

Abstract
Background
Atrial fibrosis is an important factor in initiating and maintaining atrial fibrillation. The purpose of this study was to test the hypothesis that atrial angiotensin‐converting enzyme‐2 (ACE2) overexpression might inhibit atrial collagen accumulation and improve atrial remodeling in a canine atrial pacing model.
Methods and Results
Thirty‐two mongrel dogs of both genders were divided randomly into 4 groups: sham‐operated, control, gene therapy with adenovirus‐enhanced green fluorescent protein (Ad‐EGFP), and gene therapy with Ad‐ACE2. All of the dogs in the control, Ad‐EGFP, and Ad‐ACE2 groups were paced at 450 bpm for a period of 14 days. The dogs in the sham group were instrumented without pacing. After 2 weeks, all of the dogs underwent a thoracotomy operation and received epicardial gene painting. On post–gene transfer day 21, the animals underwent electrophysiology, histology, and molecular studies. The percentage of fibrosis in the Ad‐ACE2 group was markedly lower than the percentage in the control and Ad‐EGFP groups. Compared with the other groups, ACE2 expression was increased significantly in the Ad‐ACE2 group. Compared with the sham and Ad‐ACE2 groups, the expression levels of transforming growth factor‐β1 and Smad3 were significantly higher in the Ad‐EGFP and control groups; however, the expression levels of Smad7 were lower in the atrial tissue as detected by Western blot and reverse transcription polymerase chain reaction.
Conclusions
Our results demonstrate that the overexpression of ACE2 inhibits atrial collagen accumulation and improves left atrial remodeling and function in a canine model of atrial fibrillation. Thus, targeted gene ACE2 therapy provides a promising approach for the treatment of atrial fibrillation.
Keywords: angiotensin II, angiotensin‐converting enzyme 2, atrial fibrillation, atrial fibrosis, extracellular matrix, transforming growth factor‐β1
Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia with a relevant effect on mortality and morbidity, especially in the elderly.1 The current treatment for AF includes drug therapy and ablative strategies. In patients with persistent AF, however, the relapse rate is higher.2–3 The poor curative effect in patients with persistent AF is mainly due to atrial fibrosis. Fibrosis represents excessive deposition of extracellular matrix proteins synthesized by cardiac fibroblasts or myofibroblasts upon stimulation.4 The major heart collagens, collagen type I and type III, make up almost 95% of the cardiac extracellular matrix.5–6 Transforming growth factor β‐1 (TGF‐β1) is a known profibrotic cytokine that has been demonstrated to induce selective atrial fibrosis in a transgenic mouse model, indicating the critical role of TGF‐β1 in the pathogenesis of AF.7 TGF‐β1 contributes to atrial fibrosis via the TGF‐β1‐Smad pathway.7–8 Several studies have revealed that the renin–angiotensin system (RAS) is associated with the development of AF.9–10 Angiotensin‐converting enzyme 2 (ACE2), an enzyme identified in rodents and humans with a more restricted distribution than ACE, is expressed mainly in the heart and kidney.11 ACE2 cleaves a single residue from AngI to generate Ang‐(1‐9) and cleaves AngII, the main effector of the RAS, to form the vasodilator Ang‐(1‐7).11 The results from experiments with ace2 mutant mice suggest that ACE2 negatively regulates activated RAS.12 Thus, ACE2 may have an important role in fibrosis formation during AF. However, the regulatory mechanism of ACE2 in AF is largely unexplored. Therefore, in this study, mongrel dogs were divided randomly into 4 groups: sham‐operated (sham), control, gene therapy with adenovirus‐enhanced green fluorescent protein (Ad‐EGFP) (Ad‐EGFP group), and gene therapy with Ad‐ACE2 (Ad‐ACE2 group). The purpose of this study was to determine whether ACE2 overexpression in pace‐inducing AF might inhibit atrial collagen accumulation and improve atrial remodeling and function through the AngII/AT1/TGF‐β1/Smad pathway.
Methods
Ethics Statement
All of the experiment was approved by the animal experimentation ethics committee of Chongqing Medical University, following the guidelines of the National Institutes of Health for the care and use of laboratory animals.
Adenoviral Vector Construction
Canine ACE2 cDNA was amplified by reverse transcription polymerase chain reaction (RT‐PCR) of RNA extracted from canine kidney cells. Recombinant adenoviruses carrying the canine ACE2 (Ad‐ACE2) or a control transgene (Ad‐EGFP) were prepared by Yingrun Biotechnologies (Changsha, Hunan province, China).
Animal Model and Gene Transfer
Thirty‐two mongrel dogs of both genders, weighing 20 to 30 kg, were divided randomly into 4 groups: sham‐operated (sham), control, gene therapy with Ad‐EGFP (Ad‐EGFP group), and gene therapy with Ad‐ACE2 (Ad‐ACE2 group) (n=8 per group). Baseline 12‐lead ECG and echocardiography were performed and white blood cell count, hemoglobin, serum creatinine, and electrolyte levels were measured to exclude unhealthy animals. All the dogs in the control, Ad‐EGFP, and Ad‐ACE2 groups were paced at 450 bpm for a period of 14 days. The animals were anesthetized by delivering pentobarbital sodium (20 mg/kg) into the abdominal cavity. An endocardial pacing lead (Cardiac Rhythm Management Division, St. Jude Medical, Sweden) was inserted through the right jugular vein, and the distal end of the lead was tightly positioned in the right atrium.13 The initial capture was verified by an external stimulator. Then, the proximal end of the pacing lead was connected to a programmable pacemaker (Fudan University, Shanghai, China), which was inserted into a pocket in the neck. The dogs were paced at 450 bpm with 0.2 ms square‐wave pulses at twice‐threshold current for 2 weeks. The ECG was monitored after 24 hours and then every other day to ensure continuous 1:1 atrial capture. The dogs in the sham group were instrumented without pacing. After 2 weeks, all dogs underwent a thoracotomy operation and an invasive electrophysiology study prior to receiving epicardial gene painting (LEAD2000; Sichuan Jinjiang Electronic Technology Limited Company, Chengdu, China). After sterile preparation, ventilator‐assisted breathing was implemented by endotracheal intubation, and median sternotomy was performed (Tingquan Zhou and Zhenglong Wang operated). To expose both atria, the pericardium was opened. Electrophysiology was then performed. The adenoviral vector (5×109 plaque‐forming units, 1 mL) was mixed with 20% poloxamer F407 and 0.5% trypsin and applied to the atria with a round‐bristle, flat paintbrush composed of camel hair. The 5‐mL volume of virus mixture was divided in half such that each atrium received 2.5 mL of solution. Each atrium was coated twice for 30 s each, and ≈60 s elapsed between applications to allow adsorption. After the applications, the atria were left exposed to air for 10 minutes to allow virus penetration.14 Postoperative monitoring included 30 s of 6‐lead ECG recording daily, as well as daily assessment of behavior and feeding habits. On post–gene transfer day 21, the animals underwent electrophysiological, histological, and molecular studies.
Electrophysiological Studies
For the electrophysiological studies, the EP Lab system (Lead 2000) was used with standard ablation catheters. The inducibility and duration of AF at basic pacing cycle lengths (BCLs) of 300 ms (BCL300) were observed. AF induction was defined as P‐wave disappearance and rapid atrial activation with irregular ventricular response on atrial ECG after atrial programmed stimulation (S1 to S2), which was attempted 3 times at each site. The duration of induced AF was also recorded.
Histopathology and Immunohistochemistry
After thoracotomy, the hearts were excised, rinsed briefly in PBS, fixed by immersion in 4% paraformaldehyde in PBS, and embedded in paraffin. Sections (5 μm) were stained with Masson's trichrome and picric acid–Sirius red to display the collagen components. Immunohistochemical experiments to examine collagen I and III were performed with the appropriate antibodies.
Western Blot Analysis
The expression levels of collagen I and III, Smad3, Smad7, TGF‐β1, and ACE2 proteins were assayed by Western blot. Immunoblotting was performed as described previously.15 In brief, solubilized protein was separated using electrophoresis and transferred to nitrocellulose membranes. Nonspecific binding was blocked by incubation in 5% milk in Tris‐buffered saline with Tween 20. The membranes were probed with specific antibodies and subsequently incubated with horseradish peroxidase–conjugated secondary antibodies.
Real‐Time RT‐PCR
RNA was isolated from the atrial tissue using the TRIzol reagent (Takara, Dalian, China) as directed by the manufacturer. Real‐time RT‐PCR was performed on cDNA generated with the PrimeScript RT reagent kit (Takara) and the SYBR Premix Ex Taq II kit (Takara) in a MX3005 real‐time PCR machine (Stratagene). The mixtures were heated at 50°C for 2 minutes and at 95°C for 90 s, followed by 40 cycles at 95°C for 15 s and 60°C for 30 s. All reactions were performed in triplicate, and GAPDH served as an internal control. The results were quantified as Ct values, where Ct is defined as the threshold cycle of PCR at which the amplified product is first detected, and expressed as the ratio of target to control. The expression levels of Smad3, Smad7, TGF‐β1, collagen I, collagen III, and ACE2 mRNA were quantified using RT‐PCR (Table).
Table 1.
Primer Sequences
| Primer Name | (5′ to 3′) | bp |
|---|---|---|
| GAPDH forward | ATTCCACGGCACAGTCAAG | 19 |
| GAPDH reverse | TCCACAACATACTCAGCACCA | 21 |
| ACE2 forward | TGTCCTGCTCATCTTCTCTGG | 21 |
| ACE2 reverse | ATCATCACCGCTTTGGAATC | 20 |
| Collagen I forward | TGGCAAGAACGGAGATGAC | 19 |
| Collagen I reverse | TCCAAACCACTGAAACCTCTG | 21 |
| Collagen III forward | TTCCTTTGTGGGCTGTGTCT | 20 |
| Collagen III reverse | TTGGCTTCTCTCACTTTCCAG | 21 |
| TGF‐β1 forward | CAGAATGGCTGTCCTTTGATG | 21 |
| TGF‐β1 reverse | GCAGTGTGTTATCTTTGCTGTCA | 23 |
| Smad3 forward | CGACTACAGCCATTCCATCC | 20 |
| Smad3 reverse | CTCTCCATCTTCGCTCAGGT | 20 |
| Smad7 forward | CAAGAGGCTGTGTTGCTGTG | 20 |
| Smad7 reverse | CCATCGGGTATCTGGAGTAAG | 21 |
ACE2 indicates angiotensin‐converting enzyme 2; BP, blood pressure; TGF‐β1, transforming growth factor β‐1.
Enzyme‐Linked Immunosorbent Assay
ELISA was performed to measure the levels of AngII and Ang‐(1‐7).
Statistical Analysis
All quantitative data were expressed as the mean±SEM. Shapiro‐Wilk's test was used to determine whether each variable had a normal distribution. The data analysis was evaluated using GraphPad Prism 5.0 statistical analysis software (GraphPad Software). Statistical comparisons among groups were performed by 1‐way ANOVA. If significant effects were indicated by ANOVA, a least significant difference (LSD) t test was used to evaluate the significance of differences between individual mean values. A 2‐tailed P<0.05 was considered statistically significant (SPSS 22, Chicago, IL).
Results
Electrophysiological In Vivo Studies
Electrophysiological studies were performed to determine the inducibility and the duration of AF. The inducibility of AF in the control and Ad‐EGFP groups was significantly increased compared with that in the sham and Ad‐ACE2 groups on day 35 (P<0.05). The duration of AF in the control and Ad‐EGFP groups was significantly increased compared with that in the sham or Ad‐ACE2 groups on day 35 (P<0.05) (Figure 1).
Figure 1.
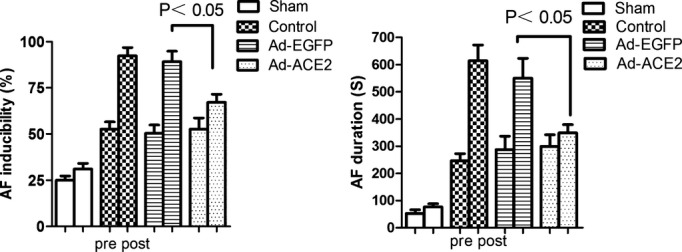
Changes in the inducibility and duration of AF before and after gene transfer. The inducibility of AF in the control and Ad‐EGFP groups was significantly increased compared with that in the sham and Ad‐ACE2 groups, P<0.05. The duration of AF in the control and Ad‐EGFP groups was significantly increased compared with that in the sham and Ad‐ACE2 groups, P<0.05. Ad‐ACE2 indicates adenovirus‐angiotensin‐converting enzyme 2; Ad‐EGFP, adenovirus‐enhanced green fluorescent protein; AF, atrial fibrillation.
Histopathology and Immunohistochemistry
Histopathology and immunohistochemistry were performed to determine the degree of fibrosis of the canine atrium. Based on Masson's Trichrome staining and picric acid–Sirius red staining, the collagen volume was larger in the Ad‐EGFP and control groups than in the Ad‐ACE2 and sham groups(P<0.01), with no significant differences between the Ad‐EGFP and control groups or between the Ad‐ACE2 and sham groups. The average collagen volume fraction in the Ad‐EGFP group was significantly higher than that in the Ad‐ACE2 group (P<0.01).
Compared with the Ad‐ACE2 group or sham group, the expression of collagen I and collagen III detected by immunohistochemistry was increased significantly in the Ad‐EGFP group and control group (P<0.01) (Figure 2).
Figure 2.

Masson's staining, picrosirius red staining, and collagen protein expression in the 4 groups of atrial tissues. Representative Masson's staining and picric‐sirius red staining of the myocardium, immunostaining of collagen I, and immunostaining of collagen III in the sham, control, adenovirus‐enhanced green fluorescent protein (Ad‐EGFP), and adenovirus‐angiotensin‐converting enzyme 2 (Ad‐ACE2) groups of canine subjects 3 weeks after gene transfer. The histograms show the quantitative analyses of the positive staining of collagen, collagen I, and collagen III. P<0.01 between the sham and control or Ad‐EGFP groups, and P<0.01 between the Ad‐ACE2 and control or Ad‐EGFP groups.
Collagen, TGF‐β1, and Smad Expression In Vivo
Western blotting and real‐time RT‐PCR were performed to determine the expression of collagen, TGF‐β1, Smad, and ACE2 in vivo. Atrial collagen deposition was decreased in the Ad‐ACE2 and sham groups compared with the Ad‐EGFP and control groups. Similarly, the protein expression levels of collagen I and collagen III were substantially lower in the Ad‐ACE2 and sham groups than in the Ad‐EGFP and control groups (P<0.01). The expression levels of collagen I and collagen III were substantially lower in the Ad‐ACE2 group than in the control group (P<0.01). Compared with the Ad‐EGFP and control groups, the protein expression levels of TGF‐β1 and Smad3 were significantly reduced in the Ad‐ACE2 and sham groups (P<0.05). The protein expression of Smad7 was higher in the Ad‐ACE2 compared with control and Ad‐EGFP groups (P<0.05) (Figure 3).
Figure 3.
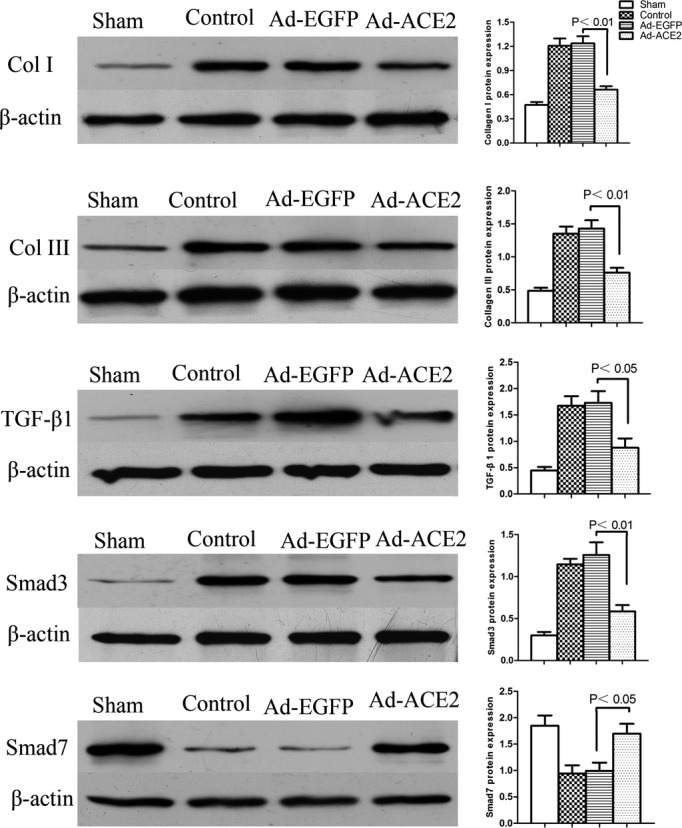
Western blot analysis of collagen I, collagen III, TGF‐β1, Smad3, and Smad7 protein levels from atrial tissues 3 weeks after gene transfer. The collagen I, collagen III, TGF‐β1, and Smad3 protein expression levels in the control and Ad‐EGFP groups were significantly increased compared with the levels in the sham subjects, P<0.05; compared with the Ad‐EGFP subjects, Smad7 protein expression was increased in the Ad‐ACE2 group, P<0.05. Ad‐ACE2 indicates adenovirus‐angiotensin‐converting enzyme 2; Ad‐EGFP, adenovirus‐enhanced green fluorescent protein; TGF‐β1, transforming growth factor β‐1.
The collagen I, collagen III, TGF‐β1, and Smad3 mRNA expression levels in the control and Ad‐EGFP groups were significantly increased compared with their levels in the sham subjects (P<0.05); additionally, the expression levels were decreased in the Ad‐ACE2 group compared with the sham group (P<0.05). Smad7 and ACE2 mRNA expression was significantly increased in the Ad‐ACE2 group compared with the control and Ad‐EGFP groups (P<0.05) (Figure 4).
Figure 4.
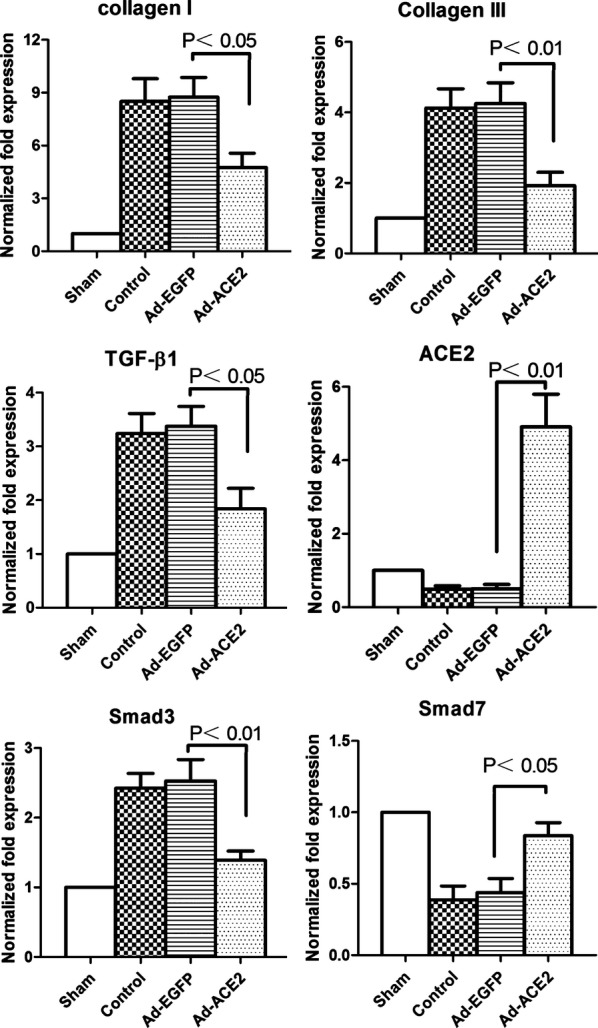
Real‐time RT‐PCR analysis of the mRNA expression of collagen I, collagen III, TGF‐β1, Smad3, Smad7, and ACE2 in atrial tissues 3 weeks after gene transfer. The collagen I, collagen III, TGF‐β1, and Smad3 mRNA expression levels in the control and Ad‐EGFP groups were significantly increased compared with the levels in the sham and ACE2 subjects, P<0.05; Smad7 and ACE2 mRNA expression was increased in the Ad‐ACE2 group compared with the control and Ad‐EGFP groups (P<0.05). Ad‐ACE2 indicates adenovirus‐angiotensin‐converting enzyme 2; Ad‐EGFP, adenovirus‐enhanced green fluorescent protein; RT‐PCR, reverse transcription polymerase chain reaction; TGF‐β1, transforming growth factor β‐1.
ACE2, AngII, and Ang‐(1‐7) Expression In Vivo
Western blot analysis showed that ACE2 expression was increased significantly in the Ad‐ACE2 group compared with the Ad‐EGFP, control, and sham groups (P<0.01).
The expression levels of AngII (as determined by ELISA) were significantly higher in the Ad‐EGFP and control groups compared with the sham and Ad‐ACE2 groups (P<0.01); however, the expression level of Ang‐(1‐7) was lower (P<0.05) (Figure 5).
Figure 5.
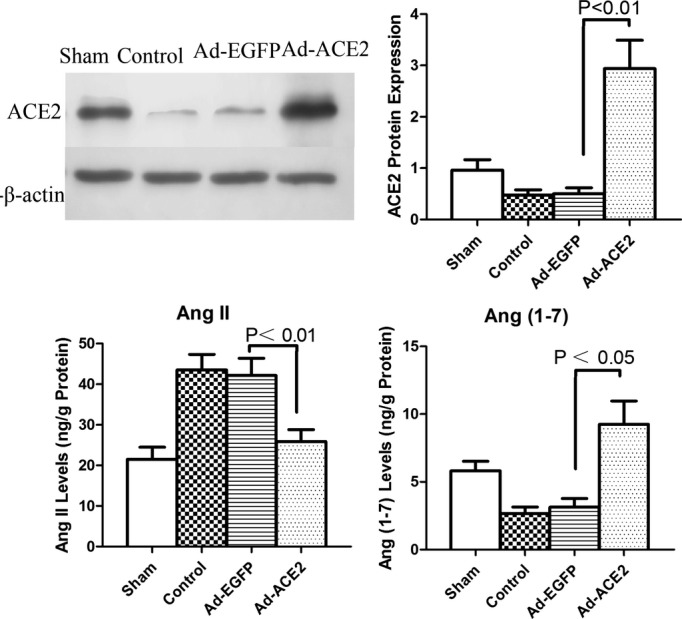
Western blot analysis of ACE2 protein levels and enzyme‐linked immunosorbent assay of AngII and Ang(1‐7). ACE2 protein expression levels in the control and Ad‐EGFP groups were significantly decreased compared with the levels in the sham group; compared with the sham group, the expression levels were increased in the Ad‐ACE2 group, P<0.01. Compared with the sham and Ad‐ACE2 groups, the expression levels of AngII by ELISA were significantly higher in the Ad‐EGFP and control groups, P<0.01; however, the expression levels of Ang(1‐7) were lower, P<0.05. Ad‐ACE2 indicates adenovirus‐angiotensin‐converting enzyme 2; Ad‐EGFP, adenovirus‐enhanced green fluorescent protein.
Discussion
The major finding of the present study was that ACE2 overexpression in vivo attenuated atrial fibrosis. To the best of our knowledge, this study is the first to report the therapeutic effects of ACE2 overexpression in a canine model of AF.
The most important pathological feature of AF is the accumulation and deposition of extracellular matrix proteins, particularly in collagen.16 Available evidence indicates that RAS plays an important role in collagen production in AF.17–19 Several studies have demonstrated that AngII increases collagen production in cultured cardiac fibroblasts in vitro. Additionally, AngII participates in the development of AF‐induced myocardial fibrosis through activation of AT1 receptors.19,18 Recent studies have demonstrated that ACE inhibitors can block AngII synthesis catalyzed by ACE but cannot block AngII synthesis catalyzed by chymase, such as that which occurs in cardiac myocytes.20 Thus, angiotensin II receptor blockers or ACE inhibitors cannot completely inhibit RAS activation in AF. In contrast, ACE2 catalyzes the conversion of pro‐proliferative AngII to antiproliferative Ang‐(1‐7) in the atrium and may provide more cardiac‐protective effects than angiotensin II receptor blockers or ACE inhibitors in AF. This possibility is supported by the results of the present study. In our study, ACE2 overexpression inhibited the production of collagen I and III proteins in the atria of canines in vivo, suggesting that the major mechanism by which ACE2 overexpression inhibits collagen accumulation involves the conversion of AngII to Ang‐(1‐7).
TGF‐β1 is a key regulator of fibrosis that enhances collagen synthesis both in vitro and in vivo, thereby affecting remodeling of the extracellular matrix.21–22 Therefore, enhancing TGF‐β1 signaling activity would result in cardiac fibrosis.23–24 AngII mediates the expression of TGF‐β1 in vitro25 and in vivo26 in various cell types, including cardiac fibroblasts. The serum level of TGF‐β1 was increased in patients with AF, and it was downregulated after defibrillation therapy.27 Changes in genes regulating TGF‐β1 function and signaling were observed in patients with permanent AF28 and in canine AF models.29 TGF‐β1 is a known profibrotic agent, and increases in the expression of TGF‐β1 have been shown to increase myocardial fibrosis.30–31 Although it has been demonstrated that the TGF‐β1/Smad pathway is involved in myocardial infarction, it is not clear how this pathway is involved in atrial fibrosis. According to previous research32 and our results, Smad7 is a negative regulator of TGF‐β1, and a decrease in Smad7 results in the hyperactivation of the TGF‐β1 Smad3 signaling pathway and increased collagen production. It was speculated that the reduced expression of Smad7 in AF patients results in enhanced activity of the TGF‐β1 profibrotic signaling pathway, which promotes atrial fibrosis in AF. In the present study, overexpression of ACE2 attenuated TGF‐β1‐induced activation of the Smad3 signaling pathway and the expression of collagens I and III in AF, demonstrating that Smad7 may be capable of inhibiting atrial fibrosis in AF.
The putative mechanisms underlying the therapeutic effects of ACE2 gene transfer are threefold. First, ACE2 overexpression inhibits collagen accumulation by converting AngII to Ang‐(1‐7). Second, the balance between ACE and ACE2 is critical for the functional state of the RAS. In the present study, ACE2 overexpression resulted in a shift in balance toward low ACE protein expression and, hence, a low AngII concentration. Third, the most important role of ACE2 in the inhibition of collagen accumulation may involve suppression of fibroblast–myocyte crosstalk for collagen production in AF.
Limitations
There are several limitations to the present study. First, we did not study the downstream molecules of Smads in atrial fibrosis. Second, AF incidence does not always correlate with atrial fibrosis; this has implications for our understanding of the molecular mechanisms of AF development. Finally, the role of the ACE2 gene in the antifibrosis pathway would be better supported by adding the knockdown of ACE2 group, which leads to altered deposition of collagen in the atria.
Conclusions
The overexpression of ACE2 inhibits atrial collagen accumulation and improves left atrial remodeling and function in a canine model of AF. Targeted gene ACE2 therapy provides a promising approach for the treatment of AF.
Sources of Funding
This study was supported by the National Natural Science Foundation of China (grant no. 81170166 and no. 31871075, Yin), and the Research Fund for the Doctoral Program of Higher Education (grant no. 20095503110002, Yin).
Disclosures
None.
References
- Magnani JW, Rienstra M, Lin HH, Sinner MF, Lubitz SA, McManus DD, Dupuis J, Ellinor PT, Benjamin EJ. Atrial fibrillation: current knowledge and future directions in epidemiology and genomics. Circulation. 2011; 124:1982-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Sanders P, Kalman JM. Catheter ablation of atrial arrhythmias: state of the art. Lancet. 2012; 380:1509-1519. [DOI] [PubMed] [Google Scholar]
- Kappenberger L. A new look at atrial fibrillation: lessons learned from drugs, pacing, and ablation therapies. Eur Heart J. 2013; 34:2739-2745. [DOI] [PubMed] [Google Scholar]
- Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002; 91:1103-1113. [DOI] [PubMed] [Google Scholar]
- Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol. 1994; 26:279-292. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano‐regulation of connective tissue remodeling. Nat Rev Mol Cell Biol. 2002; 3:349-363. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Nakajima HO, Salcher O, Dittiè AS, Dembowsky K, Jing SL, Field LJ. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor‐beta. Circ Res. 2000; 86:571-579. [DOI] [PubMed] [Google Scholar]
- Pellman J, Lyon RC, Sheikh F. Extracellular matrix remodeling in atrial fibrosis: mechanisms and implication in atrial fibrillation. J Mol Cell Cardiol. 2008; 48:461-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goette A, Arndt M, Rӧcken C, Spiess A, Staack T, Geller JC, Huth C, Ansorge S, Klein HU, Lendeckel U. Regulation of angiotensin II receptor subtypes during atrial fibrillation in humans. Circulation. 2000; 101:2678-2681. [DOI] [PubMed] [Google Scholar]
- Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol. 2003; 41:2197-2204. [DOI] [PubMed] [Google Scholar]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1‐9. Circ Res. 2000; 8:e1-e9. [DOI] [PubMed] [Google Scholar]
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki L, Scanga SE, Oliveira‐dos‐Santos AJ, Costa JD, Zhang LY, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature. 2000; 417:822-828. [DOI] [PubMed] [Google Scholar]
- Gaspo R, Bosch RF, Talajic M, Nattel S. Functional mechanisms underlying tachycardia‐induced sustained atrial fibrillation in a chronic dog model. Circulation. 1997; 96:4027-4035. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, McDonald AD, Sasano T, Donahue JK. Targeted modification of atrial electrophysiology by homogeneous transmural atrial gene transfer. Circulation. 2005; 111:264-270. [DOI] [PubMed] [Google Scholar]
- Olson ER, Naugle JE, Zhang XJ, Bomser JA, Meszaros JG. Inhibition of cardiac fibroblast proliferation and myofibroblast differentiation by resveratrol. Am J Physiol Heart Circ Physiol. 2005; 288:H1131-H1138. [DOI] [PubMed] [Google Scholar]
- Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999; 100:87-95. [DOI] [PubMed] [Google Scholar]
- Boldt A, Scholl A, Garbade J, Resetar ME, Mohr FW, Gummert JF, Dhein S. ACE‐inhibitor treatment attenuates atrial structural remodeling in patients with lone chronic atrial fibrillation. Basic Res Cardiol. 2006; 101:261-267. [DOI] [PubMed] [Google Scholar]
- Hirayama Y, Atarashi H, Kobayashi Y, Takano T. Angiotensin‐converting enzyme inhibitors are not effective at inhibiting further fibrous changes in the atria in patients with chronic atrial fibrillation: speculation from analysis of the time course of fibrillary wave amplitudes. Jpn Heart J. 2004; 45:93-101. [DOI] [PubMed] [Google Scholar]
- Chrysostomakis SI, Karalis IK, Simantirakis EN, Koutsopoulos AV, Mavrakis HE, Chlouverakis GI, Vardas PE. Angiotensin II type 1 receptor inhibition is associated with reduced tachyarrhythmia‐induced ventricular interstitial fibrosis in a goat atrial fibrillation model. Cardiovasc Drugs Ther. 2007; 21:357-365. [DOI] [PubMed] [Google Scholar]
- Kumar R, Singh VP, Baker KM. The intracellular renin‐angiotensin system: a new paradigm. Trends Endocrinol Metab. 2007; 18:208-214. [DOI] [PubMed] [Google Scholar]
- Xiao H, Lei H, Qin S, Ma K, Wang X. TGF‐beta1 expression and atrial myocardium fibrosis increase in atrial fibrillation secondary to rheumatic heart disease. Clin Cardiol. 2010; 33:149-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariscalco G, Engstrӧm KG, Ferrarese S, Cozzi G, Bruno VD, Sessa F, Sala A. Relationship between atrial histopathology and atrial fibrillation after coronary bypass surgery. J Thorac Cardiovasc Surg. 2006; 131:1364-1372. [DOI] [PubMed] [Google Scholar]
- Lei B, Hitomi H, Mori T, Nagai Y, Deguchi K, Mori H, Masaki T, Nakano D, Kobori H, Kitaura Y, Nishiyama A. Effect of efonidipine on TGF‐beta1‐induced cardiac fibrosis through Smad2‐dependent pathway in rat cardiac fibroblasts. J Pharmacol Sci. 2011; 117:98-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhu Y, Li J, Liu J, Gao Y, Ha T, Que L, Liu L, Zhu G, Chen Q, Xu Y, Li C, Li Y. Pellino1‐mediated TGF‐β1 synthesis contributes to mechanical stress induced cardiac fibroblast activation. J Mol Cell Cardiol. 2014; 79C:145-156. [DOI] [PubMed] [Google Scholar]
- Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor‐beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997; 29:1947-1958. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhang JQ, Zhang JK, Ramires FJ. Angiotensin II transforming growth factor‐beta1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998; 30:1559-1569. [DOI] [PubMed] [Google Scholar]
- Seko Y, Nishimura H, Takahashi N, Ashida T, Nagai R. Serum levels of vascular endothelial growth factor and transforming growth factor‐beta1 in patients with atrial fibrillation undergoing defibrillation therapy. Jpn Heart J. 2000; 41:27-32. [DOI] [PubMed] [Google Scholar]
- Barth AS, Merk S, Arnoldi E, Zwermann L, Kloos P, Gebauer M, Steinmeyer K, Bleich M, Kääb S, Hinterseer M, Kartmann H, Kreuzer E, Dugas M, Steinbeck G, Nabauer M. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular‐like genomic signature. Circ Res. 2005; 96:1022-1029. [DOI] [PubMed] [Google Scholar]
- Cardin S, Libby E, Pelletier P, Bouter SL, Shiroshita‐Takeshita A, Meur NL, Léger J, Demolombe S, Ponton A, Glass L, Nattel S. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res. 2007; 100:425-433. [DOI] [PubMed] [Google Scholar]
- Buxton IL, Duan D. Cyclic GMP/protein kinase G phosphorylation of Smad3 blocks transforming growth factor‐beta‐induced nuclear Smad translocation: a key antifibrogenic mechanism of atrial natriuretic peptide. Circ Res. 2008; 102:151-153. [DOI] [PubMed] [Google Scholar]
- Chen K, Mehta JL, Li D, Joseph LJ, Joseph J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004; 95:1167-1173. [DOI] [PubMed] [Google Scholar]
- Razani B, Zhang XL, Bitzer M, Gersdorff G, Bӧttinger EP, Lisanti MP. Caveolin‐1 regulates transforming growth factor TGF‐beta/SMAD signaling through an interaction with the TGF‐beta type I receptor. J Biol Chem. 2001; 276:6727-6738. [DOI] [PubMed] [Google Scholar]