

Background: ZPI-protein Z complex is a critical anticoagulant regulator of membrane-associated factor Xa.
Results: The energetics and multistep kinetics of the ZPI-protein Z interaction were characterized with fluorescently labeled K239C ZPI.
Conclusion: Ionic and hydrophobic interactions mediate ZPI-protein Z binding, and conserved ZPI Lys-239 promotes protein Z dissociation.
Significance: The studies establish the thermodynamic and kinetic basis for catalytic activation of ZPI by protein Z.
Keywords: Coagulation Factor, Kinetics, Protease, Protease Inhibitor, Serpin, ZPI, Anticoagulant, Factor Xa, Protein Z
Abstract
The anticoagulant serpin, protein Z-dependent protease inhibitor (ZPI), circulates in blood as a tight complex with its cofactor, protein Z (PZ), enabling it to function as a rapid inhibitor of membrane-associated factor Xa. Here, we show that N,N′-dimethyl-N-(acetyl)-N′-(7-nitrobenz-3-oxa-1,3-diazol-4-yl)ethylenediamine (NBD)-fluorophore-labeled K239C ZPI is a sensitive, moderately perturbing reporter of the ZPI-PZ interaction and utilize the labeled ZPI to characterize in-depth the thermodynamics and kinetics of wild-type and variant ZPI-PZ interactions. NBD-labeled K239C ZPI bound PZ with ∼3 nm KD and ∼400% fluorescence enhancement at physiologic pH and ionic strength. The NBD-ZPI-PZ interaction was markedly sensitive to ionic strength and pH but minimally affected by temperature, consistent with the importance of charged interactions. NBD-ZPI-PZ affinity was reduced ∼5-fold by physiologic calcium levels to resemble NBD-ZPI affinity for γ-carboxyglutamic acid/EGF1-domainless PZ. Competitive binding studies with ZPI variants revealed that in addition to previously identified Asp-293 and Tyr-240 hot spot residues, Met-71, Asp-74, and Asp-238 made significant contributions to PZ binding, whereas Lys-239 antagonized binding. Rapid kinetic studies indicated a multistep binding mechanism with diffusion-limited association and slow complex dissociation. ZPI complexation with factor Xa or cleavage decreased ZPI-PZ affinity 2–7-fold by increasing the rate of PZ dissociation. A catalytic role for PZ was supported by the correlation between a decreased rate of PZ dissociation from the K239A ZPI-PZ complex and an impaired ability of PZ to catalyze the K239A ZPI-factor Xa reaction. Together, these results reveal the energetic basis of the ZPI-PZ interaction and suggest an important role for ZPI Lys-239 in PZ catalytic action.
Introduction
Protein Z-dependent protease inhibitor (ZPI)2 is an anticoagulant protein of the serpin superfamily that regulates the activity of membrane-associated blood coagulation factor Xa in a manner dependent on the cofactor, protein Z (PZ) (1–4). ZPI also inhibits factor XIa efficiently independent of protein Z (3, 4). The importance of ZPI and PZ as anticoagulant regulators of factor Xa and factor XIa is suggested by the prothrombotic phenotype of PZ knock-out mice and embryonic lethality of ZPI knock-out mice harboring the factor V Leiden mutation (5, 6), consistent with the aggravated thromboembolic risk observed in factor V Leiden patients with PZ deficiency (7). Clinical studies concerning the relationship between ZPI/PZ levels and risk of venous thrombosis and thromboembolic diseases are often conflicting, although generally there are more reports supporting such association than those that do not (8, 9). ZPI and PZ may also play a role in other physiologic and pathologic processes such as inflammation (10) and cancer (11, 12).
PZ is a vitamin K-dependent plasma protein with a structure similar to that of factors VII, IX, and X and protein C but with a non-functional protease domain (13). The protein cofactor circulates as a tight complex with ZPI (14) with PZ levels limiting the amount of complex formed (3, 15). The tight binding of PZ to ZPI is thought to enable the ZPI-PZ complex to bind to a procoagulant membrane surface where it can encounter and rapidly inhibit membrane-bound factor Xa (1, 4, 16). ZPI-PZ complex structures have been solved by us and another group (16, 17). The structures show that PZ binds ZPI through extensive ionic and hydrophobic interactions centered on helix G, the C terminus of helix A, and sheet C of ZPI, a novel binding site for a serpin cofactor interaction. Our recent mutagenesis studies of ZPI residues in the contact interface with PZ have demonstrated that two of six contact residues, Asp-293 on helix G and Tyr-240 on sheet C, constitute binding hot spots and contribute the bulk of the binding energy of the protein-protein interaction (18). Based on these findings, we hypothesized that the binding hot spots could be targeted to develop novel drugs capable of disrupting the ZPI-PZ anticoagulant complex and restoring hemostasis in hemophilia bleeding disorders. To test this idea, we sought to develop a rapid and sensitive assay for ZPI-PZ complex formation for high throughput screening of small molecules that disrupt the ZPI-PZ interaction. We report here on the development of such an assay that exploits our previous finding that Lys-239 in the ZPI-PZ interface does not appear to contribute to binding PZ. By replacing Lys-239 with a cysteine and labeling with N,N′-dimethyl-N-(acetyl)-N′-(7-nitrobenz-3-oxa-1,3-diazol-4-yl)ethylenediamine (NBD) or dansyl fluorophores, we found that the fluorophore labels on ZPI underwent large fluorescence changes upon PZ binding and were only moderately perturbing.
A problem with past studies of the ZPI-PZ interaction is that they have depended on kinetic assays to infer the affinity of the interaction based on the rate-enhancing effect of PZ on ZPI inhibition of factor Xa in the presence of phospholipids and calcium because previous spectroscopic probes of the ZPI-PZ interaction have been insufficiently responsive to study the interaction under equilibrium conditions (4, 18). A detailed thermodynamic and kinetic characterization of the interaction has thus not been possible, and the true energetic contributions of individual residues to the binding interaction and the role of ZPI complex formation with factor Xa in promoting PZ release and action as a catalyst remain to be validated. Using the fluorescently labeled K239C ZPIs as probes, we now provide the first in-depth thermodynamic and kinetic characterization of the ZPI-PZ interaction, the energetic contributions of amino acid determinants to the interaction, and the effect of ZPI complexation with factor Xa on the interaction. Our results reveal an unexpected role for the highly conserved Lys-239 in promoting PZ catalytic action.
EXPERIMENTAL PROCEDURES
Proteins
Plasma-derived human factor Xa, protein Z, and factor XIa were purchased from Enzyme Research Laboratories. Recombinant wild-type and mutant ZPIs were engineered on a wild type-like ZPI background in which cysteines 169 and 264 were changed to alanine and serine, respectively (16). In the case of wild-type and K239C ZPIs, variants with the natural P1 Tyr as well as with the P1 Tyr changed to Arg were engineered to facilitate factor Xa cleavage and preparation of an RCL-cleaved form as reported previously (18). ZPI variants with Ala mutations of PZ binding site residues on a wild-type background that retained cysteines 169 and 264 were from our previous study (18). DNA mutagenesis was carried out by PCR using the QuikChange mutagenesis kit (Stratagene). All mutations were confirmed by DNA sequencing. Recombinant human ZPIs were expressed in baculovirus-infected insect cells and purified as described previously (4). To avoid oxidation of cysteine in the expression and purification of single cysteine ZPI variants, 5 mm cysteine was added to the conditioned medium of insect cells expressing the variants, and 1 mm DTT was added to buffers used for purification of the variants. Recombinant PZ lacking the γ-carboxyglutamic acid (Gla) and EGF1 N-terminal domains (residues 84–360) and with a C-terminal His tag (referred to as rPZ) was expressed in baculovirus-infected insect cells using the BD BaculoGold transfection kit (BD Biosciences). The expressed rPZ in culture medium was purified by nickel-Sepharose chromatography (GE Healthcare) as described (17). Molar concentrations of wild-type and mutant ZPIs, plasma PZ, and rPZ were determined from the absorbance at 280 nm using calculated extinction coefficients of 31,525, 62,568, and 41,940 m−1 cm−1, respectively (19). Protease concentrations were determined by standard activity assays that were calibrated based on active site titrations (20).
Fluorophore Labeling
K239C ZPI or cleaved K239C ZPI (∼30 μm) were dialyzed in 50 mm Tris, 0.1 m NaCl, pH 8.5 buffer containing 50 μm DTT, labeled with 400 μm iodoacetamido-NBD or 5-[2-(2-iodoacetamido)ethylamino]-1-naphthalenesulfonic acid (Molecular Probes) for ∼4 h at 4 °C in the dark and then purified from unreacted label as described (21). The extent of fluorophore labeling was determined from the ratio of NBD or DANS absorbance to the protein absorbance at 280 nm after correcting the latter for the contribution of the fluorophore. Extinction coefficients of 25,000 m−1 cm−1 at 478 nm for NBD and 5700 m−1 cm−1 for DANS at 330 nm were used for these calculations.
Experimental Conditions
Binding and rapid kinetic experiments were carried out at 25 °C in 50 mm Tris-HCl buffer, pH 7.1 containing 0.1 m NaCl and 0.1% PEG 8000 (I ∼ 0.15). Equilibrium binding titrations were performed in PEG 20,000-coated acrylic cuvettes. The enzyme kinetic assays for factor Xa and factor XIa inhibition were carried out in the same Tris-HCl buffer but at pH 7.4. Binding experiments at varying pH values were performed in 10 mm Mes or Tris buffers containing 0.15 m NaCl and 0.1% PEG 8000. Binding experiments were also done in 50 mm Tris-HCl buffer, pH 7.1 over a range of NaCl concentrations of 0.05–0.5 m and at 0.1 m NaCl over a range of calcium concentrations. In most experiments, ovalbumin was included at 0.1 mg/ml to improve protein stability. Binding experiments at varying temperatures were done in 50 mm sodium phosphate, 0.1 m NaCl, 0.1% PEG 8000 buffer, pH 7.1 with pH adjustments made at each temperature to ensure a constant pH.
Fluorescence Emission Spectra
Fluorescence emission spectra were measured with an SLM 8000 spectrofluorometer with excitation at 480 nm over the emission range of 500–600 nm for NBD-ZPI and at 292 or 336 nm over the emission range of 400–550 nm for DANS-ZPI. Emission was monitored in 5-nm steps with 5–10-s integrations per step. Spectra were obtained with 100 nm labeled ZPI with at least three replicate measurements averaged. Buffer or buffer plus PZ background spectra were subtracted, and dilution corrections were made for added PZ (<10%).
Equilibrium Binding of PZ to Labeled ZPIs
Equilibrium dissociation constants for fluorescently labeled ZPI-PZ interactions were measured at 25 °C by titrating PZ into solutions of labeled ZPIs (25–50 nm) and monitoring the changes in NBD fluorescence at 545 nm (λex, 480 nm) or DANS fluorescence at 480 nm (λex, 292 nm). Titrations were computer-fit by nonlinear least squares analysis using Kaleidagraph 4.1 software (Synergy) by the following quadratic binding equation.
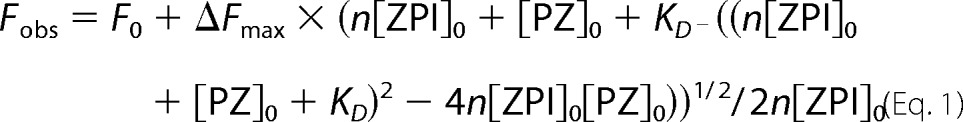 |
where Fobs, F0, and ΔFmax are the observed, initial, and maximal changes in fluorescence, respectively; [ZPI]0 and [PZ]0 are the total ZPI and PZ concentrations, respectively; KD is the dissociation constant; and n is the binding stoichiometry. F0, KD, and ΔFmax were the fitted parameters. To obtain a reliable value of n, this parameter was fitted in experiments with high concentrations of labeled ZPI (50–100 nm) in buffer containing 0.05 or 0.1 m NaCl. Fitted values of n were then assumed in experiments using 25 nm labeled ZPI to best determine KD.
The effect of ionic strength on ZPI-PZ complex affinity was analyzed by the Debye-Hückel equation (22).
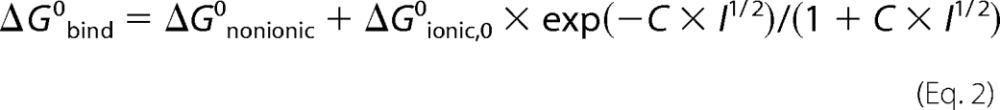 |
where ΔG0bind, ΔG0nonionic, and ΔG0ionic,0 are the standard free energies for overall binding, for the nonionic component of binding, and for the ionic component of binding at zero ionic strength, respectively; C is a constant related to the Debye-Hückel screening parameter; and I is the ionic strength. Measured KD values at different ionic strengths were converted to binding free energies by the relation ΔG0bind = −RT × ln(1/KD) where R is the gas constant and T is the absolute temperature. At 25 °C (298 K), this relation becomes ΔG0bind = 1.364 kcal/mol × log KD. ΔG0bind values as a function of the square root of the ionic strength were then fitted by the above equation with ΔG0nonionic, ΔG0ionic,0, and C, the fitted parameters. The relative contributions of nonionic and ionic interactions to the unitary binding energy were obtained by correcting ΔG0nonionic for the unfavorable entropic energy of bringing two molecules together in solution of +2.38 kcal/mol at 25 °C (23).
Calcium Effects
The dependence of ZPI-PZ complex affinity on calcium concentration was analyzed by Equation 3.
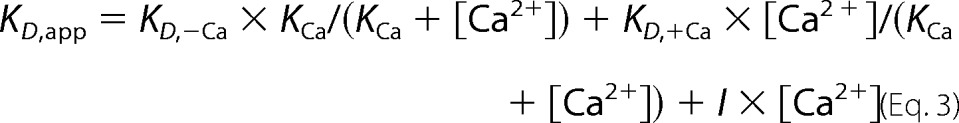 |
where KD,app is the apparent dissociation constant measured for the NBD-ZPI-PZ interaction at a given calcium concentration; KD,−Ca and KD,+Ca are dissociation constants for NBD-ZPI binding to free and calcium-saturated PZ, respectively; KCa is the dissociation constant for calcium binding to PZ (assuming equal and independent sites); and I represents a factor accounting for the ionic strength effect of calcium ions on affinity. In computer fitting of data with this equation, I was fixed based on the observed ionic strength dependence of the interaction, and KD,−Ca, KD,+Ca, and KCa were the fitted parameters.
Competitive Binding of Unlabeled and Labeled ZPIs to PZ
Binding of unlabeled wild-type and variant ZPIs to PZ was measured by competitive binding titrations in which the unlabeled ZPI was titrated into a solution of NBD-labeled ZPI-PZ complex, and the fluorescence decrease due to displacement of the labeled ZPI from the ZPI-PZ complex by unlabeled ZPI was measured. Small corrections were made for decreases in fluorescence due to scattering (<10%) in titrations of weak binding Y240A and D293A ZPI variants based on control buffer titrations. Observed relative fluorescence changes were fit by the cubic equation for competitive binding to obtain the KD for the unlabeled ZPI-PZ interaction, assuming a stoichiometry of 1 and fixing the KD and stoichiometry for the labeled ZPI interaction at their independently measured values (24).
Rapid Kinetics
Rapid kinetic studies of labeled ZPI-PZ interactions were performed with an Applied Photophysics SX-17MV stopped-flow instrument under pseudo-first order conditions in which the molar concentration of PZ was maintained at least 5-fold greater than that of NBD-labeled K239C ZPI or RCL-cleaved K239C ZPI. Reactions were monitored from NBD fluorescence changes using an excitation wavelength of 480 nm and an emission filter with a 520-nm cut-on wavelength. Data were collected over two 500-point split time frames of 2 and 50 s. Progress curves were fit by the following three-exponential equation.
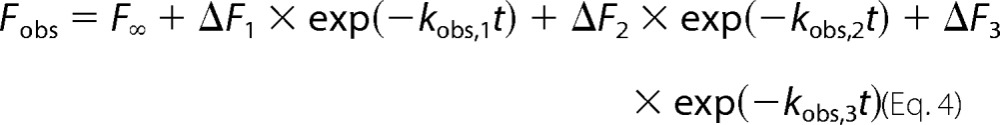 |
where Fobs is the observed fluorescence at time t; F∞, ΔF1, ΔF2, and ΔF3 are end point fluorescence and first phase, second phase, and third phase fluorescence changes, respectively; and kobs,1, kobs,2, and kobs,3 are the observed rate constants for the three kinetic phases. F∞, ΔF1, ΔF2, ΔF3, kobs,1, kobs,2, and kobs,3 were the fitted parameters. 5–10 reaction traces were averaged for each PZ concentration. Buffer and NBD-ZPI controls were done to correct observed signals for background and validate the NBD-ZPI starting fluorescence. The kinetics of ZPI-PZ complex dissociation were studied by mixing NBD-ZPI-PZ complex with buffer and increasing concentrations of unlabeled K239A to induce complex dissociation. The fluorescence decrease due to complex dissociation was fit by two instead of three exponential terms in the above equation. NBD-ZPI binding to PZ was also analyzed in the presence of increasing concentrations of unlabeled ZPI with progress curves fit by the three-exponential equation. Data were fit by numerical integration of the differential equations for a three-step binding model using Kintek Explorer 3.0 software (25). Because of variability in fluorescence amplitudes due to background scattering, corrections of observed amplitudes were made (<10%) to constrain the amplitude dependence on PZ concentration to conform to the independently measured KD in equilibrium binding titrations.
Stoichiometries of ZPI-Protease Reactions
Fixed concentrations of protease (∼100 nm factor Xa or ∼20 nm factor XIa) were incubated with increasing concentrations of ZPI ranging from a 3–10-fold molar excess in the presence of PZ equimolar with the ZPI concentration, 25 μm phospholipid vesicles (7:3 phosphatidylcholine:phosphatidylserine from Avanti prepared as described (4)), and 5 mm CaCl2 for reactions with factor Xa or in the absence of cofactors for reactions with factor XIa in reaction volumes of 25–100 μl. After allowing sufficient time to reach maximal inhibition based on measured second order association rate constants (∼3–15 min), the remaining proteolytic activity was measured by diluting the reaction mixture with 1 ml of 100 μm Spectrozyme factor Xa (American Diagnostica) for factor Xa reactions or 100 μm S2366 (Diapharma) for factor XIa reactions and monitoring the absorbance change at 405 nm. 10 mm EDTA was included in the substrate when Ca2+ was present in the reactions. Initial rates of hydrolysis were determined from second order polynomial fits of substrate hydrolysis progress curves as described previously (4). The stoichiometry of inhibition (SI) was determined from the fitted abscissa intercept of a linear plot of residual protease activity against molar ratio of inhibitor to protease.
Kinetics of ZPI-Factor Xa and ZPI-Factor XIa Reactions
The kinetics of protease inactivation by wild-type and variant ZPIs was measured under pseudo-first order conditions in which the inhibitor was in large molar excess over the protease (at least 5 times greater than the concentration of ZPI required to fully inhibit protease) in the presence of equimolar PZ, 5 mm Ca2+, and 25 μm membrane vesicles for reactions with factor Xa or in the absence of cofactors for reactions with factor XIa. Progress curves for the time-dependent inhibition of protease were measured in discontinuous assays in which samples (50–100 μl) were incubated for different times or withdrawn from reaction mixtures at varying times and diluted into 1 ml of fluorescence substrate (either 50 μm Pefafluor FXa for factor Xa inhibition or 40 μm t-butoxycarbonyl-EAR-aminomethylcoumarin for factor XIa inhibition (both from Centerchem)) (18). 10 mm EDTA was included in the substrate when Ca2+ was present in the reactions. Initial velocities of substrate hydrolysis were measured by fitting progress curves by the second order polynomial function as described previously (4). Second order association rate constants (ka) were obtained from the slopes of fits of the linear dependence of kobs on the ZPI concentration ([ZPI]0) according to the following equation.
where kd represents the rate constant for ZPI-protease complex dissociation as reported previously (4). Corrections of ka for differing SIs were made by multiplying ka by SI.
Reactions with catalytic PZ were done with fixed concentrations of 200 nm ZPI and 6 nm factor Xa in the presence of 25 μm lipid, 5 mm CaCl2, and 0–2.5 nm PZ. Progress curves were acquired as above, and kobs was measured from fits with a single exponential function. Corrections of kobs for SI were made by multiplying uncatalyzed and PZ-catalyzed contributions to kobs by the corresponding SIs for uncatalyzed and catalyzed reactions. The second order association rate constant for PZ catalysis was obtained from the slope of a plot of kobs × SI versus PZ concentration corrected for the fraction of PZ complexed with ZPI based on measured KD values.
RESULTS
Characterization of Fluorophore-labeled ZPIs as Probes of PZ Binding
To engineer a fluorescently labeled ZPI that reports PZ binding in a minimally perturbing manner, we chose to label Lys-239, a residue located on the periphery of the ZPI-PZ binding interface that appears not to be involved in binding PZ (18). Lys-239 was changed to cysteine in a cysteine-free ZPI variant in which the two wild-type Cys residues were changed to Ser or Ala. The Ser/Ala substitutions do not affect ZPI functional properties (Table 1) (16). K239C ZPI was expressed and purified with a yield similar to that of the cysteine-free parent ZPI, and the mutant was labeled with iodoacetamido-NBD or -DANS fluorophores to give final NBD/ZPI or DANS/ZPI ratios close to 1 (1.1 ± 0.1 for NBD-ZPI and 1.0 ± 0.1 for DANS-ZPI). The K239C mutation or the labeling of K239C ZPI with NBD or DANS fluorophores minimally affected the reactivity of ZPI with factor Xa in the presence of protein Z, phospholipid, and Ca2+ cofactors or with factor XIa in the absence of these cofactors (Table 1).
TABLE 1.
Kinetics of wild-type, cysteine-free “wild-type,” and K239 variant ZPI inhibition of factor Xa and factor XIa
Rate constants and stoichiometries for ZPI-factor Xa and ZPI-factor XIa reactions in I 0.15, pH 7.4 Tris buffer at 25 °C were measured in the presence of PZ, calcium, and membrane cofactors for factor Xa reactions and in the absence of cofactors for factor XIa reactions as described under “Experimental Procedures.” Apparent second order rate constants were obtained from the slopes of the linear dependence of pseudo-first order rate constants on ZPI concentration in the range of 15–300 nm. The product of the apparent association rate constant (kapp) and SI represents the corrected association rate constant for reaction through the inhibitory pathway.
| ZPI | Factor Xa reactions |
Factor XIa reactions |
||||
|---|---|---|---|---|---|---|
| kapp | SI | kapp × SI | kapp | SI | kapp × SI | |
| m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | |||
| Wild typea | 3.1 ± 0.1 × 106 | 2.8 ± 0.2 | 8.7 ± 0.9 × 106 | 14.0 ± 0.1 × 104 | 8.0 ± 0.3 | 11.0 ± 0.1 × 105 |
| C169A/C264S | 4.7 ± 0.1 × 106 | 3.7 ± 0.1 | 17.4 ± 0.8 × 106 | 6.4 ± 0.9 × 104 | 9.4 ± 0.3 | 6.0 ± 1.0 × 105 |
| +K239C | 4.8 ± 0.5 × 106 | 3.5 ± 0.1 | 16.8 ± 2.2 × 106 | 11.0 ± 0.1 × 104 | 10.4 ± 0.1 | 11.4 ± 0.2 × 105 |
| +K239C-NBD | 2.6 ± 0.5 × 106 | 3.1 ± 0.2 | 8.1 ± 2.1 × 106 | 6.2 ± 0.6 × 104 | 11.0 ± 0.2 | 6.8 ± 0.8 × 105 |
| +K239C-DANS | 2.8 ± 0.1 × 106 | 2.9 ± 0.1 | 8.1 ± 0.6 × 106 | 9.5 ± 1.5 × 104 | 9.4 ± 0.2 | 8.9 ± 1.6 × 105 |
| K239Aa | 2.4 ± 0.1 × 106 | 4.2 ± 0.1 | 10.1 ± 0.7 × 106 | 14.0 ± 0.1 × 104 | 8.7 ± 0.4 | 12.2 ± 0.6 × 105 |
a Taken from Ref. 18. The K239A variant retains the two wild-type cysteines.
Addition of a molar excess of PZ to NBD-labeled ZPI resulted in a large enhancement and 3-nm blue shift in the NBD fluorescence emission spectrum (Fig. 1A). Titration of the fluorescence enhancement showed that it was saturable and well fit by the equation for tight equilibrium binding (Fig. 2). Fits of several titrations indicated a high affinity NBD-ZPI-PZ interaction with an average KD of 2.6 ± 0.6 nm, a binding stoichiometry of 0.9 ± 0.1, and a maximum 3–4-fold fluorescence enhancement.
FIGURE 1.
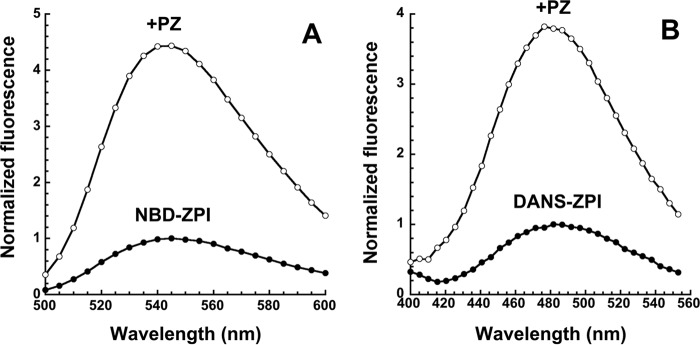
Emission spectra of fluorescently labeled ZPIs and their complexes with protein Z. A, emission spectra of 100 nm NBD-K239C ZPI in pH 7. 1, I 0.15 Tris buffer at 25 °C were taken with excitation at 480 nm in the absence and presence of 300 nm PZ. Corrections were made for background signal of buffer ±PZ and for PZ dilution, and then spectra were normalized to the maximum fluorescence of free labeled ZPI as described under “Experimental Procedures.” B, emission spectra of 100 nm DANS-K239C ZPI were taken in pH 7.1, I 0.15 buffer with excitation at 292 nm in the absence and presence of 230 nm PZ, corrected for buffer and PZ, and normalized as in A.
FIGURE 2.
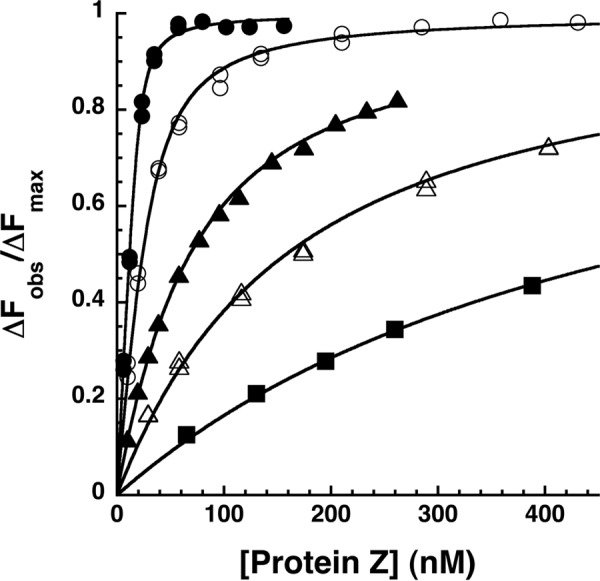
Equilibrium binding titrations of the NBD-ZPI-PZ interaction at varying ionic strengths. Fluorescence titrations of NBD-ZPI (25 nm) in pH 7. 1 Tris buffer containing 0.1 (●), 0.175 (○), 0.25 (▴), 0.35 (▵), and 0.5 (■) M NaCl at 25 °C were monitored at λex 480 nm and λem 545 nm as a function of increasing PZ concentration. Observed fluorescence values were corrected for buffer and dilution and then fit by the quadratic equilibrium binding equation (solid lines) with the binding stoichiometry fixed at values fitted from separate stoichiometric titrations as described under “Experimental Procedures.” The observed changes in fluorescence (ΔFobs) are expressed relative to the fitted maximal fluorescence change (ΔFmax), the latter showing no significant dependence on ionic strength within experimental error.
Addition of a molar excess of PZ to DANS-labeled ZPI similarly caused a 4-nm blue shift and 2–3-fold saturable enhancement in the fluorescence emission spectrum when tryptophan fluorescence was excited at 292 nm (Fig. 1B). Direct excitation of the DANS fluorophore at 336 nm resulted in the same blue shift but a smaller 1.2-fold enhancement in the emission spectrum (not shown). The larger enhancement observed with 292-nm excitation indicated that PZ binding to DANS-ZPI was accompanied by significant FRET between PZ tryptophans and the DANS label on ZPI. Equilibrium binding titrations indicated an average KD of 4 ± 1 nm and binding stoichiometry of 0.6–0.8 for the PZ-DANS-ZPI interaction, similar to that measured for the PZ-NBD-ZPI interaction.
To determine whether the fluorophore labels affected the binding affinity of PZ for ZPI, competitive binding titrations were performed in which unlabeled K239C ZPI was titrated into NBD-labeled ZPI-PZ complex, and the fluorescence decrease due to competitive displacement of labeled ZPI from the complex by unlabeled ZPI was monitored (Fig. 3). Computer fitting of data with the competitive binding equation indicated a KD for the unlabeled K239C ZPI-PZ interaction of 0.1 ± 0.1 nm, representing a >10-fold higher affinity of PZ for the unlabeled ZPI than for NBD- or DANS-labeled ZPIs. The fluorophore labels thus significantly weaken the K239C ZPI interaction with protein Z. Surprisingly, competitive binding titrations with wild-type unlabeled ZPI (Fig. 3) indicated a KD of 2 ± 1 nm, which was similar to the value measured for the NBD-labeled ZPI interaction (Table 2). This suggested that the wild-type Lys-239 residue impaired PZ binding relative to Cys at this position to an extent similar to that of the NBD-labeled Cys-239 residue and thus that PZ affinity for ZPI is sensitive to the size of the side chain at position 239. Competitive binding titrations with a K239A ZPI mutant confirmed that replacing Lys-239 with a smaller alanine side chain resulted in a subnanomolar KD (0.1 ± 0.1 nm) indistinguishable from that measured for the K239C substitution (Table 2). NBD and DANS labels thus significantly perturb K239C ZPI interactions with PZ to similar extents, and replacing Cys-239 with the wild-type Lys residue produces a comparable perturbation.
FIGURE 3.
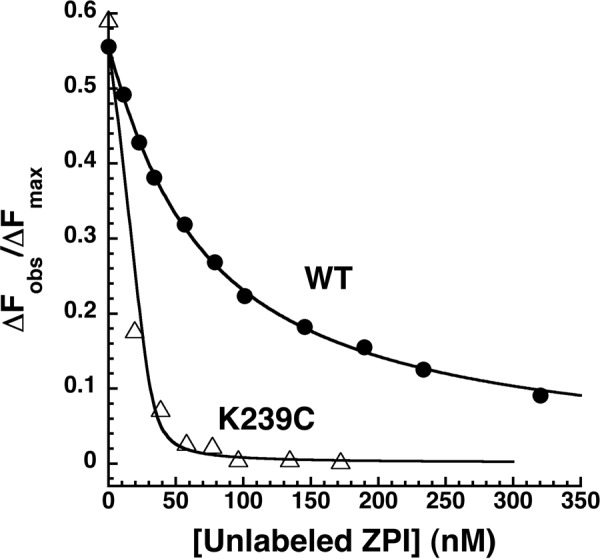
Competitive equilibrium binding titrations of the NBD-ZPI-PZ complex with wild-type and K239C ZPI. NBD-ZPI-PZ complex formed with 53 nm NBD-ZPI and 32 nm PZ was titrated with increasing concentrations of unlabeled wild-type and K239C ZPIs in pH 7. 1, I 0.15 Tris buffer at 25 °C, and the observed fluorescence was monitored at 545 nm as in Fig. 2. Fluorescence data corrected for buffer and dilution were fit by the cubic competitive binding equation with the binding stoichiometry and KD for the labeled ZPI interaction fixed at independently measured values and assuming a 1:1 binding stoichiometry for the unlabeled ZPIs as described under “Experimental Procedures.” Observed changes in fluorescence are expressed relative to the fitted maximal fluorescence change of the NBD-ZPI-PZ interaction (ΔFmax).
TABLE 2.
Binding constants for unlabeled wild-type and variant ZPI interactions with PZ
Dissociation constants (KD) for ZPI-PZ interactions were determined in pH 7.1 Tris buffer containing 0.1 m NaCl (I 0.15) or 0.25 m NaCl (I 0.30) at 25 °C from competitive binding titrations of NBD-ZPI-PZ with unlabeled ZPIs as shown in Fig. 7 and described under “Experimental Procedures.” All recombinant ZPIs contained the two wild-type cysteine residues.
| ZPI variant | KD I 0.15 | KD I 0.30 | ΔG0 I 0.15 | ΔΔG0 I 0.15 | Unitary binding free energya I 0.15 |
|---|---|---|---|---|---|
| nm | nm | kcal/mol | kcal/mol | % | |
| WT ZPI | 2.1 ± 1.0 | 16 ± 4 | −11.8 | ||
| WT cZPI | 5.2 ± 0.2 | 23 ± 2 | −11.3 | ||
| M71A | 17 ± 4 | −10.6 | 1.2 | 8.7 | |
| D74A | 84 ± 41 | −9.6 | 2.2 | 15 | |
| D238A | 4.2 ± 0.8 | 52 ± 11 | −11.4 | 0.4 | 2.9 |
| K239A | 0.1 ± 0.1 | 0.3 ± 0.2 | −13.9 | −2.0 | |
| K239C | 0.1 ± 0.1 | 1.1 ± 0.3 | −13.6 | −1.8 | |
| Y240A | 330 ± 30 | −8.8 | 3.0 | 21 | |
| D293A | 1200 ± 500 | −8.1 | 3.8 | 26 |
a The unitary binding energy represents the apparent standard free energy of binding of ZPI and PZ (ΔG0) corrected for the entropic energy required to bring two molecules together in solution (2.38 kcal/mol at 25 °C) (23).
Ionic Strength Dependence of the ZPI-PZ Interaction
The x-ray structure of the ZPI-PZ complex together with previous mutagenesis studies has suggested that ionic interactions are important contributors to the ZPI-PZ interaction (16–18). We therefore investigated the ionic strength dependence of the binding of PZ to the NBD-labeled ZPI. Increasing buffer ionic strength dramatically decreased ZPI-PZ binding affinity (Fig. 2 and Table 3). The measured KD increased ∼200-fold when NaCl was increased from 0.1 to 0.5 m. Debye-Hückel analysis of the ionic strength dependence of KD according to Equation 2 under “Experimental Procedures” (Fig. 4) indicated that ionic interactions contributed 49% (6.9 kcal/mol) to the unitary binding energy at physiologic ionic strength and that nonionic interactions contributed the remaining 51% (7.2 kcal/mol).3
TABLE 3.
Ionic strength dependence of ZPI and cZPI interactions with PZ
Binding constants for NBD-ZPI and NBD-cZPI interactions with PZ were determined at varying NaCl concentrations in pH 7.1 Tris buffer at 25 °C by computer fitting of titrations such as those in Fig. 2 as described under “Experimental Procedures.”
| [NaCl] |
KD |
|
|---|---|---|
| ZPI | cZPI | |
| m | nm | |
| 0.05 | 5.9 ± 1.0 | |
| 0.075 | 2.2 ± 0.4 | 6.5 ± 2.0 |
| 0.10 | 2.6 ± 0.6 | 18 ± 3 |
| 0.175 | 9.5 ± 1.0 | 56 ± 20 |
| 0.25 | 60 ± 2 | 150 ± 10 |
| 0.35 | 140 ± 15 | 500 ± 60 |
| 0.50 | 490 ± 20 | 1000 ± 50 |
FIGURE 4.
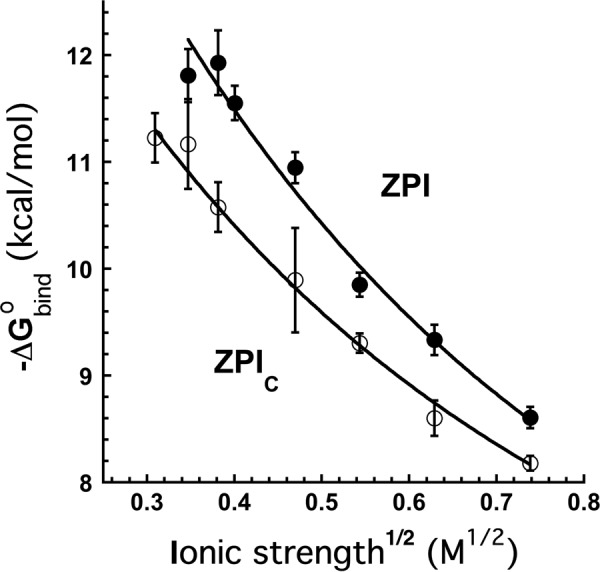
Ionic strength dependence of NBD-ZPI and NBD-cZPI interactions with PZ. The graph shows a Debye-Hückel analysis of the dependence of the binding free energy (−ΔG0bind) of NBD-labeled intact ZPI (●) and NBD-labeled cZPI (○) interactions with PZ on the square root of the ionic strength. −ΔG0bind was measured from equilibrium binding titrations of the interactions as a function of ionic strength as in Fig. 2. Solid lines are fits by Equation 2 under “Experimental Procedures.” Error bars represent ±S.E.
Given the ionic strength sensitivity of the ZPI-PZ interaction, we performed competitive binding titrations of NBD-labeled ZPI-PZ complex with unlabeled wild-type, K239C, and K239A ZPI variants to confirm their differential affinities for PZ under higher ionic strength conditions where KD values were more reliably measured. Fitting of these titrations yielded KD values of 16 ± 4 nm for wild-type, 1.1 ± 0.3 nm for K239C, and 0.3 ± 0.2 nm for K239A ZPI interactions with PZ (Table 2) as compared with the KD of 60 ± 2 nm determined for the labeled NBD-ZPI-PZ interaction at this ionic strength (Table 3). The Lys-239 side chain thus weakens affinity ≥15-fold, and the NBD label on the Cys-239 side chain weakens affinity ≥60-fold compared with Cys and Ala side chains under these higher ionic strength conditions.
Calcium and Lipid Effects
Because ZPI complexation with PZ enables ZPI to bind to a membrane through the Gla domain of PZ and this binding depends on calcium ions (16, 26), the effect of calcium ions and phospholipid membrane vesicles on the ZPI-PZ interaction was studied. PZ bound NBD-ZPI ∼8-fold more weakly in the presence of 5 mm Ca2+ than in its absence. By contrast, NBD-ZPI-PZ complex affinity decreased ∼1.5-fold when the NaCl concentration was increased to an equivalent ionic strength (Fig. 5A). Titrations of the NBD-ZPI-PZ complex with calcium ions revealed a specific calcium ion-dependent decrease in ZPI-PZ complex affinity as evidenced from the saturable decrease in fluorescence to a shallow sloping end point as the calcium concentration was increased with a midpoint at ∼1 mm Ca2+ (Fig. 5B). The sloping end point reflected the expected weakening of the complex affinity caused by calcium increases in ionic strength. The dependence of ZPI-PZ complex affinity on calcium concentration was analyzed from additional titrations at several fixed calcium concentrations (Fig. 5C). Fitting of this dependence assuming that a specific calcium ion interaction altered affinity yielded an apparent calcium concentration for half-maximal binding of ∼1 mm and a KD for the calcium-saturated ZPI-PZ complex of 15 ± 4 nm. These findings suggested that specific calcium ion interactions with either ZPI or PZ weakened ZPI-PZ complex affinity.
FIGURE 5.
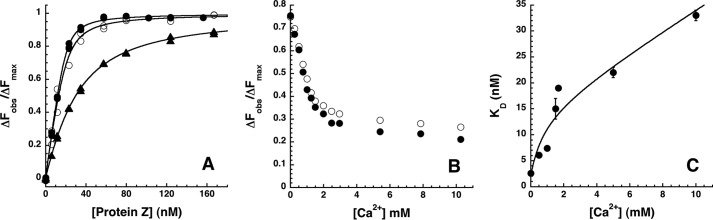
Calcium ion effects on the ZPI-PZ interaction. A, equilibrium binding titrations of 25 nm NBD-ZPI with PZ in pH 7. 1, I 0.15 Tris buffer at 25 °C (●) without or with the addition of 15 mm NaCl (○) or 5 mm CaCl2 (▴). Observed changes in fluorescence corrected for buffer and dilution are expressed relative to the fitted maximal fluorescence change. Solid lines are fits of data by the quadratic binding equation. B, titrations of NBD-ZPI-PZ complex formed with 26 nm labeled ZPI and 26 nm PZ with CaCl2 in pH 7.1, I 0.15 Tris buffer at 25 °C. Observed fluorescence changes were corrected for buffer and dilution as well as for minor scattering changes in control titrations with buffer lacking CaCl2 and expressed relative to the fitted maximal fluorescence change in separate titrations of NBD-ZPI with PZ under identical conditions. Closed and open circles represent replicate titrations. C, dependence of KD measured in equilibrium binding titrations of 25 nm NBD-ZPI with PZ in pH 7.1, I 0.15 buffer containing the indicated fixed concentrations of CaCl2. The solid line is a fit of data by Equation 3 under “Experimental Procedures” that relates specific calcium ion binding to NBD-ZPI and/or PZ and ionic strength effects of calcium to complex affinity. Error bars represent ±S.E.
Titrations of NBD-ZPI with PZ in the presence of phospholipid membrane vesicles (10 μm) and 1.5 mm calcium yielded a KD of 13 ± 1 nm, similar to the effect of calcium alone, indicating that the calcium-dependent binding of PZ to a membrane decreases ZPI affinity ∼5-fold from that in the absence of calcium and membrane as a result of specific calcium interactions with the complex. A control titration with phospholipid alone yielded a KD of 4 ± 1 nm, confirming no significant effect of membrane on the stronger NBD-ZPI-PZ interaction in the absence of calcium.
To determine whether the specific calcium ion effects on PZ binding to ZPI involved interactions with the PZ Gla domain, we examined the binding to NBD-labeled ZPI of an rPZ lacking the Gla and EGF1 domains. The truncated rPZ bound to NBD-labeled ZPI ∼5-fold more weakly than full-length plasma PZ at physiologic ionic strength (KD 12 ± 1 nm), indicating that the Gla/EGF1 domains of PZ did influence the binding interaction. However, in contrast to the NBD-ZPI-PZ interaction, the addition of calcium ions had minimal effects on rPZ binding to NBD-ZPI with the KD increasing only 1.5-fold when the calcium ion concentration was increased to 5 mm (KD 18 ± 1 nm) in accordance with that expected for the ionic strength increase caused by calcium addition. The differential sensitivities of NBD-ZPI interactions with rPZ and PZ to calcium ions were consistent with the specific calcium ion effects on the latter interaction resulting from calcium ion interactions with the PZ Gla domain.
pH and Temperature Effects
Titrations of NBD-ZPI with PZ as a function of the buffer pH at I 0.15 at 25 °C showed that binding was significantly weakened below pH 7 with the KD increasing ∼7-fold when the pH was lowered from 7.1 to 6.1 (KD 17 ± 3 nm), consistent with a role for charged acidic residues in binding. The KD changed insignificantly between pH 7.1 and 8.4 but increased significantly at pH 9.0 by ∼5-fold (KD 13 ± 2 nm), consistent with the importance of charged basic residues for binding. Varying the temperature over the range 5.0–35.0 °C resulted in no significant change in the affinity of NBD-labeled ZPI for PZ measured at pH 7.1, I 0.22 (KD 14–16 nm), implying that binding was largely entropically driven.
Effects of ZPI Cleavage
We previously provided evidence that factor Xa cleavage of ZPI at the RCL P1 residue weakens ZPI affinity for PZ and promotes PZ release and recycling as a catalytic cofactor (4, 18). To provide direct quantitation of the effect of RCL cleavage on ZPI binding to PZ under equilibrium conditions, we compared the ability of unlabeled native and RCL-cleaved wild-type ZPI to compete with NBD-labeled ZPI for binding PZ by competitive binding titrations. RCL-cleaved ZPI was prepared with a P1 Arg variant of ZPI that is preferentially cleaved by factor Xa. The KD values measured from competitive binding titrations showed that PZ bound cleaved ZPI 2.5-fold more weakly than native ZPI at physiologic ionic strength and ∼2-fold more weakly at twice this ionic strength (Table 2). To determine whether the effect of ZPI cleavage on PZ affinity was similar for both free cleaved ZPI and cleaved ZPI complexed with factor Xa, we performed competitive binding titrations in which wild-type ZPI (P1 Tyr), untreated or incubated with 1 eq of factor Xa based on the measured SI, were titrated into NBD-ZPI-PZ complex (Fig. 6). Both wild-type and K239A ZPIs were studied in these experiments. Comparable differences in KD between native ZPI and ZPI-factor Xa complexes of 2–4-fold were observed for wild-type and K239A ZPIs, indicating that similar losses in PZ affinity for ZPI result from RCL cleavage or complexation with factor Xa.
FIGURE 6.
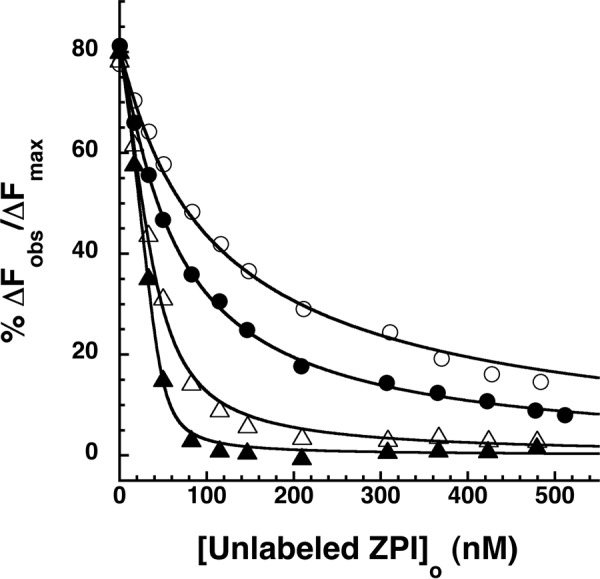
Effect of wild-type and K239A ZPI complexation with factor Xa on ZPI-PZ affinity. Competitive equilibrium binding titrations of NBD-ZPI-PZ complex formed with 55 nm NBD-ZPI and 49 nm PZ with wild-type ZPI (circles) or K239A ZPI (triangles) before (closed symbols) and after reaction with 1 SI eq of factor Xa (open symbols) are shown. Titrations were performed in pH 7.1, I 0.15 Tris buffer at 25 °C, and the observed fluorescence changes were corrected for buffer and dilution. Data were fit by the cubic competitive binding equation as in Fig. 3 (solid lines), and ΔFobs was expressed relative to the fitted maximal fluorescence change.
In a more direct approach, we prepared an RCL-cleaved NBD-labeled K239C P1 Arg ZPI variant (NBD-cZPI). PZ bound to NBD-cZPI with a fluorescence enhancement comparable with that of NBD-ZPI, a stoichiometry of 0.9 ± 0.1, and a KD of 18 ± 3 nm. The 7-fold higher KD for the NBD-cZPI-PZ interaction than the NBD-ZPI-PZ interaction suggested that PZ affinity for ZPI was reduced by RCL cleavage to a greater extent with the NBD-labeled proteins than the unlabeled proteins. Binding studies over a range of ionic strengths showed that the binding of PZ to NBD-cZPI was strongly dependent on ionic strength, similar to the NBD-ZPI-PZ interaction, and remained weaker than the NBD-ZPI-PZ interaction at all ionic strengths, although the differences in affinity progressively decreased as ionic strength was increased (Fig. 4 and Table 3). Debye-Hückel analysis showed that ionic interactions contributed significantly less unitary binding energy to the cleaved (5.4 kcal/mol) than to the uncleaved ZPI interaction (6.9 kcal/mol), whereas nonionic binding energy was not significantly different for the two interactions within experimental error (7.5 versus 7.2 kcal/mol). NBD-cZPI bound PZ and rPZ with similar affinity at physiologic ionic strength (KD values of 18 ± 3 and 20 ± 3 nm), and addition of 5 mm calcium caused similar ∼2-fold decreases in affinity (KD values of 40 ± 3 and 45 ± 2 nm), largely accountable for by increases in ionic strength due to the added calcium ions. These results suggest that calcium-Gla domain interactions do not affect the NBD-cZPI-PZ interaction as they do the NBD-ZPI-PZ interaction. Consequently, PZ affinity for NBD-ZPI is reduced somewhat less by RCL cleavage in the presence of 5 mm calcium (∼2-fold) than in its absence.
Contributions of ZPI Residues to PZ Binding
Our previous studies evaluated the relative contributions of ZPI residues to binding PZ by analyzing the effect of single Ala mutations of ZPI binding interface residues on PZ affinity through indirect kinetic titrations of the rate-enhancing effect of PZ on ZPI inhibition of membrane-bound factor Xa (18). To assess the true contributions of these ZPI residues to the binding energy of the PZ interaction under equilibrium conditions, we performed competitive binding titrations of NBD-labeled ZPI-PZ complex with the unlabeled ZPI mutants. The mutant ZPIs showed widely different abilities to displace labeled ZPI from its complex with PZ, reflecting a progressive decrease in binding affinity of the mutant ZPIs that followed the order WT > D238A > M71A > D74A > Y240A > D293A (Fig. 7 and Table 2). A clear 3-fold lower affinity of D238A ZPI than wild-type ZPI was measured at a higher ionic strength of 0.3. From the difference in standard free energy of binding (ΔΔG0) between mutant and wild-type ZPIs at physiologic ionic strength, unitary binding energy contributions of 0.3, 1.5, 2.5, 3.0 and 4.0 kcal/mol were calculated for Asp-238, Met-71, Asp-74, Tyr-240, and Asp-293 residues, respectively (Table 2).
FIGURE 7.
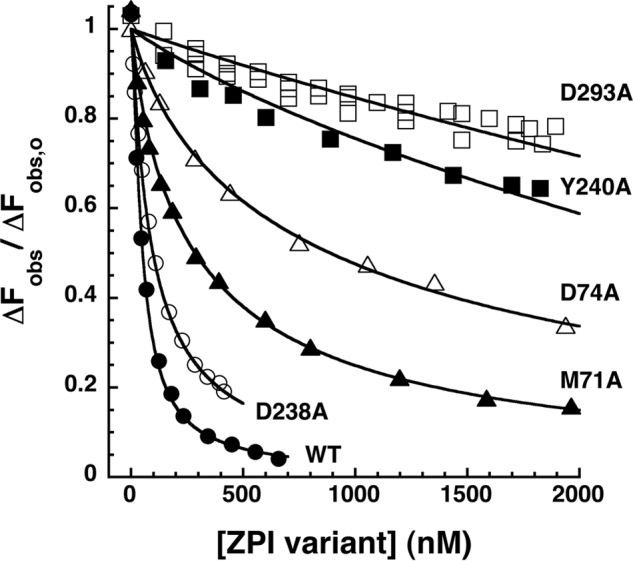
Competitive equilibrium binding titrations of NBD-ZPI-PZ complex with unlabeled mutant ZPIs. NBD-ZPI-PZ complex formed with 54 nm NBD-ZPI and 52 nm PZ was titrated with increasing concentrations of unlabeled ZPI variants with mutations in PZ binding site residues as indicated in pH 7. 1, I 0.15 Tris buffer at 25 °C, and the observed fluorescence was monitored at 545 nm as in Fig. 3. Fluorescence data corrected for buffer and dilution were fit by the cubic competitive binding equation as in Fig. 3 (solid lines). Additional details are provided under “Experimental Procedures.”
Kinetics of ZPI-PZ Interactions
The kinetics of PZ binding to NBD-labeled ZPI were studied under pseudo-first order conditions by stopped-flow fluorometry. Progress curves of the fluorescence change accompanying PZ binding to NBD-ZPI were clearly biphasic with a rapid phase accounting for ∼90% of the fluorescence change over 2 s and a slower phase accounting for the remaining ∼10% over the next 50 s (Fig. 8, A and B). Attempts to fit the kinetic data by two exponential changes in fluorescence revealed reproducible nonrandom deviations of the data from the fitted curves. However, fitting of the data with three exponential fluorescence changes resulted in random deviations of the data from the fitted curves and substantial decreases in the sum of the squares of the deviations from those observed with the two exponential fits. The observed pseudo-first order rate constants obtained from the fits for each of the three phases showed different dependences on the protein Z concentration: the dominant fast phase showed a linear dependence, the subsequent intermediate phase showed a saturable dependence, and the final very slow phase showed minimal dependence (Fig. 8C). These results suggested that PZ binding to ZPI was a three-step process with an initial bimolecular binding step causing the bulk of the fluorescence change followed by two unimolecular conformational changes in the complex that caused modest further increases in fluorescence. The slope of the linear first phase dependence of 1.6 ± 0.1 × 107 m−1 s−1 suggested a bimolecular association rate constant for the initial complex that approached the diffusion limit. The intercept of this dependence of 1.2 s−1 together with the limiting rate constant for the second phase dependence of 2–3 s−1 suggested that initial and intermediate complexes were formed in a rapid steady-state phase accounting for ∼90% of the fluorescence change. An invariant rate constant of 0.06–0.07 s−1 for the third phase implied that the intermediate complex underwent a very slow conformational change to form the final complex that accounted for the remaining ∼10% of the fluorescence change. Numerical fitting of the kinetic data by the differential equations for a three-step binding mechanism using Kintek Explorer software provided an excellent fit and confirmed the inferior fit of a two-step binding model (Table 4).
FIGURE 8.

Rapid kinetics of NBD-ZPI binding to PZ. A and B, NBD-ZPI (20 nm) was mixed with 100, 200, 300, 400, and 500 nm PZ, and the observed fluorescence changes (circles) were monitored over split time frames of 2 (A) and 50 s (B). Observed fluorescence changes from multiple averaged reaction traces were calculated by subtracting buffer background and fit by the three-exponential equation under “Experimental Procedures.” After correcting fluorescence amplitudes based on measured equilibrium KD values, ΔFobs was expressed relative to the fitted initial fluorescence. Every 10th data point is shown for clarity. Solid lines are global Kintek fits of complex association data together with dissociation data in D by a three-step sequential binding model. C, PZ concentration dependence of observed pseudo-first order rate constants for phase 1 (●), phase 2 (○), and phase 3 (▴) obtained from fits of data by the three-exponential function under “Experimental Procedures.” The solid line is a linear least squares fit of the phase 1 data, and the inset shows an expanded scale of the phase 3 data. D, kinetics of dissociation of the NBD-ZPI-PZ complex formed with 25 nm NBD-ZPI and 25 nm PZ after mixing with 200, 300, and 400 nm unlabeled K239A ZPI. Observed fluorescence changes were calculated as in A and B and fitted by the two-exponential equation under “Experimental Procedures.” After normalization based on the average fitted maximal fluorescence change, ΔFobs was expressed relative to the initial fluorescence of the complex. Solid lines are global Kintek fits of dissociation data together with the association data of A and B by the three-step binding model. The inset shows the K239A ZPI concentration dependence of kobs for phase 1 (●) and phase 2 (○).
TABLE 4.
Kinetic parameters derived from Kintek fits of progress curves of ZPI/cZPI reactions with PZ and rPZ
Fluorescence progress curves were obtained for reactions of 20 nm NBD-ZPI or NBD-cZPI with 100–600 nm PZ or rPZ in 50 mm Tris buffer, pH 7.1, I 0.15 at 25 °C such as those in Fig. 8, corrected for buffer background, and then fitted using Kintek software by a three-step sequential binding model as described under “Experimental Procedures.” Calcium was added at 1.5 mm, and lipid was added at 10 μm when present. The last column represents the calculated overall equilibrium dissociation constant obtained from the calculated dissociation constants: K1 = k−1/k1, K2 = k−2/k2, and K3 = k−3/k3, for the three binding steps.
| k+1 | k−1 | K1 | k+2 | k−2 | K2 | k+3 | k−3 | K3 | (K1K2K3)/(1 + K2K3 + K3) | |
|---|---|---|---|---|---|---|---|---|---|---|
| NBD-ZPI-PZa | 21.0 ± 0.1 (20) | 0.18 ± 0.02 (0.14) | 0.0086 ± 0.0010 | 1.3 ± 0.3 (0.52) | 2.1 ± 0.2 (0.71) | 1.6 ± 0.5 | 0.10 ± 0.02 (0.035) | 0.035 ± 0.001 (0.043) | 0.35 ± 0.08 | 0.003 |
| +Ca2+ | 12.2 ± 0.6 | 0.5 ± 0.2 | 0.04 ± 0.02 | 0.8 ± 0.8 | 1.9 ± 0.4 | 2 ± 2 | 0.10 ± 0.09 | 0.055 ± 0.005 | 0.6 ± 0.5 | 0.02 |
| +Ca2+ + lipid | 9.4 ± 0.4 | 0.5 ± 0.1 | 0.05 ± 0.01 | 1.0 ± 0.3 | 0.9 ± 0.1 | 0.9 ± 0.4 | 0.06 ± 0.02 | 0.05 ± 0.01 | 0.8 ± 0.4 | 0.01 |
| NBD-ZPI-rPZ | 15.3 ± 0.1 | 0.26 ± 0.01 | 0.017 ± 0.001 | 0.09 ± 0.02 | 0.56 ± 0.08 | 6 ± 2 | 0.15 ± 0.04 | 0.09 ± 0.01 | 0.6 ± 0.2 | 0.01 |
| NBD-cZPI-PZ | 17.4 ± 0.3 | 0.40 ± 0.05 | 0.023 ± 0.003 | 0.3 ± 0.3 | 1.9 ± 0.2 | 6 ± 7 | 0.06 ± 0.05 | 0.092 ± 0.003 | 2 ± 2 | 0.02 |
| NBD-cZPI-rPZ | 20 ± 2 | 0.64 ± 0.03 | 0.032 ± 0.005 | 0.2 ± 0.1 | 0.8 ± 0.7 | 4 ± 6 | 0.3 ± 0.2 | 0.13 ± 0.05 | 0.4 ± 0.4 | 0.02 |
a Global fit of association and dissociation kinetic data of Fig. 8. Values in parentheses are parameters obtained from fitting just association kinetic data.
To corroborate this multistep binding mechanism, we analyzed the kinetics of dissociation of the NBD-ZPI-PZ complex by mixing preformed complex with a molar excess of unlabeled K239A ZPI that binds ZPI >50-fold more tightly than the wild-type serpin. Complex dissociation was monitored from decreases in NBD probe fluorescence. Progress curves in this case were biexponential with ∼30% of the fluorescence decrease occurring in a fast exponential phase and the remainder occurring in a slower phase (Fig. 8D). K239A ZPI concentrations ranging from a 1–5-fold molar excess over NBD-ZPI resulted in a progressive convergence of dissociation progress curves, indicating that the higher K239A ZPI concentrations were sufficient to rapidly capture all PZ released from the complex and cause PZ dissociation from the NBD-ZPI-PZ complex to become rate-limiting. Limiting kobs values of 0.09 and 0.02 s−1 were measured for the rapid and slow phases for dissociation of the NBD-ZPI-PZ complex, consistent with a rapid dissociation phase reflecting dissociation of PZ from initial and intermediate complexes and a slow dissociation phase reflecting dissociation of PZ from a final more tightly bound complex. Fitting of the data by the three-step model yielded excellent fits and fair agreement of fitted kinetic parameters with those determined from association kinetic experiments. Global fitting of both association and dissociation kinetic data to the three-step binding model with relative fluorescence amplitudes constrained based on the measured overall equilibrium KD yielded excellent fits of all data (Fig. 8, A, B, and D). Such fits indicated that an initial complex with KD of 9 ± 1 nm was formed with an association rate constant of 2.1 ± 0.1 × 107 m−1 s−1 and dissociation rate constant of 0.18 ± 0.02 s−1 and then underwent two successive conformational changes that tightened affinity ∼3-fold (Table 4).
Similar kinetic studies were performed for (i) NBD-ZPI binding to PZ in the presence of calcium and phospholipid vesicles, (ii) NBD-ZPI binding to rPZ, and (iii) NBD-cZPI binding to PZ and rPZ. Progress curves resembled those observed for the NBD-ZPI-PZ reaction in all cases in showing a dominant fast phase fluorescence increase complete in ∼2 s followed by a slower fluorescence increase over ∼50 s and again required three exponential terms to provide good fits with nonrandom residuals. Kintek fitting of progress curves for each reaction by the three-step binding model suggested that the affinity differences for each of the binding interactions resulted largely from effects on the initial binding step with subsequent conformational change steps resulting in similar tightening of the complexes (Table 4).
To assess whether the kinetics of PZ binding to unlabeled ZPI resembled those for binding to labeled ZPI, we examined the kinetics of PZ binding to NBD-ZPI in the absence and presence of increasing concentrations of unlabeled wild-type, RCL-cleaved, and K239A ZPIs. Analysis of kobs for the dominant fast binding phase showed a linear increase in kobs as the concentration of competitor ZPIs was increased (Fig. 9). All three unlabeled competitor ZPIs gave indistinguishable linear increases in kobs with a slope of 1.2 ± 0.1 × 107 m−1 s−1, which is close to that for PZ binding to labeled ZPI, suggesting that all competitors bound PZ with similar kinetics of association and thus that the different PZ affinities of the competitors resulted from differences in the kinetics of complex dissociation.
FIGURE 9.
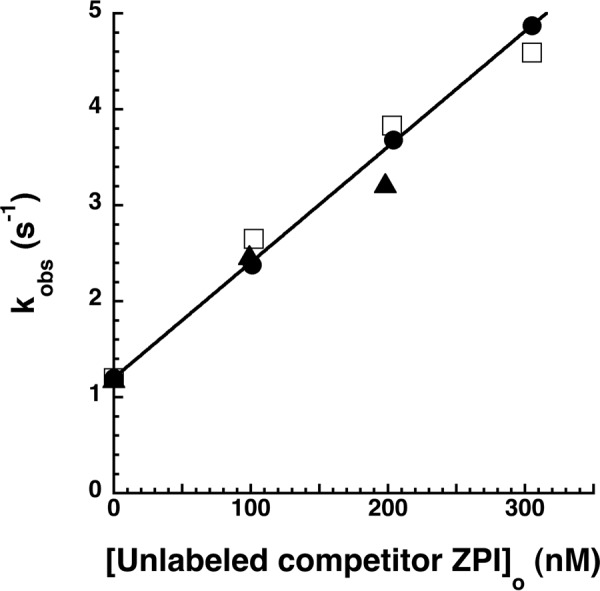
Kinetics of competitive binding of unlabeled and NBD-labeled ZPIs to PZ. NBD-ZPI (100 nm) was mixed with 20 nm PZ in the absence and presence of increasing concentrations of unlabeled wild-type intact (●) or cleaved (□) ZPI or K239A ZPI (▴). Averaged reaction traces were fit by the three-exponential equation, and kobs for phase 1 was plotted as a function of unlabeled competitor ZPI concentration. The solid line is a linear least squares fit of the wild-type intact ZPI data.
Correlation of ZPI-PZ Dissociation Rates with PZ Catalytic Action
To determine whether the differing rates of PZ dissociation from wild-type and K239A ZPI-PZ complexes affected the ability of PZ to act as a catalyst in the ZPI-factor Xa reaction (4, 18), we compared the kinetics of wild-type and K239A ZPI inhibition of factor Xa in the presence of catalytic levels of PZ, calcium, and phospholipid under pseudo-first order conditions. We also examined the D74A ZPI variant reaction because the lower affinity of PZ for this variant was expected to correlate with a faster rate of dissociation from the variant relative to the wild-type ZPI-PZ complex. Progress curves for ZPI inhibition of factor Xa in the absence and presence of PZ were well fit by single exponential functions (Fig. 10). Observed pseudo-first order rate constants increased linearly as the concentration of PZ was increased in a substoichiometric range (0.5–2.5 nm) relative to ZPI and factor Xa. Notably, the K239A ZPI-factor Xa reaction showed a ∼5-fold lower slope than either wild-type or D74A ZPI reactions. Correction for differences in SI yielded a 3.2–3.5-fold lower second order rate constant for PZ catalysis of 2.1 ± 0.3 × 106 m−1 s−1 compared with values of 7.4 ± 0.5 × 106 and 6.7 ± 0.4 × 106 m−1 s−1 for wild-type and D74A ZPI reactions, respectively. By contrast, similar second order rate constants were measured for wild-type, D74A, and K239A ZPI-factor Xa reactions in the presence of calcium and phospholipid at stoichiometric levels of PZ (18).
FIGURE 10.
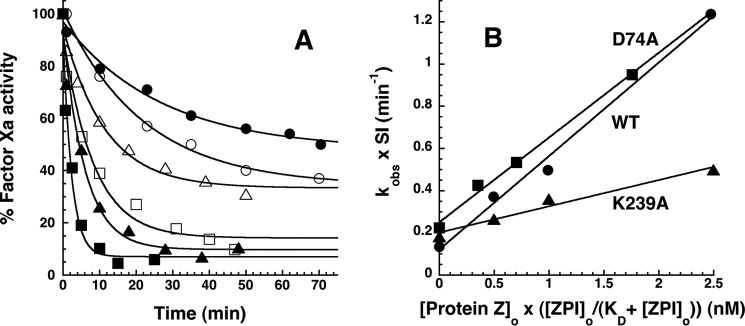
Effect of catalytic PZ on the kinetics of wild-type and variant ZPI-factor Xa reactions. A, progress curves are shown for reactions of 200 nm wild-type (closed symbols) or K239A ZPI (open symbols) with 6 nm factor Xa in the absence (circles) and presence of 1 (triangles) and 2. 5 nm (squares) PZ at 25 °C in pH 7.4 Tris buffer, I 0.15 containing 5 mm calcium and 25 μm phospholipid. Solid lines are fits by a single exponential function with nonzero end point. The end points progressively decrease with increasing PZ concentration because acylation and deacylation rates for formation and dissociation of the ZPI-PZ complex are comparable in the absence of PZ, whereas acylation is favored in the presence of PZ (4). B, dependence of SI-corrected kobs values for reactions of 200 nm wild-type (●), K239A (▴), and D74A (■) ZPIs with 6 nm factor Xa on PZ concentration corrected for the fraction of PZ bound to ZPI based on KD values reported in Table 2. kobs was obtained from exponential fits of the reaction progress curves in A and others not shown for the indicated ZPI-factor Xa reactions. SI corrections of kobs were made by summing the products, kobs × SI, for uncatalyzed and PZ-catalyzed contributions to the measured kobs based on SIs reported in Table 1 and Ref. 18. Solid lines are linear regression fits from which second order association rate constants for PZ catalysis of the ZPI-factor Xa reactions were obtained.
DISCUSSION
We have shown that replacing ZPI Lys-239 on the periphery of the PZ binding site with a Cys and labeling with NBD or DANS fluorophores yield a ZPI derivative that is a sensitive, moderately perturbing reporter of PZ binding. This has allowed us to characterize in unprecedented detail the thermodynamics and kinetics of wild-type and variant ZPI interactions with PZ under equilibrium conditions for the first time.
A surprising finding was that unlabeled K239C and K239A ZPIs bound PZ with 1–2 orders of magnitude greater affinities than unlabeled wild-type or fluorophore-labeled K239C ZPIs. The observation that ZPI affinities for PZ increased in the order K239A > K239C > wild type > NBD-K239C suggested a dependence of ZPI affinity on the length of the side chain at position 239, implying that the wild-type Lys-239 residue is detrimental to PZ binding. Lys-239 neighbors Tyr-240, a residue that we previously found to be a hot spot for PZ binding as a result of its interaction with a hydrophobic cavity in PZ formed by the juxtaposition of the pseudocatalytic and EGF2 domains of the cofactor (16, 18). The wild-type Lys and the fluorophore labels thus appear to similarly interfere sterically with the Tyr-240 side chain interaction with the PZ binding pocket.
Calcium acts as an essential cofactor to promote ZPI-PZ complex inhibition of factor Xa on a membrane surface by mediating the binding of PZ and factor Xa to the membrane through their Gla domains (1, 16). It was thus unexpected to find that calcium ions caused a membrane-independent ∼5-fold reduction in NBD-ZPI-PZ complex affinity. The differential effects of calcium on NBD-ZPI interactions with intact and Gla/EGF1-domainless PZ suggested that specific calcium interactions with the PZ Gla domain were responsible for the loss of complex affinity. Similar calcium-Gla domain interactions affect the reactions of factor Xa with its serpin inhibitors, ZPI and antithrombin, in the presence of the cofactor heparin (27, 28). In this case, calcium is thought to displace the Gla domain from an intramolecular interaction with the heparin binding site in the protease catalytic domain that blocks heparin bridging of the serpin-protease interactions. A similar intramolecular interaction of the PZ Gla domain with the pseudocatalytic or EGF2 domain in the absence of calcium could thus indirectly enhance the affinity of PZ for ZPI.
The membrane-independent KD of 15 nm measured for the NBD-ZPI-PZ interaction in the presence of physiologic calcium levels provides a close approximation of the affinity for the circulating ZPI-PZ complex. Interestingly, this affinity is about an order of magnitude weaker than the apparent affinity measured by kinetic titrations in the presence of calcium and membrane cofactors in past studies (KD ∼ 1 nm) (18). The stronger “kinetic” affinity suggests that the ZPI-PZ complex is effectively formed irreversibly in the reaction with factor Xa due to the membrane-bound ZPI-PZ complex associating with membrane-bound factor Xa before the ZPI-PZ complex can dissociate.
The ability to accurately quantitate the affinity of the ZPI-PZ interaction under equilibrium conditions prompted us to reevaluate the effects of mutations in the PZ binding site of ZPI on PZ binding affinity by competitive binding titrations. Although our findings confirm that Asp-293 and Tyr-240 are binding hot spots for PZ (18), they reveal that Asp-238, Met-71, and Asp-74 residues make more important contributions to PZ binding than suggested previously. The contribution of Asp-74 in particular approaches that of the hot spot residue Tyr-240. Assuming additivity of the interactions, Asp-238, Met-71, Asp-74, Tyr-240, and Asp-293 together account for 74% of the total unitary binding energy. The ionic binding energy represents 44% of the total and appears to derive mostly from two salt bridge clusters formed by Asp-74 interactions of ZPI with Arg-350 of PZ and Asp-293 interactions of ZPI with His-210 and Arg-298 of PZ. This is in agreement with our finding of a marked sensitivity of the binding interaction to ionic strength and to pH in the acid and alkaline ranges and 49% contribution of ionic interactions to the unitary binding energy indicated by Debye-Hückel analysis. The 51% contribution of nonionic binding energy is mostly accounted for by hydrophobic interactions of Met-71 and Tyr-240 with binding pockets in PZ. Additional nonionic binding energy may derive from the hydrophobic and van der Waals interactions of other ZPI residues that are buried and contribute to the complementary binding surface of ZPI with PZ (16, 17). The observed weak temperature dependence of the ZPI-PZ interaction is in keeping with the interaction being governed principally by ionic and hydrophobic interactions in view of the fact that such interactions are largely entropically driven.
The kinetics of PZ binding to and dissociation from NBD-ZPI revealed a three-step binding mechanism wherein an initial high affinity ZPI-PZ complex was formed at a close to diffusion limited rate of 2 × 107 m−1 s−1 and underwent two subsequent conformational changes that tightened the complex affinity a further ∼3-fold and reduced the limiting rate of complex dissociation to ∼0.02 s−1. These findings are consistent with our previous studies that found that Cys residues or fluorophore probes at positions distant from the PZ binding site underwent significant changes in reactivity or fluorescence upon PZ binding, consistent with global structural changes being induced in ZPI by PZ binding (16). However, such structural changes were found to account for at most 2-fold of the ∼1000-fold PZ rate enhancement of ZPI inhibition of membrane-associated factor Xa; the majority of the enhancement resulted from membrane bridging of the ZPI-PZ complex interaction with factor Xa. The structural changes may reflect successive induced fit alterations in the binding interface that minimize the negative effect of Lys-239 on the interaction and that are transmitted to other parts of the molecule. A similar three-step binding mechanism was indicated for the NBD-ZPI interaction with PZ in the presence of the physiologic cofactors, calcium, and membrane vesicles.
We reported previously that RCL cleavage of ZPI reduces PZ affinity and enables PZ to act catalytically in promoting ZPI inhibition of membrane-associated factor Xa (4, 18). We now confirm that cleaved ZPI, either free or in the ZPI-factor Xa complex, exhibits a 2–4-fold reduced affinity for PZ relative to native ZPI under equilibrium conditions. Comparisons of the ionic strength dependence of NBD-labeled cleaved and intact ZPI interactions with PZ showed that ionic interactions made a lesser contribution to PZ binding for cleaved ZPI than intact ZPI, whereas nonionic interactions were essentially the same. These findings are in keeping with the structural changes in cleaved ZPI reported previously that implicate an altered interaction of ZPI Asp-74 on helix A with PZ in the cleaved serpin (18).
The association rate constants for PZ binding to labeled or unlabeled ZPI, cZPI, and K239A ZPI were all similar, implying that the differences in PZ affinity for these ZPIs derive entirely from different rates of complex dissociation. The rate of PZ dissociation from ZPI must clearly limit the ability of the cofactor to act catalytically, and thus the increased rate of PZ dissociation from cleaved ZPI compared with ZPI would appear to be critical for PZ to function as a catalyst. Conversely, the lower rate of PZ dissociation from K239A ZPI than wild type suggests that PZ catalytic action might be impaired with this ZPI variant. This expectation was borne out by our finding that catalytic but not stoichiometric levels of PZ showed a ∼4-fold reduced accelerating effect on K239A ZPI inhibition of membrane-associated factor Xa compared with wild-type or D74A ZPI reactions. The reduced rate of PZ dissociation from K239A ZPI-PZ complex thus appears to limit PZ catalytic recycling, whereas faster PZ dissociation rates from wild-type and D74A ZPI-PZ complexes no longer limit PZ catalysis. This may explain why Lys-239 is highly conserved in ZPIs from lungfish to mammals. PZ levels are limiting in vivo (3), and hence the ability to release PZ from ZPI once the serpin forms inhibitory complexes with factors Xa and XIa may be important to maintain levels of PZ sufficient for anticoagulant regulation.
The importance of hot spot residues in mediating the ZPI-PZ interaction supports the feasibility of blocking the interaction with small molecules as a potential means of treating hemophilias. The sensitivity of our present fluorescence assay for detecting ZPI-PZ complex formation suggests its usefulness as a screening assay for small molecule inhibitors of the interaction. The importance of ZPI-PZ as a natural anticoagulant is clear from human clinical and animal knock-out studies. Preliminary data have shown that disrupting the ZPI-PZ complex in normal and hemophilic plasmas significantly increases thrombin formation and has the potential to ameliorate the bleeding of hemophilia by restoring a procoagulant/anticoagulant balance sufficient to achieve normal hemostasis. Studies are in progress to validate this potential.
Acknowledgments
We thank Matthew Anderson for assistance in early stages of this study and Dr. Peter G. W. Gettins of the Department of Biochemistry and Molecular Genetics at the University of Illinois at Chicago for critical comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R37 HL39888 (to S. T. O.). This work was also supported by American Heart Association Scientist Development Grant SDG 48880022 (to X. H.).
The unitary binding energy represents the apparent standard free energy of binding of ZPI and PZ (ΔG0) corrected for the entropic energy required to bring two molecules together in solution (2.38 kcal/mol at 25 °C) (23).
- ZPI
- protein Z-dependent protease inhibitor
- cZPI
- reactive center loop-cleaved ZPI
- PZ
- protein Z
- rPZ
- recombinant Gla/EGF1-domainless protein Z
- NBD
- N,N′-dimethyl-N-(acetyl)-N′-(7-nitrobenz-3-oxa-1,3-diazol-4-yl)ethylenediamine
- DANS
- dansyl (5-dimethylaminonaphthalene-1-sulfonyl)
- RCL
- reactive center loop
- SI
- stoichiometry of inhibition
- Gla
- γ-carboxyglutamic acid.
REFERENCES
- 1. Han X., Fiehler R., Broze G. J., Jr. (1998) Isolation of a protein Z-dependent plasma protease inhibitor. Proc. Natl. Acad. Sci. U.S.A. 95, 9250–9255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Han X., Huang Z. F., Fiehler R., Broze G. J., Jr. (1999) The protein Z-dependent protease inhibitor is a serpin. Biochemistry 38, 11073–11078 [DOI] [PubMed] [Google Scholar]
- 3. Han X., Fiehler R., Broze G. J., Jr. (2000) Characterization of the protein Z-dependent protease inhibitor. Blood 96, 3049–3055 [PubMed] [Google Scholar]
- 4. Huang X., Swanson R., Broze G. J., Jr., Olson S. T. (2008) Kinetic characterization of the protein Z-dependent protease inhibitor reaction with blood coagulation factor Xa. J. Biol. Chem. 283, 29770–29783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yin Z. F., Huang Z. F., Cui J., Fiehler R., Lasky N., Ginsburg D., Broze G. J., Jr. (2000) Prothrombotic phenotype of protein Z deficiency. Proc. Natl. Acad. Sci. U.S.A. 97, 6734–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J., Tu Y., Lu L., Lasky N., Broze G. J., Jr. (2008) Protein Z-dependent protease inhibitor deficiency produces a more severe murine phenotype than protein Z deficiency. Blood 111, 4973–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kemkes-Matthes B., Nees M., Kühnel G., Matzdorff A., Matthes K. J. (2002) Protein Z influences the prothrombotic phenotype in factor V Leiden patients. Thromb. Res. 106, 183–185 [DOI] [PubMed] [Google Scholar]
- 8. Bafunno V., Santacroce R., Margaglione M. (2011) The risk of occurrence of venous thrombosis: focus on protein Z. Thromb. Res. 128, 508–515 [DOI] [PubMed] [Google Scholar]
- 9. Vasse M. (2011) The protein Z/protein Z-dependent protease inhibitor complex. Systemic or local control of coagulation? Hamostaseologie 31, 155–158, 160–164 [DOI] [PubMed] [Google Scholar]
- 10. Girard T. J., Lasky N. M., Tuley E. A., Broze G. J., Jr. (2013) Protein Z, protein Z-dependent protease inhibitor (serpin A10) and the acute-phase response. J. Thromb. Haemost. 11, 375–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sierko E., Wojtukiewicz M. Z., Ostrowska-Cichocka K., Zimnoch L. (2010) Protein Z-dependent protease inhibitor (ZPI) is present in loco in human breast cancer tissue. Thromb. Haemost. 104, 183–185 [DOI] [PubMed] [Google Scholar]
- 12. Sierko E., Wojtukiewicz M. Z., Zimnoch L., Tokajuk P., Ostrowska-Cichocka K., Kisiel W. (2012) Colocalization of protein Z, protein Z-dependent protease inhibitor and coagulation factor X in human colon cancer tissue: Implications for coagulation regulation on tumor cells. Thromb. Res. 129, e112-e118 [DOI] [PubMed] [Google Scholar]
- 13. Sejima H., Hayashi T., Deyashiki Y., Nishioka J., Suzuki K. (1990) Primary structure of vitamin K-dependent human protein Z. Biochem. Biophys. Res. Com. 171, 661–668 [DOI] [PubMed] [Google Scholar]
- 14. Tabatabai A., Fiehler R., Broze G. J., Jr. (2001) Protein Z circulates in plasma in a complex with protein Z-dependent protease inhibitor. Thromb. Haemost. 85, 655–660 [PubMed] [Google Scholar]
- 15. Miletich J. P., Broze G. J., Jr. (1987) Human plasma protein Z antigen: range in normal subjects and the effect of warfarin therapy. Blood 69, 1580–1586 [PubMed] [Google Scholar]
- 16. Huang X., Dementiev A., Olson S. T., Gettins P. G. (2010) Basis for the specificity and activation of the serpin protein Z-dependent proteinase inhibitor (ZPI) as an inhibitor of membrane-associated factor Xa. J. Biol. Chem. 285, 20399–20409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wei Z., Yan Y., Carrell R. W., Zhou A. (2009) Crystal structure of protein Z-dependent inhibitor complex shows how protein Z functions as a cofactor in the membrane inhibition of factor X. Blood 114, 3662–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang X., Yan Y., Tu Y., Gatti J., Broze G. J., Jr., Zhou A., Olson S. T. (2012) Structural basis for catalytic activation of protein Z-dependent protease inhibitor (ZPI) by protein Z. Blood 120, 1726–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 20. Olson S. T., Swanson R., Raub-Segall E., Bedsted T., Sadri M., Petitou M., Hérault J.-P., Herbert J.-M., Björk I. (2004) Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb. Haemost. 92, 929–939 [DOI] [PubMed] [Google Scholar]
- 21. Swanson R., Raghavendra M. P., Zhang W., Froelich C., Gettins P. G., Olson S. T. (2007) Serine and cysteine proteases are translocated to similar extents upon formation of covalent complexes with serpins. J. Biol. Chem. 282, 2305–2313 [DOI] [PubMed] [Google Scholar]
- 22. Stone S. R., Dennis S., Hofsteenge J. (1989) Quantitative evaluation of the contribution of ionic interactions to the formation of the thrombin-hirudin complex. Biochemistry 28, 6857–6863 [DOI] [PubMed] [Google Scholar]
- 23. Lewis S. D., Shields P. P., Shafer J. A. (1985) Characterization of the kinetic pathway for liberation of fibrinopeptides during assembly of fibrin. J. Biol. Chem. 260, 10192–10199 [PubMed] [Google Scholar]
- 24. Lindahl P., Raub-Segall E., Olson S. T., Björk I. (1991) Papain labelled with fluorescent thiol-specific reagents as a probe for characterization of interactions between cysteine proteinases and their protein inhibitors by competitive titrations. Biochem. J. 276, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson K. A., Simpson Z. B., Blom T. (2009) Global Kinetic Explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 26. McDonald J. F., Shah A. M., Schwalbe R. A., Kisiel W., Dahlbäck B., Nelsestuen G. L. (1997) Comparison of naturally occurring vitamin K-dependent proteins: correlation of amino acid sequences and membrane binding properties suggests a membrane contact site. Biochemistry 36, 5120–5127 [DOI] [PubMed] [Google Scholar]
- 27. Huang X., Rezaie A. R., Broze G. J., Jr., Olson S. T. (2011) Heparin is a major activator of the anticoagulant serpin, protein Z-dependent protease inhibitor. J. Biol. Chem. 286, 8740–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rezaie A. R. (1998) Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by a template mechanism: evidence that calcium alleviates Gla domain antagonism of heparin binding to factor Xa. J. Biol. Chem. 273, 16824–16827 [DOI] [PubMed] [Google Scholar]