

Macroautophagy (here autophagy) is a catabolic mechanism responsible for the degradation of bulk cytoplasm, long-lived proteins and organelles. During autophagy, the cargos are engulfed by double-membrane structures named phagophores, which expand to form the autophagosomes. Subsequently, these autophagosomes fuse with lysosomes, in which the cytoplasmic cargos are degraded. Autophagy is a constitutive process, which plays an important role in cellular homeostasis. In primary neurons autophagosome formation occurs continuously and preferentially at the distal end of axons. On the other hand, autophagy is increased by different stresses, and its dysregulation or excessive induction may lead to detrimental effects. Many neurological disorders have been associated with alterations in the autophagic pathway and an increase in autophagy during axonal degeneration was described.
After a spinal cord injury (SCI), damaged axons degenerate forming what Ramon y Cajal described as “retraction bulbs”. Damage to the spinal cord currently cannot be sufficiently repaired by any therapy. Failure to preserve axonal integrity is one reason for limited functional regeneration following traumatic lesions. Therefore, understanding the molecular mechanisms of axonal degeneration after SCI could provide new insights that could be exploited for the development of new therapeutic approaches. Interestingly, autophagy has been shown to be increased after SCI. The autophagosome marker LC3 is upregulated in neurons, astrocytes and oligodendrocytes after SCI and an ultrastructural analysis showed an increase in autophagosome number in the damaged neural cell bodies (Kanno et al., 2011). However, the characterization of the role of autophagy during axonal degeneration after SCI is still incomplete. In a recent study we therefore approached this question by studying levels of autophagy-related proteins in a mammalian model of SCI (Ribas et al., 2014).
A key set of autophagy-related genes (Atg) was identified in mammals. In our study we focused on four autophagy proteins: ULK1, Atg7, Atg5 and LC3 (Ribas et al., 2014). The ULK1 protein is a serine/threonine kinase that regulates the very initial steps of autophagy. The activity of ULK1 is blocked by mTOR, which is a master negative regulator of autophagy. During autophagy induction, ULK1 localizes at the autophagosome formation site. Atg7 is a key autophagy protein, which has dual functions. Atg7 acts like an E1 enzyme, which is working together with Atg10, conjugating Atg5 to Atg12 and together with Atg3 activating LC3 protein by adding a phosphatidylethanolamine (PE) group, both steps being necessary for autophagosome formation. The Atg5 protein works in a complex; it is covalently attached to Atg12 by Atg7 and Atg10 and further to Atg16. This complex is localized in the membrane of the phagophore and dissociates from it after autophagosome complete development, having an essential role in autophagosome development. In addition, the Atg5-Atg12-Atg16 complex has also a role in LC3 lipidation. The lipidated active form of LC3 (LC3-II) becomes inserted into the inner and outer membranes of the phagophore and the autophagosome until its fusion with the lysosome. LC3 regulates autophagosome development and is used extensively as an autophagosome marker. Thus, these four proteins have important functions in the process of autophagy and regulate distinct steps in this tightly regulated homeostatic mechanism. To learn more about the involvement of these proteins in the regulation of autophagy following SCI, we examined their expression in the rubrospinal tract axons on both sides of the lesion at different time points after SCI.
We found that ULK1, which localizes in the autophagosome formation site during autophagy induction, accumulates in the retraction bulbs early after SCI (Figure 1A). This suggests that ULK1 might be an early initiating protein of the autophagy cascade in the spinal cord and that this activation may be triggered initially in the retraction bulbs. ULK1 activation was previously shown to be induced by calcium influx. In addition, our group showed that accumulation of autophagosomes in lesioned optic nerve axons is dependent on calcium influx. Our results showed that the calpain-mediated spectrin fragment accumulates also in the retraction bulbs early after SCI, suggesting that calcium influx may occur in this region. Thus, we hypothesize that the accumulation of ULK1 and consequent autophagy activation in retraction bulbs could be induced by lesion-induced calcium influx to this region.
Figure 1.
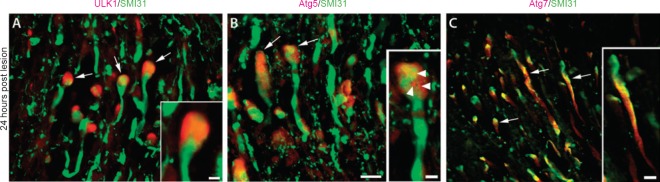
Representative photomicrographs of spinal cord longitudinal sections showing the rubrospinal tract region in the rostral side 24 hours after spinal cord injury.
The injured spinal cord immunostained for the axonal marker SMI31 (green) and for the autophagy proteins (red) ULK1 (A), Atg5 (B) or Atg7 (C). Arrows indicate examples of immunopositive axons. Scale bars: 20 μm. Insets: Higher magnification showing a retraction bulb stained for ULK1 (A), a retraction bulb with puncta-like structures positive for Atg5 (B; arrowheads) and an axon stained diffusely for Atg7 (C). Scale bars: 5 μm. Reproduced with permission from Brain Pathology, 2014, doi:10.1111/bpa.12170.
The Atg5 protein, which is conjugated to Atg12-Atg16 during autophagy induction and localizes in the membrane of the phagophore, is upregulated in damaged axons after SCI. We found a high expression of Atg5 in the axons at longer distances from the injury site. The pattern was more diffuse and not puncta-like, suggesting that this widespread Atg5 distribution might represent its inactive monomeric form. Nevertheless, the major increase of Atg5 occurred in retraction bulbs, similar to ULK1. In addition, we identified the formation of some puncta-like structures showing Atg5 expression (Figure 1B), indicating the presence of phagophores in the retraction bulbs. The accumulation of ULK1 and Atg5 in the retraction bulbs after SCI points to the retraction bulb as the site where autophagosome biogenesis occurs after axonal damage. Some studies also showed that autophagy is elevated in the distal axon in disease models. For example, in a mouse model of excitotoxic neurodegeneration, autophagosomes preferentially accumulate in the distal axons of Purkinje neurons (Wang et al., 2006). Taken together, these data indicate that the autophagosome biogenesis might occur preferentially at the distal axonal tips during axonal degeneration.
The key autophagy protein Atg7 is upregulated very early in degenerating axons after SCI and is localized diffusely in the axon (Figure 1C). During autophagy induction, the Atg7 protein does not accumulate in any specific compartment and has a dual role (to mediate the conjugation reactions of Atg5-Atg12 and LC3). Although the autophagy cascade may be activated initially in the retraction bulbs, the widespread increase in Atg7 expression suggests that the autophagic cascade might be rapidly propagated to more distal axonal compartments and that Atg7 may play an important role in this propagation along the axon. The diffusion of Atg7 along the axon could facilitate the conjugation of Atg5 to Atg12 and also the lipidation of LC3.
The most protracted response was observed in the number of axonal LC3-positive autophagosomes, which started to increase as early as 30 minutes after SCI, but remained at a high level even at 6 weeks after SCI. The number of LC3-positive autophagosomes was increased throughout the entire evaluated axon, suggesting that although the autophagosome biogenesis might be triggered initially in the retraction bulbs, the autophagosomes may be transported through the axons in a similar manner as shown in primary neurons. The maximum number of LC3-positive autophagosomes occurred as long as 2 weeks after SCI, which was long after the maximum increase observed for ULK1, Atg7 and Atg5 that occurred at 24 hours. In contrast to other analyzed autophagy proteins, which are mostly involved in the initial steps of the autophagic cascade, LC3 is inserted only later in the membrane of the phagophore and autophagosome until its fusion with lysosomes. This might explain why the increase of LC3 occurred in a more prolonged time frame. LC3 expression thus follows the widespread expression of Atg7 and Atg5 with a temporal delay. We thus hypothesize that sustained activation of autophagy contributes to chronic effects on axonal degeneration after SCI.
Our ultrastructural analysis showed an enrichment of axonal bulbs containing a high number of double-membrane vesicles most likely autophagosomes in damaged axons, corroborating the immunohistochemical LC3 analysis. Interestingly, in all sections examined we did not observe marked differences between the rostral and caudal axons. Previous in vivo life imaging studies using the SCI model showed that in the first 30 hours after the lesion the axons degenerate in a similar manner on both sides of SCI, with characteristics that include the formation of bulb-like structures in axonal tips and the activation of calpain (Kerschensteiner et al., 2005). In our study, the most prominent alterations in autophagy levels occurred in the first 24 hours in a similar manner on both sides of the lesion. Thus, we speculate that autophagy activation might be an additional executive mechanism of axonal self-destruction, which is activated during axonal degeneration on both sides of the lesion (Figure 2).
Figure 2.
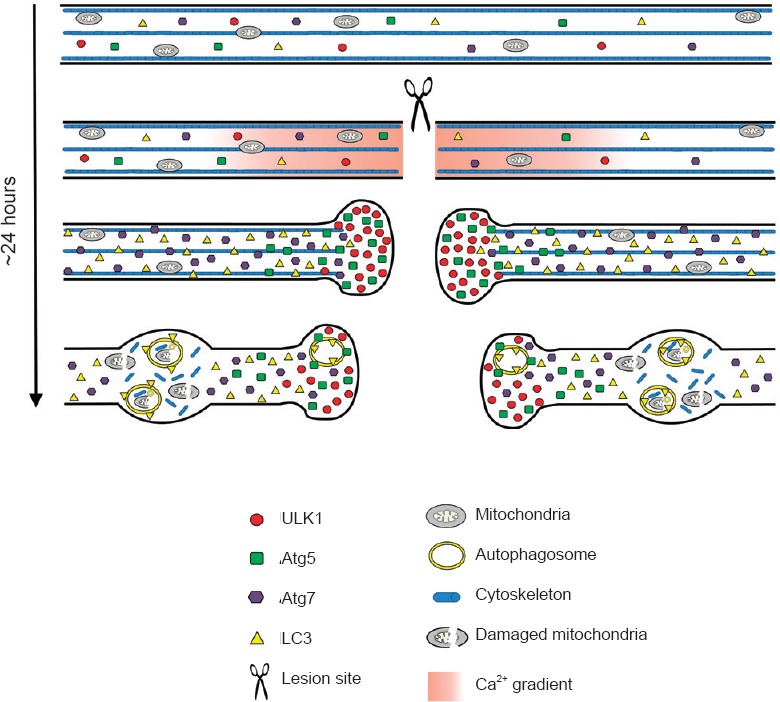
Scheme of the proposed morphological events that occurs in damaged axons after spinal cord injury.
Before lesion, the basal axonal levels of the autophagy-related proteins ULK1, Atg7, Atg5 and LC3 are very low. Injury to the axons of the spinal cord induces a rapid influx of calcium that could activate the autophagy cascade through ULK1 stimulation, which strongly accumulates in the retraction bulbs. Atg5 also accumulates in the retraction bulbs showing puncta-like structures, indicating the presence of phagophores. The accumulation of ULK1 and Atg5 in retraction bulbs suggests that autophagosome biogenesis may take place in this region after the lesion. The Atg7 protein shows an increased expression throughout the damaged axons and could contribute to the spatial propagation of the autophagy cascade. Finally, the diffuse increase of LC3-positive autophagosome suggests that they may be transported through the axons and accumulate in axonal swellings on both sides, rostral and caudal, to the lesion site. Reproduced with permission from Brain Pathology, 2014, doi:10.1111/bpa.12170.
Recently, studies showed that the modulation of the autophagy pathway influences recovery after SCI. For example, melatonin administration appears to contribute to motor recovery by downregulation of, among others, LC3-II and Beclin-1 after SCI (Park et al., 2012). Bisperoxovanadium treatment promoted significant neuroprotection correlated with enhanced signaling of mTOR and reduced autophagic activity (Walker et al., 2012). The activation of mTOR and consequent inhibition of autophagy by treatment with basic fibroblast growth factor improved recovery and increased the survival of neurons after SCI, while the autophagy activator rapamycin partially abolished the protective effects of basic fibroblast growth factor (Zhang et al., 2013). To emphasize mTOR as an important target in this context, forced upregulation of mTOR activity by conditional deletion of PTEN promotes a robust regenerative response of injured axons after SCI (Liu et al., 2010). Despite the fact that in this study the autophagy pathway was not analyzed, it is likely that autophagy is decreased after PTEN deletion. Taken together, these results indicate that autophagy inhibition could be beneficial for recovery after SCI. On the other hand, it has been shown that rapamycin treatment leads to higher expression levels of LC3 and Beclin 1 and this resulted in reduced neuronal loss and improved locomotor recovery after SCI (Sekiguchi et al., 2011). It is, however, important to note that most of these studies were performed using synthetic substances, which can affect other cellular process. In addition, these chemicals could influence autophagy not only in neurons but also in other cell types. The precise role of specific autophagy proteins in these cell types during axonal degeneration remains thus unclear and requires further investigation.
Taken together, there is evidence to suggest that autophagy is an early and sustained process in degenerating axons after SCI, suggesting that it is an important initiating and executive step for axonal degeneration. These kinetics also point to autophagy-related proteins as promising therapeutic targets for the modulation of axonal degeneration following traumatic spinal cord lesions and other axonal pathologies.
VTR is a fellow of the National Council for Scientific and Technological Development (CNPq), Brazil. PL was funded by the International Foundation for Research in Paraplegia (IRP-P 112) and the Deutsche Forschungsgemeinschaft (DFG-LI 1308/3-1) and the Else Kröner-Fresenius-Stiftung.
References
- Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Itoi E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine (Phila. Pa. 1976) 2011;36:E1427–E1434. doi: 10.1097/BRS.0b013e3182028c3a. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Lee SK, Park K, Lee Y, Hong Y, Lee S, Jeon JC, Kim JH, Lee SR, Chang KT, Hong Y. Beneficial effects of endogenous and exogenous melatonin on neural reconstruction and functional recovery in an animal model of spinal cord injury. J Pineal Res. 2012;52:107–119. doi: 10.1111/j.1600-079X.2011.00925.x. [DOI] [PubMed] [Google Scholar]
- Ribas VT, Schnepf B, Challagundla M, Koch JC, Bähr M, Lingor P. Early and sustained activation of autophagy in degenerating axons after spinal cord injury. Brain Pathol. 2014 doi: 10.1111/bpa.12170. doi: 10.1111/bpa.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi A, Kanno H, Ozawa H, Yamaya S, Itoi E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J Neurotrauma. 2012;29:946–956. doi: 10.1089/neu.2011.1919. [DOI] [PubMed] [Google Scholar]
- Walker CL, Walker MJ, Liu NK, Risberg EC, Gao X, Chen J, Xu XM. Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS One. 2012;7:e30012. doi: 10.1371/journal.pone.0030012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Wang ZG, Wu FZ, Kong XX, Yang J, Lin BB, Zhu SP, Lin L, Gan CS, Fu XB, Li XK, Xu HZ, Xiao J. Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Mol Neurobiol. 2013;48:452–464. doi: 10.1007/s12035-013-8432-8. [DOI] [PubMed] [Google Scholar]