

Abstract
Proteogenomics is an area of research at the interface of proteomics and genomics. In this approach, customized protein sequence databases generated using genomic and transcriptomic information are used to help identify novel peptides (not present in reference protein sequence databases) from mass spectrometry-based proteomic data; in turn, the proteomic data can be used to provide protein-level evidence of gene expression and to help refine gene models. In recent years, owing to the emergence of next generation sequencing technologies such as RNA-Seq and dramatic improvements in the depths and throughput of mass spectrometry-based proteomics, the pace of proteogenomics research has greatly accelerated. Here I review the current state of proteogenomics methods and applications, including computational strategies for building and using customized protein sequence databases. I also draw attention to the challenge of false positives in proteogenomics, and provide guidelines for analyzing the data and reporting the results of proteogenomics studies.
Introduction
Proteomics is the comprehensive, integrative study of proteins and their biological functions. The goal of proteomics is often to produce a complete and quantitative map of the proteome of a species, including defining protein cellular localization, reconstructing their interaction networks and complexes, and delineating signaling pathways and regulatory post-translational protein modifications 1.
Proteomic data is generally obtained using a combination of liquid chromatography (LC) and tandem mass spectrometry (MS/MS) 2, also referred to as shotgun proteomics. A key step in proteomics is how peptides are identified from acquired MS/MS spectra (Figure 1). Unlike genomics technologies, in which the DNA or RNA fragments are actually sequenced, in proteomics, peptides are most commonly identified by matching MS/MS spectra against theoretical spectra of all candidate peptides represented in a reference protein sequence database 3. The underlying assumption is that all protein-coding sequences in the genome are known and accurately annotated as a collection of gene models, and that all protein products of these gene models are present in a reference protein sequence database such as Ensembl, RefSeq, or UniProtKB used for peptide identification (Box 1). Much of the subsequent data analysis and interpretation, including inference of the protein identity 4 and protein quantification using the sequences and abundances of the identified peptides, are based on this assumption.
Figure 1. Peptide and protein identification in shotgun proteomics.

A) Overview of shotgun proteomics. Proteins are digested into peptides, then separated using liquid chromatography coupled online to a mass spectrometer, then analyzed by the mass spectrometer which generates tandem mass (MS/MS) spectra. B) Peptides are most commonly identified using a sequence database search approach. Traditionally, experimental MS/MS spectra are matched with theoretical spectra predicted for each peptide contained in a protein sequence database. Sequence tag-assisted database searching starts with extraction of short tags followed by database searching in which the list of candidate peptides is restricted to those peptides only that contain one of the extracted sequence tags, allowing for mutations in the sequences of candidate database peptides. Peptide sequence can also be extracted directly from the spectrum using de novo sequencing (extracted sequences can then be searched in a protein sequence database to find the exact or a homologous peptide).
Box 1. Reference protein sequence databases.
Ensembl
Ensembl is an automatic annotation system that generates gene models via integration of data from multiple sources, including gene prediction algorithms, comparative analysis of genomic sequences across multiple organisms, and mapping of transcriptional (cDNA) or translational evidence (protein sequence from UniProtKB categories 1 and 2, see below, and RefSeq) to the DNA sequence. In addition, annotations are imported from the organism-specific databases such as FlyBase, WormBase and SGD, each of which themselves provide reference protein sequences. The annotated gene models are divided into categories based on their functional potential and the type of supporting evidence available. The locus level categories (“biotypes”) include ‘protein-coding gene’, ‘long noncoding RNA (lncRNA) gene’, or ‘pseudogene’. At the transcript level, additional biotypes are introduced reflecting known or suspected functionality of that transcript (or lack of thereof), e.g. ‘protein-coding’ or ‘subject to nonsense mediated decay (NMD)’. In addition, a “status” is assigned at both the gene locus and transcript level: ‘known’ (represented in the HUGO Gene Nomenclature Committee (HGNC) database and RefSeq); ‘novel’ (not currently represented in HGNC or RefSeq databases, but supported by transcript evidence or evidence from a paralogous or orthologous locus); or ‘putative’ (i.e. supported by transcript evidence of lower confidence). For human and more recently mouse - the organisms with the high quality-finished genomes and where gene annotation efforts are most extensive - the GENCODE consortium provides refined gene annotations by integrating Ensembl automated predictions and the Human and Vertebrate Genome Analysis and Annotation (HAVANA) manual annotations. For these two organisms, the GENCODE annotations are steadily supplementing or replacing the Ensembl automatic annotations. Both Ensemble and GENCODE provide transcript and protein sequence databases available for download (in FASTA format supported by all MS/MS database search tools), along with annotation information and classification of entries into different categories.
RefSeq and Entrez Protein
The National Center for Biotechnology Information (NCBI) produces two databases suitable for MS-based proteomics: the Reference Sequence (RefSeq) database and Entrez Protein database. RefSeq is a result of manual curation of a collection of publicly available data for organisms with sufficient amount of data available, with an emphasis on cDNA data. It provides separate records for the genomic DNA, the transcripts, and the proteins sequences corresponding to those transcripts. Entrez Protein is a much larger database containing sequences from multiple sources, including RefSeq and UniProtKB/SwissProt protein sequences, but also translations of the GenBank transcripts and records from other sources.
UniProtKB
The UniProt Knowledgebase (UniProtKB) is an extensive effort to collect all sources of functional information on proteins. In addition to providing the database of protein sequences for each organism, it aims to supplement each sequence with rich annotation. This includes biological ontologies such as Gene Ontology, sequence classifications and annotation of the secondary structure, cross-references to other resources and databases (protein interaction data, biological pathways, etc.). It also uses a classification scheme to indicate five degree of evidence supporting each protein entry (1: evidence at protein level; 2: evidence at transcript level; 3: inferred from homology; 4: predicted; 5: uncertain). The database itself consists of two parts. The UniProtKB/Swiss-Prot subset contains manually-annotated records, whereas UniProtKB/TrEMBL subset contains automatically annotated records awaiting manual analysis. In generating the Swiss-Prot database, protein sequences arising from the same gene are merged into a single UniProtKB/Swiss-Prot entry (‘canonical sequence’). The extended database that includes, in addition to Swiss-Prot and TrEMBL, manually reviewed isoform sequences (e.g. alternative splice forms, polymorphisms, or sequence conflicts) can be also be downloaded in FASTA format.
A problem with this assumption is that many peptides are not present in a particular reference protein sequence database, or any reference database. Peptides may contain mutations and may represent novel protein-coding loci and alternative splice forms. One strategy to account for peptides with mutations is to use sequence tag-based database searching, where several short peptide sequence tags are extracted from the spectrum, and the list of candidate peptides is restricted to those peptides only that contain one of the extracted sequence tags 5. This allows for mutations in the sequences of candidate database peptides. Another strategy is to perform de novo sequencing 6, but this approach is computationally inefficient and error prone for large-scale studies.
An alternative, more comprehensive approach to identify novel peptides is termed proteogenomics. The term was first introduced in the literature in 2004 7, and was initially used to describe studies in which proteomic data is used for improved genome annotation and characterization of the protein-coding potential. The term has since been broadened to include any type of application where a proteogenomics-like approach is used to interpret MS/MS spectra. In a proteogenomics approach, novel peptides are identified by searching MS/MS spectra against customized protein sequence databases containing predicted novel protein sequences and sequence variants; such databases are generated using genomic and transcriptomic sequence information. Proteogenomics therefore not only provides protein-level validation of gene expression and gene model refinement, but also enables the improvement of protein sequence databases (Figure 2).
Figure 2. The concept of proteogenomics.
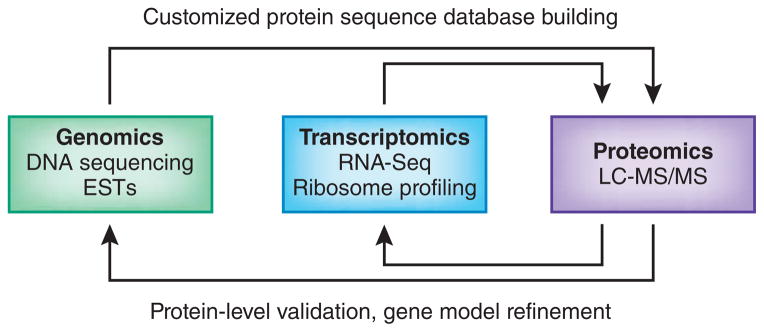
In a proteogenomics approach, genomics (DNA sequencing, expressed sequence tags (ESTs) and transcriptomics (RNA-Seq, ribosome profiling) data is used to generate customized protein sequence databases to help interpret proteomics (LC-MS/MS) data. In turn, the proteomics data provides protein-level validation of the gene expression data, as well as helping to refine gene models. The enhanced gene models can help improve protein sequence databases for traditional proteomics analysis.
In recent years, the pace of proteogenomics research has greatly accelerated. Substantial improvements in the depth and throughput of mass spectrometry-based proteomics technologies have been achieved. The development of proteomics data repositories (Box 2) has also improved access to published large-scale proteomic datasets. Additionally, next generation sequencing technologies have dramatically changed the genome characterization landscape. As a result, proteogenomics is being increasingly applied to organisms with previously unsequenced or partially sequenced genomes – organisms for which proteomic and next generation DNA sequencing data can now be acquired in rapid and cost-effective manner. For many organisms, especially human and model organisms, a tremendous amount of the next generation transcriptome sequencing (RNA-Seq) 8 data is now available in the public domain. More recently, RNA-Seq technology has been extended to global analysis of translational products (ribosome profiling) 9. These data suggest the presence of thousands of novel transcripts on top of the reference transcripts defined by the ongoing genome annotation efforts such as RefSeq or GENCODE (Ensembl). Publicly available proteomic data may be mined to obtain protein-level evidence of expression of the novel transcripts nominated by genomics and transcriptomics technologies 10. Furthermore, as generation of both transcriptomic and proteomic data in parallel is becoming increasingly common, there is an emerging trend of identifying peptides and proteins using proteomic data by matching MS/MS spectra against sample-specific protein sequence databases generated with the help of RNA-Seq (and/or ribosome profiling data) from the same samples11–16.
Box 2. Mass spectrometry data repositories.
An increasing number of proteomic datasets are now available in public repositories such as PRIDE (http://www.ebi.ac.uk/pride/archive/) and PeptideAtlas (www.peptideatlas.org), the two major repositories within the larger ProteomeXchange consortium (proteomexchange.org). Other existing data repositories include Proteomics DB (https://www.proteomicsdb.org), MassIVE (proteomics.ucsd.edu/ProteoSAFe) which includes data rescued after the collapse of the previously commonly used Tranche data repository, and Chorus (chorusproject.org). CPTAC data is released through a dedicated data portal (https://cptac-data-portal.georgetown.edu/cptacPublic/). PeptideAtlas and also GPMdb (thegpm.org) provide results of a uniform re-analyzes of raw data submitted to proteomics data repositories, including data generated as part of the cHPP project (http://www.peptideatlas.org/hupo/c-hpp/). While GPMdb itself does not store raw data, it serves as a useful resource for identifying relevant datasets available in the public domain.
Proteogenomic approaches have been brought into the spotlight with recent large-scale human proteome studies reporting high numbers of identification of novel peptides and peptide variants 17–19. At the same time, there is a growing concern that the scale of these studies challenges our ability to accurately process the data and to estimate false discovery rates (FDR), especially for novel peptides. It is therefore an opportune time to review the current state of proteogenomics, including the computational strategies and error rate estimation methods that are central to this area of research.
Proteogenomics technology
Type of peptides identified in proteogenomics
Different classes of peptides identified in proteogenomics map to different locations on the genome (Figure 3). Peptides can be classified into intergenic (i.e. mapping to regions located between annotated gene models) or intragenic (mapping to genomic regions contained within or in close proximity to an annotated gene model). Intragenic peptides can be further categorized based on the annotation of the corresponding gene model (e.g. ‘protein-coding gene’, ‘long noncoding RNA (lncRNA) gene’, and ‘pseudogene’ 20 when using Ensembl (GENCODE) as reference; see Box 1). The majority of peptides identified in proteogenomics studies (at least for commonly studied, well annotated organisms) are known peptides that map to a protein coding gene. In eukaryotes (with an intron-exon structure of genes), most of these peptides are localized within an exon, and the remaining peptides - typically less than twenty percent - span an exon-exon junction. Novel peptides not found in any reference protein database include those that identify previously undiscovered protein-coding loci (intergenic peptides) and variant peptides (e.g. single amino acid variants, SAVs). Depending on the organism, these may also include peptides mapping to untranslated regions (3′ or 5′ UTR) or introns, peptides spanning the boundary between the coding region and the neighboring intron region (exon extensions), peptides spanning un-annotated (alternative) splice junctions, and out of frame peptides. Novel peptides may also provide evidence of protein expression for chimeric transcripts, transcripts thought to be non-coding RNAs, gene fusions, and RNA editing events, although such events are expected to be rare in proteomic datasets.
Figure 3. Type of peptides identified in proteogenomics.

Peptides identified by searching customized protein sequence databases are mapped on the genome. Intergenic peptides map to regions located between annotated gene models, whereas intragenic peptides map to genomic regions contained within or in close proximity to an annotated gene model. Intragenic peptides can be further categorized based on the annotation of the corresponding gene model (e.g. ‘protein-coding gene’, ‘long noncoding RNA (lncRNA) gene’, and ‘pseudogene’). The majority of peptides map to a protein coding gene, and can be divided into Exon and exon-exon junction (Junction) peptides. Novel peptides include peptides mapping to untranslated regions (3′ or 5′ UTR peptides) or Intron peptides, peptides spanning the boundary between the coding sequence region and the neighboring UTR or intron region (Exon boundary), peptides spanning un-annotated (alternative) splice junctions (Alt junction), and out of frame peptides (Alt frame).
Generation of customized protein sequence databases
The key step in proteogenomics is peptide identification by matching acquired MS/MS spectra against a customized protein sequence database. Here we describe in more detail different strategies and data sources used to generate such databases. The final database is typically created by combining predicted protein sequences with an equal number of decoy sequences for subsequent FDR analysis, then appending known sequences (i.e. a reference protein sequence database) and the corresponding set of decoys. As a note of caution, proteogenomics users need to balance database comprehensiveness with the increased search time and elevated FDR that comes by searching larger databases (see below). The optimal choice is dependent on the goals of the experiments, and more specifically on the type(s) of novel peptides the study seeks to identify.
Six-frame translation of the genome
Predicted protein sequences can be generated using six-frame translation of the genomic sequence 21–25 using e.g., the getorf program in the EMBOSS package. Limitations of this strategy are the extremely large size of the resulting database (consisting of mostly artificially created non-existing protein sequences) and failure to capture exon-exon junction peptides (in eukaryotes). For example, direct translation of the human genome (UCSC v 19) results in a ~ 3.2 Gb protein sequence database 23, a 70 fold increase compared to ~ 45 Mb size of the corresponding Ensembl reference protein sequence database. Several computational strategies can be used to eliminate less likely sequences, including selection of the most likely frame based on homology to known coding sequences, using predictions of the coding potential, and possibly excluding translated sequence that are shorter than a certain minimum length (e.g. 30 amino acids) 26.
Ab initio gene prediction
Instead of direct six-frame translation of the genome, protein coding regions can be identified with the help of ab initio gene prediction algorithms, e.g. Augustus or Gene-ID, as was done in e.g. 25, 27, 28. Empirical information such as cDNA sequence data can be utilized as part of the gene prediction process as well 25 (for a review of gene prediction algorithms see 29). One advantage of using predicted exons is the knowledge of the reading frame. Identification of exon-exon junction peptides (including novel junction peptides) is also possible by generating theoretical junction peptides connecting all predicted exon sequences within a gene 27. The resulting protein sequence databases can still be very large, e.g. a 10 fold increase (selecting the gene prediction parameters allowing for maximum sensitivity) over the size of a typical reference protein sequence database 27. With the knowledge of the genomic coordinates of predicted exons, the computational efficiency of the peptide identification process can be improved by creating a compact representation of all possible splice junctions using the exon splice graph approach 25, 28, 30. This process merges transcripts with shared sequence, so that every predicted exon appears only once in the graph.
EST data
Protein sequences can be predicted using six-frame translation of EST data, which provides experimental evidence of transcription, including information about intron-exon structure and splicing. While EST data is already used as part of gene annotation pipelines, it can be re-analyzed independently to predict a larger set of protein sequences 28, 31–33. Compared to the ab initio gene prediction strategy, ESTs provide a more direct way to generate peptide sequence candidates, including novel junction peptides and SAVs. The drawback is again a substantial increase in the size of the resulting database (~300 times the size of the reference protein sequence databases in human 32) due to unknown translation frame and the number of accumulated ESTs (e.g. almost nine million sequences in the human dbEST database at this time). For more efficient computational analysis, ESTs can be processed to generate a compressed protein sequence database, effectively eliminating most of the redundant sequences 32. Several additional filtering steps can be applied such as requiring that the ESTs map to the vicinity of a known gene, keeping only translated peptide sequence of a certain minimum length, and requiring that all peptide sequences are confirmed by multiple ESTs (to minimize sequencing errors). ESTs can also be clustered to generate a set of putative exons and introns, followed by generation of a compact protein sequence database using the splice graph approach 28.
Annotated RNA transcripts
Protein sequences can be generated using three-frame translation of annotated RNA transcripts from e.g. Ensembl (GENCODE) or RefSeq, i.e. going beyond the annotated coding sequence and translation frame. This allows identification of alternative translation initiation sites (TIS) and out-of-frame peptides, but without the sequence space explosion typical of other strategies. For example, translation of the human GENCODE v 7 annotated transcripts (mRNA of 84,408 annotated protein sequences), results in a ~200 MB database size 23, i.e. only 4.5 fold increase compared to the size of the corresponding reference protein sequence database. Customized protein sequence databases generated this way can include translations of RNA transcripts annotated as pseudogenes or lncRNAs 34. Given the explicit knowledge of the exon-intron structure, mRNA transcript annotations can be used to generate theoretical peptides corresponding to all combinatorial exon-exon junction possibilities.
RNA-Seq data
Customized protein sequence databases can be generated based on transcript information from RNA-Seq data. Transcript reconstruction using RNA-Seq data starts with read mapping, i.e. alignment of short reads to the reference genome using e.g. a popular Bowtie/TopHat combination or ultra-fast aligners such as STAR (reviewed in 35). The splice junctions reported by the aligner (keeping junctions supported by a certain minimum number of reads only) can be extended into the exon regions on both sides of the junction and then translated to generate a comprehensive database of splice junction peptide sequences 11, 13. Full transcriptome reconstruction (assembly) can be achieved using e.g. Cufflinks (for a review of transcriptome assemblers see 36). Genome-guided assembly approach is recommended for organisms with referenced genomes, whereas genome-independent (de novo) strategy can be applied to any organism but requires more advanced expertise and bioinformatics infrastructure (evaluated for proteogenomics applications in 37). Reconstructed transcripts are aligned using BLAST or compared using genomic coordinates to the reference transcripts to remove redundant sequence, and additionally filtered requiring a certain minimum level of abundance (estimated using mapped read counts). Remaining transcript are translated, again optionally keeping predicted protein sequences of a certain minimum length 15. Ribosome profiling data can be used in essentially the same way 12. The process can be automated using several recently described bioinformatics tools. Using Galaxy-P system the users can convert input RNA-Seq data into three types of protein sequence databases suitable for proteogenomics analysis: databases containing novel single amino acid polymorphisms, databases containing novel splice junction sequences, and reduced databases only containing proteins corresponding to transcripts above a certain minimum level of expression 38. CustomProDB 39 performs similar tasks and also incorporates variant sequences extracted from public databases (see below). Protein sequence databases can also be generated from large scale RNA-Seq data aggregated from multiple studies using the splice graph approach 25, 40.
Variant sequences
Protein sequences in a reference database can be extended to include protein changing variants (SAVs, but also single amino acid deletions and insertions) catalogued in various public resources. For each variant, the reference sequence is modified accordingly and a larger sequence region covering the variant site is added as an independent entry to the customized database 41. SAVs can be downloaded from the NCBI dbSNP database, and supplemented with known disease mutations from the Online Mendelian Inheritance in Man (OMIM) and the Protein Mutant Database (PMD) 41. When building customized databases from RNA-Seq data, customProDB 35 can combine SAVs and short insertion and deletions (identified from RNA-Seq) with known SAVs extracted from the dbSNP database. RNA editing events can be detected via the bioinformatics comparison of RNA and DNA data from the same samples using publicly available tools 42.
Other specialized databases
Reference RNA transcripts can also be supplemented with predicted transcripts from more specialized databases. These include: the ECgene database, which applies less stringent procedures for construction of gene models and transcript assembly to encompass a larger number of alternative splicing events (see e.g. 43); the Pseudogene Database (http://pseudogene.org/), the non-coding RNA sequence database NONCODE 44 (used in 17) or the Broad Institute collection of lincRNAs and transcripts of unknown coding potential (TUCPs) 45 (used in 18). The ChiTaRS database of chimeric RNAs transcripts 46 represents read-through events and gene fusions identified in the literature and using computational analysis of ESTs and RNA-Seq data (used in 47).
Peptide identification using customized protein sequence databases
In proteogenomics, and in proteomics in general, successful peptide identification depends on the completeness of the protein sequence database, the sensitivity and specificity (error rates) associated with a particular peptide identification strategy, the computational time and resources necessary for processing the data, and the ability to interpret the resulting findings in a biological context. Many of these issues I have reviewed previously 3; below, I focus on these aspects from a proteogenomics perspective.
Effect of the database size
The ability to identify the correct peptide sequence that generated an experimental MS/MS spectrum using the database search approach depends on multiple factors. First, it requires that the peptide sequence is present in the searched protein sequence database. However, the more candidates there are to be scored against an experimental MS/MS spectrum, the higher the likelihood of the best scoring match to the spectrum to be incorrect, and also the more difficult it becomes to distinguish between true and false identifications 3. As a result, while searching MS/MS spectra against large proteogenomic databases may result in a (relatively small) number of novel peptide identifications, the total number of identified peptides may drop substantially compared to conventional reference sequence database searching 26, 48 (e.g. 30% or higher when using six-frame genome translation 26). Searching larger databases also increases the computational time, and requires additional modifications to common data analysis workflows, e.g. splitting the searched database into multiple chunks 24. Thus, a key consideration in proteogenomics is the selection of the most optimal strategy for generating the custom sequence database, i.e. finding the right balance between the completeness of the database and its size.
Strategies for improving the sensitivity of peptide identification
Strategies known in proteomics to increase the number of identified peptides include application of multiple database search tools to the same dataset 49 and post-database search re-scoring of peptide identifications via combining multiple sources of information using machine learning techniques 3. One complementary strategy to reduce search space is to fractionate peptides prior to LC-MS/MS analysis based on a certain physico-chemical or sequence property of the peptides. For example, using isoelectric focusing (IEF), MS/MS spectra from a particular IEF fraction can be scored only against candidate peptides having a predicted pI value expected for that fraction (pI-restricted database search) 50.
Improved sensitivity of peptide identification in proteogenomics can also be achieved using a multi-stage data analysis strategy. This type of analysis can start by searching a reference protein sequence database to interpret a majority of MS/MS spectra, and then proceed to searching larger databases to interpret the MS/MS spectra that remain unidentified after the initial search 33, 51, 52. In this manner, the results of the initial search are used to refine the customized database used in subsequent searches. For example, the most likely frame of translation can be inferred with the help of high scoring peptides identified in the initial search 26. Similarly, the search for alternative TIS can be restricted to genomic regions containing a protein coding gene identified by high confidence known peptides; a novel alternative splice junction can be considered only if both corresponding exons are supported by high scoring exon mapping peptides.
Estimating identification confidence
Accurate statistical assessment of the identification confidence for different classes of peptides is a crucial step in proteogenomics. As with proteomics in general, to prevent accumulation of error rates when going from peptide to spectrum match (PSM) level to unique peptide ion level 3, redundant PSMs should be collapsed and represented by the highest-scoring PSM. Identifications of the same peptide sequence from multiple peptide ions (e.g. doubly and triply charged ions) or in multiple forms (e.g. unmodified and oxidized methionine forms) should also be collapsed (as a conservation approach), or treated probabilistically 53. In multi-stage strategies, in which the searched protein sequence database is constructed based on the results of a previous search, it is imperative to generate and include in the customized database an appropriate number of decoy sequences at each stage of the analysis 3. In addition to the estimation of global error rates (FDR), it is important to estimate the confidence in each individual event (e.g. posterior probability of true peptide identification; for a discussion on this and related statistical concepts in MS based proteomics see e.g. 3). When a particular protein or, in proteogenomics, a particular ‘event’ (such as a novel coding region or a splice junction) is identified from multiple peptide ions and or multiple unique peptides, the posterior probabilities of the supporting identifications can in principle be combined to calculate the probability score for that event 53, 54. Such models, however, have not yet been tested on proteogenomics data.
Class-specific analysis and FDR estimation
When estimating the posterior probabilities for individual peptides and the specific events that they define (e.g. novel coding regions) it is necessary to take into consideration the difference in the likelihood of identifying different classes of peptides11, 50. The direct analogy in conventional proteomics data analysis is performing enzyme unconstrained MS/MS database searches (i.e. allowing non-tryptic peptides) when analyzing data generated from trypsin digested protein samples. As non-tryptic peptides are required to have stronger supporting evidence (e.g. database search scores) compared to tryptic peptides to obtain the same level of confidence 3, novel peptides identified using proteogenomics approaches should be required to have stronger evidence compared to known peptides. Further, among the novel peptides, peptides identifying very rare events (e.g. intergenic peptides suggesting the presence of novel protein-coding loci) should require stronger supporting evidence than those identifying more common events (e.g. alternative TIS for known protein coding regions). Thus, when using the target-decoy strategy for FDR estimation, the analysis should be done separately for each class of peptides (at least known vs. novel, but ideally also separately for different categories of novel peptides) to compute the class-specific FDR (Figure 4). Similarly, when using more advanced approaches involving computation of posterior probabilities using e.g. the model-based approach of PeptideProphet 3, the underlying statistical models should explicitly incorporate the peptide class. Note that in many published proteogenomics studies, including two recent large-scale studies in human 17, 18, the same database search score cutoffs were applied across all categories of peptides, known or novel. Thus, it is likely that the error rates among the novel peptides reported in these studies are substantially higher than acknowledged.
Figure 4. Statistical assessment of peptide identifications in proteogenomics.

MS/MS spectra are searched against a customized protein sequence database that includes target sequences for the organism of interest, i.e. a reference protein database and predicted protein sequences (containing novel peptides). In addition, two decoy databases (e.g. reversed sequences) of the same sizes as the target reference and predicted databases are appended to the target databases. The best database peptide match for each spectrum is selected for further analysis. Peptide identifications are classified as known or novel (for a decoy peptide the class - ‘known’ or ‘novel’ – is determined based on the class of the corresponding target sequence from which the decoy was generated). When using simple database search score based filtering, the numbers of target and decoy peptide identifications passing a certain score threshold are counted and used to estimate FDR corresponding to that threshold. FDR analysis should be done separately for known and novel peptides (class-specific FDR) due to difference in the number of known and novel sequences in the searched customized sequence database, and due to lower likelihood of correctly identifying a novel peptide than known peptide. When using more advanced methods based on computing posterior peptide probabilities, both the database search scores and the peptide class (known or novel) should be taken into consideration.
False peptide identifications of non-random nature
Incorrect peptide identifications result from two different sources: random high scoring matches of MS/MS spectra to unrelated sequences and matches to peptides homologous to the true peptides. Regardless of how the decoy database is generated (e.g. reversing or randomizing target protein sequences), false identifications of the second kind are likely to be underestimated3, especially when using large customized protein sequence databases. A common scenario is false identification of a novel peptide from an MS/MS spectrum acquired on a chemically modified, highly abundant peptide ion with a mass shift introduced by the chemical modification equaling the mass difference between the novel and the unmodified known peptide 33, 55. As a general guideline, it is advisable to compare, e.g. using BLAST, the sequence of each identified novel peptide against the sequences of all peptides in the reference database to detect and eliminate (or at least clearly mark) all identifications of novel peptides with a high degree of homology to a known sequence. When it is important to keep such peptides (e.g. when specifically searching for SAVs), it is necessary to check that the observed mass shift between the novel peptide and the closest homologue(s) in the reference database does not match the mass of one of the common chemical or post-translational modifications (e.g. oxidation, deamidation, carbamylation, acetylation, etc.) 25, 33, 41, 50, 51, 55. The sample-specific list of the most common chemical modifications for a particular biological sample can be established using ‘blind’ modification search tools 56. Furthermore, I/L substitutions cannot be distinguished using mass spectrometry, and thus such peptides should not be included in the list of identified peptide variants.
Levels of data summarization and inference of novel events
In proteomics, results are typically presented as a list of identified proteins (protein-level summary), or genes (gene-level summary), along with a list of identified unique peptides, with FDR estimated at these levels. In proteogenomics, these levels of data summarization are not sufficient and should explicitly include the type(s) of events that a particular proteogenomics study seeks to identify. For example, ‘Novel coding region’ or ‘TIS’ identification events should be provided as separate lists, in addition to the protein/gene levels of data summarization and the supporting unique peptide evidence. The same peptide sequence may arise from multiple different genomic locations (e.g. gene paralogues, or a protein-coding gene and a pseudogene). Such ‘shared’ peptides (‘multi-mapped’ in the language used in the transcriptome literature) do not provide unambiguous evidence of protein expression at a particular locus 4. Furthermore, a novel peptide mapping to single location in the genome could also have multiple (ambiguous) interpretations, e.g. as supporting one event type for one transcript of a gene and another even type for another transcript of the same gene (e.g. out of frame peptide and intronic/UTR peptide). Thus, the principle of parsimony in creating the summary lists described previously for proteomics 4, i.e. presenting multiple protein sequence database entries identified by the same peptides as indistinguishable groups, should be extended and applied to proteogenomics studies as well.
Defining novel peptides
The results of a proteogenomics analysis, and in particular the peptides reported as novel, depend on the choice of the reference database selected in that study, and even the specific version of that database. As discussed above, multiple reference protein sequence databases exist for many organisms, and these databases vary in terms of their completeness and annotation quality. Furthermore, all major reference databases are constantly updated, with new entries added and some removed with each new version. Therefore, in proteogenomics studies, peptides identified using customized protein sequence databases should be mapped to all major reference databases available for the organism under investigation and to most common sample contaminants, and protein annotations available for the closest matches in those databases should be reported as part of the final output.
Proteogenomics applications
The feasibility of various proteogenomics applications has been discussed and demonstrated in multiple studies in human and in many model organisms, including in Plasmodium falciparum 57, C. elegans 58, Drosophila melanogaster 22, Arabidopsis thaliana 21, 30, and Anopheles gambiae 59 (Supplementary Table 1). Only few of these studies, however, exhibited attributes of a directed, comprehensive proteogenomics project - a consistent and coordinated effort on the part of both genomics and proteomics groups, with a functioning feedback mechanism in which the sequences of the identified peptides are passed to (and used by) the genome annotators. The ENCODE project, and its sub-project GENCODE, made attempts to include proteomic data in their work on the improved annotation of the mouse 27 and human 23 genomes. The limited extent of these and other early proteogenomics studies could be attributed in part to a lack of sufficient amount of proteomics data, low sensitivity of the previous generations of proteomic technologies, and lack of understanding of proteomic data by the genomic community. As a result, most proteogenomics studies in human and model organisms, including recent large-scale studies 17, 18, focused on a less ambitious but nevertheless important task of providing protein-level ‘validation’, i.e. confirmation of the protein-level expression of putative gene models or sequence variants predicted from the genome sequence and often supported by transcriptional evidence.
Recent improvements in proteomics technologies, coupled with wide availability of next-generation sequencing data, have led to a resurgence of proteogenomics studies. In human and mouse, the focus of many such studies has shifted toward detection of abnormal protein variants (e.g. SAVs) across cohorts of cancer tissue samples 60, exemplified by the recent publication from the Cancer Proteomic Tumor Analysis Consortium (CPTAC) 19. Several common proteogenomics applications, and the type of new information they seek to obtain, are discussed in more detail below.
Novel protein-coding regions
The possibility of using MS/MS proteomics data for discovery of novel protein-coding regions, and refinement of gene boundaries for previously annotated ones has been discussed since the early days of proteomics 31, 61. This is most commonly achieved by searching MS/MS spectra against customized databases generated using direct six-frame genome translation, three frame translations of protein coding sequences predicted using initio gene prediction algorithms, or using six-frame translation of the transcripts reconstructed from the EST or RNA-Seq data (three-frame if strand specific RNA-Seq). The efforts to discover novel protein-coding regions are likely to be most fruitful for less studied, non-model organisms 25, 30, 62 – the organisms that have not benefitted from extensive genome annotation efforts. Even for well annotated higher eukaryote organisms, recent studies report a substantial numbers of novel identifications. For example, a deep proteome profiling study (to the depth of 13,078 human and 10,637 mouse proteins) reported the identification of 98 and 52 previously undiscovered protein-coding loci in human and mouse, respectively, using the using six-frame genome translation approach 50.
Short open reading frames and new translation initiation sites
The computational prediction of short open reading frames (sORFs), and frames which use non-AUG initiation codons, is particularly difficult 62. It has been suggested that sORFs may account for an additional 10% of the number of protein-coding elements in eukaryote genomes 63. These sORFs may be located within a genomic region of an annotated transcript, (e.g. in the 5′ UTR located upstream of a known open reading frame (uORFs) or can result from a frame-shift within the coding sequence of the ORF), or within unannotated transcripts or intergenic regions thought to be lacking protein-coding capacity. With the advent of ribosome profiling methods, strong experimental evidence for the existence of protein-coding sORFs and non-AUG translation initiation sites have emerged (reviewed in 62). While ribosome profiling and conventional RNA-Seq demonstrate the protein-coding potential of these sORFs, proteogenomics provides additional evidence for the production of a stable protein product encoded by them12, 64, 65. The search for peptides confirming sORFs and novel TIS is likely to be most fruitful with one or several additional sample preparation steps. The likelihood of identifying a peptide from a sORFs is greatly increased with fractionation of protein samples prior to LC-MS/MS analysis to enrich for low molecular weight proteins 65. Detection of novel TIS generally requires conclusive identification of N-terminal peptides which can be enriched prior to MS analysis using protein N-terminus labeling approaches (N-terminal proteomics 66).
In a recent study, N-terminal proteomics data was analyzed using a customized protein sequence database created using publicly available ribosome profiling data. The study revealed, via the identification of peptides mapping to 5′ UTR regions or to downstream in-frame AUG codons, a large number of proteins or protein isoforms with different (compared to the reference annotation) N-terminal extensions or truncations 67. It has been noted that in human and mouse proteomes up to 20% of all identified protein N-termini point to alternative TIS, incorrect assignments of the translation start codon, the use of translation initiation at near-cognate start codons, or alternative splicing events resulting in a different N terminal protein sequence 67.
Alternative splicing
Alternative splicing is a major source of cell-specific and tissue-specific protein variation in higher eukaryotes 20, 68. Most early proteogenomics studies were based on the analysis of EST data, with MS/MS spectra searched either directly against six-frame translation of the EST sequences 32, or against customized databases of alternative splice transcripts, e.g. the ECgene database constructed using the EST data with a help of gene-modeling algorithms 69. In recent years, the analysis of alternative splicing using proteomic data has been increasingly relying on the availability of RNA-Seq transcriptome data for generation of customized databases from six-frame translations of reconstructed transcripts 14, 37 and predicted splice junctions 11, 13. A recent proteogenomic analysis of the HeLa cell line using a custom splice junction database created from the sample-specific RNA-Seq data resulted in the detection of 57 novel splice junction peptides (out of 24,834 novel transcript junctions identified in RNA-Seq data), representing an array of different splicing events, including skipped exons and alternative donors and acceptors sites 13.
Sequence variants
Identification of sequence variants using genomics technologies, including disease associated variants, is a long standing area of research. As with alternative splicing, due to an overwhelming number of variants detected in the genome and transcriptome data, understanding which of those variants are functional is a challenging task 70. Detection of these variants at the protein level provides an opportunity to reduce the set of candidates for subsequent investigation of their functional role or clinical relevance. The ability to search for all possible amino-acid mutations using MS/MS data has been implemented in several commonly used database search tools (e.g. Mascot, X! Tandem), but such an approach is computationally inefficient (sequence-tag based database search strategies, also shown in Figure 1, allow minor speed improvement 5, 55). Even more important than search speed is that any strategy that considers all possible amino-acid mutations quickly loses the sensitivity due to a very large increase in the search space, and the error rates become a serious concern. Sequence variants can be identified in a more targeted way by searching against translated ESTs 32, but more commonly used approach now involves building customized databases of protein sequences explicitly incorporating known variant peptides.
In one such recent example, 81 distinct variant peptides were identified in proteomic data from three colon cancer cell lines, and 204 variants in three lung cancer tissues, by searching MS/MS spectra against a custom CanProVar database 41. As with other applications discussed above, when the same samples are profiled using both RNA-Seq and proteomics, the custom sequence database for variant peptide identification can be generated from the RNA-Seq data 14, 37, 71, 72. Using this strategy and one of the most comprehensive proteomic datasets to date, 38 and 88 nonsynonymous variants were detected at the peptide level in two different strains of rat 14. Furthermore, the identification of more than a thousand of variant peptides were reported in a recent comprehensive study using proteomics data from a large cohort of the Cancer Genome Atlas (TCGA) initiative cancer tissue samples 19. While this number is impressive, it is necessary to keep in mind the difficulty with estimating FDR for variant peptides as discussed above.
Other sources of genome variation
There are multiple other sources of genome variation of high biological significance potentially resulting in novel or variant protein-coding transcripts. These include RNA-editing, which occurs during post-translation processing and whose role and biological significance has yet to be fully understood. The extent to which these events are present in the transcriptome is being debated (reviewed in 73), but it is likely to be less than what was initially thought 74. Proteogenomics may provide valuable protein-level evidence for some of these putative RNA editing events 14. It may also provide evidence of protein expression for novel gene fusions and chimeric transcripts 47 and transcripts annotated as pseudogenes 27. The function and coding potential of transcripts annotated as long non-coding RNAs is another very active area of research 75. If expressed, these proteins are likely to be at a very low level, meaning that these events have a very low likelihood of being detected in a typical proteomic dataset 76. Thus, extra caution should always be applied with respect to FDR estimation when looking for evidence of such peptides in proteomic data.
Non-model organisms
While human and model organisms have received extensive attention regarding their genome annotation, this is clearly not the case for organisms with unsequenced or partially sequenced genomes where consistent gene annotation efforts are lacking. Thus, proteogenomics can be very impactful for non-model organisms. Until recently, the genome and cDNA sequencing data for non-model organisms were scarce. As a result, the reference protein sequence databases for these organisms were incomplete and poorly annotated. This gave rise to homology-based proteomic data analysis strategies - a combination of de novo peptide sequencing (Figure 1) and sequence similarity searching against protein databases of homologous organisms 77, 78.
More recently, with the advent of next-generation sequencing technologies, it has become possible to rapidly and cost-effectively determine the genome sequence of any species of interest. While these data can then be analyzed using computational gene annotation pipelines, automated approaches make a relatively high number of annotation errors. Importantly, in the absence of expert manual curation, especially for organisms for which related sequences are not available (e.g. many microorganisms) to allow homology-guided annotation, proteomic data often provides the only source of experimental evidence confirming the protein expression of computationally predicted gene models. The significance of proteogenomics for non-model organisms has been illustrated using microbial organisms and plants. These efforts have been recently reviewed in 79 (for an extended list of published proteogenomics efforts for a variety of organisms see Supplementary Table 1). Prokaryotes are particularly amenable to proteogenomics analysis due to their smaller genomes, single-cell organization (i.e. intraindividual and interindividual homogeneity), and lower dynamic range of their proteomes which allows generation of fairly complete proteome profiles with less effort. The benefits of performing simultaneous proteogenomics analysis of data from multiple related species, termed comparative proteogenomics, have also been discussed 80, 81.
Metaproteomics
Proteogenomics has a potential to make substantial contributions in the analysis of community samples such as microbial communities studied in environmental genomics and microbiomics 82, 83. This area of research, referred to as metaproteomics or community proteogenomics 79, 84 is concerned with untangling the interplay between many different organisms contained within the analyzed communities. At the same time, metaproteomics presents great challenges. The organisms that constitute such communities are typically poorly annotated (and thus their known reference proteomes are very incomplete), but moreover, the presence of multiple highly homologous organisms presents a challenge for conclusive protein identification and quantification 85. Several recent studies highlighted the importance of the proper generation of customized protein sequences databases for metaproteomics 85–87 using next-generation sequencing data obtained for single microorganisms, along with protein sequences parsed from RefSeq and UniProtKB at different taxonomic levels. The large size of the customized protein sequence databases typical of metaproteomics studies requires very substantial computational resources, reduces the sensitivity of peptide identification, and increases the rate of misidentifications. Furthermore, because the ability to quantitative compare the abundances of proteins from different organisms within the community samples is very important, it is necessary to apply label-free protein quantification strategies designed to accurately deal with a large number of shared peptides mapping to multiple homologous proteins in different organisms 88.
Concluding remarks
Early proteogenomics efforts were hampered by technical challenges resulting from an overall low sensitivity of proteomics technology. The last several years, however, have witnessed great improvements in MS instrumentation, including new instrument types, alternative fragmentation mechanisms, and advanced data acquisition strategies 1, 2. These developments, coupled with improvements in protein separation and enrichment methods for proteomics applications, have resulted in a substantial increase in the depth of protein detection, approaching that of global transcriptome profiling studies 1. Significant advances have also been achieved in the area of top-down proteomics – a technology that offers complementary information useful for proteogenomic characterization 89. A substantial challenge in proteogenomics has been lack of a sufficient amount of proteomic data in the public domain necessary to make a significant contribution to genome annotation efforts, in part due to the “data hoarding” mentality prevalent in the proteomics community in the early days. There has been a clear shift toward more open data sharing in proteomics, further strengthen by new requirements from funding agencies 90. As a result, an increasing number of proteomic datasets are now available in public repositories 91.
Although the depth of RNA-Seq data is still greater than that of proteomic data, transcriptome data contains elements not expected to comprise mature proteins (e.g. a large number of nonfunctional transcripts) 92. Ribosome profiling data provides evidence of translational activity, and thus can be used to identify novel transcripts that are more likely to be protein-coding. Still, these data do not provide direct evidence of expression of a stable, functional protein. Thus, despite the clear success of RNA-Seq and related technologies in uncovering the previously uncharacterized diversity of the genome, proteomic data plays a critical role in identifying functional transcripts among the many novel transcripts nominated by genomics and transcriptomics technologies. Joint analyses using multi-omics data should be particularly informative when done in close collaboration between the genomics and proteomics group, with biological experiments carefully designed to generate paired genomics/proteomics datasets.
Incorrect peptide and protein identifications have been a long standing problem in proteomics 3. In the early days, many datasets with very high FDR were published, prompting the calls for establishing robust data analysis and publication guidelines 93. Proteogenomics presents additional challenges that are not yet fully acknowledged. I have highlighted the most significant sources of false discoveries in proteogenomics, including application of the same filtering thresholds to both known and novel peptides, incorrect identification of novel peptides highly homologous to known sequences, and making unsupported conclusions based on shared peptides. Future efforts should focus on establishing data analysis guidelines for proteogenomic studies, extending some general guidelines I present here (Box 3).
Box 3. Minimum guidelines for proteogenomics studies.
Here, I suggest some guidelines for reporting novel peptides identified in a proteogenomics analysis.
Customized protein sequence databases used to identify novel peptides should be made available upon publication.
Peptides identified using customized protein database should be queried against all major reference databases available for the organism of interest (e.g. RefSeq, UniProtKB, and Ensembl, and also common sample contaminants). For each peptide reported as novel, the closest reference peptide sequence(s) should be listed, along with the accession numbers of the corresponding proteins.
FDR estimation procedure applied to novel peptides, and how it is different than that applied to known peptides, should be clearly described. To the degree possible, different categories of novel peptides should be analyzed separately.
When reporting novel peptides homologous to a reference sequence, efforts taken to eliminate the most likely sources of false positives (e.g. common post-translational and chemical modifications, errors in mass measurements, etc.) should be described.
Peptides mapping to multiple genome locations should be clearly marked. The same peptide(s) should not be used as evidence for multiple different proteins/ protein forms.
Proteogenomics is playing a central role in two ongoing large-scale initiatives. The community-driven, chromosome-centric Human Proteome Project (cHPP) has a broad goal of characterizing the parts list of the human proteome 94, whereas the NIH funded CPTAC 95 project specifically aims to improve the understanding of the molecular basis of cancer via proteomics characterization of common cancer specimens obtained though the TCGA initiative. These initiatives provide not only rich proteomic datasets for proteogenomic analysis, but also present an opportunity for the development of advanced data integration and modeling strategies across the entire spectrum of omics data. There is a need for making proteomics data in general, and the results of proteogenomics analyses in particular, more accessible and useful to a broader scientific community. A good start would be the development of a computational infrastructure for querying specific novel peptides of interest to a particular laboratory across a large collection of publicly available proteomic data, including cHPP and CPTAC data, with a goal to obtain and visualize protein-level evidence of their expression.
Looking at proteogenomics in a broader context, questions remain as to what fraction of novel alternative splice forms are translated into stable functional proteins vs. those that are prone to nonsense mediated decay or protein degradation immediately following translation. Further analysis can help identify the differences between confirmed (at the protein level) and unconfirmed splice forms in terms of their secondary structure and sequence properties 96, 97. Recent studies also suggest that SAVs could affect protein stability 14, possibly explaining the lower than expected rate of detection of such variants in proteomic data 41. Furthermore, somatic variants have been found to have reduced protein abundance compared to germline variants 19. These and other recent studies 98–100 involving quantitative analysis of transcripts and protein expression data and integration with DNA variation provide valuable insights into how the proteome is regulated using genetic effects. In summary, there is every indication that the field of proteogenomics will remain an active area of research for the foreseeable future.
Supplementary Material
Acknowledgments
This work has been funded in part with US National Institute of Health grant R01-GM-094231. The author would like to acknowledge Andy Kong, Brendan Veeneman, Avinash Kumar, and Gil Omenn for useful discussions.
Footnotes
Competing Financial Interests
The author declares no competing financial interests.
References
- 1.Mann M, Kulak NA, Nagaraj N, Cox J. The coming age of complete, accurate, and ubiquitous proteomes. Mol Cell. 2013;49:583–590. doi: 10.1016/j.molcel.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 2.Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–965. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 3.Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. Journal of proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data - The protein inference problem. Molecular & Cellular Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Dasari S, et al. TagRecon: High-throughput mutation identification through sequence tagging. Journal of Proteome Research. 2010;9:1716–1726. doi: 10.1021/pr900850m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma B, Johnson R. De novo sequencing and homology searching. Mol Cell Proteomics. 2012;11:O111 014902. doi: 10.1074/mcp.O111.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaffe JD, Berg HC, Church GM. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics. 2004;4:59–77. doi: 10.1002/pmic.200300511. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15:205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- 10.Desiere F, et al. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biology. 2005;6:R9. doi: 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ning K, Nesvizhskii AI. The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: a preliminary assessment. Bmc Bioinformatics. 2010;11 (Suppl 11):S14. doi: 10.1186/1471-2105-11-S11-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menschaert G, et al. Deep proteome coverage based on ribosome profiling aids MS-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Molecular & Cellular Proteomics. 2013;12:1780–1790. doi: 10.1074/mcp.M113.027540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheynkman GM, Shortreed MR, Frey BL, Smith LM. Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Molecular & Cellular Proteomics. 2013;12:2341–2353. doi: 10.1074/mcp.O113.028142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low TY, et al. Quantitative and qualitative proteome characteristics extracted from in-depth integrated genomics and proteomics analysis. Cell Reports. 2013;5:1469–1478. doi: 10.1016/j.celrep.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 15.Wu P, et al. Discovery of Novel Genes and Gene Isoforms by Integrating Transcriptomic and Proteomic Profiling from Mouse Liver. Journal of Proteome Research. 2014;13:2409–2419. doi: 10.1021/pr4012206. [DOI] [PubMed] [Google Scholar]
- 16.Omasits U, et al. Directed shotgun proteomics guided by saturated RNA-seq identifies a complete expressed prokaryotic proteome. Genome Research. 2013;23:1916–1927. doi: 10.1101/gr.151035.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim MS, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilhelm M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrow J, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baerenfaller K, et al. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science. 2008;320:938–941. doi: 10.1126/science.1157956. [DOI] [PubMed] [Google Scholar]
- 22.Brunner E, et al. A high-quality catalog of the Drosophila melanogaster proteome. Nature Biotechnology. 2007;25:576–583. doi: 10.1038/nbt1300. [DOI] [PubMed] [Google Scholar]
- 23.Khatun J, et al. Whole human genome proteogenomic mapping for ENCODE cell line data: identifying protein-coding regions. BMC Genomics. 2013;14:141. doi: 10.1186/1471-2164-14-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fermin D, et al. Novel gene and gene model detection using a whole genome open reading frame analysis in proteomics. Genome Biology. 2006;7:R35. doi: 10.1186/gb-2006-7-4-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castellana NE, et al. An automated proteogenomic method uses mass spectrometry to reveal novel genes in Zea mays. Molecular & Cellular Proteomics. 2014;13:157–167. doi: 10.1074/mcp.M113.031260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blakeley P, Overton IM, Hubbard SJ. Addressing statistical biases in nucleotide-derived protein databases for proteogenomic search strategies. Journal of Proteome Research. 2012;11:5221–5234. doi: 10.1021/pr300411q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brosch M, et al. Shotgun proteomics aids discovery of novel protein-coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome. Genome Research. 2011;21:756–767. doi: 10.1101/gr.114272.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanner S, et al. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007;17:231–239. doi: 10.1101/gr.5646507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brent MR. Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat Rev Genet. 2008;9:62–73. doi: 10.1038/nrg2220. [DOI] [PubMed] [Google Scholar]
- 30.Castellana NE, et al. Discovery and revision of Arabidopsis genes by proteogenomics. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:21034–21038. doi: 10.1073/pnas.0811066106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choudhary JS, Blackstock WP, Creasy DM, Cottrell JS. Interrogating the human genome using uninterpreted mass spectrometry data. Proteomics. 2001;1:651–667. doi: 10.1002/1615-9861(200104)1:5<651::AID-PROT651>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 32.Edwards NJ. Novel peptide identification from tandem mass spectra using ESTs and sequence database compression. Molecular Systems Biology. 2007;3:102. doi: 10.1038/msb4100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesvizhskii AI, et al. Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data - Toward more efficient identification of post-translational modifications, sequence polymorphisms, and novel peptides. Molecular & Cellular Proteomics. 2006;5:652–670. doi: 10.1074/mcp.M500319-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Research. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engstrom PG, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. 2013;10:1185–1191. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steijger T, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10:1177–1184. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans VC, et al. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nature Methods. 2012;9:1207–U1111. doi: 10.1038/nmeth.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheynkman G, et al. Using Galaxy-P to leverage RNA-Seq for the discovery of novel protein variations. Bmc Genomics. 2014;15:703. doi: 10.1186/1471-2164-15-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Zhang B. customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics. 2013;29:3235–3237. doi: 10.1093/bioinformatics/btt543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woo S, et al. Proteogenomic database construction driven from large scale RNA-seq data. J Proteome Res. 2014;13:21–28. doi: 10.1021/pr400294c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, et al. A bioinformatics workflow for variant peptide detection in shotgun proteomics. Molecular & Cellular Proteomics. 2011;10:M110.006536. doi: 10.1074/mcp.M110.006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Picardi E, Pesole G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics. 2013;29:1813–1814. doi: 10.1093/bioinformatics/btt287. [DOI] [PubMed] [Google Scholar]
- 43.Menon R, et al. Identification of novel alternative splice isoforms of circulating proteins in a mouse model of human pancreatic cancer. Cancer Research. 2009;69:300–309. doi: 10.1158/0008-5472.CAN-08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie C, et al. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Research. 2014;42(Database issue):D98–103. doi: 10.1093/nar/gkt1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & Development. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frenkel-Morgenstern M, et al. ChiTaRS: a database of human, mouse and fruit fly chimeric transcripts and RNA-sequencing data. Nucleic Acids Res. 2013;41:9. doi: 10.1093/nar/gks1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frenkel-Morgenstern M, et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22:1231–1242. doi: 10.1101/gr.130062.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krug K, et al. Deep coverage of the Escherichia coli proteome enables the assessment of false discovery rates in simple proteogenomic experiments. Molecular & Cellular Proteomics. 2013;12:3420–3430. doi: 10.1074/mcp.M113.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shteynberg D, Nesvizhskii AI, Moritz RL, Deutsch EW. Combining results of multiple search engines in proteomics. Molecular & Cellular Proteomics. 2013;12:2383–2393. doi: 10.1074/mcp.R113.027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Branca RM, et al. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat Methods. 2014;11:59–62. doi: 10.1038/nmeth.2732. [DOI] [PubMed] [Google Scholar]
- 51.Ning K, Fermin D, Nesvizhskii AI. Computational analysis of unassigned high-quality MS/MS spectra in proteomic data sets. Proteomics. 2010;10:2712–2718. doi: 10.1002/pmic.200900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helmy M, Sugiyama N, Tomita M, Ishihama Y. Mass spectrum sequential subtraction speeds up searching large peptide MS/MS spectra datasets against large nucleotide databases for proteogenomics. Genes to Cells. 2012;17:633–644. doi: 10.1111/j.1365-2443.2012.01615.x. [DOI] [PubMed] [Google Scholar]
- 53.Shteynberg D, et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics. 2011;10:M111 007690. doi: 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Castellana N, Bafna V. Proteogenomics to discover the full coding content of genomes: A computational perspective. Journal of proteomics. 2010;73:2124–2135. doi: 10.1016/j.jprot.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abraham P, Adams RM, Tuskan GA, Hettich RL. Moving away from the reference genome: evaluating a peptide sequencing tagging approach for single amino acid polymorphism identifications in the Genus Populus. Journal of Proteome Research. 2013;12:3642–3651. doi: 10.1021/pr400192r. [DOI] [PubMed] [Google Scholar]
- 56.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Identification of post-translational modifications by blind search of mass spectra. Nat Biotechnol. 2005;23:1562–1567. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 57.Lasonder E, et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002;419:537–542. doi: 10.1038/nature01111. [DOI] [PubMed] [Google Scholar]
- 58.Merrihew GE, et al. Use of shotgun proteomics for the identification, confirmation, and correction of C. elegans gene annotations. Genome Research. 2008;18:1660–1669. doi: 10.1101/gr.077644.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaerkady R, et al. A proteogenomic analysis of Anopheles gambiae using high-resolution Fourier transform mass spectrometry. Genome Research. 2011;21:1872–1881. doi: 10.1101/gr.127951.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kislinger A, Boutros T. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods. 2014;11:XX–XX. doi: 10.1038/nmeth.3138. [DOI] [PubMed] [Google Scholar]
- 61.Kuster B, Mortensen P, Andersen JS, Mann M. Mass spectrometry allows direct identification of proteins in large genomes. Proteomics. 2001;1:641–650. doi: 10.1002/1615-9861(200104)1:5<641::AID-PROT641>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 62.Yang XH, et al. Discovery and annotation of small proteins using genomics, proteomics, and computational approaches. Genome Research. 2011;21:634–641. doi: 10.1101/gr.109280.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frith MC, et al. The abundance of short proteins in the mammalian proteome. PLoS Genet. 2006;2:e52. doi: 10.1371/journal.pgen.0020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oyama M, et al. Diversity of translation start sites may define increased complexity of the human short ORFeome. Molecular & Cellular Proteomics. 2007;6:1000–1006. doi: 10.1074/mcp.M600297-MCP200. [DOI] [PubMed] [Google Scholar]
- 65.Slavoff SA, et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 2013;9:59-+. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hartmann EM, Armengaud J. N-terminomics and proteogenomics, getting off to a good start. Proteomics. 2014 doi: 10.1002/pmic.201400157. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 67.Van Damme P, Gawron D, Van Criekinge W, Menschaert G. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Molecular & Cellular Proteomics. 2014;13:1245–1261. doi: 10.1074/mcp.M113.036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Menon R, Omenn GS. In: Data Mining in Proteomics: From Standards to Applications. Hamacher M, Eisenacher M, Stephan C, editors. Vol. 696. 2011. pp. 319–326. [Google Scholar]
- 70.Stunnenberg HG, Hubner NC. Genomics meets proteomics: identifying the culprits in disease. Hum Genet. 2014;133:689–700. doi: 10.1007/s00439-013-1376-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheynkman GM, Shortreed MR, Frey BL, Scalf M, Smith LM. Large-scale mass spectrometric detection of variant peptides resulting from nonsynonymous nucleotide differences. Journal of Proteome Research. 2013;13:228–240. doi: 10.1021/pr4009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X, et al. Protein identification using customized protein sequence databases derived from RNA-Seq data. J Proteome Res. 2012;11:1009–1017. doi: 10.1021/pr200766z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stepanova VV, Gelfand MS. RNA editing: Classical cases and outlook of new technologies. Molecular Biology. 2014;48:11–15. [PubMed] [Google Scholar]
- 74.Li MY, et al. Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2011;333:53–58. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–251. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Banfai B, et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Research. 2012;22:1646–1657. doi: 10.1101/gr.134767.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Junqueira M, et al. Protein identification pipeline for the homology-driven proteomics. Journal of proteomics. 2008;71:346–356. doi: 10.1016/j.jprot.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Renard BY, et al. Overcoming species boundaries in peptide identification with Bayesian information criterion-driven error-tolerant peptide search (BICEPS) Mol Cell Proteomics. 2012;11:M111 014167. doi: 10.1074/mcp.M111.014167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Armengaud J, et al. Non-model organisms, a species endangered by proteogenomics. J Proteomics. 2014;105:5–18. doi: 10.1016/j.jprot.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 80.Gupta N, et al. Comparative proteogenomics: Combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Research. 2008;18:1133–1142. doi: 10.1101/gr.074344.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tovchigrechko A, Venepally P, Payne SH. PGP: parallel prokaryotic proteogenomics pipeline for MPI clusters, high-throughput batch clusters and multicore workstations. Bioinformatics. 2014;30:1469–1470. doi: 10.1093/bioinformatics/btu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lo I, et al. Strain-resolved community proteomics reveals recombining genomes of acidophilic bacteria. Nature. 2007;446:537–541. doi: 10.1038/nature05624. [DOI] [PubMed] [Google Scholar]
- 83.Delmotte N, et al. Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proceedings of the National Academy of Sciences. 2009;106:16428–16433. doi: 10.1073/pnas.0905240106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seifert J, et al. Bioinformatic progress and applications in metaproteogenomics for bridging the gap between genomic sequences and metabolic functions in microbial communities. Proteomics. 2013;13:2786–2804. doi: 10.1002/pmic.201200566. [DOI] [PubMed] [Google Scholar]
- 85.Muth T, Benndorf D, Reichl U, Rapp E, Martens L. Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Mol Biosyst. 2013;9:578–585. doi: 10.1039/c2mb25415h. [DOI] [PubMed] [Google Scholar]
- 86.Tanca A, et al. Evaluating the impact of different sequence databases on metaproteome analysis: insights from a lab-assembled microbial mixture. Plos One. 2013;8:e82981. doi: 10.1371/journal.pone.0082981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Souza GA, et al. Proteogenomic analysis of polymorphisms and gene annotation divergences in prokaryotes using a clustered mass spectrometry-friendly database. Mol Cell Proteomics. 2011;10:M110 002527. doi: 10.1074/mcp.M110.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Penzlin A, et al. Pipasic: similarity and expression correction for strain-level identification and quantification in metaproteomics. Bioinformatics. 2014;30:i149–i156. doi: 10.1093/bioinformatics/btu267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Albright JC, Goering AW, Doroghazi JR, Metcalf WW, Kelleher NL. Strain-specific proteogenomics accelerates the discovery of natural products via their biosynthetic pathways. J Ind Microbiol Biotechnol. 2014;41:451–459. doi: 10.1007/s10295-013-1373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rodriguez H, et al. Recommendations from the 2008 International Summit on Proteomics Data Release and Sharing Policy: the Amsterdam principles. J Proteome Res. 2009;8:3689–3692. doi: 10.1021/pr900023z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vizcaino JA, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mudge JM, Frankish A, Harrow J. Functional transcriptomics in the post-ENCODE era. Genome Res. 2013;23:1961–1973. doi: 10.1101/gr.161315.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carr S, et al. The need for guidelines in publication of peptide and protein identification data - Working group on publication guidelines for peptide and protein identification data. Molecular & Cellular Proteomics. 2004;3:531–533. doi: 10.1074/mcp.T400006-MCP200. [DOI] [PubMed] [Google Scholar]
- 94.Omenn GS. The strategy, organization, and progress of the HUPO Human Proteome Project. J Proteomics. 2014;100:3–7. doi: 10.1016/j.jprot.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ellis MJ, et al. Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013;3:1108–1112. doi: 10.1158/2159-8290.CD-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ezkurdia I, et al. Comparative proteomics reveals a significant bias toward alternative protein isoforms with conserved structure and function. Molecular Biology and Evolution. 2012;29:2265–2283. doi: 10.1093/molbev/mss100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Leoni G, Le Pera L, Ferre F, Raimondo D, Tramontano A. Coding potential of the products of alternative splicing in human. Genome Biology. 2011;12:R9. doi: 10.1186/gb-2011-12-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu L, et al. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Albert FW, Treusch S, Shockley AH, Bloom JS, Kruglyak L. Genetics of single-cell protein abundance variation in large yeast populations. Nature. 2014;506:494–497. doi: 10.1038/nature12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Picotti P, et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013;494:266–270. doi: 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.