

Abstract
Background
The results of recent studies suggest that dipeptidyl‐peptidase‐4 inhibitors have antiatherogenic effects. However, whether or not dipeptidyl‐peptidase‐4 inhibitors could suppress arterial inflammation and intimal hyperplasia after injury remains undetermined. The present study aims to clarify the anti‐inflammatory effects of the dipeptidyl‐peptidase‐4 inhibitor, alogliptin (AGP), on the arteries of atherogenic low‐density lipoprotein receptor‐deficient (LKO) mice.
Methods and Results
We compared intimal hyperplasia in LKO mice 2 weeks after femoral artery injury using an external vascular cuff model. All mice received oral injection of AGP (20 mg/kg per day) or normal saline (control) once daily for 14 days. Fasting blood sugar levels, serum cholesterol levels, or blood pressure did not significantly differ between the 2 groups. Plasma levels of active glucagon‐like peptide‐1 were higher in the AGP than in the control LKO mice (22.2±1.9 versus 15.6±0.9 pg/mL; P<0.05). Compared with saline, AGP significantly reduced intimal hyperplasia (1087±127 versus 1896±140 μm2; P<0.001) as well as the intima/media ratio (0.08±0.01 versus 0.16±0.02; P<0.001). Immunostaining showed that AGP reduced proliferating cells (proliferating cell nuclear antigen–positive nuclei; P<0.001), percent smooth‐muscle cell area (α‐SMA‐positive cells; P<0.001), inflammatory cells infiltration (lymphocyte antigen 6 complex–positive cells; P<0.05), tumor necrosis factor‐α expression (P<0.05), and percent phospho‐NF‐κB‐positive cell compared with saline. Levels of tumor necrosis factor ‐α (0.5‐fold P<0.05), monocyte chemoattractant protein 1 (0.3‐fold P<0.01), and interleukin‐1β (0.2‐fold P<0.05) mRNA were lower in the injured arteries of the AGP than in the control group.
Conclusions
AGP appeared to suppress neointimal formation by inhibiting inflammation, independently of its effects on glucose or cholesterol metabolism in atherogenic LKO mice.
Keywords: cytokine, dipeptidyl‐peptidase‐4 inhibitor, inflammation, intimal hyperplasia, smooth muscle cell
Introduction
Dipeptidyl peptidase‐4 (DPP‐4) inhibitors are novel oral antihyperglycemic agents for treating type 2 diabetes mellitus patients. Recent studies suggest that several DPP‐4 inhibitors exert antiatherosclerotic effects though suppressing inflammatory reactions of monocytes and smooth‐muscle cell (SMC) proliferation in vitro.1–3 However, whether or not DPP‐4 inhibitors suppress arterial inflammation and intimal hyperplasia after injury remains undetermined.
Alogliptin (2‐({6‐[(3R)‐3‐aminopiperidinyl‐1‐yl]‐3‐methyl‐2,4‐dioxo‐3,4‐dihydropyrimidin‐1(2H)yl} methyl)benzonitrile monobenzoate) (AGP) is a selective DPP‐4 inhibitor that has also improves glycemic control.4–7 Shah et al indicated that AGP suppresses inflammatory chemokine expression and the migration of bone marrow–derived monocytes in vitro.1 However, it remains unknown whether AGP has anti‐inflammatory effects and reduces neointimal hypertrophy in an artery injury model.
Methods
Animals
The study was performed using low‐density lipoprotein receptor‐deficient (LKO) mice (8 to 10 weeks of age), which are known to have atherogenic properties in a cuff‐injury model.8 LKO mice were fed with standard chow. The experimental protocol proceeded under the approval of the National Defense Medical College and Juntendo University Boards for Studies in Experimental Animals.
DPP‐4 Inhibitor
The DPP‐4 inhibitor, alogliptin benzonate (Takeda Pharmaceutical Company, Tokyo; 136 mg) was dissolved in distilled water to a final concentration of 1 mg/mL, stored at 4°C, and warmed to room temperature before use.
Femoral Artery Injury and AGP Administration
Only male mice were studied to exclude gender differences. The mice were anesthetized by intraperitoneal injection of pentobarbital (50 mg/kg), and we dissected the left femoral artery from its surrounding, as demonstrated previously.9 Vascular injury was inflicted by placing a nonocclusive polyethylene cuff (length 2 mm; internal diameter 0.56 mm; Becton Dickinson, Mountain View, CA) around the femoral artery. We administered AGP (20 mg/kg per day) or normal saline by oral injection once daily to the mice for 14 days.
Blood Pressure Measurement
Systolic blood pressure was measured in nonanesthetized mice at 13 days postinjury by the tail‐cuff method (MK‐2000; Muromachi Kikai).
Levels of Plasma Lipid, Glucose, and Active Glucagon‐Like Peptide‐1
Mice were fasted for 12 hours and blood samples were collected from the tail veins of both groups at 14 days postinjury. Levels of plasma total cholesterol, low‐density lipoprotein cholesterol, and high‐density lipoprotein cholesterol were measured using high‐performance liquid chromatography (Skylight Biotech Inc, Akita, Japan) as described previously.10 Levels of plasma fasting blood glucose were measured using Free Style Freedom (Nipro Co, Osaka, Japan). Levels of active glucagon‐like peptide‐1 (GLP‐1) were measured in plasma from nonfasted mice using ELISA kits (Shibayagi Co, Gunma, Japan) at 14 days postinjury. Blood samples for measurement of active GLP‐1 were immediately collected into tubes containing 1% AGP (10 mmol/L) vol/vol.
Arterial Harvest and Morphometric Analysis
After blood collection, the animals were euthanized by pentobarbital injection and the vascular tree was perfused with 0.9% NaCl followed by 4% paraformaldehyde. After the perfusion procedure, the femoral artery was harvested and fixed with 10% neutral‐buffered formalin for 48 hours, embedded in paraffin, and sectioned (each 5‐μm thickness).We used equally spaced (200‐μm interval) 10 cross‐sections to qualify a neointimal lesion for each mouse. The samples were stained with elastica van Gieson, and then photographed using an ECLIPS LV100 microscope (Nikon, Tokyo, Japan). The luminal, neointimal, and medial areas were calculated using NIH Image J 1.42 (National Institutes of Health, public domain software).
Immunohistochemistry
Smooth muscle cells were visualized using α‐smooth muscle cell actin staining (N1584; DAKO, Tokyo, Japan), cell proliferation was investigated using proliferating cell nuclear antigen staining (N1529; DAKO, Tokyo, Japan), and inflammatory cells were detected by lymphocyte antigen 6 complex staining (N550291; BD Pharmingen in Japan, Tokyo, Japan). The proliferating cell nuclear antigen and the lymphocyte antigen 6 complex indexes were calculated as ratios of stained areas per total intimal area of injured arteries. In addition, anti‐tumor necrosis factor‐α (TNF‐α) staining (ab‐6671; Abcam, Cambridge, MA) was used to detect cytokine expressions and the extent of the TNF‐α, and monocyte chemoattractant protein 1 (MCP‐1) antibody (sc‐1785; Santa Cruz Biotechnology) was used for detection of MCP‐1 expression and evaluation for positive area per total intimal and medial area of injured arteries. Nuclear translocation of nuclear factor‐κB (NF‐κB) was detected by phospho‐NF‐κB p65 (ser276) (pNF‐κB) staining (#3037; Cell Signaling Technology). Activation of pNF‐κB was evaluated for percentage of positive nuclei in the intima and media or in the adventitia within a 50 μm radius from the external elastic lamina of injured arteries.
Quantitative Real‐Time Polymerase Chain Reaction (PCR)
Total RNA from the injured femoral artery was isolated at 3 days postinjury using the TRI reagent (Sigma‐Aldrich) (2 vessels for each sample). Complementary DNA was prepared from total RNA (500 ng) using reverse transcriptase according to the manufacturer's instructions (High Capacity cDNA Reverse Transcription Kit; Applied Biosystems). Quantitative mRNA expression was assessed by real‐time PCR with Power SYBR Green PCR Master Mix (Applied Biosystems) using primers specific for TNF‐α (forward: TCC CAG GTT CTC TTC AAG GGA, reverse: GGT GAG GAG CAC GTA GTC GG), MCP‐1 (forward: CCT GGA TCG GAA CCA AAT GA, reverse: CGG GTC AAC TTC ACA TTC AAA G), interleukin (IL)‐1β (forward: TGG TGT GTG ACG TTC CCA TT, reverse: CAG CAC GAG GCT TTT TTG TTG), and GAPDH (forward: GTC ATT GAG AGC AAT GCC AG, reverse: GTG TTC CTA CCC CCA ATG TG). Samples were run on the 7500 Fast Real‐Time PCR system (Applied Biosystems). Data were analyzed by 7500 Software (Applied Biosystems), and relative expression levels of target mRNA of the AGP treatment group was compared with those of the control group.
Statistical Analysis
Statistical analyses were carried out using the Statistical Package for the Social Science (SPSS) software program, version 22.0 (SPSS Inc, Chicago, IL). Results are shown as the means±SE. Mean values of continuous variables were compared between groups using Student t test according to whether normally distributed as tested by the Shapiro–Wilk test, and P<0.05 was regarded as significant.
Results
LKO mice were treated with normal saline (control) or AGP for 14 days after cuff injury (Figure 1A). Blood pressure, fasting blood sugar, and serum cholesterol levels were similar at 2 weeks after cuff injury between the AGP‐ and saline‐treated groups (Figure 1B). The nonfasting plasma levels of active GLP‐1 were higher in mice treated with AGP than with saline (22.2±1.9 versus 15.6±0.9 pg/mL; P<0.05) (Figure 1C).
Figure 1.

Study protocol, blood pressure, and blood sample findings at 14 days after injury. A, Schematic of study protocol. B, Systolic blood pressure (SBP) (AGP: n=9, control: n=8), fasting blood sugar levels (FBS) (AGP: n=7, control: n=8), and serum levels of non‐HDL and HDL cholesterol (n=4 for each group) in mice at 14 days after injury. Data are expressed as means±SEM (n=4 to 9). C, Serum levels of active GLP‐1 in AGP treated and control mice at 14 days after injury. Data are expressed as means±SEM (AGP: n=8, control: n=7), *P<0.05. AGP indicates alogliptin; GLP‐1, glucagon‐like peptide‐1; HDL, high‐density lipoprotein; LDLR KO, low‐density lipoprotein receptor knockout; NS, not significant.
Neointimal Formation After Cuff Injury
We investigated the effect of AGP treatment on arterial inflammation in LKO mice after cuff‐induced injury. Figure 2A shows representative cross‐sections of femoral arteries harvested from AGP‐ and saline‐treated LKO mice at 14 days postinjury. Quantitative analysis demonstrated that AGP significantly reduced the amount of intimal hyperplasia (1087±127 versus 1896±140 μm2; P<0.001) (Figure 2B) and the intima/media ratio (0.08±0.01 versus 0.16±0.02; P<0.001) (Figure 2C).
Figure 2.
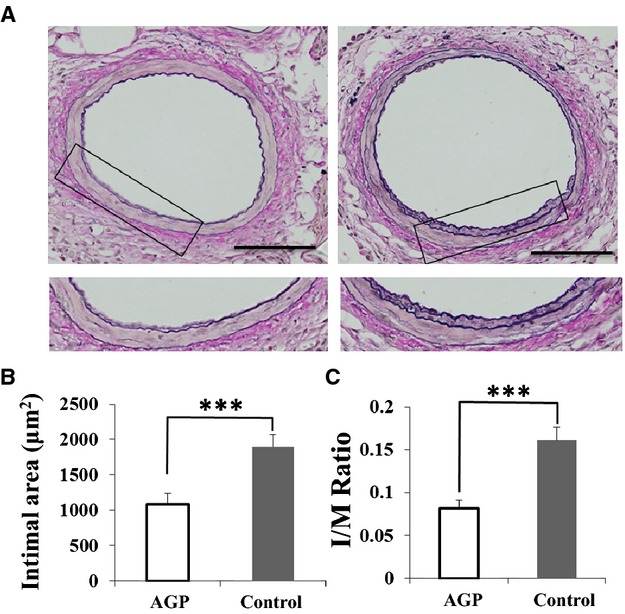
Neointimal formation at 14 days after injury is significantly decreased in alogliptin (AGP)‐treated compared with saline‐treated (control) mice. A, Elastica van Gieson staining of cuffed femoral arteries from AGP‐treated (left) and control (right) mice at 14 days postinjury (bars=100 μm). Boxed areas are shown in panels below. Bar graphs show the neointimal area (B) and intima/media (I/M) ration (C) of cuff‐injured arteries. Data are expressed as means±SEM (AGP: n=9, control: n=8), ***P<0.001.
Proliferation of SMCs and Inflammatory Cells After Cuff Injury
We evaluated the effect of AGP on cell proliferation at 7 days after injury by staining for α‐ smooth muscle cell actin, proliferating cell nuclear antigen, and lymphocyte antigen 6 complex. The α‐smooth muscle cell actin–positive areas in the neointima were significantly decreased in mice treated with AGP (percent α‐smooth muscle cell actin area within external elastic lumina: 30.8±3.1 versus 45.4±6.0%, P<0.001) (Figure 3A and 3B). The percent proliferating cell nuclear antigen–positive nuclei were significantly decreased in the neointima of mice treated with AGP compared with saline (1.5±0.2 versus 11.5±0.8%; P<0.001) (Figure 4). The lymphocyte antigen 6 complex–positive areas in the neointima were significantly decreased in mice treated with AGP compared with saline (4.3±0.5 versus 10.9±2.8%; P=0.012) (Figure 5).
Figure 3.
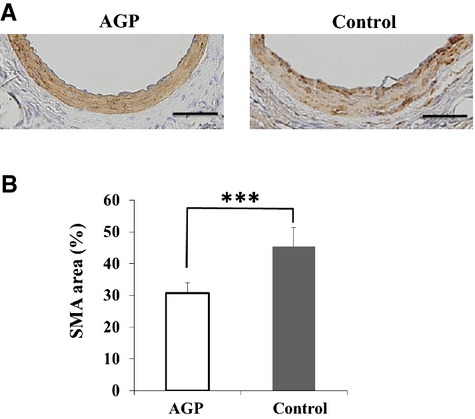
AGP decreases α‐SMA‐positive areas in the neointima of mice after injury. A, Staining for α‐SMA in cuffed femoral arteries from AGP‐treated (left) and control (right) mice 14 days postinjury (bars=50 μm). B, Quantitative analysis of α‐SMA positive areas in cuffed‐arteries of AGP‐treated and control mice at 14 days after injury. Data are expressed as means±SEM (AGP: n=9, Control: n=8), ***P<0.001. AGP indicates alogliptin; SMA, smooth muscle cell area.
Figure 4.
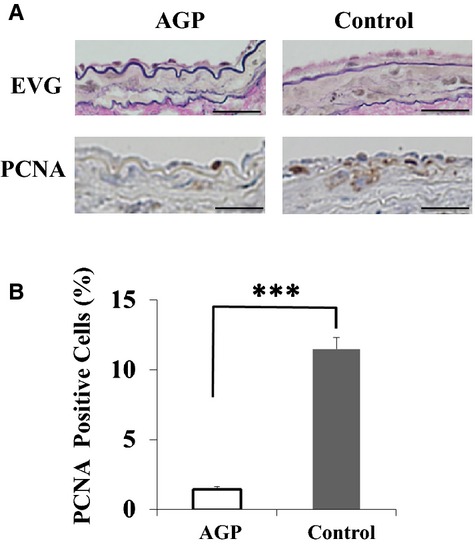
AGP decreases PCNA‐positive areas in neointima of mice after injury. A, Elastica van Gieson (EVG) and PCNA staining of cuffed femoral arteries from mice at 7 days postinjury (bars=25 μm). B, Quantitative analysis of PCNA‐positive nuclei in the intima of cuffed‐arteries from mice at 7 days after injury. Data are expressed as means±SEM (n=3 per group), ***P<0.001. AGP indicates alogliptin; PCNA, proliferating cell nuclear antigen.
Figure 5.
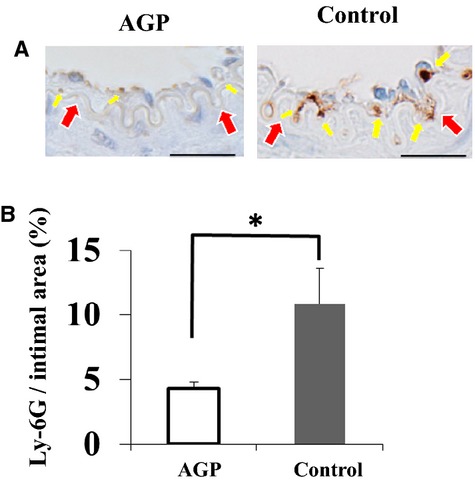
Alogliptin (AGP) decreases numbers of inflammatory cells in the injured arteries of mice. A, Staining for lymphocyte antigen 6 complex (Ly‐6G) staining in cuffed femoral arteries from mice at 7 days postinjury (bars=25 μm). Yellow arrows indicate the inflammatory cells and red arrows indicate internal elastic lamina, respectively. B, Quantitative analysis of Ly‐6G‐positive nuclei in the intima of cuffed arteries from mice at 7 days after injury. Data are expressed as means±SEM (n=3 for each group), *P<0.05.
Expression of Inflammatory Cytokine
Figure 6 shows the expression of the proinflammatory cytokine TNF‐α and MCP‐1 after cuff injury determined using immunohistochemistry and real‐time PCR. Figure 6B shows the areas that expressed TNF‐α and MCP‐1 at 7 days after cuff injury in LKO mice. The percent positive area of TNF‐α expression was significantly lower in the intima and media of mice treated with AGP than with saline (TNF‐α: 1.4±0.6 versus 4.8±1.0%; P<0.05, MCP‐1: 2.5±0.5 versus 5.4±0.4%; P<0.05) (Figure 6B).
Figure 6.
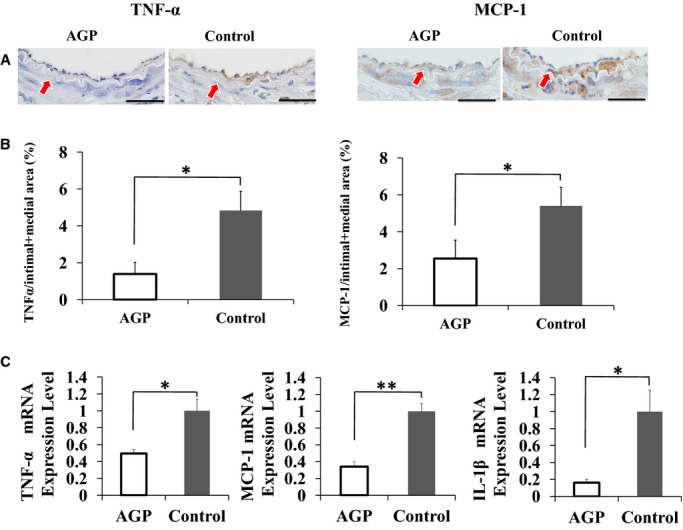
AGP decreases TNF‐α expression in the injured arteries of LKO mice. A, Staining for TNF‐α and MCP‐1 in cuffed femoral arteries from AGP‐treated (left) and control (right) mice at 7 days postinjury, respectively (bars=25 μm). Red arrows indicate internal elastic lamina. B, Quantitative analysis of TNF‐α and MCP‐1 positive areas in intima+media of AGP‐treated and control mice at 7 days after injury. Data are expressed as means±SEM (n=3 for each group), *P<0.05. C, Bar graphs show the mRNA expression of TNF‐α, MCP‐1, and IL‐1β in injured arteries from AGP‐treated and control mice at 3 days after injury. Degree of change in gene expression is based on the mean amount of expression in control LKO mice. Data are expressed as means±SEM (n=4 per group), *P<0.05, **P<0.01. AGP indicates alogliptin; IL, interleukin; LKO, low‐density lipoprotein receptor knockout; MCP‐1, monocyte chemoattractant protein 1; TNF, tumor necrosis factor.
Real‐time PCR of the injured femoral artery revealed significantly decreased mRNA expression levels of TNF‐α, MCP‐1, and IL‐1β in the AGP group compared with the control group (TNF‐α: 0.49±0.05 versus 1.00±0.13; P<0.05, MCP‐1: 0.34±0.06 versus 1.00±0.09; P<0.01, IL‐1β: 0.16±0.03 versus 1.00±0.25; P<0.05) (Figure 6C).
Evaluation of NF‐κB Activation
To examine postinjury NF‐κB activation in the arteries of both AGP and control groups, we performed pNF‐κB staining in the injured arteries of both groups at 7 days postinjury. Fewer pNF‐κB‐positive cells were observed in the intima and media (Figure 7A), and adventitia (Figure 7C) of AGP treated mice compared with controls. The percentage of pNF‐κB‐positive cells in the arteries of the AGP group was significantly lower than in the control group (11.9±1.1 versus 20.6±1.7%; P<0.05) (Figure 7B). Furthermore, the percentage of pNF‐κB‐positive cells in the adventitia of the AGP group decreased by nearly half compared with the control group (18.0±4.4 versus 33.3±2.3%; P<0.05) (Figure 7D).
Figure 7.
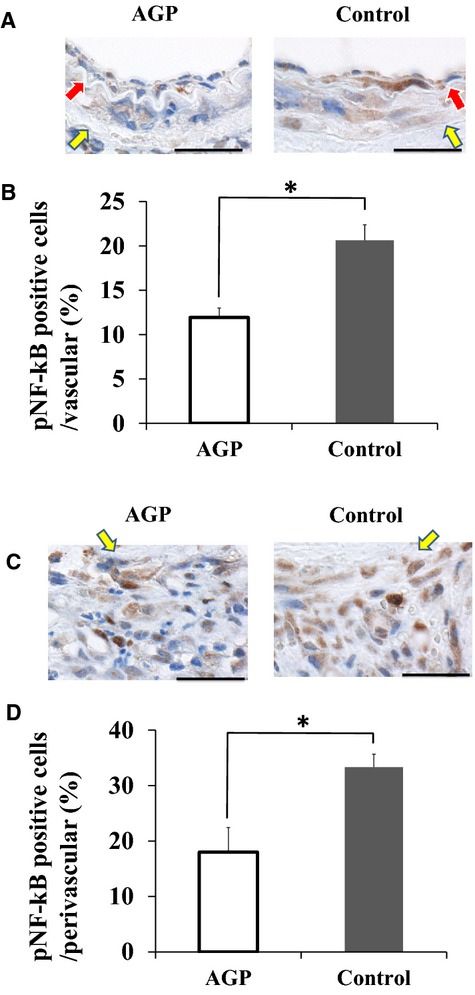
AGP suppresses NF‐kB activation in the injured arteries of LKO mice. A, Staining for phosphor‐NF‐kB (pNF‐kB) in the intima and media from AGP‐treated (left) and control (right) mice at 7 days postinjury, respectively (bars=25 μm). Red arrows indicate internal elastic lamina. Yellow arrows indicate external elastic lamina. B, The bar graphs show the percentage of pNF‐kB‐positive cells in the intima and media of AGP‐treated and control mice at 7 days after injury. Data are expressed as means±SEM (n=3 for each group), *P<0.05. C, Staining for pNF‐kB in the adventitia from AGP‐treated (left) and control (right) mice at 7 days postinjury respectively (bars=25 μm). Yellow arrows indicate external elastic lamina. D, The bar graphs show the percentage of pNF‐kB‐positive cells in the adventitia of AGP‐treated and control mice at 7 days after injury. Data are expressed as means±SEM (n=3 for each group), *P<0.05. AGP indicates alogliptin; LKO, low‐density lipoprotein receptor knockout; NF‐κB, nuclear factor‐κB.
Discussion
This is the first investigation into how AGP affects injured arteries in vivo. AGP significantly reduced inflammation, SMC proliferation, and the TNF‐α and MCP‐1 levels in the injured arteries of LKO mice. Levels of fasting blood sugar and serum cholesterol did not significantly differ between AGP and control groups of LKO mice. These results suggest that AGP reduced neointimal formation by suppressing inflammation and smooth muscle proliferation independently of lipid and glycemic profiles. Recent studies have identified anti‐atherosclerogenic effects of AGP in noninjured aortic arteries from low‐density lipoprotein receptor‐deficient and apolipoprotein E‐deficient mice.1,3 AGP also decreased mRNA levels of the inflammatory cytokines (TNF‐α, IL‐6, and IL‐1β) in mononuclear cells and attenuated monocyte migration activity that was stimulated by TNF‐α. These findings support our results. Although we also evaluated plasma levels of IL‐6 and TNF‐α in each group using an ELISA kit (Invitrogen) at 7 days postinjury, the results showed that the plasma levels of TNF‐α and IL‐6 were not significantly different between AGP‐treated mice and saline‐treated mice (IL‐6: 17.4±7.5 versus 24.8±7.7 pg/mL; P=0.52, TNF‐α: 4.4±0.7 versus 3.9±0.7 pg/mL; P=0.46 [n=4 to 5 per group]), suggesting that the cuff injury could induce local inflammation around the vascular, but not systemic inflammation. Thus, we believe the cuff‐injury model cannot be used for the evaluation of the effect of AGP on the suppression of systemic inflammation. Other models are needed for evaluation of the systemic anti‐inflammatory effect of AGP.
TNF‐α activates NF‐κB, which regulates macrophage migration and chemokine expression as well as SMC proliferation and migration.11–14 We previously showed that a TNF‐α receptor 1 antagonist attenuated intimal hyperplasia, indicating that TNF‐α signaling plays a critical role in the development of intimal hyperplasia after injury.15 Here, AGP suppressed TNF‐α expression, which subsequently attenuated neointimal formation in the injured artery.
Our previous report showed that NF‐κB activation and translocation are increased in the response to cuff injury.15 Furthermore, our preliminary study revealed that an inhibitor of IkBα phosphorylation (BAY11‐7082, Wako) prevented intimal hyperplasia completely in injured arteries of both AGP‐treated and saline‐treated mice (data not shown), suggesting that NF‐κB activation is a major contributor to neointimal hyperplasia in cuff‐injured arteries. Then, we performed pNF‐κB staining in the injured arteries of both groups at 7 days postinjury. The results revealed that AGP treatment reduced NF‐κB activation in the intima, media, and adventitia of injured arteries significantly compared with saline treatment. Taken together, AGP suppresses inflammation and neointimal hyperplasia in the cuff‐injured artery by partially inhibiting NF‐κB activation.
Recent reports have described the anti‐atherogenic effects of DPP‐4 inhibitors. Lim et al showed that sitagliptin suppressed neointimal formation after carotid artery balloon injury in rats.16 Matsubara et al and Ervinna et al reported that both sitagliptin and anagliptin exerted anti‐atherogenic effects in ApoE‐deficient mice fed with a standard diet.2,17 Several investigators have postulated how AGP exerts such anti‐inflammatory effects. GLP‐1 is rapidly degraded and inactivated by DPP‐4, and thus DPP‐4 inhibitors increase serum concentrations of GLP‐1.18–19 A recent study has found that exendin‐4 (GLP‐1 receptor agonist) modulated monocyte adhesion to endothelial cells and attenuated atherosclerosis in mice and that these effects might contribute to the inhibition of p65 nuclear translocation in macrophages by means of cAMP levels that are increased by GLP‐1 receptor activation.20 Another study found that exendin‐4 suppresses SMC proliferation in arteries after wire injury.21 Furthermore, Matsubara et al showed that DPP‐4 inhibitors and GLP‐1 produce anti‐inflammatory effects that are followed by increases in cytosolic cAMP levels and decreases in extracellular signal‐regulated kinase (ERK) 1/2 and c‐jun N‐terminal kinase (JNK) phosphorylation as well as NF‐κB activation in vitro.17
The cAMP/Protein kinase A (PKA) pathway attenuates TNF‐α production in macrophages,22 and the present study found that AGP suppresses TNF‐α expression in the injured artery. These findings suggest that GLP‐1 levels increased by AGP significantly contribute to the anti‐inflammatory effects of AGP though activating cAMP/PKA signaling. However, it is not clear whether the anti‐inflammatory effects of DPP‐4 inhibitors depend on GLP‐1. Levels of active GLP‐1 after AGP administration in the present study were much lower than those of the GLP‐1 analog (exendin‐4) that exerted anti‐atherosclerogenic effect in another study21 (GLP‐1 in our study: 5 to 7 pmol/L to GLP‐1 analogue: 10 nmol/L), suggesting that DPP‐4 may directly suppress atherosclerosis.
SMC proliferation participates in both the early and late phase of arterial diseases.23 NF‐κB is a common regulator of proinflammatory genes. SMC proliferation and CARD11 mediate factor‐specific activation of NF‐κB.24 Interaction between DPP‐4 and CARD11 leads to NF‐κB activation in T‐cells.25 Such activation was reduced by DPP‐4 or CARD11 knockdown, and SMCs proliferation was dose‐dependently attenuated by sitagliptin via suppressing NF‐κB activation in vitro.16 In the present study, we show that AGP also attenuated SMC proliferation by suppressing NF‐κB activation in vivo.
The ERK pathway is also an important signaling cascade in inflammation.26 An in vitro study found that AGP reduced SMCs proliferation and attenuated ERK phosphorylation in SMCs, and that AGP reduces TNF‐α production and NF‐κB activation in monocytes.2 These findings suggest that inflammation and SMC proliferation might be suppressed by DPP‐4 inhibitors directly, and not via GLP‐1‐dependent pathways.
DPP‐4 inhibitors exert cardiovascular protective effects via other pathways. Several reports showed that AGP improved endothelial function through a GLP‐1 independent pathway that includes NO release via the Src‐Akt‐eNOS pathway.27–28 Levels of stromal‐derived factor‐1α (SDF‐1α), which is another physiological substrates of DPP‐4, are increased by DPP‐4 inhibitors and this might affect endothelial progenitor cells that mediate cardiovascular repair.29–30
Levels of fasting blood sugar levels and serum cholesterol did not significantly differ between AGP‐ and saline‐treated LKO mice. These findings are consistent with those of a study showing that AGP administration for 12 weeks did not change blood pressure or the lipid and glycemic profiles in LKO mice with a diet containing normal amounts of fat.1 Clinical data also suggest that AGP exerts minor effects on lipid profiles.4,6
In conclusion, AGP suppressed intimal hyperplasia caused by vascular inflammation such as atherosclerosis and helped to reduce the frequency of cardiovascular events.
Acknowledgments
We thank the members of the Animal Institute for assistance in animal care.
Sources of Funding
This study was supported by National Defense Medical College grants (H14 [Isoda] and H18 [Isoda]), Grant‐in‐Aid for Scientific Research (C) (JSPS KAKENHI grant number 25461142) (Isoda) and the grant from MEXT (Ministry of Education, Culture, Sports, Science and Technology)‐Supported Program for Strategic Research Foundation at Private Universities (Shimada).
Disclosures
Takeda Pharmaceutical Company (Tokyo) supplied the DPP‐4 inhibitor, alogliptin benzonate. Kazunori Shimada received lecture fees from Takeda Pharmaceutical Company. Hiroyuki Daida received reserch funds and lecture fees from Takeda Pharmaceutical Company.
References
- Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z, Xu X, Lu B, Moffatt‐Bruce S, Durairaj R, Sun Q, Mihai G, Maiseyeu A, Rajagopalan S. Long‐term dipeptidyl‐peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation. 2011; 124:2338-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervinna N, Mita T, Yasunari E, Azuma K. Anagliptin, a DPP‐4 inhibitor, suppresses proliferation of vascular smooth muscles and monocyte inflammatory reaction and attenuates atherosclerosis in male apo E‐deficient mice. Endocrinology. 2013; 154:1260-1270. [DOI] [PubMed] [Google Scholar]
- Ta NN, Schuyler CA, Li Y, Lopes‐Virella MF, Huang Y. DPP‐4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E‐deficient mice. J Cardiovasc Pharmacol. 2011; 58:157-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino Y, Yabe D. Alogliptin benzoate for the treatment of type 2 diabetes. Expert Opin Pharmacother. 2014; 15:851-863. [DOI] [PubMed] [Google Scholar]
- Jarvis CI, Cabrera A, Charron D. Alogliptin: a new dipeptidyl peptidase‐4 inhibitor for type 2 diabetes mellitus. Ann Pharmacother. 2013; 47:1532-1539. [DOI] [PubMed] [Google Scholar]
- Andukuri R, Drincic A, Rendell M. Alogliptin: a new addition to the class of DPP‐4 inhibitors. Diabetes Metab Syndr Obes. 2009; 2:117-126. [PMC free article] [PubMed] [Google Scholar]
- Moritoh Y, Takeuchi K, Asakawa T, Kataoka O, Odaka H. Chronic administration of alogliptin, a novel, potent, and highly selective dipeptidyl peptidase‐4 inhibitor, improves glycemic control and beta‐cell function in obese diabetic ob/ob mice. Eur J Pharmacol. 2008; 588:325-332. [DOI] [PubMed] [Google Scholar]
- von der Thüsen JH, van Berkel TJC, Biessen EAL. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E‐deficient and low‐density lipoprotein receptor‐deficient mice. Circulation. 2001; 103:1164-1170. [DOI] [PubMed] [Google Scholar]
- Isoda K, Shiigai M, Ishigami N, Matsuki T, Horai R, Nishikawa K, Kusuhara M, Nishida Y, Iwakura Y, Ohsuzu F. Deficiency of interleukin‐1 receptor antagonist promotes neointimal formation after injury. Circulation. 2003; 108:516-518. [DOI] [PubMed] [Google Scholar]
- Isoda K, Sawada S, Ayaori M, Matsuki T, Horai R, Kagata Y, Miyazaki K, Kusuhara M, Okazaki M, Matsubara O, Iwakura Y, Ohsuzu F. Deficiency of interleukin‐1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. J Biol Chem. 2005; 280:7002-7009. [DOI] [PubMed] [Google Scholar]
- Collins T, Cybulsky MI. NF‐kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001; 107:255-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Connell MC, MacEwan DJ. TNFR1‐induced NF‐kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF‐induced ICAM‐1 and VCAM‐1 expression on endothelial cells. Cell Signal. 2007; 19:1238-1248. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999; 340:115-126. [DOI] [PubMed] [Google Scholar]
- Ip JH, Fuster V, Badimon L, Badimon J, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990; 15:1667-1687. [DOI] [PubMed] [Google Scholar]
- Kitagaki M, Isoda K, Kamada H. Novel TNF‐alpha receptor 1 antagonist treatment attenuates arterial inflammation and intimal hyperplasia in mice. J Atheroscler Thromb. 2012; 19:36-46. [DOI] [PubMed] [Google Scholar]
- Lim S, Choi S, Shin H, Cho B, Park H. Effect of a dipeptidyl peptidase‐IV inhibitor, des‐fluoro‐sitagliptin, on neointimal formation after balloon injury in rats. PLoS One. 2012; 7:e35007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara J, Sugiyama S, Sugamura K, Nakamura T, Fujiwara Y, Akiyama E, Kurokawa H, Nozaki T, Ohba K, Konishi M, Maeda H, Izumiya Y, Kaikita K, Sumida H, Jinnouchi H, Matsui K, Kim‐Mitsuyama S, Takeya M, Ogawa H. A dipeptidyl peptidase‐4 inhibitor, des‐fluoro‐sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E‐deficient mice. J Am Coll Cardiol. 2012; 59:265-276. [DOI] [PubMed] [Google Scholar]
- Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev. 2012; 33:187-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheen AJ. Cardiovascular effects of gliptins. Nat Rev Cardiol. 2013; 10:73-84. [DOI] [PubMed] [Google Scholar]
- Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, Fujitani Y, Hirose T, Kawamori R, Watada H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon‐like peptide‐1 receptor agonist, exendin‐4. Diabetes. 2010; 59:1030-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Nomiyama T, Mita T, Yasunari E, Azuma K, Komiya K, Arakawa M, Jin WL, Kanazawa R, Fujitani Y, Watada H. Exendin‐4, a glucagon‐like peptide‐1 receptor agonist, reduces intimal thickening after vascular injury. Biochem Biophys Res Commun. 2011; 405:79-84. [DOI] [PubMed] [Google Scholar]
- Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, Liu J, Driver A, Bao XR, Stemweis PC, Simon MI, Fraser ID. Suppression of LPS‐induced TNF‐alpha production in macrophages by cAMP is mediated by PKA‐AKAP95‐p105. Sci Signal. 2009; 2:ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008; 28:812-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz JL, Denny EM, Baltimore D. CARD11 mediates factor‐specific activation of NF‐kappaB by the T cell receptor complex. EMBO J. 2002; 21:5184-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma K, Uchiyama M, Yamochi T, Nishibashi K, Hosono O, Takahashi N, Kina S, Tanaka H, Lin X, Dang NH, Morimoto C. Caveolin‐1 triggers T‐cell activation via CD26 in association with CARMA1. J Biol Chem. 2007; 282:10117-10131. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007; 1773:1263-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda Y, Miyoshi T, Oe H, Ohno Y, Nakamura K, Toh N, Kohno K, Morita H, Kusano K, Ito H. Alogliptin ameliorates postprandial lipemia and postprandial endothelial dysfunction in non‐diabetic subjects: a preliminary report. Cardiovasc Diabetol. 2013; 12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah Z, Pineda C, Kampfrath T, Maiseyeu A, Ying Z, Racoma I, Deiuliis J, Xu X, Sun Q, Moffatt‐Bruce S, Villamena F, Rajagopalan S. Acute DPP‐4 inhibition modulates vascular tone through GLP‐1 independent pathways. Vascul Pharmacol. 2011; 55:2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Boscaro E, Albiero M, Menegazzo L, Frison V, de Kreutzenberg S, Agostini C, Tiengo A, Avogaro A. The oral dipeptidyl peptidase‐4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal‐derived factor‐1alpha. Diabetes Care. 2010; 33:1607-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaruba M‐M, Theiss HD, Vallaster M, Mehl U, Brunner S, David R, Fischer R, Krieg L, Hirsch E, Huber B, Nathan P, Israel L, Imhof A, Herbach N, Assmann G, Wanke R, Mueller‐Hoecker J, Steinbeck G, Franz WM. Synergy between CD26/DPP‐IV inhibition and G‐CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell. 2009; 4:313-323. [DOI] [PubMed] [Google Scholar]