- GltPh
- aspartate transporter from P. horikoshii
- PE
- phosphatidylethanolamine
- TM
- transmembrane domain
- HP
- hairpin loop
- MTSET
- 2-(trimethylammonium)ethyl methanethiosulfonate
- TCEP
- tris(2-carboxyethyl)phosphine
- PG
- phosphatidylglycerol
- CL
- cardiolipin
- RSO
- right-side out
- ISO
- inside out
- AEBSF
- 4(2aminoethyl)benzenesulfonyl fluoride.
REFERENCES
- 1. van Meer G., Voelker D. R., Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee A. G. (2004) How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 1666, 62–87 [DOI] [PubMed] [Google Scholar]
- 3. Valiyaveetil F. I., Zhou Y., MacKinnon R. (2002) Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry 41, 10771–10777 [DOI] [PubMed] [Google Scholar]
- 4. Andersen O. S., Koeppe R. E. (2007) Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 36, 107–130 [DOI] [PubMed] [Google Scholar]
- 5. Vandenberg R. J., Ryan R. M. (2013) Mechanisms of glutamate transport. Physiol. Rev. 93, 1621–1657 [DOI] [PubMed] [Google Scholar]
- 6. Yernool D., Boudker O., Jin Y., Gouaux E. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818 [DOI] [PubMed] [Google Scholar]
- 7. Boudker O., Ryan R. M., Yernool D., Shimamoto K., Gouaux E. (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393 [DOI] [PubMed] [Google Scholar]
- 8. Reyes N., Ginter C., Boudker O. (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Verdon G., Boudker O. (2012) Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 19, 355–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryan R. M., Mindell J. A. (2007) The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 14, 365–371 [DOI] [PubMed] [Google Scholar]
- 11. Mulligan C., Mindell J. A. (2013) Mechanism of transport modulation by an extracellular loop in an archaeal excitatory amino acid transporter (EAAT) homolog. J. Biol. Chem. 288, 35266–35276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Compton E. L., Taylor E. M., Mindell J. A. (2010) The 3–4 loop of an archaeal glutamate transporter homolog experiences ligand-induced structural changes and is essential for transport. Proc. Natl. Acad. Sci. U.S.A. 107, 12840–12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ryan R. M., Compton E. L., Mindell J. A. (2009) Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J. Biol. Chem. 284, 17540–17548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geertsma E. R., Nik Mahmood N. A., Schuurman-Wolters G. K., Poolman B. (2008) Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc. 3, 256–266 [DOI] [PubMed] [Google Scholar]
- 15. Kaplan R. S., Pedersen P. L. (1985) Determination of microgram quantities of protein in the presence of milligram levels of lipid with amido black 10B. Anal. Biochem. 150, 97–104 [DOI] [PubMed] [Google Scholar]
- 16. Bastug T., Heinzelmann G., Kuyucak S., Salim M., Vandenberg R. J., Ryan R. M. (2012) Position of the third Na+ site in the aspartate transporter GltPh and the human glutamate transporter, EAAT1. PLoS One 7, e33058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hakizimana P., Masureel M., Gbaguidi B., Ruysschaert J. M., Govaerts C. (2008) Interactions between phosphatidylethanolamine headgroup and LmrP, a multidrug transporter: a conserved mechanism for proton gradient sensing? J. Biol. Chem. 283, 9369–9376 [DOI] [PubMed] [Google Scholar]
- 18. Gustot A., Smriti, Ruysschaert J. M., McHaourab H., Govaerts C. (2010) Lipid composition regulates the orientation of transmembrane helices in HorA, an ABC multidrug transporter. J. Biol. Chem. 285, 14144–14151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai M.-F., Fang Y., Miller C. (2012) Sided functions of an arginine-agmatine antiporter oriented in liposomes. Biochemistry 51, 1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ryan R. M., Vandenberg R. J. (2002) Distinct conformational states mediate the transport and anion channel properties of the glutamate transporter EAAT-1. J. Biol. Chem. 277, 13494–13500 [DOI] [PubMed] [Google Scholar]
- 21. Simister P. C., Banfield M. J., Brady R. L. (2002) The crystal structure of PEBP-2, a homologue of the PEBP/RKIP family. Acta Crystallogr. D Biol. Crystallogr. 58, 1077–1080 [DOI] [PubMed] [Google Scholar]
- 22. Serre L., Vallée B., Bureaud N., Schoentgen F., Zelwer C. (1998) Crystal structure of the phosphatidylethanolamine-binding protein from bovine brain: a novel structural class of phospholipid-binding proteins. Structure 6, 1255–1265 [DOI] [PubMed] [Google Scholar]
- 23. de Planque M. R., Bonev B. B., Demmers J. A., Greathouse D. V., Koeppe R. E., 2nd, Separovic F., Watts A., Killian J. A. (2003) Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry 42, 5341–5348 [DOI] [PubMed] [Google Scholar]
- 24. Tunuguntla R., Bangar M., Kim K., Stroeve P., Ajo-Franklin C. M., Noy A. (2013) Lipid bilayer composition can influence the orientation of proteorhodopsin in artificial membranes. Biophys. J. 105, 1388–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reyes N., Oh S., Boudker O. (2013) Binding thermodynamics of a glutamate transporter homolog. Nat. Struct. Mol. Biol. 20, 634–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wadiche J. I., Kavanaugh M. P. (1998) Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J. Neurosci. 18, 7650–7661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brew H., Attwell D. (1987) Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature 327, 707–709 [DOI] [PubMed] [Google Scholar]
- 28. Szatkowski M., Barbour B., Attwell D. (1990) Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348, 443–446 [DOI] [PubMed] [Google Scholar]
- 29. Hänelt I., Wunnicke D., Bordignon E., Steinhoff H.-J., Slotboom D. J. (2013) Conformational heterogeneity of the aspartate transporter GltPh. Nat. Struct. Mol. Biol. 20, 210–214 [DOI] [PubMed] [Google Scholar]
- 30. Akyuz N., Altman R. B., Blanchard S. C., Boudker O. (2013) Transport dynamics in a glutamate transporter homologue. Nature 502, 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Erkens G. B., Hänelt I., Goudsmits J. M., Slotboom D. J., van Oijen A. M. (2013) Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters. Nature 502, 119–123 [DOI] [PubMed] [Google Scholar]
- 32. Lezon T. R., Bahar I. (2012) Constraints imposed by the membrane selectively guide the alternating access dynamics of the glutamate transporter GltPh. Biophys. J. 102, 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. daCosta C. J., Wagg I. D., McKay M. E., Baenziger J. E. (2004) Phosphatidic acid and phosphatidylserine have distinct structural and functional interactions with the nicotinic acetylcholine receptor. J. Biol. Chem. 279, 14967–14974 [DOI] [PubMed] [Google Scholar]
- 34. Baenziger J. E., Morris M.-L., Darsaut T. E., Ryan S. E. (2000) Effect of membrane lipid composition on the conformational equilibria of the nicotinic acetylcholine receptor. J. Biol. Chem. 275, 777–784 [DOI] [PubMed] [Google Scholar]
- 35. daCosta C. J., Baenziger J. E. (2009) A lipid-dependent uncoupled conformation of the acetylcholine receptor. J. Biol. Chem. 284, 17819–17825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schiller D., Ott V., Krämer R., Morbach S. (2006) Influence of membrane composition on osmosensing by the betaine carrier BetP from Corynebacterium glutamicum. J. Biol. Chem. 281, 7737–7746 [DOI] [PubMed] [Google Scholar]
- 37. Kapri-Pardes E., Katz A., Haviv H., Mahmmoud Y., Ilan M., Khalfin-Penigel I., Carmeli S., Yarden O., Karlish S. J. (2011) Stabilization of the α2 isoform of Na,K-ATPase by mutations in a phospholipid binding pocket. J. Biol. Chem. 286, 42888–42899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]