

Background: Mutation of the aspartate associated with Na+ site III of Na+,K+-ATPase causes neurological disorders.
Results: A second-site mutation distant from Na+ site III increases Na+ affinity, ATPase activity, and cellular K+ uptake in mutants with the replacement of the aspartate.
Conclusion: The defective pump function is rescued.
Significance: The rescue represents a novel principle for improvement of Na+,K+ pump function of disease mutants.
Keywords: Membrane Transport; Na+/K+-ATPase; Neurological Disease; Site-directed Mutagenesis; Sodium Transport; Na+,K+ Pump; P-type ATPase; Alternating Hemiplegia of Childhood; Rapid-onset Dystonia Parkinsonism; Second-site Revertant
Abstract
The Na+,K+-ATPase binds Na+ at three transport sites denoted I, II, and III, of which site III is Na+-specific and suggested to be the first occupied in the cooperative binding process activating phosphorylation from ATP. Here we demonstrate that the asparagine substitution of the aspartate associated with site III found in patients with rapid-onset dystonia parkinsonism or alternating hemiplegia of childhood causes a dramatic reduction of Na+ affinity in the α1-, α2-, and α3-isoforms of Na+,K+-ATPase, whereas other substitutions of this aspartate are much less disruptive. This is likely due to interference by the amide function of the asparagine side chain with Na+-coordinating residues in site III. Remarkably, the Na+ affinity of site III aspartate to asparagine and alanine mutants is rescued by second-site mutation of a glutamate in the extracellular part of the fourth transmembrane helix, distant to site III. This gain-of-function mutation works without recovery of the lost cooperativity and selectivity of Na+ binding and does not affect the E1-E2 conformational equilibrium or the maximum phosphorylation rate. Hence, the rescue of Na+ affinity is likely intrinsic to the Na+ binding pocket, and the underlying mechanism could be a tightening of Na+ binding at Na+ site II, possibly via movement of transmembrane helix four. The second-site mutation also improves Na+,K+ pump function in intact cells. Rescue of Na+ affinity and Na+ and K+ transport by second-site mutation is unique in the history of Na+,K+-ATPase and points to new possibilities for treatment of neurological patients carrying Na+,K+-ATPase mutations.
Introduction
Na+,K+-ATPase is a ubiquitous, membrane-bound enzyme responsible for creating vital gradients for Na+ and K+ across cell membranes. This enzyme belongs to the family of P-type ATPases, also including the closely related H+,K+-ATPase, Ca2+-ATPases, heavy metal-transporting ATPases, and ATPases with lipid substrates and unknown substrates. The common characteristic of these ATPases is the formation of a covalent phosphoenzyme intermediate as part of the energy transduction mechanism. For each ATP hydrolyzed by the Na+,K+-ATPase, 3 internal Na+ are exchanged for 2 external K+ in a reaction cycle (Scheme 1) involving sequential binding and release of Na+ and K+ and transition between two major conformational states, the so-called E1 (Na+-selective) and E2 (K+-selective) forms (1, 2). The Na+ and K+ binding sites are located in the membrane domain of the α-subunit of the protein, comprising 10 transmembrane helices (M1–M10). The three-dimensional structures of Na+,K+-ATPase in Na+- and K+-bound forms (3–5) show that one Na+ ion is bound at a unique, Na+-specific site (site III), whereas the other two Na+ ions bind at sites that overlap substantially with the K+ sites (so-called sites I and II) (Fig. 1). Site III interacts functionally with the C terminus of the protein (4, 6). The three Na+ ions bind cooperatively, and the crystal structure predicts that the first Na+ binds at site III with site I and II being filled consecutively thereafter, the occupation of site II leading to activation of phosphoryl transfer from bound ATP (3). Mutations in the α3-isoform of Na+,K+-ATPase have been found associated with the neurological disorders rapid-onset dystonia parkinsonism (RDP)2 and alternating hemiplegia of childhood (AHC), which likely are different phenotypical manifestations of the same disease (7–9). Some of these disease mutations reduce Na+ affinity (9–11), a defect that in intact cells leads to increase of the intracellular Na+ concentration, which may be instrumental in the development of disease (11). A mutation replacing the aspartate in M8 with asparagine (D923N) is present in some RDP and AHC patients (12–14) and causes as much as ∼200-fold reduction of the affinity for Na+ without impairment of K+ binding, thus associating this residue with the Na+-specific site III (10). It is unclear whether this huge effect reported for the neurological disease mutation is unique for the asparagine substituent or for the α3-isoform. In fact the high-resolution (2.8 Å) crystal structures of the pig α1 Na+,K+-ATPase in the E1(Na3)·AlF4−·ADP state (3) do not provide an unambiguous answer to the question whether the M8 aspartate (Asp-926/Asp-928 in pig/rat α1, Asp-930 in human α2, and Asp-923 in human α3) is a critical Na+-coordinating residue. Hence, the distance between the closest M8 aspartate side chain oxygen and the Na+ ion at site III differs between the two protomers, A and B, in the asymmetric unit, and also depends on the presence of oligomycin bound to the enzyme (3), the distances being 3.6 and 4.2 Å for the two protomers without oligomycin and 3.3 and 4.2 Å with oligomycin (Fig. 1). The protomers A and B might represent different snapshots of the Na+ binding process with Na+ at slightly different positions. Only the shortest distances of 3.3 and 3.6 Å, corresponding to protomer B, with and without oligomycin, respectively, can be considered sufficiently small to represent direct coordination of the Na+ ion by the aspartate, and even these distances are too large to be compatible with very tight interaction. Furthermore, it is only one of the two oxygen atoms of the M8 aspartate side chain that according to the crystal structure could be directly involved in the binding of Na+, contrary to some of the other aspartates of the Na+ binding sites that contribute both of their side-chain oxygen atoms to Na+ binding (Fig. 1). Hence, it is not immediately clear from the high-resolution crystal structures that mutation of the M8 aspartate should reduce Na+ affinity to the extent observed for the RDP/AHC mutation D923N in α3. Another crystal structure of the Na+-bound pig α1 Na+,K+-ATPase shows a 2.4 Å distance between Na+ at site III and the nearest Asp-926 side-chain oxygen in both protomers (15), but due to its low resolution (4.3 Å), the uncertainty is significant in this case. These ambiguities have led us to seek more information about the role of the aspartate in Na+ binding and the exact reason for the dramatic reduction of Na+ affinity in the α3-D923N mutant by studying the effect of replacing the aspartate with asparagine in other isoforms as well as the effects of other substituents.
SCHEME 1.

Post-Albers model of the Na+,K+-ATPase reaction cycle. E1 and E2 represent the main conformational states of the enzyme. Occluded Na+ and K+ ions are shown in brackets, and free ions are labeled c and e for cytoplasmic and extracellular, respectively. P indicates phosphorylation. Boxed ATP indicates ATP bound in a non-phosphorylating mode, enhancing the rate of K+ deocclusion, and accompanying E2-E1 conformational change.
FIGURE 1.
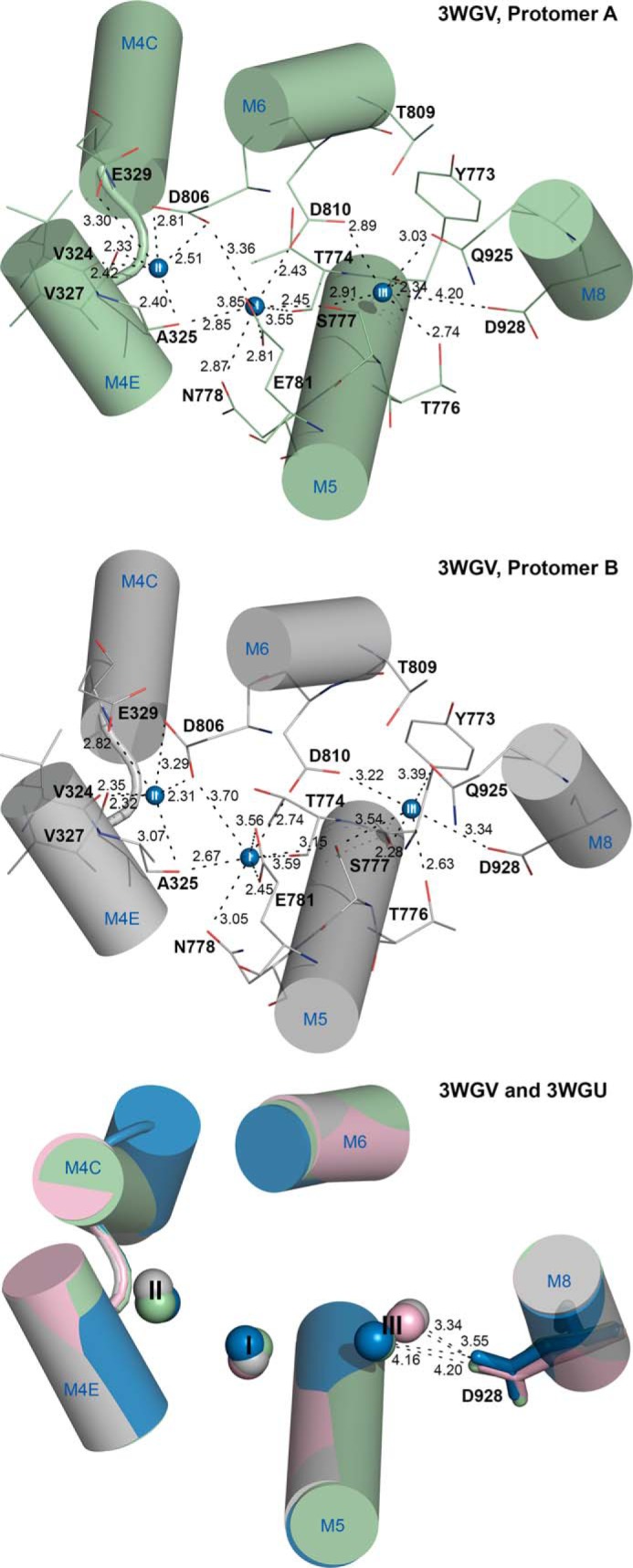
Disposition of Na+ binding residues in protomers of Na+,K+-ATPase E1(Na3)·AlF4−·ADP crystal structures. Shown is the view from the extracellular side. Relevant transmembrane helices are shown as cylinders with blue letters and numbers (M4C and M4E, cytoplasmic and extracellular parts of M4), and Na+ ions as spheres numbered I, II, and III. Broken lines with numbers indicate distances in Å. Atomic models (Protein Data Bank codes 3WGV (with oligomycin) and 3WGU (without oligomycin) (3)) were aligned with the M7–M10 transmembrane helices. Upper and middle panels show protomers A and B, respectively, of the 3WGV structure. Selected residues are shown as lines colored according to the elements (carbon, green or gray; oxygen, red; nitrogen, blue) and numbered according to the rat α1-isoform. The lower panel shows all four different protomers superposed, with the side chain of Asp-928 in stick representation. Color codes: green, 3WGV protomer A; gray, 3WGV protomer B; blue, 3WGU protomer A; pink, 3WGU protomer B.
During the course of this work, we unexpectedly discovered that the reduced affinity for Na+ caused by Asp-928 mutations could be compensated by introduction of a second-site mutation, E314D, of a glutamate located in the extracellular part of transmembrane helix M4, far away from Asp-928. Such a rescuing effect on Na+ binding by second-site mutation has not previously been described for the Na+,K+-ATPase and provides essential insight in the functioning of the Na+ sites.
EXPERIMENTAL PROCEDURES
Mutations were introduced by PCR into cDNA encoding either the rat α1-isoform or an ouabain-resistant version of the human α2-isoform of Na+,K+-ATPase (11). Mutants and wild type were expressed in COS-1 cells under ouabain selection pressure (16, 17). The cDNA was full-length sequenced before transfection as well as following isolation of stable transfectants, where it had become integrated in the genome. Functional analysis was carried out on the plasma membrane fraction of the cells made leaky by use of either sodium deoxycholate or alamethicin to allow access of ions and ATP from both sides of the membrane.
ATPase activity was determined at 37 °C by following the liberation of Pi within the linear time range (up to 20 min) using the colorimetric method of Baginski et al. (17–19). The various incubation media allowing determination of Na+, K+, ATP, and vanadate concentration dependences are detailed in the figure legends. In each case, the Na+,K+-ATPase activity associated with the expressed exogenous enzyme was calculated by subtracting the ATPase activity measured at 10 mm ouabain (inhibiting all Na+,K+-ATPase activity) from that measured at 10 μm ouabain (inhibiting the endogenous COS-1 cell Na+,K+-ATPase).
The transport activity was evaluated by measuring cellular K+ uptake using the K+ congener 86Rb+ as described previously (11). In short, COS-1 cells were grown to confluence in culture medium containing 5 μm ouabain and were incubated for 5 or 10 min at 37 °C and 5% CO2 in serum-free medium containing 154 mm Na+ and 5.3 mm K+, to which had been added 86Rb+ (0.5 μCi/ml) and 5 μm ouabain (to inhibit the endogenous Na+,K+-ATPase) or 10 mm ouabain (to inhibit all Na+,K+-ATPase). 86Rb+ uptake was terminated by washing the cells with an ice-cold stop solution (11). Following incubation in 0.2 m NaOH for 2 h at 4 °C, the cells were scraped off, and the amount of 86Rb+ in the suspension was determined by liquid scintillation counting. K+ uptake was calculated by multiplying the relative 86Rb+ uptake per mg of protein by the K+ concentration in the uptake medium. For direct comparison of the wild type and various mutants, the uptake data were normalized to represent identical expression levels as calculated from the active site concentration determination (see below).
Phosphorylation from [γ-32P]ATP was determined on the leaky plasma membrane preparation in the presence of oligomycin to inhibit dephosphorylation and thus allow accumulation of phosphoenzyme. For determination of the Na+ concentration dependence of phosphorylation, the enzyme was mixed with [γ-32P]ATP at 0 °C in medium containing various concentrations of Na+ as detailed in the figure legends, followed by acid quenching 10 s later, using manual mixing. For determination of phosphorylation rate by transient kinetic studies, at 25 °C a QFM-5 quenched-flow module (Bio-Logic Instruments, Claix, France) was used for rapid mixing as described previously (20). By varying the flow rate and the volume of the delay line, reaction times between 10 and 250 ms were obtained. Following acid quenching, the precipitated 32P-labeled phosphoenzyme was washed twice by centrifugation, and SDS-PAGE was performed under acid conditions (pH 6.0), ensuring stability of the acyl-phosphoryl bond during electrophoresis. The radioactivity associated with the separated Na+,K+-ATPase band was quantified by phosphor-imaging using a Cyclone storage phosphor system (PerkinElmer Life Sciences) (10). To inhibit the endogenous COS-1 cell Na+,K+-ATPase, the membranes were preincubated with 10 μm ouabain, which was also present during the phosphorylation reaction. For every assay, the level of background phosphorylation was determined in a reaction mix containing a high concentration of K+ (50 mm) instead of Na+, as K+ prevents phosphorylation by reversing the reaction cycle, and the K+ background (only a tiny fraction of the total 32P incorporation) was subtracted before calculation of the phosphorylation level. The active site concentration was obtained as the maximum phosphoenzyme level corresponding to stoichiometric phosphorylation as determined in the presence of saturating Na+ concentration.
The results are reported as mean ± S.E. values (shown by error bars in figures, seen only when larger than the size of the symbols). In all cases, the number of independent experiments was ≥3. Data points were fitted by non-linear regression using the SigmaPlot program (SPSS, Inc.), and the best fits are shown as lines in the figures. For fitting of the Na+ dependence of phosphorylation and ATP dependence of ATPase activity, the Hill equation for binding of an activating ligand (L) was used (Equation 1).
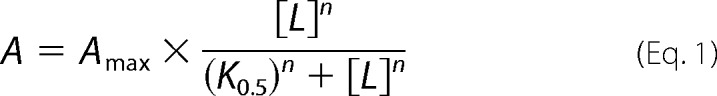 |
For fitting of the vanadate dependence of ATPase activity, the Hill equation modified to describe binding of an inhibiting ligand was used. (Equation 2).
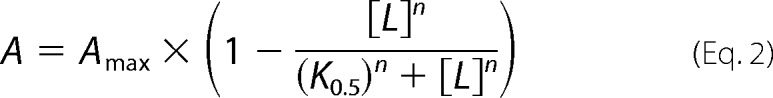 |
where Amax is the maximum level of phosphorylation or ATPase activity (set as 100% in the graphs), K0.5 is the ligand concentration giving half-maximum effect (“apparent affinity”), and n is the Hill coefficient. For fitting of the time dependence of phosphorylation, a mono-exponential “rise to max” function was used (Equation 3).
where EP is the phosphorylation level, EPmax is the maximum phosphorylation level reached at infinite time (set as 100% in the graphs), and kphos is the phosphorylation rate constant.
Structural figures were prepared with PyMOL software.
RESULTS
Expression of Mutants
We examined the functional properties of a series of rat Na+,K+-ATPase α1-isoform mutants with alterations to Asp-928 in transmembrane helix M8 (D928N, D928A, D928L, D928E, D928H) as well as the human α2 equivalent of D928N, D930N (henceforth denoted α2-D930N). Furthermore, we studied two double mutations, E314D/D928A and E314D/D928N, and the single mutation E314D, none of which was originally planned (see below). The mutants and the corresponding wild types were expressed in COS-1 cells using the ouabain selection method, which allows stable expression of mutants retaining significant function (16, 17). The expression levels of the mutants were generally similar to those of the wild types.
Interaction of E1 with Na+
To trigger the phosphorylation of Na+,K+-ATPase from ATP, all three Na+ binding sites of the E1 form need to be occupied by Na+, binding from the cytoplasmic side (Scheme 1). As seen in Fig. 2A, the Na+ dependence of phosphorylation from [γ-32P]ATP was examined to determine the apparent Na+ affinity (K0.5). For D928N and α2-D930N, a dramatic reduction of the apparent Na+ affinity (increase of K0.5) was observed corresponding to 78- and 41-fold, respectively, relative to the wild type (Table 1). A much more modest, but still significant reduction of the apparent Na+ affinity was seen for D928A, D928L, D928E, and D928H (5-, 10-, 4-, and 7-fold, respectively). The Na+ activation profiles also allowed extraction of the cooperativity parameter (Hill coefficient), nH, which for the α1 and α2 wild types ranges between 2 and 3, consistent with three Na+ ions being bound cooperatively. All the mutants showed nH values close to 1 (0.8 and 0.9 for D928N and α2-D930N, respectively), indicating the complete loss of cooperativity (Table 1).
FIGURE 2.
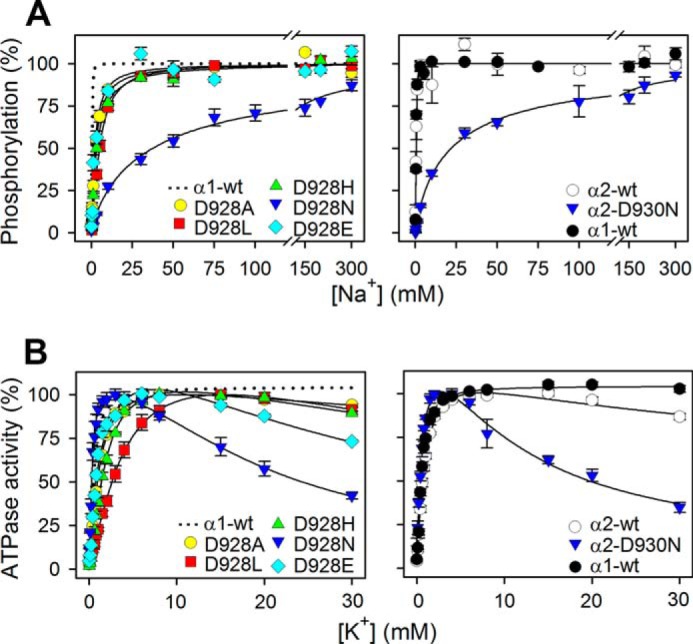
Na+ and K+ dependences of M8 aspartate mutants. A, phosphorylation was carried out for 10 s at 0 °C in 20 mm Tris (pH 7.5), 3 mm MgCl2, 1 mm EGTA, 2 μm [γ-32P]ATP, 10 μm ouabain, 20 μg oligomycin/ml, and the indicated concentration of Na+, added as NaCl with various concentrations of N-methyl-d-glucamine to maintain the ionic strength. Each line represents the best fit of a Hill function (see Equation 1 of “Experimental Procedures”) with extracted K0.5 values and Hill coefficients being listed in Table 1. B, ATPase activity was measured at 37 °C in 40 mm NaCl, 3 mm ATP, 3 mm MgCl2, 30 mm histidine (pH 7.4), 1 mm EGTA, 10 μm ouabain, and K+ concentrations as indicated, added as KCl. K0.5 values for the rising parts of the curves are listed in Table 1. For direct comparison, the dotted line in the left panel reproduces the data for α1 wild type from the corresponding right panel. Error bars indicate mean ± S.E. (seen only when larger than the size of the symbols).
TABLE 1.
Functional analysis of mutants
Standard errors are indicated. The number of independent determinations in each case: n ≥ 3. -Fold change of K0.5 relative to corresponding wild type is indicated by number with arrow.
| Turnover ratea | Na+ dependence |
kphosb | K0.5(K+) | K0.5(VO43−) | K0.5(ATP) | ||
|---|---|---|---|---|---|---|---|
| K0.5(Na+) | Hill coefficient | ||||||
| min−1 | μm | s−1 | μm | μm | μm | ||
| α1 wild type | 8474 ± 165 | 483 ± 18 | 2.4 ± 0.2 | 20 ± 2 | 624 ± 13 | 2.5 ± 0.1 | 432 ± 25 |
| D928A | 7172 ± 253 | 2557 ± 266 (5.3×↑) | 1.1 ± 0.1 | NDc | 953 ± 30 (1.5×↑) | 3.8 ± 0.1 (1.5×↑) | 280 ± 18 (1.5×↓) |
| D928L | 7953 ± 218 | 4959 ± 182 (10×↑) | 1.4 ± 0.1 | 18 ± 1 | 2537 ± 223 (4.1×↑) | 9.7 ± 0.3 (3.9×↑) | 97 ± 23 (4.5×↓) |
| D928N | 3345 ± 160 | 37,413 ± 6322 (78×↑) | 0.8 ± 0.1 | 7 ± 1 | 357 ± 41 (1.7×↓) | 5.4 ± 0.2 (2.2×↑) | 224 ± 35 (1.9×↓) |
| D928E | 9912 ± 540 | 2138 ± 334 (4.4×↑) | 1.2 ± 0.2 | 13 ± 1 | 774 ± 19 (1.2×↑) | 3.4 ± 0.1 (1.4×↑) | 325 ± 73 (1.3×↓) |
| D928H | 6127 ± 238 | 3412 ± 290 (7.1×↑) | 1.1 ± 0.1 | NDc | 1426 ± 70 (2.3×↑) | 3.5 ± 0.1 (1.4×↑) | 170 ± 10 (2.5×↓) |
| E314D/D928A | 9947 ± 478 | 391 ± 29 (1.2×↓) | 1.1 ± 0.1 | NDc | 1032 ± 66 (1.7×↑) | 4.1 ± 0.1 (1.6×↑) | 212 ± 16 (2.0×↓) |
| E314D/D928N | 5692 ± 398 | 3898 ± 950 (8.1×↑) | 0.7 ± 0.1 | 17 ± 1 | 409 ± 20 (1.5×↓) | 3.7 ± 0.1 (1.5×↑) | 264 ± 32 (1.6×↓) |
| E314D | 7733 ± 290 | 152 ± 15 (3.2×↓) | 1.6 ± 0.2 | 22 ± 2 | 375 ± 11 (1.7×↓) | 4.3 ± 0.2 (1.7×↑) | 307 ± 12 (1.4×↓) |
| α2 wild type | 7163 ± 319 | 541 ± 44 | 2.1 ± 0.4 | 22 ± 1 | 698 ± 27 | 17.8 ± 0.4 | 144 ± 7 |
| α2-D930N | 1521 ± 72 | 21,968 ± 2630 (41×↑) | 0.9 ± 0.1 | 7 ± 1 | 399 ± 59 (1.7×↓) | 39 ± 2.4 (2.2×↑) | 64 ± 16 (2.3×↓) |
| α3 wild typed | 8199 ± 209 | 570 | 1.7 | 23 ± 1 | 560 | 4.7 | NDc |
| α3-D923Nd | 843 ± 27 | 124,000 (218×↑) | 1.0 | 3 ± 0.3 | 170 (3.3×↓) | 41 (8.7×↑) | NDc |
a The Na+,K+-ATPase activity was determined at 37 °C in the presence of 30 mm histidine buffer (pH 7.4), 130 mm NaCl, 20 mm KCl, 3 mm ATP, 3 mm MgCl2, 1 mm EGTA, and 10 μm ouabain. The catalytic turnover rate was calculated as the ratio between the Na+,K+-ATPase activity and the active site concentration (maximum phosphorylation from [γ-32P]ATP measured at 0 °C in the presence of 150 mm NaCl and oligomycin).
b Phosphorylation rate constant.
c ND, not determined.
d Data were reproduced from Ref. 10 except the phosphorylation rates, which were determined in the present study.
K+ Interaction
At submillimolar concentration, K+ activates the Na+,K+-ATPase by stimulating dephosphorylation through binding at the sites on E2P that in the intact cell face the extracellular side (Scheme 1). D928E showed wild type-like apparent affinity for K+ activation. D928L displayed a significant 4-fold reduction (increase of K0.5) relative to wild type, and D928A and D928H displayed slight 1.5- and 2.3-fold reductions of the apparent affinity for K+ activation, respectively, whereas D928N and α2-D930N showed slight 1.5- and 1.7-fold increases of apparent K+ affinity (Fig. 2B and Table 1). In addition to the K+ activation phase, an inhibition phase was observed at high K+ concentrations for the mutants. This inhibition is much more pronounced for D928N and α2-D930N than for the other mutants and indicates the loss of Na+ selectivity, with K+ binding in place of Na+ at site III (see “Discussion”).
E1-E2 Distribution
In the wild type Na+,K+-ATPase, the E1 conformation is Na+-specific, and the E2 conformation is K+-specific. Hence, the apparent affinities for Na+ and K+ are influenced by the E1-E2 conformational equilibrium. We analyzed the E1-E2 distribution by determining the ATP and vanadate concentration dependences of the Na+,K+-ATPase activity (Fig. 3). Vanadate inhibits the enzyme by binding to the E2 form, whereas ATP binds with high affinity to E1 and only with low affinity to E2. As seen in Table 1, we found a good match between the inverse changes in apparent affinities for ATP and vanadate, D928L exhibiting an ∼4-fold increase in ATP affinity and a similar decrease in vanadate affinity, indicating a shift of the E1-E2 distribution in favor of E1, which seems to explain the 4-fold reduction of apparent K+ affinity without the need to assume a change of the intrinsic K+ affinity of the E2 form. Likewise, the slight reductions of K+ affinity in mutants D928A and D928H are accounted for by a shift toward E1.
FIGURE 3.
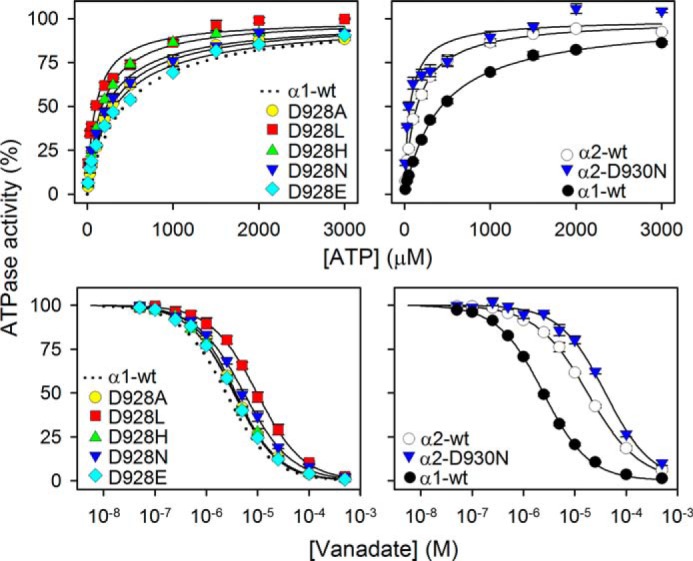
ATP and vanadate dependences of Na+,K+-ATPase activity of M8 aspartate mutants. ATPase activity was measured at 37 °C in 130 mm NaCl, 20 mm KCl, 3 mm MgCl2, 30 mm histidine (pH 7.4), 1 mm EGTA, 10 μm ouabain, and the indicated ATP concentrations (upper panels) or 3 mm ATP and the indicated vanadate concentrations (lower panels). Each line represents the best fit of a Hill function (see Equations 1 and 2 of “Experimental Procedures”) with Hill coefficient ranging between 0.9 and 1.1. Extracted K0.5 values are listed in Table 1. The dotted line in the left panel reproduces the data corresponding to wild type from the corresponding right panel for direct comparison in each panel. Error bars indicate mean ± S.E. (seen only when larger than the size of the symbols).
Mutation E314D Suppresses the Deleterious Effects of Asp-928 Mutations on Na+ Affinity
In the first attempt to introduce mutation D928A, the sequencing of the full-length DNA revealed an unintended base substitution in the DNA, probably a PCR error, which resulted in the presence of an additional mutation, E314D, together with D928A. Surprisingly, the expressed enzyme with this double mutation showed wild type-like apparent Na+ affinity in contrast to the 5-fold reduction seen for the mutant with correct single D928A substitution made subsequently (Fig. 4A). The finding that the E314D mutation suppressed the deleterious effect of D928A on the apparent Na+ affinity led us to examine whether the dramatic 78-fold reduction of the apparent Na+ affinity of D928N could also be compensated by combining D928N with E314D. Indeed, mutant E314D/D928N exhibited only 8-fold lower apparent Na+ affinity as compared with the wild type, indicating a substantial, 10-fold increase of Na+ affinity relative to that of D928N (Fig. 4A). In comparison, only a 3-fold increase of the apparent Na+ affinity was seen for the E314D single mutation, relative to that of the wild type (Fig. 4A).
FIGURE 4.
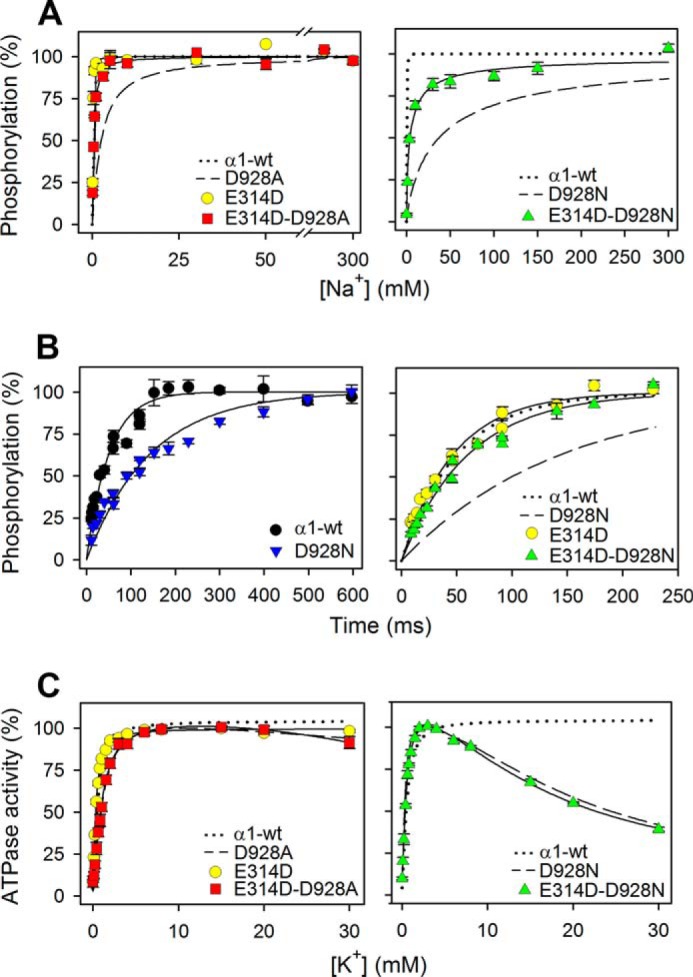
Rescue of Na+ affinity, but not of Na+ selectivity, in the double mutants E314D/D928A and E314D/D928N. A, Na+ dependence of phosphorylation was studied as described for Fig. 2A. The dotted and broken lines reproduce wild type and mutant data from Fig. 2A. B, rapid kinetic measurements of phosphorylation time course at 25 °C in the presence of 150 mm NaCl, 40 mm Tris (pH 7.5), 3 mm MgCl2, 2 μm [γ-32P]ATP, 10 μm ouabain, and 20 μg of oligomycin/ml. Each line represents the best fit of a mono-exponential rise to max function (see Equation 3 of “Experimental Procedures”). The dotted and broken lines in the right panel reproduce wild type and mutant data from the left panel. C, K+ dependence of Na+,K+-ATPase activity was studied as described for Fig. 2B. The dotted and broken lines reproduce wild type and mutant data from Fig. 2B. For all panels, the extracted parameters are listed in Table 1. Error bars indicate mean ± S.E. (seen only when larger than the size of the symbols).
Using a rapid kinetics quenched-flow module, we determined the rate coefficient of the phosphorylation reaction at 25 °C in the presence of 150 mm Na+ (Fig. 4B and Table 1). Even at this high Na+ concentration, the phosphorylation rate of D928N and the corresponding α2-D930N mutant was only one-third that of the wild type. Similar measurements carried out with α3-D923N showed an even lower rate (Table 1). However, a close to wild type-like phosphorylation rate was observed for E314D/D928N, again indicating a rescuing effect of E314D. The compensating effect of E314D on the phosphorylation rate could in principle arise either from an increase of the Na+ affinity or from a rise of the Vmax of phosphorylation (corresponding to saturating Na+). The phosphorylation rate of E314D was, however, found to be wild type-like (Fig. 4B), and because both E314D and the wild type are saturated with respect to Na+ at 150 mm, the measured rates correspond to Vmax in these cases. It may therefore be concluded that the effect of E314D is to enhance Na+ affinity rather than the Vmax of phosphorylation.
Contrary to the affinity for Na+ and the phosphorylation rate at high Na+ concentration, the cooperativity of Na+ binding was not rescued by the presence of E314D in the double mutants. Hence, the nH value was 1.1 for E314D/D928A as well as for D928A and 0.7 for E314D/D928N, versus 0.8 for D928N, which should be compared with 2.4 for the wild type (Table 1). The difference in cooperativity between E314D/D928A and wild type is clearly revealed by the crossing of the respective hyperbolic and sigmoidal curves seen in Fig. 5. Moreover, the E314D single mutation reduced the cooperativity to some extent, resulting in an nH value of 1.6 (Table 1). Also of note, the marked inhibition of the Na+,K+-ATPase activity by high K+ concentrations in D928N was unchanged upon the addition of the E314D mutation (Fig. 4C), thus showing that the Na+ selectivity of site III was not rescued.
FIGURE 5.
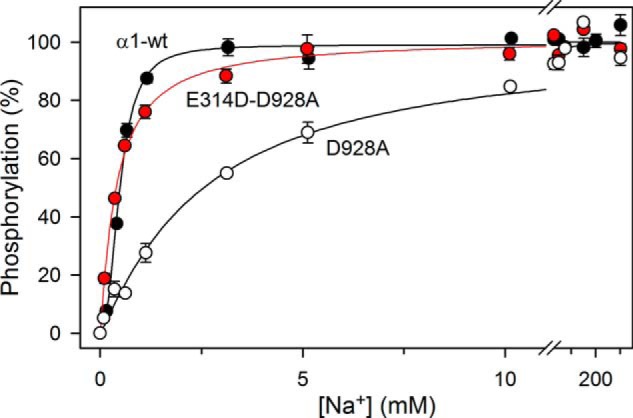
The cooperativity is not rescued by E314D. Data and fitted lines from Figs. 2A and 4A are shown on an expanded abscissa scale to demonstrate the difference in cooperativity but similar apparent Na+ affinities of the wild type and E314D/D928A. As in the other figures, the error bars indicating S.E. values are seen only when larger than the size of symbols.
We wondered whether the rescuing effect of E314D on the apparent Na+ affinity could be a consequence of displacement of the E1-E2 equilibrium in favor of the Na+ binding E1 form. However, the apparent vanadate and ATP affinities indicated rather similar E1-E2 distributions for E314D/D928A, E314D/D928N, E314D, D928A, and D928N, with roughly the same slight shift in favor of E1, relative to the wild type (Fig. 6 and Table 1). The E1 shift is certainly not larger for E314D/D928N as compared with D928N, thus indicating that the rescue of Na+ affinity is caused by a “true” change in the intrinsic Na+ affinity of the E1 form.
FIGURE 6.
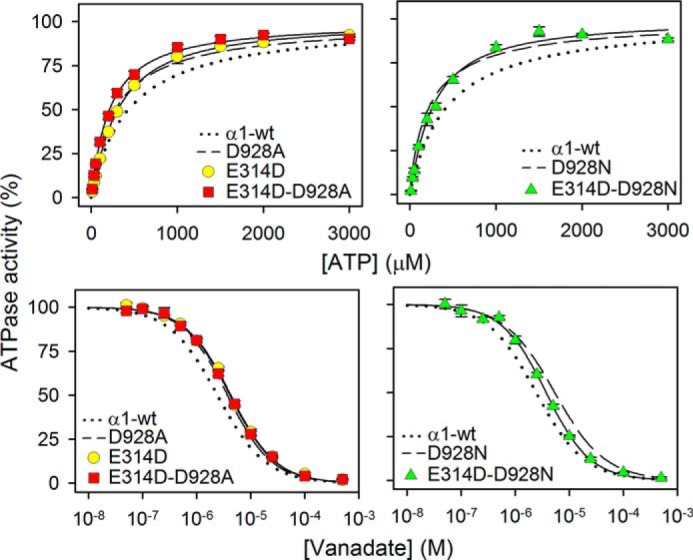
ATP and vanadate dependences of Na+,K+-ATPase activity of E314D/D928A and E314D/D928N. Data were obtained and analyzed as for Fig. 3. Extracted K0.5 values are listed in Table 1. The dotted and broken lines reproduce wild type and mutant data from Fig. 3 for direct comparison. Error bars indicate mean ± S.E. (seen only when larger than the size of the symbols).
Na+,K+-ATPase Catalytic Turnover Rate
The catalytic turnover rate at 130 mm Na+, 20 mm K+, and 3 mm ATP was calculated by relating the ATPase activity determined under these conditions to the active site concentration ascertained as the maximum capacity for phosphorylation (Table 1). All the Asp-928 mutants except D928N showed catalytic turnover rates >70% that of the wild type (85% for D928A). For D928N, the number was considerably lower (39%), as was also the case for α2-D930N (21%) and α3-D923N (10% (10)). Relative to the respective mutants with single substitutions, D928A and D928N, the catalytic turnover rates of E314D/D928A and E314D/D928N were enhanced 1.4- and 1.7-fold by the rescuing effect of E314D.
Na+ Interaction at Externally Facing Sites
To examine Na+ interaction at the sites that in the intact cell face the extracellular side, the Na+ dependence of the ATPase activity was determined in the absence of K+ (so-called Na+-ATPase activity). Under these conditions, the enzyme still hydrolyzes ATP, but at a lower rate than in the presence of K+, because the efficacy as activator of E2P dephosphorylation is much less for Na+ than for K+ (1). In the wild type enzyme, the Na+-ATPase activity decreases at high Na+ concentrations because Na+ binding from the external side with low affinity drives the E1P → E2P transition backwards to E1P (21, 22) (Scheme 1). As seen in Fig. 7, in all the mutants with single substitution of Asp-928, the inhibition at high Na+ concentrations was abolished, corresponding to a significant reduction of the affinity for Na+ binding from the external side to the phosphoenzyme. E314D/D928N and E314D/D928A showed partial inhibition at high Na+ concentrations, indicating that the Na+ affinity at one or more externally facing sites is rescued to some extent by E314D. The single E314D mutation led to an increased affinity for inhibitory Na+ as compared with the wild type. Furthermore, the E314D mutation increased the maximal Na+-ATPase activity, both when present in the double mutants and as a single mutation, which may be explained by enhanced efficacy of Na+ as activator of E2P dephosphorylation.
FIGURE 7.
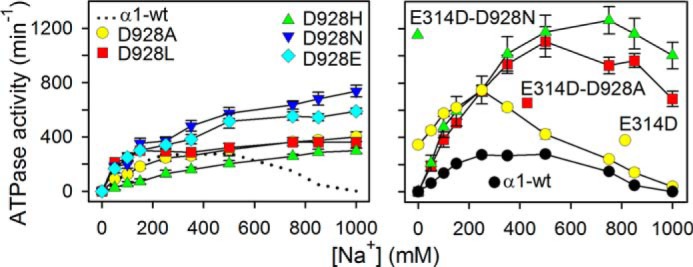
Na+ dependence of ATPase activity in absence of K+. ATPase activity was measured at 37 °C in 3 mm ATP, 3 mm MgCl2, 30 mm histidine (pH 7.4), 1 mm EGTA, 10 μm ouabain, and the indicated concentrations of Na+ added as NaCl. The catalytic turnover rate shown on the ordinate was calculated as the ratio between the specific ATPase activity and the active site concentration. For direct comparison, the dotted line in the left panel reproduces the data corresponding to wild type in the right panel. Error bars indicate mean ± S.E. (seen only when larger than the size of the symbols).
Transport Measurements in Intact Cells
The above described results obtained with the isolated leaky plasma membrane fractions raised the question whether the E314D mutation would also exert a rescuing effect under physiological conditions in intact cells. This question was addressed by using the K+ congener 86Rb+ to measure K+ uptake in cells expressing mutants or wild type. For direct comparison, the uptake data were corrected for variation of expression level. As seen in Fig. 8, the specific rate of K+ uptake of mutant D928N is only half that of the wild type, whereas in E314D/D928N, the K+ uptake rate is slightly higher than that of the wild type, indicating a full rescue under physiological conditions. The uptake rate of E314D was very similar to that of the double mutant.
FIGURE 8.
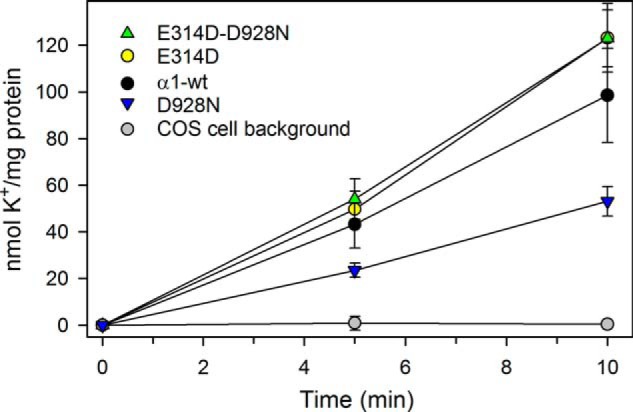
K+ uptake in intact cells under physiological conditions. Uptake of the K+ congener 86Rb+ at 37 °C in intact COS-1 cells stably expressing either wild type or mutants was determined following incubation for the indicated time intervals. K+ uptake was calculated by multiplying the relative 86Rb+ uptake per mg of protein by the K+ concentration in the uptake medium (see “Experimental Procedures”). The uptake mediated by expressed mutant or wild type was calculated by subtracting data obtained at 10 mm ouabain from those obtained at 5 μm ouabain. The ordinate shows the data normalized to represent identical expression levels of wild type and mutants. The indicated background corresponds to non-transfected COS-1 cells. Error bars indicate mean ± S.E. (seen only when larger than the size of the symbols).
DISCUSSION
The Disproportionate Effect of D928N
The replacement of the M8 aspartate with asparagine caused a disproportionate reduction of the affinity of the E1 form for Na+ in comparison with the much smaller reductions seen for the other substituents examined, including alanine, thus explaining the association of the asparagine mutation with the RDP/AHC disorder(s). However, the severity of the disturbance did not deviate much between Na+,K+-ATPase isoforms (Fig. 2A and Table 1). For all the Asp-928 mutants, a selective effect on the Na+-specific site III is indicated by the lack of impairment of K+ binding to E2. As compared with the conspicuous 78-fold reduction of the apparent Na+ affinity seen for D928N, the 5-fold reduction observed for D928A is diminutive, indicating that most of the disturbance seen for D928N is caused by the introduction of the amide group, rather than by the removal of a Na+-coordinating carboxylate oxygen. In fact, the D928A mutation did not reduce the apparent Na+ affinity much more than D928E (4-fold reduction), in which the carboxylate group is preserved. Fig. 9 illustrates likely positions of the substituent side chains in the Asp-928 mutants based on energy minimizations of the orientation of the substituted side chains in the high-resolution crystal structure of the wild type. The amide hydrogen bond-donating potential introduced with the asparagine likely interferes with the hydrogen-bonding network involving two other Na+-coordinating residues at site III, Thr-776 and Gln-925. Hence, a probable rotamer of the asparagine side chain has its terminal amide nitrogen within 2.2 Å distance from Gln-856, which in the wild type interacts with Thr-776 and Gln-925 (Fig. 9, A and D). The D928L mutant showed a larger reduction (10-fold) of the apparent Na+ affinity than D928A, probably due to the bulkiness of leucine resulting in collision with the other residues that are part of the hydrogen-bonding network of site III, including Gln-856 (Fig. 9C), but the reduction of the apparent Na+ affinity was considerably smaller for D928L as compared with D928N. It is furthermore of note that the replacement of Asp-928 with histidine reduced the apparent Na+ affinity only 7-fold, although the histidine is similar in bulkiness to leucine, suggesting that the potential of the histidine side chain as hydrogen bond acceptor as well as cation ligand (ϵ2-N and imidazole ring π-interaction, Fig. 9F) plays a role.
FIGURE 9.

Interaction network around Asp-928 and residues replacing Asp-928 in mutants studied. Shown is the view from the cytoplasmic side. This figure illustrates likely positions based on the high-resolution crystal structure of the wild type (Protein Data Bank code 3WGV) with Asp-928 being replaced. Selected residues (rat α1 numbering) are shown as sticks and colored according to the elements (carbon, gray; oxygen, red; nitrogen, blue), except that in mutants, the substituent is highlighted by the yellow color of the carbon atoms. The Na+ ions bound at sites I and III are shown as blue spheres. Broken lines with numbers indicate distances in Å. A, wild type. B, D928A. C, D928L. D, D928N. E, D928E. F, D928H. For each mutant, the most probable rotamer of the introduced substituted side chain is shown based on energy minimization of the side-chain orientation carried out using the PyMOL software.
The relatively minor role of Asp-928 in Na+ binding indicated by the present results is in line with the high-resolution crystal structures of the E1(Na3)·AlF4−·ADP form (3) showing a location of the aspartate in the periphery of Na+ site III (Fig. 1). The observation of the huge effect of the α3-D923N mutation on the apparent Na+ affinity led to the proposal of a C-terminal Na+ entry pathway from the cytoplasm to Na+ site III, between M5, M7, M8, and M10, with a role for the aspartate in receiving Na+ at the bottom of this half-channel (10). However, with the reduction of the apparent Na+ affinity caused by alanine replacement being only 5-fold, there is no longer any need for such speculation, and because the C-terminal pathway is closed in the E1(Na3)·AlF4−·ADP crystal structures, it seems more likely that cytoplasmic Na+ enters site III from the N-terminal side of the protein by a pathway through sites I and II (3).
It has also been suggested that the M8 aspartate plays a crucial role in Na+/H+ exchange at the cytoplasmic side as an obligatory part of the Na+,K+ transport mechanism (23). Although such an exchange involving the aspartate might occur in the wild type enzyme under certain conditions (24), it is certainly not obligatory for Na+,K+ pump function because we observed a close to wild type-like turnover rate for the D928A mutant and an even higher turnover rate for E314D/D928A (Table 1), as well as slightly higher transport activity of E314D/D928N as compared with the wild type (Fig. 8).
The Rescuing Effect of E314D on Na+ Affinity
A most intriguing finding is the rescue of the apparent Na+ affinity obtained by combining D928N or D928A with E314D. Hence, in the phosphorylation experiments monitoring Na+ binding at the cytoplasmically facing sites, the double mutant E314D/D928N displayed a 10-fold higher apparent Na+ affinity than D928N and a wild type-like phosphorylation rate at 150 mm Na+, and the other double mutant, E314D/D928A, displayed a wild type-like, 5-fold higher apparent Na+ affinity than D928A. Importantly, the rescuing effect of E314D also encompassed transport activity determined as K+ uptake in intact cells under physiological conditions (Fig. 8). As described under “Results,” the increases of the apparent Na+ affinity are not caused indirectly by a shift of the E1-E2 equilibrium in favor of E1 or by an increase of the Vmax of phosphorylation, but are probably intrinsic to the Na+ binding pocket. The increased Na+ affinity also manifested as a significant inhibition by high Na+ of the Na+-ATPase activity (Fig. 7), i.e. binding of Na+ from the external side. The correlation between the changes of apparent affinity for Na+ on the two sides of the membrane suggests that the effects involve the Na+ occluded state in E1P, reached irrespective of the side from which Na+ binds.
The rescue of Na+ affinity by E314D is a long-range effect, Glu-314 being located in the M4E helix, at the extracellular surface and far away from Asp-928 (Fig. 10). In the Na+-bound E1 form, Glu-314 might form a hydrogen bond to an aspartate/asparagine, which in the crystal structure is located within 3.3 Å distance in M2 (Asn-124 in Fig. 10A). This is also the asparagine involved in ouabain binding, which in the ouabain-insensitive rodent Na+,K+-ATPases (16) is replaced by aspartate (also able to form a hydrogen bond with glutamate, depending on the protonation state). In the E2 form, Glu-314 and Asn-124 have moved apart (Fig. 10B). The shortening of the Glu-314 side chain by replacement with aspartate prevents interaction between these residues in E1, which might be the reason for the observed effect of the mutation on Na+ affinity. The breakage of the hydrogen bond could possibly lead to a reorientation of the M4E helix, thereby affecting the binding of Na+ at site II by the backbone carbonyl oxygen atoms of Val-324, Ala-325, and Val-327 at the other end of M4E and by the side chain of Glu-329 in M4C. Ala-325 also forms a bond to Na+ at site I (Figs. 1 and 10A), and thus a significant tightening of the binding pocket might occur even with a small movement of M4E.
FIGURE 10.

Structural relations of Glu-314 and Asn-124 to M4 and the Na+ and K+ binding region. Shown is the side view of the transmembrane domain, cytoplasmic side upwards (left panels) and view from the extracellular side toward the membrane surface. Transmembrane helices are shown as cylinders with blue letters and numbers, with selected residues indicated (rat α1 numbering; M4C and M4E refer to cytoplasmic and extracellular parts of M4, respectively). Broken lines with numbers indicate distances between Glu-314 and Asn-124 in Å. Putative bonds between selected residues and Na+ or K+ ions are also shown as broken lines in the right panels, but without numbers. A, Na+-bound form (E1(Na3)·AlF4−·ADP, Protein Data Bank code 3WGV (3)). Na+ ions are shown as blue spheres numbered I, II, and III. B, K+-bound form (E2(K2)·MgF42−, Protein Data Bank code 2ZXE (5)). K+ ions are shown as purple spheres numbered I and II.
In addition, a remarkable finding was the loss of cooperativity of Na+ binding upon mutation of the M8 aspartate. The largest effect was seen for D928N (reduction of the Hill coefficient to 0.8 from the wild type value of 2.4), but also the other Asp-928 mutants showed nH values close to 1. The cooperativity is thought to arise from the sharing of coordinating groups/side chains between the three Na+ sites and the resulting effect of the occupation of one site on the availability and affinity of the next site in the consecutive binding mechanism (3). Based on the crystal structure, it was proposed that Na+ site III is filled first, thereby creating site I, the filling of which creates site II (3). The present result indicates that the hydrogen-bonding network around Asp-928 in site III is an essential part of the structural basis for the cooperativity, possibly because of its importance for the positioning of the two residues that are shared between sites III and I, Ser-777 and Asp-810 (Figs. 1 and 10).
Importantly, the cooperativity of Na+ binding was not rescued by the E314D mutation, thus raising the question whether Na+ binding at site III is normalized at all in the double mutants. The apparent affinity with which Na+ activates phosphorylation may to a large extent depend on the cooperative interaction because the phosphorylation is triggered by the occupation of site II by the last Na+ ion bound (3). Hence, the reduced apparent affinity seen for the Asp-928 mutants may primarily be because of the loss of cooperativity rather than a reduced intrinsic affinity of site III. This means that the rescue of apparent affinity for Na+ caused by mutation E314D may result from an increased intrinsic affinity of site II without “repair” of site III. In other words, the effect on site II exerted by the second-site mutation E314D fulfils what the occupation of site III cannot accomplish due to the missing cooperativity.
Related to the inability of E314D to rescue the cooperativity of Na+ binding is the finding that the anomalous inhibition by K+ at high concentration occurs to the same extent in E314D/D928N as in D928N (Fig. 4C). This inhibition is caused by K+ binding to the E1 form in place of one or more of the Na+ ions and indicates the loss of selectivity for Na+. Structural analysis has indicated that K+ binding at site III becomes feasible if the cytoplasmic half of the M5 helix (M5C) is allowed to incline ∼10° relative to its normal position (3). Given that site III is formed almost on M5C, with M5C residues Tyr-773, Thr-776, and Ser-777 donating Na+ ligands at site III (Tyr-773 donating both backbone and side-chain ligands), it is conceivable that the perturbation of the hydrogen-bonding network of site III introduced by the replacement of Asp-928, particularly with asparagine, could lead to the minimal movement of M5C required to allow K+ to bind at site III. Hence, the inability of E314D to rescue Na+ selectivity provides another piece of evidence that the rescuing effect of E314D does not involve a repair of site III.
Conclusion and Perspectives
Here we have identified a second-site mutation, E314D, leading to gain of function with respect to the apparent Na+ affinity of D928A and D928N, but not with respect to the cooperativity and selectivity of Na+ binding in these mutants. The underlying mechanism may be a tightening of Na+ binding at site II, the last of the three Na+ sites to be filled in the consecutive and cooperative binding process that activates phosphorylation from ATP. This effect represents a new principle that may be exploited in the future in the development of medications for treatment of patients with defective Na+ binding, as reported for several cases of RDP/AHC (9–11). The 10-fold enhancement of Na+ affinity and 1.7-fold increase of catalytic turnover rate obtained by introducing E314D as a second-site mutation in the presence of the disease-causing D928N mutation is expected to vastly improve the rate of Na+ and K+ transport under physiological conditions in the cell, where the cytoplasmic Na+ sites are far from being saturated even in the wild type. Indeed, this prediction was fulfilled by our experiments demonstrating that the second-site mutation causes an ∼2-fold rise of the pumping activity of D928N to a wild type-like level in intact cells.
Acknowledgments
We thank Kirsten Lykke Pedersen, Janne Petersen, Randi Scheel, and Nina Juste for expert technical assistance and Professor C. Toyoshima, University of Tokyo, for discussion.
This work was supported in part by grants from the Danish Medical Research Council, the Novo Nordisk Foundation, the Lundbeck Foundation, and L'Oréal Denmark.
- RDP
- rapid-onset dystonia parkinsonism
- AHC
- alternating hemiplegia of childhood.
REFERENCES
- 1. Post R. L., Hegyvary C., Kume S. (1972) Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 247, 6530–6540 [PubMed] [Google Scholar]
- 2. Albers R. W., (1967) Biochemical aspects of active transport. Annu. Rev. Biochem. 36, 727–756 [DOI] [PubMed] [Google Scholar]
- 3. Kanai R., Ogawa H., Vilsen B., Cornelius F., Toyoshima C. (2013) Crystal structure of a Na-bound Na,K-ATPase preceding the E1P state. Nature 502, 201–206 [DOI] [PubMed] [Google Scholar]
- 4. Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., Nissen P. (2007) Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 5. Shinoda T., Ogawa H., Cornelius F., Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4 Ä resolution. Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 6. Toustrup-Jensen M. S., Holm R., Einholm A. P., Schack V. R., Morth J. P., Nissen P., Andersen J. P., Vilsen B. (2009) The C terminus of Na+,K+-ATPase controls Na+ affinity on both sides of the membrane through Arg935. J. Biol. Chem. 284, 18715–18725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Carvalho Aguiar P., Sweadner K. J., Penniston J. T., Zaremba J., Liu L., Caton M., Linazasoro G., Borg M., Tijssen M. A., Bressman S. B., Dobyns W. B., Brashear A., Ozelius L. J. (2004) Mutations in the Na+/K+-ATPase α3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175 [DOI] [PubMed] [Google Scholar]
- 8. Rosewich H., Thiele H., Ohlenbusch A., Maschke U., Altmüller J., Frommolt P., Zirn B., Ebinger F., Siemes H., Nürnberg P., Brockmann K., Gärtner J. (2012) Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol. 11, 764–773 [DOI] [PubMed] [Google Scholar]
- 9. Heinzen E. L., Arzimanoglou A., Brashear A., Clapcote S. J., Gurrieri F., Goldstein D. B., Jóhannesson S. H., Mikati M. A., Neville B., Nicole S., Ozelius L. J., Poulsen H., Schyns T., Sweadner K. J., van den Maagdenberg A., Vilsen B., and ATP1A3 Working Group (2014) Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 13, 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Einholm A. P., Toustrup-Jensen M. S., Holm R., Andersen J. P., Vilsen B. (2010) The rapid-onset dystonia parkinsonism mutation D923N of the Na+, K+-ATPase α3 isoform disrupts Na+ interaction at the third Na+ site. J. Biol. Chem. 285, 26245–26254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toustrup-Jensen M. S., Einholm A. P., Schack V. R., Nielsen H. N., Holm R., Sobrido M. J., Andersen J. P., Clausen T., Vilsen B. (2014) Relationship between intracellular Na+ concentration and reduced Na+ affinity in Na+,K+-ATPase mutants causing neurological disease. J. Biol. Chem. 289, 3186–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anselm I. A., Sweadner K. J., Gollamudi S., Ozelius L. J., Darras B. T. (2009) Rapid-onset dystonia-parkinsonism in a child with a novel ATP1A3 gene mutation. Neurology 73, 400–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roubergue A., Roze E., Vuillaumier-Barrot S., Fontenille M. J., Méneret A., Vidailhet M., Fontaine B., Doummar D., Philibert B., Riant F., Nicole S. (2013) The multiple faces of the ATP1A3-related dystonic movement disorder. Mov. Disord. 28, 1457–1459 [DOI] [PubMed] [Google Scholar]
- 14. Sasaki M., Ishii A., Saito Y., Morisada N., Iijima K., Takada S., Araki A., Tanabe Y., Arai H., Yamashita S., Ohashi T., Oda Y., Ichiseki H., Hirabayashi S., Yasuhara A., Kawawaki H., Kimura S., Shimono M., Narumiya S., Suzuki M., Yoshida T., Oyazato Y., Tsuneishi S., Ozasa S., Yokochi K., Dejima S., Akiyama T., Kishi N., Kira R., Ikeda T., Oguni H., Zhang B., Tsuji S., Hirose S. (2014) Genotype-phenotype correlations in alternating hemiplegia of childhood. Neurology 82, 482–490 [DOI] [PubMed] [Google Scholar]
- 15. Nyblom M., Poulsen H., Gourdon P., Reinhard L., Andersson M., Lindahl E., Fedosova N., Nissen P. (2013) Crystal structure of Na+, K+-ATPase in the Na+-bound state. Science 342, 123–127 [DOI] [PubMed] [Google Scholar]
- 16. Price E. M., Lingrel J. B. (1988) Structure-function relationships in the Na,K-ATPase α subunit: site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry 27, 8400–8408 [DOI] [PubMed] [Google Scholar]
- 17. Vilsen B. (1992) Functional consequences of alterations to Pro328 and Leu332 located in the 4th transmembrane segment of the α-subunit of the rat kidney Na+,K+-ATPase. FEBS Lett. 314, 301–307 [DOI] [PubMed] [Google Scholar]
- 18. Baginski E. S., Foà P. P., Zak B. (1967) Microdetermination of inorganic phosphate, phospholipids, and total phosphate in biologic materials. Clin. Chem. 13, 326–332 [PubMed] [Google Scholar]
- 19. Jørgensen P. L., Petersen J. (1982) High-affinity 86Rb-binding and structural changes in the α-subunit of Na+,K+-ATPase as detected by tryptic digestion and fluorescence analysis. Biochim. Biophys. Acta 705, 38–47 [DOI] [PubMed] [Google Scholar]
- 20. Toustrup-Jensen M., Hauge M., Vilsen B. (2001) Mutational effects on conformational changes of dephospho- and phospho-forms of the Na+,K+-ATPase. Biochemistry 40, 5521–5532 [DOI] [PubMed] [Google Scholar]
- 21. Kaplan J. H., Hollis R. J. (1980) External Na dependence of ouabain-sensitive ATP:ADP exchange initiated by photolysis of intracellular caged-ATP in human red cell ghosts. Nature 288, 587–589 [DOI] [PubMed] [Google Scholar]
- 22. Lee K. H., Blostein R. (1980) Red cell sodium fluxes catalysed by the sodium pump in the absence of K+ and ADP. Nature 285, 338–339 [DOI] [PubMed] [Google Scholar]
- 23. Poulsen H., Khandelia H., Morth J. P., Bublitz M., Mouritsen O. G., Egebjerg J., Nissen P. (2010) Neurological disease mutations compromise a C-terminal ion pathway in the Na+/K+-ATPase. Nature 467, 99–102 [DOI] [PubMed] [Google Scholar]
- 24. Vedovato N., Gadsby D. C. (2014) Route, mechanism, and implications of proton import during Na+/K+ exchange by native Na+/K+-ATPase pumps. J. Gen. Physiol. 143, 449–464 [DOI] [PMC free article] [PubMed] [Google Scholar]