

Background: Toxins such as LvIA can help elucidate the physiological roles of nAChR subtypes.
Results: Three residues in the β2 subunit were identified as critical to LvIA binding.
Conclusion: The β complementary subunit plays a crucial role in the subtype selectivity of α-conotoxin LvIA.
Significance: This study provides new insights into the unique selectivity of LvIA and more broadly into toxin-receptor interactions.
Keywords: Docking, Receptor, Receptor Structure-Function, Receptor-interacting Protein (RIP), Toxin, β subunit contribution, α-Conotoxin LvIA, α3β2 nAChR, Mutant α3β2 Subtype.
Abstract
α-Conotoxin LvIA (α-CTx LvIA) is a small peptide from the venom of the carnivorous marine gastropod Conus lividus and is the most selective inhibitor of α3β2 nicotinic acetylcholine receptors (nAChRs) known to date. It can distinguish the α3β2 nAChR subtype from the α6β2* (* indicates the other subunit) and α3β4 nAChR subtypes. In this study, we performed mutational studies to assess the influence of residues of the β2 subunit versus those of the β4 subunit on the binding of α-CTx LvIA. Although two β2 mutations, α3β2[F119Q] and α3β2[T59K], strongly enhanced the affinity of LvIA, the β2 mutation α3β2[V111I] substantially reduced the binding of LvIA. Increased activity of LvIA was also observed when the β2-T59L mutant was combined with the α3 subunit. There were no significant difference in inhibition of α3β2[T59I], α3β2[Q34A], and α3β2[K79A] nAChRs when compared with wild-type α3β2 nAChR. α-CTx LvIA displayed slower off-rate kinetics at α3β2[F119Q] and α3β2[T59K] than at the wild-type receptor, with the latter mutant having the most pronounced effect. Taken together, these data provide evidence that the β2 subunit contributes to α-CTx LvIA binding and selectivity. The results demonstrate that Val111 is critical and facilitates LvIA binding; this position has not previously been identified as important to binding of other 4/7 framework α-conotoxins. Thr59 and Phe119 of the β2 subunit appear to interfere with LvIA binding, and their replacement by the corresponding residues of the β4 subunit leads to increased affinity.
Introduction
Nicotinic acetylcholine receptors (nAChRs),3 which comprise many different molecular subtypes, are ligand-gated ion channels that are activated by the endogenous neurotransmitter acetylcholine (ACh) or exogenous nicotine (1, 2). nAChRs are found in the neuromuscular junction, and in peripheral and central nervous systems throughout the animal kingdom, and play important roles in regulating synaptic transmission (3–9). Neuronal nAChRs are pentameric membrane-bound proteins, which are made up of α (α2–α10) and β (β2–β4) subunits (10). Pharmacological properties of the heteromeric nAChRs are influenced by the presence of β2 and/or β4 subunits (11). This study is part of an ongoing effort to elucidate the physiological role of each subtype of nAChR and the key binding residue determinants for selective ligands (12).
α-Conotoxins (α-CTxs) are a rich source of highly selective ligands that discriminate among different nAChR subtypes (13–15). α-CTxs contain two conserved disulfide bridges and are classified into several structural subfamilies according to the number of residues in the two backbone loops bracketed by cysteine residues, the largest subfamilies being the 4/7, 4/6, 4/5, 4/4, 4/3, and 3/5 α-CTxs. The 4/7 α-CTxs are able to discriminate between diverse neuronal α-β nAChR subunit combinations and stoichiometries (16, 17).
Previous research revealed that some of the 4/7 α-CTxs, such as α-CTx MII, PnIA, and BuIA, bind to a small conserved cleft of the α3β2 nAChR, and the β2 subunit contributes to binding and selectivity (18, 19). This cleft contains the ligand-accessible residues β2 Leu121, Val111, Phe119, and Thr59, which act as a common binding site/pocket for 4/7 α-CTxs (18, 19). α-CTx MII from Conus magus is a potent antagonist of α3β2 and α6β2* (* indicates the other subunit) nAChRs (20). The α-CTx PnIA is selective on the α3β2 and α7 nAChRs (21). α-CTx BuIA from Conus bullatus blocks both β2* (* indicates the other subunit) and β4* (* indicates the other subunit) nAChRs, and kinetically distinguishes between them with long and short off-times (22). The 4/7 α-CTx LtIA targets a novel microsite and has a shallow binding site on the α3β2 nAChR that includes β2 Lys79 outside of the cleft (23). This indicates that different key residues of the β2 subunit are targeted by different α-CTxs to block the α3β2 nAChR (19, 24).
α-CTx LvIA from Conus lividus was recently characterized and has high affinity for α3β2 nAChRs, with an IC50 of 8.7 nm (25). LvIA is notable for its ability to selectively block α3β2 versus α6/α3β2β3 or α3β4 nAChRs. The residues in the β2 subunit that contribute to α-CTx LvIA binding to the α3β2 nAChR remain unknown. We therefore performed a mutational study of the α3β2 nAChR in which we assessed the influence of residues that line the β2 subunit on the binding of α-CTx LvIA.
EXPERIMENTAL PROCEDURES
Materials
Reagents for peptide synthesis were from GL Biochem (Shanghai, China). Reversed-phase HPLC analytical Vydac C18 column (5 μm, 4.6 × 250 mm) and preparative C18 Vydac column (10 μm, 22 × 250 mm) were from Grace Vydac (Hesperia, CA). Clones of rat α3, β2, and β4 cDNAs were kindly provided by S. Heinemann (Salk Institute, San Diego, CA).
Peptide Synthesis
A two-step oxidation protocol was used to synthesize α-CTx LvIA as described previously (25). Because this protocol worked well, we did not attempt a simpler one-step oxidation approach. In this protocol, linear (see Fig. 1A) and folded (see Fig. 1B) peptides were purified by HPLC on a reversed-phase C18 Vydac column. HPLC elution conditions included a linear gradient of 0–40% solvent B over 40 min. Solvent A was 0.075% trifluoroacetic acid in H2O. Solvent B was 0.05% TFA, 90% acetonitrile in H2O. Absorbance was monitored at 214 nm.
FIGURE 1.

Reduced α-CTx LvIA synthetic intermediate (A) and oxidized, folded α-CTx LvIA (B) sequences and corresponding HPLC chromatograms (C and D). A, sequence of a synthetic intermediate of α-CTx LvIA with Cys2 and Cys4 residues protected with S-acetamidomethyl (Acm), and Cys1 and Cys3 residues with free -SH (mercapto) before initial cleavage. The first and third cysteine residues were initially protected with acid-labile groups (trityl), which were removed after cleavage from the resin. B, folded peptide sequences with disulfide connectivity 1–3, 2–4. *, C-terminal carboxamide. C, HPLC chromatograms of the synthetic intermediate shown in A. D, HPLC chromatograms of oxidized and folded α-CTX LvIA. Peptides were analyzed on a reversed-phase analytical Vydac C18 HPLC using a linear gradient of 0–40% Solvent B over 40 min, where Solvent A = 0.075% TFA and Solvent B = 0.05% TFA, 90% acetonitrile in H2O. Absorbance was monitored at 214 nm. Flow rate was 0.75 ml/min. AU, absorbance units.
Mutagenesis and Construction of Chimeric β2 Point Mutation Receptors
Point mutants of nAChR β2 subunit cDNA (see Table 1) were created using PCR and the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Primers that contained the desired point mutation as well as at least 15 bases on either side of the mutation were synthesized. The mutagenic primers were extended and incorporated by PCR. DpnI was then used to digest the methylated, non-mutated parental cDNA. The point mutated DNA was inserted in the pSP65 or pGEMHE vector, which was transformed into DH5α-competent cells. All the PCR mutations were sequenced to confirm incorporation of the desired mutation (19). The nomenclature for the point mutants lists the naturally occurring residue position followed by the change made, e.g. α3β2[V111I] is a β2 subunit with the valine residue at position 111 position replaced by an isoleucine residue.
TABLE 1.
IC50 and Hill slope values for block of nAChRs by α-CTx LvIA
| Subtypes | IC50a | Ratiob | Hill slope |
|---|---|---|---|
| nm | |||
| α3β2 | 8.67 (6.9–11.0) | 1 | 1.17 (0.88–1.46) |
| α3β4 | 148 (103–213) | 17 | 1.14 (0.72–1.55) |
| α3β2[F119Q] | 0.58 (0.44–0.76) | 0.07 | 1.12 (0.79–1.44) |
| α3β2[T59K] | 0.96 (0.56–1.65) | 0.11 | 0.80 (0.47–1.13) |
| α3β2[T59L] | 2.03 (1.52–2.69) | 0.23 | 1.07 (0.77–1.37) |
| α3β2[Q34A] | 8.64 (4.80–15.5) | 1.0 | 0.90 (0.22–1.58) |
| α3β2[K79A] | 10.8 (6.44–18.0) | 1.3 | 0.86 (0.43–1.30) |
| α3β2[T59I] | 15.2 (9.71–23.9) | 1.8 | 1.15 (0.43–1.86) |
| α3β2[V111I] | 126 (97.2–163) | 15 | 1.31 (0.66–1.96) |
a Numbers in parentheses are 95% confidence intervals.
b IC50/wild-type α3β2 IC50.
cRNA Preparation and Injection into Xenopus laevis Oocytes
Capped cRNA was synthesized in vitro following linearization of the plasmid containing template DNA encoding the rat α3, β2, and β4 subunits, as well as the various mutant subunits using the mMESSAGE mMACHINE in vitro transcription kit (Applied Biosystems/Ambion, Austin, TX), as described previously (20). The cRNA was purified using the Qiagen RNeasy kit (Qiagen). Their concentration was determined by absorbance at 260 nm. Mature X. laevis frogs were anesthetized by submersion in 0.1% 3-aminobenzoic acid ethyl ester, and oocytes were surgically removed. Two collagenase treatments lasting 1 h were performed at room temperature to remove follicle cells. RNA transcripts of wild-type α3 subunit with either wild-type β2 or mutant β2 subunit were mixed at a molar ratio of 1:1. Fifty nl of this mixture with ∼10 ng of each cRNA was injected into each Xenopus oocyte and incubated at 17 °C. Oocytes were injected within 1 day of harvesting.
Voltage-clamp Recording
Voltage-clamp recordings were performed 1–4 days after cRNA injection. All recordings were done at ∼22 °C room temperature. Briefly, oocytes were voltage-clamped at −70 mV and exposed to ACh and peptide in a 30-μl cylindrical oocyte recording chamber, which was gravity-perfused at a rate of 2 ml/min with ND-96 buffer. The ND-96 buffer consisted of 96.0 mm NaCl, 2.0 mm KCl, 1.8 mm CaCl2, 1.0 mm MgCl2, 1 μm atropine, 5 mm HEPES, 0.1 mg/ml bovine serum albumin, pH 7.1–7.5. The oocyte was subjected once a minute to a 1-s pulse of 100 μm ACh. Once a stable baseline was achieved, either ND-96 alone or ND-96 containing varying concentrations of the α-CTx LvIA was manually pre-applied in a static bath for 5 min prior to the addition of the agonist ACh pulse.
Data Analysis
Three to five ACh responses were averaged for the baseline responses of ND-96 after a 5-min incubation just preceding a test response, which was used to normalize evoked responses as a percentage of control response. The percentage of response of the toxin was divided by the pre-toxin baseline value to yield a percentage of response. The dose-response data were fitted to the equation: % of response = 100/(1 + ([toxin]/IC50)nH), where nH is the Hill coefficient, using GraphPad Prism (GraphPad Software, San Diego, CA). Each data point of a dose-response curve is the average ± S.E. from at least three oocytes. IC50 values were determined by nonlinear regression analysis using Graph-Pad Prism.
Molecular Modeling
A molecular model of the interaction between LvIA and the ligand-binding domain of α3β2 nAChR was built by homology using the NMR solution structure of LvIA (Protein Data Bank (PDB) identifier 2mdq) and the crystal structure of the complex between acetylcholine-binding protein (AChBP) and conotoxin PnIA variant (PDB identifier 2br8) as templates, as described previously (25). The molecular model was refined by a 30-ns explicit water molecular dynamics simulation carried out with the GROMACS 4.6.5 (26) package and the ff03 force field (27), using a procedure described previously (28, 29). All the models of complexes involving β2 subunit mutants were generated by substituting residue side chains using Modeler 9v14 (30). This procedure refines the positions of the substituted side chain atoms as well as of those of the neighbor residues using a conjugate gradient minimization followed by a short molecular dynamics simulation. The molecular models were refined by a 2-ns explicit water molecular dynamics simulation, and the simulations of the T59K, V111I, and F119Q mutants were extended to 10 ns.
RESULTS
Chemical Synthesis of α-CTx LvIA
α-CTx LvIA linear peptide (Fig. 1A) was successfully synthesized with Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry, in which Cys residues were orthogonally protected using acid-labile S-trityl and acid-stable S-acetamidomethyl groups. The acid-labile groups (trityl) were first removed after cleavage of the assembled peptide chain from the resin. The linear peptide after the initial cleavage was purified by HPLC with a retention time of 27.7 min (Fig. 1C). Ferricyanide was used to close the first disulfide bridge, and iodine oxidation was used to subsequently close the second disulfide bridge. The fully folded peptide of α-CTx LvIA with Cys1–Cys3 and Cys2–Cys4 disulfide bonds (Fig. 1B) was purified again by HPLC, with a retention time of 27.9 min (Fig. 1D). The mass of the α-CTx LvIA matched that of the amidated sequence (calculated average mass, 1679.9 Da; observed, 1679.7). This synthesized fully folded peptide was utilized in all subsequent experiments.
Effect of Mutations of the β2 Subunit on Block by α-CTx LvIA
Previous studies using molecular modeling of related toxins suggested the nAChR positions that form the ligand-binding pocket of α-conotoxins (23, 31, 32). The residue positions of the β2 and β4 subunits that were suggested to form the LvIA-binding pocket in a previous modeling study (25) are highlighted in Fig. 2. We created point mutations of the β2 subunit where residues in this pocket were replaced with those found in the homologous position of the β4 subunit. These mutant receptors were then tested to determine toxin potency differences (Table 1). Seven nAChR β2 mutants were created, including Q34A, T59I, T59K, T59L, K79A, V111I, and F119Q. The concentration-response block by α-CTx LvIA on α3β4 nAChR and wild-type and mutant α3β2 nAChRs was investigated (Table 1 and Fig. 3). The potency at wild-type α3β2 nAChR was 17-fold greater than that at wild-type α3β4 nAChR. Among the seven α3β2 mutants, LvIA had the lowest activity on α3β2[V111I], with an IC50 of 126 nm, which is similar to the 148 nm IC50 on wild-type α3β4. Thus, the β2 subunit mutation V111I reduced the binding of LvIA to α3β2 nAChR by 15-fold. There were no significant differences in LvIA potency on mutants α3β2 Q34A, K79A, or T59I when compared with activity at wild-type α3β2 nAChR (Table 1, Fig. 3).
FIGURE 2.
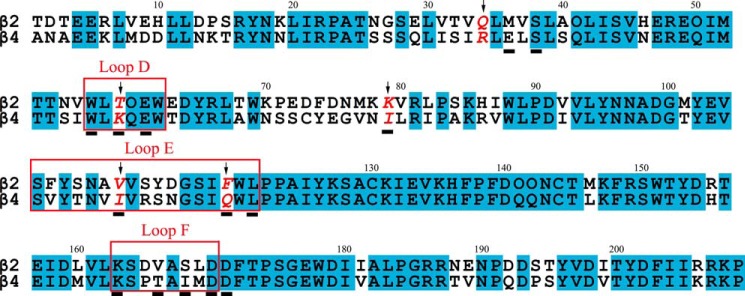
Amino acid sequence alignment of rat β2 and β4 nAChR subunits, which have 68.4% sequence identity in their ligand-binding domains. The positions that were mutated in this study, i.e. positions 34, 59, 79, 111, and 119, are indicated with arrows. Rectangles indicate the agonist-binding domain loops D, E, and F (3). The subunit positions that were shown to contact LvIA according to a previous molecular modeling study (Luo et al. (25)) are underlined.
FIGURE 3.

α-CTx LvIA dose-response curves for wild-type and mutant α3β2 nAChRs. All seven mutant receptors exhibited similar sensitivity for ACh to wild-type α3β2 nAChR. Values are mean ± S.E. from a recording made using 5–9 separate oocytes. Results are summarized in Table 1.
Increased potency of α-CTx LvIA was observed at α3β2[F119Q], α3β2[T59K], and α3β2[T59L] nAChRs. LvIA potently blocked ACh-evoked currents of these nAChRs with IC50 values of 0.58, 0.96, and 2.03 nm, respectively (Table 1). The ratio between the IC50 values of wild-type and mutant α3β2 nAChRs was 15 and 11 for α3β2[F119Q] and α3β2[T59K], respectively. The mutation T59L resulted in only a small increase (∼4.3-fold) of LvIA potency. The most LvIA-sensitive mutant, α3β2[F119Q], had a potency 2 orders of magnitude higher than that of the least sensitive mutant, α3β2[V111I] (Table 1, Fig. 3B).
LvIA is 255-fold less potent at α3β4 than at α3β2[F119Q], and 154-fold less potent at α3β4 than at α3β2[T59K]. Although introducing the β4 residues Lys and Gln at positions 119 and 59, respectively, increased the potency of LvIA for α3β2, this CTx is more potent at wild-type α3β2 than α3β4. A synergistic effect of binding site positions displaying different residues between the two subtypes might explain why LvIA is more active at α3β2 than at α3β4.
Mutations of the β2 Subunit Affect Recovery Time after Block by α-CTx LvIA
The α3β2 receptor mutants affected not only the potency of LvIA but also its recovery (Table 2, Fig. 4). α-CTx LvIA (10 nm) blocked wild-type α3β2 nAChRs versus mutant receptors α3β2[F119Q], α3β2[T59K], and α3β2[V111I] to different degrees (Fig. 4). α-CTx LvIA at 10 nm blocked ∼55% current of wild-type α3β2 but produced little or no block of α3β2[V111I] (Fig. 4, A and B). In contrast, complete block of ACh-evoked currents was obtained with 10 nm α-CTx LvIA on mutant receptors α3β2[F119Q] and α3β2[T59K] (Fig. 4, C and D).
TABLE 2.
Recovery time after block by α-CTx LvIA
| nAChR subtype | T95a | nAChR subtype | T95a |
|---|---|---|---|
| min | min | ||
| α3β2 | <2 | α3β2[T59L] | 6–9 |
| α3β2[K79A] | <1 | α3β2[F119Q] | 10–12 |
| α3β2[V111I] | <1 | α3β4 | 20–26 |
| α3β2[Q34A] | 2–3 | α3β2[T59K] | ≫20b |
| α3β2[T59I] | 2–3 |
a Time to 95% recovery after toxin washout.
b < 3% recovery after 20 min; concentration of α-CTx LvIA was 10 μm.
FIGURE 4.

α-CTx LvIA differentially blocks wild-type α3β2 nAChR (A) and mutant receptors α3β2[V111I] (B), α3β2[F119Q] (C), and α3β2[T59K] (D). The nAChRs display different reversibility kinetics after block. C indicates control responses to ACh. Oocytes were exposed to 10 nm peptide for 5 min followed by repetitive application of ACh. α-CTx LvIA at 10 nm blocked ∼55% current of wild-type α3β2 nAChR with rapid reversibility (A), but did not block α3β2[V111I] nAChR (B). LvIA at 10 nm blocked ∼100% current of mutant receptors α3β2[F119Q] nAChR with slow reversibility (C) and α3β2[T59K] nAChR with slowest reversibility (D).
We compared the recovery time (>95% initial current) after block by 10 μm toxin for the wild-type and mutant receptors (Table 2). Recovery of wild-type α3β2 nAChR was complete within 2 min after toxin washout. Thus, the t½ was estimated to be <30 s, and this time scale is beyond the resolution of the experimental setup. Similarly the off-rates for α3β2[T59I], α3β2[K79A], α3β2[V111I], and α3β2[Q34A] were also rapid (full recovery in 1–3 min). The recovery time of wild-type α3β4 nAChR was 20–26 min, which is slower than that of α3β2[F119Q] (10–12 min), but much faster than α3β2[T59K].
The three α3β2 mutations, T59K, T59L, and F119Q, affected the off-rates of LvIA significantly, as evidenced by the corresponding receptors having much slower reversible block by LvIA than wild-type α3β2 nAChR or mutants T59I, K79A, V111I, and Q34A. The recovery times of mutants α3β2[T59L] and α3β2[F119Q] were 6–9 and 10–12 min, respectively (Fig. 4C, Table 2), whereas α3β2[T59K] displayed the slowest recovery time, with less than 3% recovery 20 min after washout (Table 2). Even at low concentrations of LvIA (10–100 nm), α3β2[T59K] recovered very slowly from block. At 10 nm LvIA concentration, α3β2[T59K] recovered to 28 ± 3.5% current 20 min after washout, and at 100 nm concentration, only 13 ± 2% current was recovered 20 min after washout (Fig. 4D).
Molecular Modeling
A molecular model of the interactions between LvIA and the wild-type α3β2 nAChR showed that the β2 subunit positions considered for mutations are all potentially in contact with the conotoxin with the exception of position 34, as shown in Fig. 5A. LvIA had similar activity at wild-type and α3β2 Q34A nAChR, in agreement with the absence of interaction of this position. The wild-type β2 residue Lys79 can form a surface salt bridge with LvIA Asp11, and this interaction was found to be stable over a 30-ns molecular dynamics simulation (Fig. 5B). The three other substituted positions, i.e. positions 59, 111, and 119, are at least partly buried at the interface with LvIA. The change of activity of LvIA correlated with a change of buried surface solvent-accessible surface area at the interface for mutants of positions 59 and 119 (Fig. 5C). The molecular model did not provide a simple explanation for the decreased activity of LvIA at the V111I mutant, and we propose that this mutation could potentially result in conformational changes that cannot be modeled using short molecular dynamics simulations. The other mutated positions, i.e. 59 and 119, are located in β-strands, and the residues chosen for substitutions are not likely to disrupt this β-strand secondary structure. The 10-ns simulations of complexes incorporating the mutations T59K, V111I, or F1119Q resulted in similar binding modes with no change of binding site conformation (Fig. 6).
FIGURE 5.
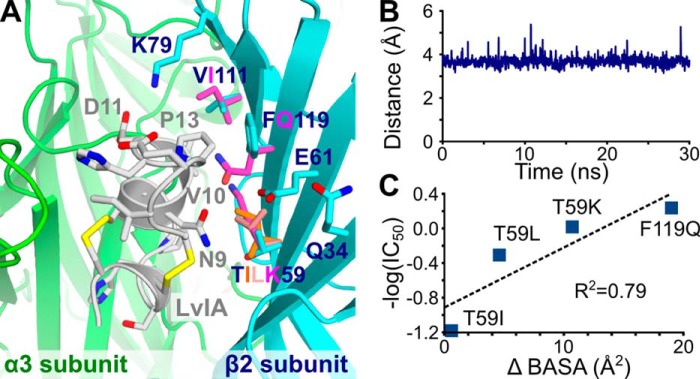
Molecular modeling of the interaction between LvIA and α3β2 wild-type and mutant nAChR. A, binding of LvIA (white) into the rat wild-type α3β2-binding pocket, which comprises the α3 principal subunit (green) and the β2 complementary subunit (blue). The conformation of the side chains of the β2 positions that were mutated are displayed overlaid with those of the wild-type receptor. The side chains of the mutants are shown in different colors from those used for the wild-type structure. B, distance between the NZ atom of β2 Lys79 and the CG atom of LvIA Asp11 over a 2-ns molecular dynamics simulation. C, correlation between the differences of buried solvent-accessible surface area between wild-type and mutant complexes (Δ BASA) and the IC50 for the α3β2 mutants.
FIGURE 6.
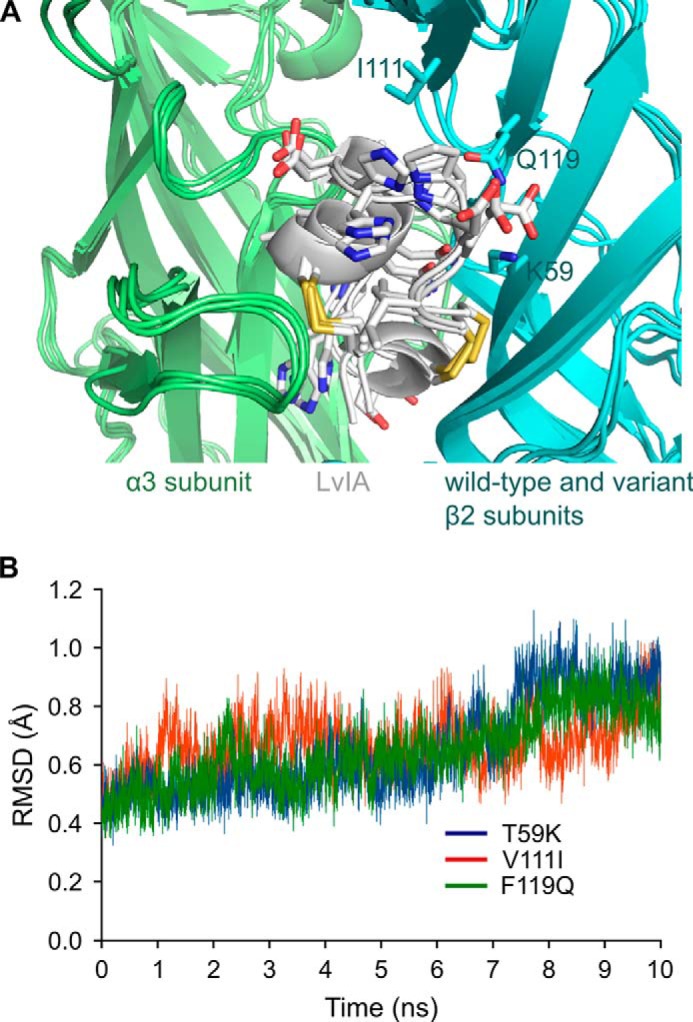
Molecular dynamics simulation of LvIA/α3β2 incorporating β2 subunit mutations T59K, V111I, or F119Q. A, overlay of the conformation of the binding sites after 10-ns simulations, with the α3 subunit in green, the β2 subunit in blue, and conotoxin LvIA in white. The side chains of LvIA as well as the mutated side chains Lys59, Ile111, and Gln119 are in stick representation. B, backbone root mean square deviation (RMSD) over the 10-ns simulations from the starting conformation of the β2 subunit binding site. This binding site is defined here as including the β1 (positions 32–40), β2 (positions 57–63), β5′ (positions 109–113), and β6 (positions 116–120) strands.
DISCUSSION
Neuronal nAChRs are widely expressed in the CNS and peripheral nervous system in adults and during development, but the identification of which subtype is expressed in which nervous cell is challenging (33–38). LvIA is the first ligand to be highly specific for α3β2 nAChR, and it could potentially be used in physiological studies of this receptor (25). We sought here to gain further insights into the binding interactions of LvIA at this receptor through mutations of positions that have been shown to be important for the binding of other α-CTxs and ligands (24, 39). These studies have shown that competitive nicotinic ligands of nAChRs generally bind to both the α subunits and the β subunits that form a ligand-binding interface (18, 32).
We investigated the influence of seven α3β2 nAChR mutants on the binding of α-CTx LvIA. These residues were chosen based on previous findings with the related α-CTx LtIA (23). Four mutants, α3β2[F119Q], α3β2[T59K], α3β2[T59L] and α3β2[V111I], had significantly different sensitivity than the wild-type receptor to α-CTx LvIA (Tables 1 and 2; Figs. 3 and 4). The three other mutations, Q34A, K79A and T59I, had little or no detectable effect on α-CTx LvIA activity (Tables 1 and 2; Figs. 3 and 4). α-CTx LvIA at 10 nm blocked ∼55% of the current of wild-type α3β2 with rapid reversibility but blocked >95% of the current of α3β2[F119Q] and α3β2[T59K]; the block of the latter two nAChRs had much slower reversibility after toxin washout when compared with that observed for the wild-type α3β2 nAChR (Fig. 4). The substitution of Phe119 of the β2 subunit by Gln, which is present in the homologous position of the β4 subunit (α3β2[F119Q]), resulted in a 15-fold increase in α-CTx LvIA potency. The mutation T59K caused an 11-fold increase sensitivity for α-CTx LvIA, partly due to a decrease in off-rate (Fig. 4D). A similar finding has been reported for the 4/4 α-CTx BuIA (19). The potency of BuIA at α3β2[F119Q] and α3β2[T59K] increased 8- and 20-fold, respectively, when compared with wild-type α3β2, with very slow off-rates. However, BuIA had a faster off-rate but similar IC50 at α3β2[V111I] versus wild-type α3β2, in contrast to LvIA, which has a 15-fold decrease in potency at α3β2[V111I] when compared with wild-type α3β2 (Table 3) (19). Thus, we suggest that BuIA and LvIA have overlapping, yet distinct binding interactions with the receptor. Overall our data suggest that the three positions on the receptor, 59, 111, and 119, are key to LvIA binding. Of course, because we examined only a finite number of mutations, we cannot exclude the possibility that other positions might also be important.
TABLE 3.
IC50 of α-conotoxins on nAChR β2 subunit mutants
a Conserved amino acids are shaded in light grey. Conserved cysteine residues are boldface and boxed. Disulfide connectivity of these α-conotoxins is Cys1–Cys3 and Cys2–Cys4. * = C-terminal amide.
b Data for LtIA, BuIA, and MII are from previous studies of these toxins tested on the indicated receptors expressed in Xenopus oocytes.
c IC50 in nm.
d Ratio of IC50 of mutant α3β2 nAChR/IC50 wild-type α3β2.
As far as ligand residues contributing to binding are concerned, the highly conserved Ser-Xaa-Pro motif in the first loop of α-CTxs contains a small α-helix important for nAChR binding. α-CTx LtIA is atypical because it lacks this Ser-Xaa-Pro motif and has been suggested to bind a novel microsite on the α3β2 nAChR (19). α-CTx LtIA potentially interacts with β2 Phe119 and β2 Lys79 because the Phe119 and Lys79 mutants disrupted LtIA binding (19), but mutations of these positions were without effect for activity of α-CTxs MII, PnIA, and GID (18). By contrast, the mutation F119Q increased affinity of LvIA, and the mutation K79A did not affect LvIA activity. The mutation V111I in the β2 subunit was previously reported to have only a small effect on the activity of 4/7 α-CTxs MII, PnIA, GID (18), and LtIA (23) (Table 3). By contrast, this mutation decreased LvIA activity by 15-fold. LvIA displays the conserved Ser-Xaa-Pro motif in its first loop, suggesting that it adopts a similar binding mode to most 4/7 α-CTxs. The sequence in the second loop of α-CTx LvIA is therefore probably a determinant of its unique selectivity and distinct binding site. Molecular models indeed suggest that this second loop, especially residues Asn9, Val10, Asp11, and Pro13, interacts with the β2 subunit. Interestingly, the 4/7 α-CTx PeIA also potently blocks α3β2 nAChRs and has similar residues in its second loop (31).
The mutant K79A does not show a significant difference in activity from the wild-type nAChR, but the molecular models suggest that the Lys79 residue establishes a stable charge interaction with LvIA Asp11 (Fig. 5B). It has been proposed that surface salt bridges can have little contribution to binding affinity because the favorable charge-charge interaction can be counterbalanced by the negative entropic effect of restraining the conformation of the side chain (40). This compensation of enthalpy for entropy components between apo and bound states is a potential explanation for the innocuous nature of the K79A substitution. Indeed, the Lys79 side chain is highly exposed to the solvent and should therefore have considerable conformational freedom in the absence of the toxin. The side chain of Lys79 was restrained during the molecular simulations of the bound toxin, suggesting a significant entropic cost to the immobilization of the side chain.
The molecular models suggest that substitutions at positions 119 and 59 increase the solvent-accessible surface area buried at the interface, and this increase correlates with higher affinities of LvIA observed experimentally (Fig. 5C). In particular, the three mutations, T59I, T59L, and T59K, incrementally introduce longer side chains at position 59, and they result in increasing inhibitory potency of LvIA. The introduction of a positively charged Lys at position 59 results in the burial of a positively charged group, which could be detrimental to binding, but this residue can potentially interact with the negatively charged β2 subunit Glu61, which is proximally located (Fig. 5A). The F119Q mutation resulted in better complementarity at the interface by creating further interactions, especially with LvIA residues Val10 and Pro13, resulting in the largest buried surface area among all mutants in this study, in agreement with the highest inhibitory activity of LvIA among all mutants.
It is interesting to compare the trends in LvIA binding to α3β2 versus α3β4 relative to the individual residue substitutions at the three key positions of 59, 111, and 119. In principle, the decreases in IC50 values associated with the T59K and F119Q substitutions should more than compensate for the increased IC50 associated with the V111I substitution. Nevertheless LvIA is 17-fold more potent at α3β2 than at α3β4. The non-additivity of the single point mutant effects can probably be explained by the spatial organization of these positions because the side chain at position 119 is sandwiched by those of positions 111 and 59. The β4 subunit, which displays bulkier side chains at these positions than the β2 subunit, should present a different interface to LvIA than the β2 subunit single point mutants.
In conclusion, we have identified three residues in the nAChR β2 subunit that are key to the binding interaction of LvIA with the α3β2 nAChR. Furthermore, molecular modeling indicates that the sequence of residues in the second loop of LvIA is particularly important for high affinity for the α3β2 nAChR. These findings help provide insights into the unique selectivity profile of this toxin. Understanding interactions between different α-CTxs and α3β2 nAChR should further help to elucidate the molecular pharmacology of this subtype.
Acknowledgments
We thank Olena Filchakova, Layla Azam, Baldomero Olivera, and Doju Yoshikami for advice and help.
This work was supported, in part, by the National Natural Science Foundation of China (Grants 81420108028 and 41366002), State High-Tech Research and Development Project (863) of the Ministry of Science and Technology of China Grant 2012AA021706, Program for International Science and Technology Cooperation Program of China Grant 2011DFR31210, Changjiang Scholars and Innovative Research Team in University Grant PCSIRT, IRT1123. This work was also supported by National Institutes of Health Grants GM48677 and GM103801, University of Queensland Early Career Research Grant (2013002338), and Australian Research Council Grant 1093115.
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- CTx
- conotoxin.
REFERENCES
- 1. Yakel J. L. (2013) Cholinergic receptors: functional role of nicotinic ACh receptors in brain circuits and disease. Pflugers Arch. 465, 441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wallace T. L., Bertrand D. (2013) Importance of the nicotinic acetylcholine receptor system in the prefrontal cortex. Biochem. Pharmacol. 85, 1713–1720 [DOI] [PubMed] [Google Scholar]
- 3. Corringer P. J., Le Novère N., Changeux J. P. (2000) Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 40, 431–458 [DOI] [PubMed] [Google Scholar]
- 4. Rahman S. (2013) Nicotinic receptors as therapeutic targets for drug addictive disorders. CNS Neurol. Disord. Drug Targets 12, 633–640 [DOI] [PubMed] [Google Scholar]
- 5. Somm E. (2014) Nicotinic cholinergic signaling in adipose tissue and pancreatic islets biology: revisited function and therapeutic perspectives. Arch. Immunol. Ther. Exp. (Warsz.) 62, 87–101 [DOI] [PubMed] [Google Scholar]
- 6. Schaal C., Chellappan S. P. (2014) Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol Cancer Res. 12, 14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen J. X., Qin D., Wang H., Wu C., Shi F. D., Wu J. (2013) Roles of nicotinic acetylcholine receptors in stem cell survival/apoptosis, proliferation and differentiation. Curr. Mol. Med. 13, 1455–1464 [DOI] [PubMed] [Google Scholar]
- 8. Posadas I., López-Hernández B., Ceña V. (2013) Nicotinic receptors in neurodegeneration. Curr. Neuropharmacol. 11, 298–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poorthuis R. B., Mansvelder H. D. (2013) Nicotinic acetylcholine receptors controlling attention: behavior, circuits and sensitivity to disruption by nicotine. Biochem. Pharmacol 86, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 10. Albuquerque E. X., Pereira E. F., Alkondon M., Rogers S. W. (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jensen A. A., Frølund B., Liljefors T., Krogsgaard-Larsen P. (2005) Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 48, 4705–4745 [DOI] [PubMed] [Google Scholar]
- 12. Rossman A. C. (2011) The physiology of the nicotinic acetylcholine receptor and its importance in the administration of anesthesia. AANA J. 79, 433–440 [PubMed] [Google Scholar]
- 13. Muttenthaler M., Akondi K. B., Alewood P. F. (2011) Structure-activity studies on α-conotoxins. Curr. Pharm. Des. 17, 4226–4241 [DOI] [PubMed] [Google Scholar]
- 14. Armishaw C. J. (2010) Synthetic α-conotoxin mutants as probes for studying nicotinic acetylcholine receptors and in the development of novel drug leads. Toxins (Basel) 2, 1471–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lebbe E. K., Peigneur S., Wijesekara I., Tytgat J. (2014) Conotoxins targeting nicotinic acetylcholine receptors: an overview. Mar. Drugs 12, 2970–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lewis R. J., Dutertre S., Vetter I., Christie M. J. (2012) Conus venom peptide pharmacology. Pharmacol. Rev. 64, 259–298 [DOI] [PubMed] [Google Scholar]
- 17. Azam L., McIntosh J. M. (2009) α-Conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 30, 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dutertre S., Nicke A., Lewis R. J. (2005) β2 subunit contribution to 4/7 α-conotoxin binding to the nicotinic acetylcholine receptor. J. Biol. Chem. 280, 30460–30468 [DOI] [PubMed] [Google Scholar]
- 19. Shiembob D. L., Roberts R. L., Luetje C. W., McIntosh J. M. (2006) Determinants of α-conotoxin BuIA selectivity on the nicotinic acetylcholine receptor β subunit. Biochemistry 45, 11200–11207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cartier G. E., Yoshikami D., Gray W. R., Luo S., Olivera B. M., McIntosh J. M. (1996) A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J. Biol. Chem. 271, 7522–7528 [DOI] [PubMed] [Google Scholar]
- 21. Luo S., Nguyen T. A., Cartier G. E., Olivera B. M., Yoshikami D., McIntosh J. M. (1999) Single-residue alteration in α-conotoxin PnIA switches its nAChR subtype selectivity. Biochemistry 38, 14542–14548 [DOI] [PubMed] [Google Scholar]
- 22. Azam L., Dowell C., Watkins M., Stitzel J. A., Olivera B. M., McIntosh J. M. (2005) α-Conotoxin BuIA, a novel peptide from Conus bullatus, distinguishes among neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 280, 80–87 [DOI] [PubMed] [Google Scholar]
- 23. Luo S., Akondi K. B., Zhangsun D., Wu Y., Zhu X., Hu Y., Christensen S., Dowell C., Daly N. L., Craik D. J., Wang C. I., Lewis R. J., Alewood P. F., McIntosh J. M. (2010) Atypical α-conotoxin LtIA from Conus litteratus targets a novel microsite of the α3β2 nicotinic receptor. J. Biol. Chem. 285, 12355–12366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harvey S. C., McIntosh J. M., Cartier G. E., Maddox F. N., Luetje C. W. (1997) Determinants of specificity for α-conotoxin MII on α3β2 neuronal nicotinic receptors. Mol Pharmacol 51, 336–342 [DOI] [PubMed] [Google Scholar]
- 25. Luo S., Zhangsun D., Schroeder C. I., Zhu X., Hu Y., Wu Y., Weltzin M. M., Eberhard S., Kaas Q., Craik D. J., McIntosh J. M., Whiteaker P. (2014) A novel α4/7-conotoxin LvIA from Conus lividus that selectively blocks α3β2 vs. α6/α3β2β3 nicotinic acetylcholine receptors. FASEB J. 28, 1842–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 [DOI] [PubMed] [Google Scholar]
- 28. Yu R., Craik D. J., Kaas Q. (2011) Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 7, e1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu R., Kompella S. N., Adams D. J., Craik D. J., Kaas Q. (2013) Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J. Med. Chem. 56, 3557–3567 [DOI] [PubMed] [Google Scholar]
- 30. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 31. Celie P. H., Kasheverov I. E., Mordvintsev D. Y., Hogg R. C., van Nierop P., van Elk R., van Rossum-Fikkert S. E., Zhmak M. N., Bertrand D., Tsetlin V., Sixma T. K., Smit A. B. (2005) Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an α-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 12, 582–588 [DOI] [PubMed] [Google Scholar]
- 32. Dutertre S., Lewis R. J. (2004) Computational approaches to understand α-conotoxin interactions at neuronal nicotinic receptors. Eur. J. Biochem. 271, 2327–2334 [DOI] [PubMed] [Google Scholar]
- 33. Garza A., Huang L. Z., Son J. H., Winzer-Serhan U. H. (2009) Expression of nicotinic acetylcholine receptors and subunit messenger RNAs in the enteric nervous system of the neonatal rat. Neuroscience 158, 1521–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. D'hoedt D., Bertrand D. (2009) Nicotinic acetylcholine receptors: an overview on drug discovery. Expert Opin. Ther. Targets 13, 395–411 [DOI] [PubMed] [Google Scholar]
- 35. Dani J. A., Bertrand D. (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 47, 699–729 [DOI] [PubMed] [Google Scholar]
- 36. Tsuneki H., Kimura I., Dezaki K., Kimura M., Sala C., Fumagalli G. (1995) Immunohistochemical localization of neuronal nicotinic receptor subtypes at the pre- and postjunctional sites in mouse diaphragm muscle. Neurosci. Lett. 196, 13–16 [DOI] [PubMed] [Google Scholar]
- 37. Arredondo J., Chernyavsky A. I., Marubio L. M., Beaudet A. L., Jolkovsky D. L., Pinkerton K. E., Grando S. A. (2005) Receptor-mediated tobacco toxicity: regulation of gene expression through α3β2 nicotinic receptor in oral epithelial cells. Am. J. Pathol. 166, 597–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Quik M., Vailati S., Bordia T., Kulak J. M., Fan H., McIntosh J. M., Clementi F., Gotti C. (2005) Subunit composition of nicotinic receptors in monkey striatum: effect of treatments with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or l-DOPA. Mol. Pharmacol. 67, 32–41 [DOI] [PubMed] [Google Scholar]
- 39. Antil-Delbeke S., Gaillard C., Tamiya T., Corringer P. J., Changeux J. P., Servent D., Ménez A. (2000) Molecular determinants by which a long chain toxin from snake venom interacts with the neuronal α7-nicotinic acetylcholine receptor. J. Biol. Chem. 275, 29594–29601 [DOI] [PubMed] [Google Scholar]
- 40. Sun D. P., Sauer U., Nicholson H., Matthews B. W. (1991) Contributions of engineered surface salt bridges to the stability of T4 lysozyme determined by directed mutagenesis. Biochemistry 30, 7142–7153 [DOI] [PubMed] [Google Scholar]