

Stroke management exerts insurmountable societal and economic burden on the patient as well as their caregivers. In the year 2010 alone, the direct and indirect costs of stroke care amounted to 36.5 billion dollars (Go et al., 2014). Despite concentrated efforts to develop a safe, effective drug for stroke, we have not discovered one since the introduction of recombinant tissue plasminogen activator (rtPA)—the standalone FDA-approved therapy for stroke. While rtPA is highly effective, it needs to be given within 3–4.5 hours of the onset of stroke symptoms (Zivin, 2009). This is often complicated by the delay in the commencement of treatment due to preliminary inclusion parameters that are required to be ascertained before rtPA administration.
This ushered in research on molecules intended to protect cells undergoing disruptive changes following the reduction of blood supply in an effort to prevent their demise and thus, loss of brain function. These potential neuroprotective agents have been the mainstay of drug discovery efforts for many years. While some of these agents have shown promise in the pre-clinical level, they have largely failed in the clinical setting (Gladstone et al., 2002). An attribute common to most of these failed leads is that they acted on a single molecular target, often ion channels/receptors whose modulation was ineffective because their window of action did not match the window during which the drug treatment was initiated in the trial (Gladstone et al., 2002). Moreover, the lag time between the onset of stroke symptoms and the onset of treatment in most patients—a factor not often controlled for in pre-clinical studies—rules out many potential molecular targets that are vulnerable to the immediate effects of stroke. Therefore, drugs designed against such early-acting targets would be ineffective in the clinic.
It is necessary, therefore, to realign our pre-clinical strategies to reflect the lessons learnt from the clinical studies. A topic that has gained interest in this regard is the search for drugs that act on multiple cellular mechanisms, such that these mechanisms span the temporal spectrum of stroke pathogenesis. This would broaden the scope of the drug's action beyond the acute period so far targeted by rtPA and increase the chances of a clinical success. These drugs could be envisaged to act in conjunction with rtPA, although not necessarily so. Increasingly, such strategies are being applied for targets that act in the repair phase of stroke. Repair processes after stroke are triggered by endogenous mechanisms and often initiate several days after stroke and continue until weeks or even months after it (Gladstone et al., 2002). Therefore, drugs acting to potentiate repair have the advantage of relatively relaxed therapeutic time windows and are subjected to dose manipulations without perturbing the riskier, acute phase of stroke.
Repair mechanisms after stroke mainly involve neurogenesis, angiogenesis, and synaptic plasticity (Font et al., 2010). Neurogenesis or the formation of new neurons is a complex cascade of processes beginning with the proliferation of neuronal precursor cells (NPC) and their migration to the site of injury, which in our case, is the infarct site. It is not until the NPCs differentiate into mature neurons and integrate into the existing neuronal network that they elicit functional recovery. Although stroke itself induces neurogenesis, few NPCs survive to differentiate into mature neurons; therefore, any intervention that could augment this process would improve stroke recovery. There is no clear evidence from human studies that supports the role of neurogenesis in stroke recovery and further studies are necessary to substantiate this idea.
Neurogenesis is not a solitary process; the vasculature provides a migratory route and supports the survival of NPCs. In fact, the proliferation of new blood vessels—i.e., angiogenesis— is essential for neurogenesis. Pro-angiogenic factors like the vascular endothelial growth factor (VEGF) promote both angiogenesis and neurogenesis (Ruiz de Almodovar et al., 2009). Both these processes culminate in successful recovery only when they integrate into the existing neurovascular niche to re-form the severed connections. This is achieved by inducing synaptogenesis and activating silent synapses to compensate for the lost ones (Font et al., 2010).
These repair mechanisms, however, face a major obstacle to their progress. Stroke injury triggers the expression of molecules that pose an inhibitory environment to axonal sprouting and eventual neuronal regeneration. These molecules belong to three classes: myelin associated proteins like NogoA and myelin-associated glycoprotein; extracellular matrix proteins like chondroitin sulfate proteoglycans and tenascin; developmentally associated axonal guidance molecules like ephrin A & B and semaphorins (Carmichael, 2008). Their inhibitory cues persist for a long time after the stroke insult and could curtail any endogenous repair processes in play. Hence, effective recovery can only result after the inhibitory signals are overcome. Drugs that neutralize the repulsive cues and bolster repair mechanisms would therefore be ideal candidates for regenerative therapy.
Natural products have been productive sources of drug leads in past decades. They possess unique chemical diversity and many of them exhibit polypharmacology—the ability to modulate several molecular targets simultaneously. This is a desirable quality in the complex pathophysiology of stroke, wherein multiple cell death pathways are activated simultaneously. In this regard, the constituents of the herb Withania somnifera have recently generated interest for their effects on stroke and other neurodegenerative conditions (Raghavan and Shah, 2014).
Withania somnifera (WS), also commonly known as ‘Ashwagandha’ or Winter cherry, has been used for centuries to treat ailments in the ayurvedic as well as indigenous systems of medicine as an aphrodisiac, nerve-tonic, anti-inflammatory and anti-cancer agent (Kulkarni and Dhir, 2008). Chemically, WS is mainly composed of steroidal lactones also called withanolides, which are responsible for most of its biological effects. In addition to stroke, extracts and individual components of WS have been tested in various models of neurological disorders like Alzheimer's disease (AD), Parkinson's disease (PD). Huntington's disease (Kuboyama et al., 2006), epilepsy, stress disorders with successful results (Kulkarni and Dhir, 2008). Along with the potent anti-oxidant, anti-inflammatory, and anti-apoptotic properties, some of these have also unearthed the neuroregenerative properties of WS.
In a model of AD, withanoside-IV (W-IV), a withanolide component of WS, was able to reverse the synaptic loss and neurite atrophy characteristic of the disease (Kuboyama et al., 2006). This effect also translated to a better functional outcome of reduced cognitive deficits. The authors attributed the observed effects to increased neurite outgrowth, synaptogenesis, and synaptic integration induced by W-IV. Another withanolide—withanolide A (W-A)—was also able to improve outcomes in an amyloid-beta (Aβ) mouse model of AD. W-A improved axonal and dendritic regeneration and synaptic integration, thereby showing strong regenerative potential in AD (Kuboyama et al., 2006).
We, and others have investigated the effects of WS in stroke models (Raghavan and Shah, 2014). While the previous studies evaluated the effect of WS when given as a pre-treatment before the onset of stroke, we tested an extract of WS in both pre- and post-treatment paradigms. Since ischemic strokes constitute 87% of all strokes (Go et al., 2014), and the permanent occlusion model of ischemic stroke is considered to be reliable, we utilized the permanent middle cerebral artery occlusion (pMCAO) model for our experiment. Mice administered with an aqueous root extract of WS (200 mg/kg) daily for 7 days before pMCAO and a survival period of 7 days exhibited significantly lower infarct volumes when compared to the vehicle treated group. Moreover, the WS treated mice also exhibited significantly better short-term functional outcomes in terms of locomotor activity (Raghavan and Shah, 2014). The same trend was witnessed in mice treated with WS (200 mg/kg) 4 hours after pMCAO followed by daily doses for 7 days. Thus, both pre- and post-treatment with WS improves histological outcomes (infarct volume) and short-term functional outcomes (locomotor activity). The ability to improve functional outcomes after the onset of stroke is a good indicator of the functional recovery. Although we were not able to establish the effect of WS on long-term functional recovery, these results are nevertheless promising and further longitudinal studies might reveal long-term benefits of WS.
This led us to evaluate the molecular underpinnings of the observed results. We found that WS induced the expression of the anti-oxidant enzyme hemoxygenase-1 (HO-1) in the pre-treatment group. It also inhibited the caspase-independent mechanism of apoptosis evidenced by the inhibition of poly (ADP-ribose) polymerase-1 mediated release of apoptosis inducing factor (Raghavan and Shah, 2014). Hence, we concluded that the combined anti-oxidant and anti-apoptotic effects and possible crosstalk between the two is responsible for at least part of its protective effects. Next, we wanted to extend this idea and test if WS could have modulated repair mechanisms to elicit the observed response.
We looked at levels of semaphorin 3A (Sema3A), a member of the semaphorin family of negative guidance cues. Sema3A is known to cause axonal repulsion and growth cone collapse. It acts via the receptor neuropilin-1 (NRP-1), a member of the neuropilin family of receptors that function with plexins and vascular endothelial growth factor receptor as co-receptors (Acevedo et al., 2008). Both Sema3A and NRP-1 levels remain elevated for a long time after stroke damage, preventing the neurons from regenerating and integrating into the circuit. Therefore, reducing levels of Sema3A would be beneficial in attenuating the inhibitory signals that thwart repair. When we assessed sema3A levels, we found that mice pre-treated with WS showed dramatically lower levels of Sema3A (statistically significant) when compared to the vehicle group (Raghavan and Shah, 2014). This suggests that WS is able to reduce sema3A-mediated repulsive cues, thereby indirectly promoting repair. Another interesting effect of sema3A is its anti-angiogenic effect, particularly against VEGF-mediated angiogenesis (Acevedo et al., 2008). Thus, the inhibition of sema3A could also reduce its anti-angiogenic influence, leading to indirect VEGF-mediated angiogenesis (Figure 1).
Figure 1.
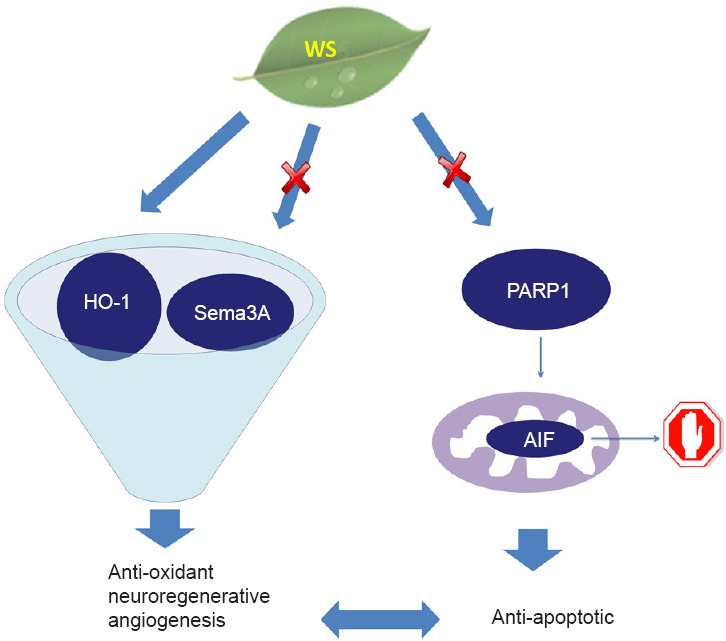
This schematic depicts the probable mechanisms of action of an aqueous extract of Withania somnifera (WS) in ischemic stroke.
We confirmed the neuroprotective effects of WS in both pre- and post-treatment models of permanent ischemic stroke in mice. We believe that part of its observed effects could be due to the induction of expression of hemoxygenase-1 (HO-1), thereby providing an anti-oxidant effect. We believe that WS-mediated attenuation of the expression of Semaphorin 3A (Sema3A) could promote neuronal regeneration. Moreover, HO-1 mediated vascular endothelial growth factor (VEGF) induction and the antagonistic effects of Sema3A and VEGF could have the combined result of higher VEGF levels and a resulting pro-angiogenic effect. WS was also found to reduce levels of poly (ADP-ribose) polymerase 1 (PARP1), which prevents translocation of anti-apoptotic factor (AIF) from the mitochondria to the nucleus. The PARP1-AIF pathway is a prime mediator of caspase-independent apoptosis, which is prevented by WS in our model of stroke. We believe that there is a strong possibility of cross talk between the anti-oxidant, possible pro-angiogenic and the anti-apoptotic properties exhibited by WS.
Since angiogenesis is essential for neurogenesis, our next step was to evaluate the neurogenic potential of WS. The Wnt pathway is a prime mediator of NPC proliferation and differentiation steps of neurogenesis. It functions via two pathways: β-catenin dependent/canonical pathway and β-catenin independent/non-canonical pathway, of which the former is activated after ischemia (Shruster et al., 2012). When evaluated for Wnt ligand levels, we found no significant difference between the WS pre-treated and vehicle groups. We also tested the levels of markers involved in both canonical and non-canonical pathways, but found no statistically significant difference between the treatment groups (Raghavan and Shah, 2014). Thus, we concluded that WS does not considerably perturb either of the Wnt pathways to mediate its neuroprotective effects. This warrants further research into other neurogenesis mechanisms triggered by ischemia before the lack of influence of WS on neurogenesis could be confidently established. Additionally, any crosstalk between the WS's molecular mechanism need to be confirmed to discover potential synergistic effects. For example, the relationship between HO-1 and Sema3A is worth investigation. It is known that HO-1 induces the expression of VEGF and Sema3A is antagonistic to VEGF. Thus, the HO-1 induction and Sema3A inhibition would have the combined effect of VEGF induction and its concomitant pro-angiogenic effects (Raghavan and Shah, 2014). Based on the study outcomes, WS works as a neuroprotectant and also shows effect on the regenerative and recovery pathways, however, a thorough longitudinal study analyzing major components of recovery processes could further substantiate our findings on the role of WS in stroke recovery.
Ischemic stroke is a complex disorder that needs novel approaches aimed at its various facets. The combined neuroprotective and neuroregenerative potential exhibited by WS is one such strategy that might aid recovery after stroke by preventing cell death as well as stimulating repair. However, this is just the surface of WS's potential as well the promise of neuroregenerative therapies in stroke.
References
- Acevedo LM, Barillas S, Weis SM, Gothert JR, Cheresh DA. Semaphorin 3A suppresses VEGF-mediated angiogenesis yet acts as a vascular permeability factor. Blood. 2008;111:2674–2680. doi: 10.1182/blood-2007-08-110205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST. Themes and strategies for studying the biology of stroke recovery in the poststroke epoch. Stroke. 2008;39:1380–1388. doi: 10.1161/STROKEAHA.107.499962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font MA, Arboix A, Krupinski J. Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr Cardiol Rev. 2010;6:238–244. doi: 10.2174/157340310791658802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladstone DJ, Black SE, Hakim AM. Heart, Stroke Foundation of Ontario Centre of Excellence in Stroke R (2002) Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboyama T, Tohda C, Komatsu K. Withanoside IV and its active metabolite, sominone, attenuate Abeta(25-35)-induced neurodegeneration. Eur J Neurosci. 2006;23:1417–1426. doi: 10.1111/j.1460-9568.2006.04664.x. [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Dhir A. Withania somnifera: an Indian ginseng. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1093–1105. doi: 10.1016/j.pnpbp.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Raghavan A, Shah ZA. Withania somnifera improves ischemic stroke outcomes by attenuating PARP1-AIF-mediated caspase-independent apoptosis. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-8907-2. doi: 10.1007/s12035-014-8907-2. [DOI] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of VEGF in the nervous system. Physiol Rev. 2009;89:607–648. doi: 10.1152/physrev.00031.2008. [DOI] [PubMed] [Google Scholar]
- Shruster A, Ben-Zur T, Melamed E, Offen D. Wnt signaling enhances neurogenesis and improves neurological function after focal ischemic injury. PLoS One. 2012;7:e40843. doi: 10.1371/journal.pone.0040843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivin JA. Acute stroke therapy with tissue plasminogen activator (tPA) since it was approved by the U.S. Food and Drug Administration (FDA) Ann Neurol. 2009;66:6–10. doi: 10.1002/ana.21750. [DOI] [PubMed] [Google Scholar]