

Abstract
In addition to causing polymalformative syndrome, 22q11.2 deletion can lead to various neuropsychiatric disorders including mental retardation, psychosis, and epilepsy. However, few reports regarding epilepsy-related psychosis in 22q11.2 deletion syndrome (22q11.2DS) exist. We describe the clinical characteristics and course of 22q11.2DS in a Japanese patient with comorbid mild mental retardation, childhood-onset localization-related epilepsy, and adult-onset, interictal schizophrenia-like psychosis. From a diagnostic viewpoint, early detection of impaired intellectual functioning and hyperprolinemia in patients with epilepsy with 22q11.2DS may be helpful in predicting the developmental timing of interictal psychosis. From a therapeutic viewpoint, special attention needs to be paid to phenytoin-induced hypocalcemia in this syndrome.
Keywords: 22q11.2 deletion syndrome, Hyperprolinemia, Mental retardation, Epilepsy, Interictal psychosis, Phenytoin-induced hypocalcemia, DiGeorge syndrome
1. Introduction
22q11.2 deletion syndrome (22q11.2DS), also known as DiGeorge or velocardiofacial syndrome, is one of the most common recurrent genomic disorder, with an estimated prevalence of 1 in 4000 live births [1], [2]. It results from a hemizygous microdeletion of approximately 1.5 to 3 Mb on the long arm of chromosome 22 [3]. Its phenotypic expression is highly variable and ranges from severe, life-threatening conditions to only a few subtle features. Physical phenotypes include cardiovascular malformations, palatal anomalies, immune deficiency, hypocalcemia, hyperprolinemia, and dysmorphic facial features [4]. Neuropsychiatric phenotypes include attention deficit hyperactivity disorder, autism spectrum disorders, schizophrenia, mood disorders, anxiety disorders, intellectual disabilities, and epilepsy [5]. Although psychosis and epilepsy may coexist in adult patients with 22q11.2DS, few reports have discussed the clinical characteristics of epilepsy-related psychosis in 22q11.2DS. Here, we report a case of 22q11.2DS in an adult patient with comorbid interictal schizophrenia-like psychosis.
2. Case report
A 40-year-old man was admitted to our hospital for his first episode of psychosis, which occurred after a 2-month seizure-free period and persisted for nearly 3 months.
The patient was born with polyhydramnios after an uncomplicated full-term pregnancy. Tetralogy of Fallot was diagnosed at 8 months and treated surgically at the age of 4 years. Other than a delay in learning to walk on his own, no remarkable developmental delays were mentioned during his preschool age. Unprovoked generalized tonic–clonic seizures (GTCSs) during sleep appeared for the first time at the age of 13 years and subsequently occurred biweekly without other seizure types. At the age of 14 years, the patient was diagnosed with localization-related epilepsy and started on phenytoin monotherapy. At the age of 20 years, his seizure frequency increased in conjunction with new-onset hypocalcemia. At that time, he was diagnosed with idiopathic hypoparathyroidism. Following this diagnosis, he maintained a combination therapy of phenytoin, phenobarbital, and alfacalcidol. Through his 20s and 30s, he had only a few part-time jobs and withdrew from social activities; however, his seizures were well controlled, except when he forgot to take his medications. He had no family history of epilepsy or psychiatric disorders.
At the time of admission, his height was 153 cm, and his weight was 56 kg. He had swollen eyelids, short palpebral fissures, flat cheeks, a bulbous nose tip, a broad nasal root, and low-set ears. Physical examination revealed clear consciousness, hypernasal speech, and a deviation of the nasal septum. Neurological examination was unremarkable. A mental status examination revealed a psychotic state including paranoid delusions of being pursued, watched, and killed by someone. In addition, the patient presented with auditory hallucinations of unfamiliar voices commanding him to stab someone to death with a knife, intermittent psychomotor excitement, and lack of insight. Neuropsychological assessment revealed a full-scale IQ of 64, a verbal IQ of 71, and a performance IQ of 61 on the Wechsler Adult Intelligence Scale, 3rd edition. Blood count was normal without any sign of infection. Serum calcium level was 8.8 mg/dL, and serum phosphorous level was 3.7 mg/dL. Thyroid function tests and intact parathyroid hormone were normal. Serum IgG, IgA, and IgM levels were normal. Amino acid analysis showed severe hyperprolinemia (748.3 μmol/L). Serum phenytoin level was 12.5 μg/mL, and serum phenobarbital level was 14.8 μg/mL. Brain CT and MRI scans showed bilateral basal ganglion calcification without other cerebral malformations (Fig. 1). An ECG showed a complete right bundle branch block. Interictal EEG showed high-voltage, bilateral centroparietal 2- to 4-Hz spike-and-wave and sharp-and-wave complexes, which were accentuated by hyperventilation (Fig. 2). The 22q11.2 deletion was confirmed via a fluorescence in situ hybridization probe. To narrow down the breakpoints, single nucleotide polymorphism arrays were performed to reveal a deletion of about 2.5 Mb (spanning the most commonly deleted region, presumably containing 10 pathogenic genes between PRODH and LZTR1).
Fig. 1.
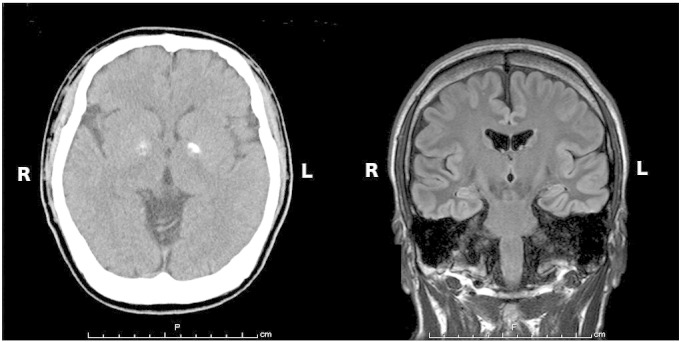
Brain CT (left) and MRI (right) scans showed bilateral basal ganglion calcification without other cerebral malformations.
Fig. 2.

Interictal EEG showed high-voltage, bilateral centroparietal 2- to 4-Hz spike-and-wave and sharp-and-wave complexes, which were accentuated by hyperventilation.
Based on these findings, the patient was diagnosed as having 22q11.2DS, localization-related epilepsy, mild mental retardation, and interictal schizophrenia-like psychosis. Risperidone (3 mg daily) was effective against his psychotic symptoms and was well tolerated. After his psychotic symptoms were alleviated, his phenytoin (200 mg po daily) was successfully switched to levetiracetam (1000 mg po daily) without seizure relapse. He was discharged from the hospital after remaining seizure-free on the 81st day after the hospital admission.
3. Discussion
This is the first case report to our knowledge to demonstrate the clinical characteristics of 22q11.2DS in a patient with trimorbidity: mild mental retardation, childhood-onset localization-related epilepsy, and adult-onset interictal schizophrenia-like psychosis.
Hypocalcemic seizures and epilepsy have been associated with 22q11.2DS [6]. Although hypocalcemic seizures should be distinguished from unprovoked seizures, the clinical manifestations of phenytoin-induced hypocalcemia in patients with epilepsy with 22q11.2DS can be indistinguishable from those of preexisting GTCSs, making diagnosis difficult. Phenytoin increases the metabolism of vitamin D and its active metabolites by hepatic enzyme induction, which lowers calcium absorption from the gut and causes hypocalcemia [7]. If a hypocalcemic seizure occurs during phenytoin therapy, an increase in phenytoin dose results in a paradoxical increase in seizure frequency. Indeed, our patient's experience of a sudden reduction in seizure control at the age of 20 years could be explained by phenytoin-induced hypocalcemia. Because it can be paradoxically epileptogenic in the presence of hypocalcemia, phenytoin should be avoided or used carefully in patients with 22q11.2DS.
Two cross-sectional studies have focused on the comorbidity of psychosis and epilepsy associated with 22q11.2DS [8], [9]. Raux et al. investigated the genotype–phenotype correlations in 8 children with type I hyperprolinemia and 92 adults or adolescents with 22q11.2DS [8]. The authors found an inverse correlation between plasma proline levels and IQ scores and revealed that severe hyperprolinemia (> 550 μmol/L) was associated with mental retardation, psychotic disorder, and epilepsy. In addition, they suggested that patients with hyperprolinemia with 22q11.2DS bearing the Met-COMT low-activity allele had a high risk of psychosis. Although there was no description of epilepsy classification in this study, their published data demonstrated that the group with epilepsy with 22q11.2DS had a significantly higher rate of comorbid psychotic disorder (as defined in the DSM-III-R) compared with the group without epilepsy with 22q11.2DS (18.5% versus 4.6%). Sporn et al. compared the clinical variables among 75 patients with childhood-onset schizophrenia (COS) and 870 patients with adult-onset schizophrenia (AOS) [9]. The authors found that patients with COS had a significantly higher prevalence of 22q11.2DS compared with patients with AOS (5.3% versus 0.46%) and that 75% of the patients with COS with 22q11.2DS developed epilepsy. The results from the two studies support the close relationship between epilepsy and psychosis in the population with 22q11.2DS.
Our patient showed a 27-year-interval between the onset of epilepsy and that of the first psychotic episode. Adachi et al. reported that the mean interval between the onset of epilepsy and that of psychosis was 14.4 years; the authors also suggested that mental retardation and localization-related epilepsy were considered risk factors for late-onset interictal psychosis [10]. Applying this relationship to our case, it is possible to consider that the comorbidity of mild mental retardation and localization-related epilepsy may have contributed to the development of middle-age-onset interictal psychosis in our patient. If so, early detection of impaired intellectual functioning, as well as hyperprolinemia, in patients with epilepsy with 22q11.2DS may be helpful in predicting the developmental timing of interictal psychosis.
The effectiveness and safety of antipsychotics in the treatment of psychosis associated with 22q11.2DS (22q11.2DS-P) are not well established. 22q11.2DS-P is relatively unresponsive to currently used antipsychotic drugs [11]. Müller and Fellgiebel reported that 22q11.2DS-P in a 41-year-old female patient with a congenital heart defect was successfully treated with quetiapine [12]. Carandang and Scholten reported that aripiprazole-resistant 22q11.2DS-P was successfully treated with metyrosine in a 17-year-old female patient with a congenital heart defect and low COMT activity [13]. Several researchers also reported that clozapine was effective against 22q11.2DS-P but increased the risk of provoked seizures [14], [15]. Our patient's 22q11.2DS-P symptoms improved on a low dose of risperidone, after failing to respond to quetiapine. When choosing an antipsychotic drug, it is important to assess the associated risk of worsening comorbid conditions and lowering the patient's quality of life.
4. Conclusions
In summary, a patient with hyperprolinemia with 22q11.2DS developed mild mental retardation, childhood-onset localization-related epilepsy, and adult-onset interictal schizophrenia-like psychosis. From a diagnostic viewpoint, early detection of impaired intellectual functioning and hyperprolinemia in patients with epilepsy with 22q11.2DS may be helpful in predicting the developmental timing of interictal psychosis. From a therapeutic viewpoint, special attention needs to be paid to phenytoin-induced hypocalcemia in this syndrome.
Conflict of interest
None.
References
- 1.Kobrynski L.J., Sullivan K.E. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 2.Botto L.D., May K., Fernhoff P.M., Correa A., Coleman K., Rasmussen S.A. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- 3.Shprintzen R.J. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev. 2008;14:3–10. doi: 10.1002/ddrr.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryan A.K., Goodship J.A., Wilson D.I., Philip N., Levy A., Seidel H. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider M., Debbané M., Bassett A.S., Chow E.W., Fung W.L., van den Bree M. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry. 2014;171:627–639. doi: 10.1176/appi.ajp.2013.13070864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kao A., Mariani J., McDonald-McGinn D.M., Maisenbacher M.K., Brooks-Kaya A.R., Zackai E.H. Increased prevalence of unprovoked seizures in patients with a 22q11.2 deletion. Am J Med Genet A. 2004;129A:29–34. doi: 10.1002/ajmg.a.30133. [DOI] [PubMed] [Google Scholar]
- 7.Ali F.E., Al-Bustan M.A., Al-Busairi W.A., Al-Mulla F.A. Loss of seizure control due to anticonvulsant-induced hypocalcemia. Ann Pharmacother. 2004;38:1002–1005. doi: 10.1345/aph.1D467. [DOI] [PubMed] [Google Scholar]
- 8.Raux G., Bumsel E., Hecketsweilerl B., van Amelsvoort T., Zinkstok J., Manouvrier-Hanu S. Involvement of hyperprolinemia in cognitive andpsychiatric features of the 22q11 deletion syndrome. Hum Mol Genet. 2007;16:83–91. doi: 10.1093/hmg/ddl443. [DOI] [PubMed] [Google Scholar]
- 9.Sporn A., Addington A., Reiss A.L., Dean M., Gogtay N., Potocnik U. 22q11 deletion syndrome in childhood onset schizophrenia: an update. Mol Psychiatry. 2004;9:225–226. doi: 10.1038/sj.mp.4001477. [DOI] [PubMed] [Google Scholar]
- 10.Adachi N., Akanuma N., Ito M., Kato M., Hara T., Oana Y. Epileptic, organic and genetic vulnerabilities for timing of the development of interictal psychosis. Br J Psychiatry. 2010;196:212–216. doi: 10.1192/bjp.bp.108.056721. [DOI] [PubMed] [Google Scholar]
- 11.Vogels A., Verhoeven W.M.A., Tuinier S., DeVriendt K., Swillen A., Curfs L.M. The psychopathological phenotype of velocardiofacial syndrome. Ann Genet. 2002;45:89–95. doi: 10.1016/s0003-3995(02)01114-0. [DOI] [PubMed] [Google Scholar]
- 12.Müller U.J., Fellgiebel A. Successful treatment of long-lasting psychosis in a case of 22q11.2 deletion syndrome. Pharmacopsychiatry. 2008;41:158–159. doi: 10.1055/s-2008-1062700. [DOI] [PubMed] [Google Scholar]
- 13.Carandang C.G., Scholten M.C. Metyrosine in psychosis associated with 22q11.2 deletion syndrome: case report. J Child Adolesc Psychopharmacol. 2007;17:115–120. doi: 10.1089/cap.2006.0013. [DOI] [PubMed] [Google Scholar]
- 14.Yacoub A., Aybar M. Response to clozapine in psychosis associated with velo-cardio-facial syndrome. Psychiatry. 2007;5:14. [PMC free article] [PubMed] [Google Scholar]
- 15.Gladston S., Clarke D.J. Clozapine treatment of psychosis associated with velo-cardio-facial syndrome: benefits and risks. J Intellect Disabil Res. 2005;49:567–570. doi: 10.1111/j.1365-2788.2005.00708.x. [DOI] [PubMed] [Google Scholar]