

Abstract
Background
Shb is a signaling protein downstream of vascular endothelial growth factor receptor-2 and Shb deficiency has been found to restrict tumor angiogenesis. The present study was performed in order to assess metastasis in Shb deficiency using B16F10 melanoma cells.
Methods
B16F10 melanoma cells were inoculated subcutaneously on wild type or Shb +/− mice. Primary tumors were resected and lung metastasis determined after tumor relapse. Lung metastasis was also assessed after bone marrow transplantation of wild type bone marrow to Shb +/− recipients and Shb +/− bone marrow to wild type recipients. Primary tumors were subject to immunofluorescence staining for CD31, VE-cadherin, desmin and CD8, RNA isolation and isolation of vascular fragments for further RNA isolation. RNA was used for real-time RT-PCR and microarray analysis.
Results
Numbers of lung metastases were increased in Shb +/− or −/− mice and this coincided with reduced pericyte coverage and increased vascular permeability. Gene expression profiling of vascular fragments isolated from primary tumors and total tumor RNA revealed decreased expression of different markers for cytotoxic T cells in tumors grown on Shb +/− mice, suggesting that vascular aberrations caused altered immune responses.
Conclusions
It is concluded that a unique combinatorial response of increased vascular permeability and reduced recruitment of cytotoxic CD8+ cells occurs as a consequence of Shb deficiency in B16F10 melanomas. These changes may promote tumor cell intravasation and metastasis.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-015-1269-y) contains supplementary material, which is available to authorized users.
Keywords: Shb, Melanoma, Vascular permeability, Metastasis, CD8+ cells, Pericytes
Background
Angiogenesis inhibition [1] has become a clinically accepted treatment of numerous malignant diseases (www.cancer.gov/cancertopics/factsheet/Therapy/angiogenesis-inhibitors). The primary targets of anti-angiogenic therapy so far have been the main angiogenic factor VEGF (vascular endothelial growth factor) or its receptor VEGFR-2 [2,3]. Despite these advances, this therapeutic regimen is clearly not as straightforward as initially thought and certain reports suggest that some tumors may become more aggressive showing increased invasiveness and metastatic spread as a consequence of anti-angiogenic treatment [4,5].
SHB (Src homology domain containing protein B) is an adaptor protein [6] operating downstream of VEGFR-2 [7], the receptor active in VEGF’s angiogenic response [8]. The Shb knockout mouse phenotype was found to be pleiotropic with aberrations in female reproduction [9,10], glucose homeostasis [11], the T lymphocyte response to T cell receptor stimulation [12,13] and the vasculature [14-16]. In particular, the vasculature displayed reduced angiogenesis and vascular permeability in response to VEGF [14,16]. Consequently, absence of one Shb-allele conferred a restriction on tumor growth of Lewis lung carcinoma and T241 fibrosarcoma cells [14] and of inheritable RT2 (rat insulin promoter-SV 40 T antigen) insulinomas [15]. In addition to the ameliorating effects of Shb-deficiency on pathological angiogenesis, Shb knockout mice displayed vascular abnormalities that resulted in impaired recovery after ischemic injury [16]. Shb knockout endothelial cells show reduced responsiveness to VEGF-stimulation with respect to ERK (extracellular-signal regulated kinase), Akt, FAK (focal adhesion kinase), Rac1 and myosin light chain kinase [14,17]. In concert, this abnormal signaling signature affects endothelial cell migration and adherens junction dissolution in response to VEGF [14,16,17], explaining the vascular dysfunction in vivo.
Melanomas are highly invasive cancers that metastasize at an early stage [18,19]. Since Shb-deficiency appears to reduce tumor growth by restricting the angiogenic expansion of the vasculature, the question of whether this will cause increased tumor invasiveness and metastasis or not remains unanswered. In the RT2 insulinoma model, no evidence for increased liver metastasis was obtained [15]. However, melanomas have a high inherent propensity for metastasis and for that reason, B16F10 melanoma cells were grown in Shb-deficient mice and the numbers of lung metastases determined. Indeed, it was observed that melanoma metastasis was increased in Shb-deficient mice because of a defective vasculature showing elevated vascular permeability and diminished recruitment of CD8+ cells to vascular structures.
Methods
Animals
Shb +/+ and +/− mice were bred on the C57Bl6 background for 8 generations (F8). Alternatively, Shb −/− and +/+ mice bred for four generations (F4) on that strain of mice were used. It was previously shown that Shb −/− mice cannot be obtained after breeding for more than 4 generations onto the C57Bl6 background [10]. All animal experiments had been approved by the local animal ethics committee at the Uppsala County Court.
Tumor cell injections
B16F10 melanoma cells (2 x 105) were injected subcutaneously in the subscapular region. When the tumor reached a size of 0.5 – 1 cm3 (determined by a caliper) the tumor was resected under anesthesia. Excised tumors were weighed for size determination. The mice were housed for an additional 10–19 days (commonly, but not always, there was a tumor relapse deciding the end-point of the experiment) after which the mice were sacrificed. Some of the mice were injected with 2 mg/kg FITC-conjugated Dextran-70000 (46945, Sigma, St. Louis, MO, USA) 30 minutes before sacrifice into the tail vein in order to determine blood vessel permeability. For lung seeding, 200000 B16F10 cells were injected in the tail vein and the mice maintained for three weeks before sacrifice. Lungs were excised and macroscopically visible metastases counted. The area was also inspected carefully for lymph node metastases but none were detected. The resected primary tumor was frozen on dry ice for immunofluorescence staining or stored in RNA-later (Quiagen, Hilden, Germany) for subsequent RNA preparation.
Immunofluorescence
Excised tumors were sectioned (5 μm) and subjected to immunofluorescence staining for CD31 (553370, BD Pharmingen, Franklin Lakes, NJ, USA), VE-cadherin (vascular endothelial-cadherin) (AF1002, R&D Systems, Minneapolis, MN, USA), desmin (ab6322, Abcam, Cambridge, UK) and fibrin/fibrinogen (GAM/Fbg/7S, Nordic Immunological Laboratories, Eindhoven, the Netherlands) as previously described [15].
At least five pictures were taken randomly of each tumor using a Nikon fluorescence and confocal C-1 microscope (Nikon, Japan). The area, diameter, perimeter of blood vessels, the fibrin spread area and pericyte covered length were measured with Image J software. Quantification of blood vessel permeability of FITC-conjugated Dextran was performed using Photoshop software.
Isolation of vascular fragments
Microvascular fragments were isolated from B16F10 melanomas grown on Shb +/− and control mice as previously described [20]. Briefly, tumors (0.5-1.0 cm3) were perfused with Hanks’ salt solution under anesthesia and then excised. They were then cut into small pieces and digested in 1.5 ml of 5 mg/ml Collagenase A (#103586, Roche Diagnostics, Basel, Switzerland) and 100 U/ml DNaseI (Invitrogen, Carlsbad, CA) Hanks’ solution per tumor for 15 min at 37°C. The tumor suspension was pipetted, filtered through a 70 μm diameter cell strainer (BD Bioscience, Franklin Lakes, NJ), washed and filtered a second time with a 40 μm cell strainer. After washing, the cells were incubated with CD31-coated Dynabeads. The magnetic beads (with the captured vascular fragments) were collected using a magnetic rack, washed extensively after which RNA was prepared from the captured cells using the Quiagen RNeasy Mini Kit (Quiagen, Hilden, Germany). Endothelial cells were isolated as described [14].
Gene expression
Total RNA of tumor was extracted according to RNeasy mini kit (74104; Qiagen) with RNase-Free DNase set (79254,Qiagen). One-step quantitative real-time RT-PCR was performed with QuantiTect™ SYBR®Green RT-PCR-kit (204243,Qiagen) on a LightCycler™ real-time PCR machine (lightcycler 2.0; Roche, Mannheim, Germany). Cycle threshold (Ct) values were determined with the LightCycler Software v3.5 (Qiagen). Gene expression was normalized for differences in RNA by subtracting the corresponding β-actin Ct-value. Statistical comparisons were made on normalized Ct-values. TaqMan qPCR gene expression analysis (Taqman, Life technologies, Carlsbad, CA) was used for analysis of PDGF-D (platelet-derived growth factor), CSFR2 (colony stimulating factor receptor 2), CXCL12 (chemokine C-X-C motif ligand), CXCR-4 (CXCL receptor) and CXCR-7.
Microarray analysis
High quality vascular fragment RNA from five tumors of each genotype was analyzed with Affymetrix 1.0 ST chips at the microarray core facility at Uppsala University Hospital (Uppsala Array Platform, Department of Medical Science, Science for Life Laboratory, Uppsala University Hospital, Sweden). Ingenuity software (Quiagen, Hilden, Germany) was used to perform pathway analysis on the microarray samples.
Bone marrow transplantation for generating chimeric mice
Iliac bones, femurs and tibias were collected from 8 to 10 week-old C57Bl/6 Shb +/+ and Shb +/− donor mice and processed as described [21,22]. Cell numbers were determined and 1.5 x106 cells were transplanted into congenic Shb +/+ or Shb +/− recipients by retro-orbital injection. Prior to the bone marrow transfer, the recipients were irradiated [22] with a split dose separated by two hours of 10 Gy. Peripheral blood chimerism in the recipient mice was determined 6 weeks post-transplantation by bleeding 100–200 μl blood in 0.05 mM EDTA. Total RNA was isolated from the remaining leukocytes with the RNeasy mini kit (74104; Qiagen). The Shb gene expression in the chimeric mice was compared between different Shb genotypes, following the real-time RT-PCR procedure.
In separate bone marrow transplantation experiments we assessed chimerism after bone marrow transfer by CD45.1 and CD45.2 staining after transfer of CD45.2-positive bone marrow to CD45.1-positive recipients. In such experiments, the donor bone marrow repopulated the host with more than 75% efficiency (results not shown).
Statistics
All values are given as means ± SEM. Probabilities (P) of chance differences between the groups were calculated with Mann–Whitney rank sum test (tumor metastases) or Students’ t-test (all other comparisons). Relative frequencies of metastasis were determined as follows: each mouse was categorized with category 0 having 0 metastases, category 1 having 1–5 metastases and category 2 having >5 metastases.
Results
B16F10 melanoma growth and metastasis in Shb +/− mice
Tumor growth, vasculature and metastasis was investigated using the B16F10 melanoma model [23]. In Additional file 1: Figure S1, tumor growth in Shb +/+ and Shb +/− mice is shown. No statistically significant difference was observed although 5 of 18 +/− tumors were much larger than the others on day 14 thus explaining the prominent error bar. Tumor resection was followed by a time period before animal sacrifice that occurred at the same time point for both groups of mice (Additional file 1: Figure S1). When determining lung metastases at sacrifice, the Shb +/− mice showed an increased number of lung metastases compared with controls (Figure 1A,B). Lung metastases were also categorized as zero (0 metastases), low = 1 (1–5 metastases) and high = 2 (>5 metastases) and using this scoring method, the Shb +/− mice also exhibited an increased frequency of metastasis (Figure 1C).
Figure 1.
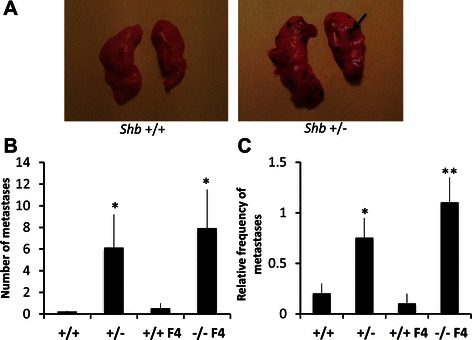
B16F10 melanoma lung metastasis inShb+/+, +/− and −/− mice. (A) Pictures show examples of lungs without (+/+) and with (+/−) B16F10 melanoma metastases. (B) At time of sacrifice, numbers of lung metastases were determined (arrow indicates a metastasis). These are given as absolute numbers for each mouse (B) or scored as relative frequencies after grouping in categories (C) (0 = 0, 1 = 1–5, 2= >5). Means ± SEM for n = 16 +/+, n = 16 +/−, n = 11 +/+ F4 and n = 11 −/− F4. * and ** indicate p < 0.05 and p < 0.01, respectively when compared with corresponding +/+ controls by Mann–Whitney U test.
Shb −/− mice can be obtained after breeding for maximally four generations onto the C57Bl6 background (F4) and we thus determined numbers and relative frequencies of metastasis in F4 Shb −/− mice, comparing them with corresponding F4 wild type controls (Figure 1B, C). Similar numbers were obtained as was seen in the F8 Shb +/− mice showing an increased absolute number of metastases, regardless of whether determined as numbers or relative frequency of metastasis when scored in categories (Figure 1B, C). For the following experiments, Shb +/− F8 mice were studied and compared with F8 +/+ controls since F4 mice are not considered inbred.
Lung seeding of tail vein-injected B16F10 melanoma cells
B16F10 melanoma cells were also injected in the tail vein to assess metastatic lung seeding of the injected cells (Additional file 2: Figure S2). No difference could be noted between wild type or Shb-deficient mice after this procedure, indicating that the ability of the target organ to seed tumor cells spread in the bloodstream was not dependent on genotype.
Shb +/− B16F10 melanoma vasculature
In previous studies, absence of one Shb allele conferred reduced tumor angiogenesis [14,15]. We currently decided to investigate the vasculature of B16F10 melanomas grown on the Shb +/− background. Surprisingly, no difference in vascular density was observed between Shb +/+ and +/− tumors (Figure 2A-C). The vasculature was unevenly distributed in both genotypes with certain areas showing a high vascular density and others not.
Figure 2.
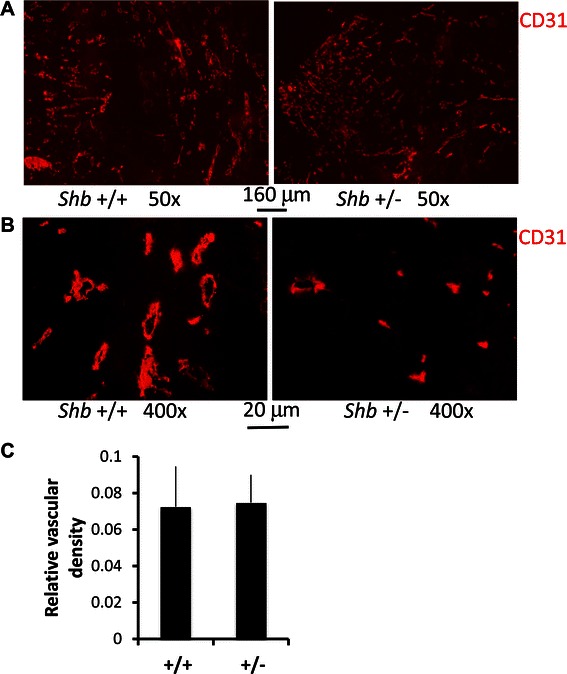
Vascular characteristics ofShb+/+ and +/− B16F10 tumors. Sections from B16F10 melanomas grown in Shb +/+ and +/− mice were stained for CD31 (red, A, B). Representative images at the magnifications indicated are shown (A-B). Relative vascular density (C, grid intersections over a red structure divided by total intersections) were quantitated and given as means ± SEM for 5 tumors each genotype and the value of each tumor was based on five images.
Shb +/− melanoma vascular pericyte coverage and permeability
Endothelial pericyte coverage and vascular permeability have been suggested as factors contributing to increased tumor metastasis [24]. Although no apparent difference in the endothelial pericyte coverage of tumors grown in Shb deficient mice was previously detected [14], morphological aberrations were noted [15]. These could relate to the abnormal endothelial cell morphology but could also reflect cell-autonomous changes of pericytes as well. We currently set out to investigate the status of pericytes in the Shb +/− melanomas by staining for the pericyte marker desmin (Figure 3A, D). We observed reduced pericyte coverage in melanomas grown in Shb +/− mice. This reduction correlated with increased fibrin deposits outside the vessel itself (Figure 3B, E), indicating increased vascular leakage. Increased vascular leakage was also noted in melanomas grown in Shb +/− mice by assessing FITC-Dextran-70000 fluorescence after injection (Figure 3C, F). Thus the combined data in Figures 2 and 3 provide evidence for a vascular phenotype showing increased vascular permeability that is supportive of B16 melanoma metastasis by allowing melanoma cell intravasation and dissemination.
Figure 3.
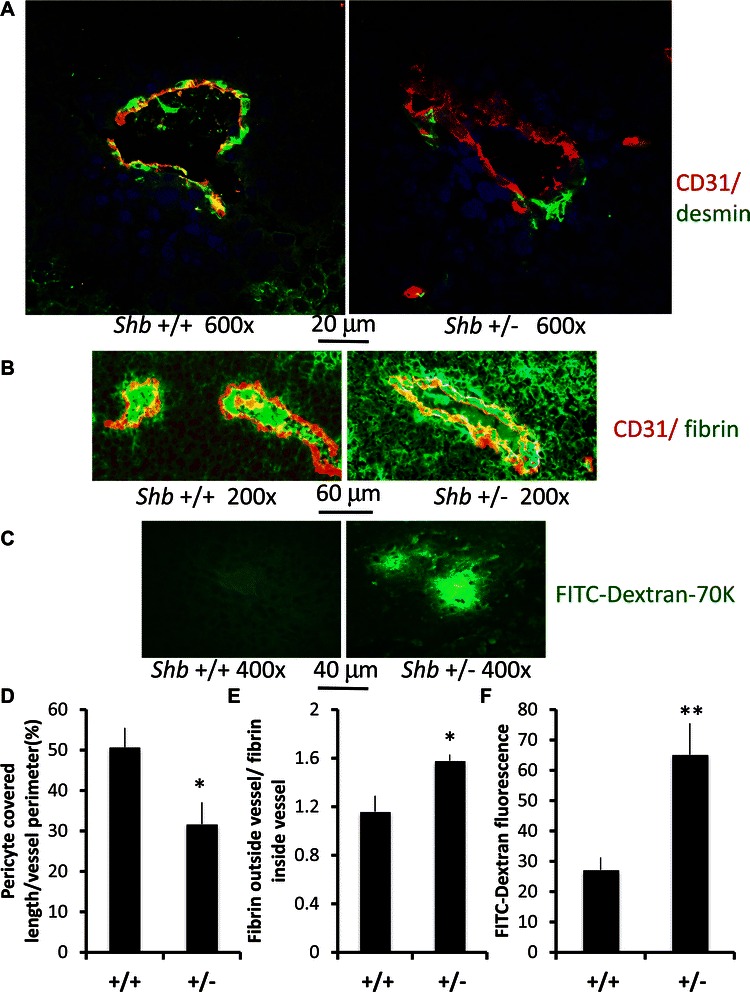
Pericyte coverage and leakage of fibrinogen in B16F10 tumors grown inShb+/+ and +/− mice. Sections from B16F10 melanomas were stained for CD31 (red) + desmin (green) (A) or CD31 (red) + fibrin (green) (B). Representative images are shown at the magnifications indicated. FITC-Dextran-70 K leakage was also determined by direct immunofluorescence of frozen sections of tumors after injection of the fluorescent dye (C). The percentage vessel coverage by pericytes was quantitated (D), as well as fibrin staining outside the CD31-lines vessel divided by the corresponding value contained within the CD31-positive structure (E). FITC-Dextran fluorescence was determined by Photoshop and given as relative values (F). Means ± SEM are given and five tumors each genotype were analyzed. The value of each tumor was based on five images. * and ** indicate p < 0.05 and 0.01, respectively, by Students’ t-test.
Gene expression in vascular fragments isolated from tumors
To understand the molecular mechanisms responsible for the observed vascular abnormalities, vascular fragments were isolated from tumors grown on Shb +/+ and +/− mice. Real-time RT-PCR for various angiogenic factors, receptors and chemokines of potential relevance for the vascular phenotype showed no changes in expression of most of these (Table 1), i.e. VEGFA, PDGF-AA, PDGF-BB, PDGF-CC, PDGF-DD, angiopoietin-1, angiopoietin-2, VEGFR1, VEGFR2, Tie-2, PDGFRB, GM-CSF, CXCL-12, CXCR-4 and CXCR-7 were unchanged. PlGF (placental growth factor) was increased and PDGFRA was reduced in Shb +/− vascular fragments. The decrease in PDGFRA expression, together with decreased vascular coverage of desmin staining, point towards reduction of the B-type pericyte [25] characterized by expression of these markers.
Table 1.
Gene expression in vascular fragments
| Gene product | Shb+/+ | Shb+/− |
|---|---|---|
| VEGFA | 1.0 ± 0.12 | 1.1 ± 0.15 |
| PlGF | 1.0 ± 0.25 | 1.8 ± 0.22 * |
| PDGF-AA | 1.0 ± 0.15 | 1.2 ± 0.11 |
| PDGF-BB | 1.0 ± 0.21 | 1.2 ± 0.32 |
| PDGF-CC | 1.0 ± 0.09 | 1.0 ± 0.08 |
| PDGF-DD | 1.0 ± 0.64 | 1.3 ± 0.22 |
| Angiopoietin-1 | 1.0 ± 0.13 | 1.1 ± 0.25 |
| Angiopoietin-2 | 1.0 ± 0.22 | 1.0 ± 0.34 |
| VEGFR1 | 1.0 ± 0.16 | 1.0 ± 0.28 |
| VEGFR2 | 1.0 ± 0.18 | 1.1 ± 0.27 |
| Tie-2 | 1.0 ± 0.22 | 1.2 ± 0.31 |
| PDGFRA | 1.0 ± 0.11 | 0.7 ± 0.09 * |
| PDGFRB | 1.0 ± 0.27 | 1.0 ± 0.30 |
| GM-CSF | 1.0 ± 0.11 | 0.8 ± 0.40 |
| CXCL-12 | 1.0 ± 0.59 | 0.9 ± 0.22 |
| CXCR-4 | 1.0 ± 0.46 | 1.3 ± 0.22 |
| CXCR-7 | 1.0 ± 0.63 | 1.4 ± 0.36 |
RNA isolated from vascular fragments was analyzed by real-time RT-PCR. Ct values were determined using the Roche Lightcycler software and beta-actin values were subtracted to normalize for differences in total amounts of RNA. Relative values shown in table were calculated by the formula 2-ΔCt (Shb+/− minus Shb+/+). *indicates p < 0.05 when compared with wild type. N = 5–7.
These findings were further supplemented with microarray analysis using Affymetrix 1.0/2.0 chips (Additional file 3: Table S1). The most striking findings were reduced expression of IL-6 (interleukin) and numerous markers for active cytotoxic CD8+ cells. These include several granzymes (Gzmc, Gzmd, Gzme, Gzmf and Gzmg), Pdcd1l2 (PD-L2) and Xcl1. One gene that showed increased expression in Shb +/− vascular fragments was Pten, which exerts an antagonistic role relative that of phosphatidylinositol 3-kinase. The analysis of the microarrays by Ingenuity software revealed that cell-to-cell signaling and interaction, inflammatory response, immune cell trafficking, cell-mediated immune response, immunological disease, and natural killer cell proliferation and development were amongst the most deregulated pathways under the “top diseases and biological functions” heading. Ingenuity analysis also confirmed that granzymes and IL-6 were significantly downregulated in the Shb +/− microvascular fragments. Collectively, microarray data points to a decrease in the recruitment of immune cells, particularly CD8+ cells, to the Shb +/− tumors as compared to wild type controls.
The possible involvement of CD8+ cells in affecting rates of metastasis was studied additionally by tumor staining for CD8a. Tumor numbers of CD8+ cells was difficult to assess due very uneven distribution of these cells within the tumors. However, the association of CD8+ cells with vascular structures could be quantified by double staining for CD8a and the vascular marker VE-cadherin showing a lower percentage CD8+ cells in direct contact with endothelial cells in Shb +/− tumors (Figure 4).
Figure 4.
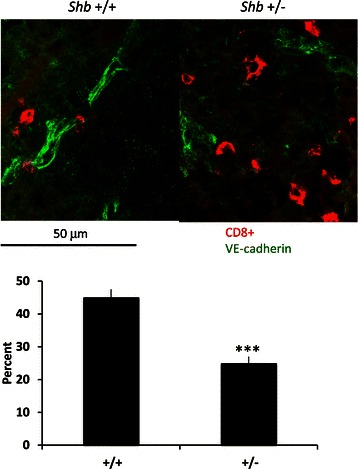
Proximity of CD8+ T cells with vascular structures. Cryosections of subcutaneous B16F10 melanomas were stained for CD8+ (red) and VE-cadherin (green). Pictures were taken at 20X (for statistical analysis) or 40X (for representative image) original magnification and the numbers of CD8+ cells immediately adjacent (direct contact) to VE-cadherin positive cells were scored in percent of total CD8+ cells for each host genotype present in the images. Representative images and statistics are shown as percent adjacent CD8+ cells (means ± SEM) for 4 tumors each genotype (5 images each tumor).
To obtain reliable estimates of tumor CD8+ cell infiltration, CD8a gene expression was determined by real-time RT-PCR of total tumor RNA and was found to be selectively decreased in Shb +/− tumors (Figure 5). IL-6 expression was similar in wild type and Shb +/− total tumor RNA. In vascular fragments, expression of CD8a as well as granzyme B and IL-6 were decreased as a consequence of Shb deficiency. Gene expression analysis of isolated endothelial cells showed significantly less IL-6 gene expression (<1%) compared with that of the vascular fragments, suggesting that passenger lymphocytes provide the main source of IL-6 mRNA in the vascular fragment preparations (results not shown).
Figure 5.
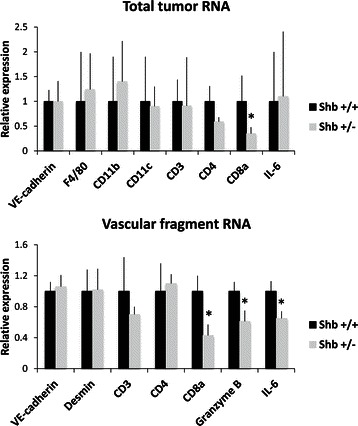
Gene expression in whole tumors and isolated vascular fragments. Total tumor RNA or RNA from isolated vascular fragments were isolated from either host genotype and analyzed by real-time RT-PCR. Ct-values were determined using the Roche Lightcycler software and beta-actin values were subtracted to normalize for differences in total amounts of RNA. Relative values shown in table were calculated by the formula 2-ΔCt (Shb+/− minus Shb+/+). *indicates p < 0.05 when compared with wild type. N = 5–7.
Bone marrow transplantation experiments
The possibility that immune cells could be responsible for the increased rate of metastasis motivated us to perform bone marrow transplantations and to follow the effect of the hematopoietic cell genotype on metastasis. Wild type bone marrow was transplanted to Shb +/− recipients and Shb +/− bone marrow was transplanted to wild type recipients. After restitution of hematopoiesis (>3 months) B16F10 melanoma cells were inoculated subcutaneously, primary tumors removed and the appearance of lung metastasis monitored. There was as in Additional file 1: Figure S1 no difference in primary tumor growth, time of resection and time point of final sacrifice between the two groups (results not shown). However, number of lung metastasis was higher (Figure 6) in the Shb +/− recipient group receiving wild type bone marrow when scored as categories (0, 1 and 2 as in Figure 1), suggesting that the main determinant responsible for the increased metastasis in Shb +/− mice is the vascular genotype and not that of hematopoietic cells.
Figure 6.
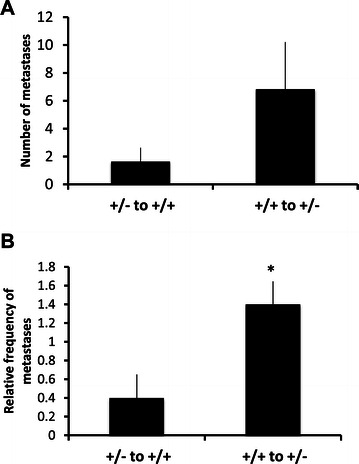
Effects of bone marrow genotype on B16F10 metastasis.Shb +/− mice received +/+ bone marrow and Shb +/+ mice received +/− bone marrow after transplantation to recipients that had been depleted of their endogenous bone marrow by irradiation. After 3 months when hematopoiesis had been restored, B16F10 tumors were gown subcutaneously, resected and the mice sacrificed when tumor recurrence occurred (average 34 ± 2 days after initial injection of cells in the +/+ recipient group and 36 ± 0 days in the +/− recipient group). Numbers of lung metastases were then scored and given as absolute values (A) or as frequencies of categories (B) where 0 = 0, 1 = 1–5 and 2= >5 metastases. Means ± SEM for 5 mice in each group are given. *indicates p < 0.05 by Students’ t-test.
Discussion
The current study elucidates the metastatic properties of melanoma cells in relation to the absence of the Shb gene. This cancer has been extensively investigated and tumor progression follows a characteristic pattern [19]. The driver mutations in melanomas have recently been mapped [26] and revealed a limited number of genetic changes in this cancer. Melanoma growth is dependent on angiogenesis [18,27,28] and numerous angiogenic factors have been linked to melanoma growth [29,30] although the role of VEGF appears modest [31]. Alternative candidates for support of melanoma growth and metastasis are inflammatory cytokines and chemokines [32]. Factors of particular importance in this context are IL-8 [29], CCL19/21 [33], CXCL12 [34], IL-6 and macrophage migration inhibitory factor [35,36]. The combined data suggest a scenario that utilizes multiple factors for melanoma growth and angiogenesis.
The B16F10 melanoma cell line is a useful model for studying melanoma metastasis in vivo [23] and consequently, we tested tumor metastasis in Shb deficient mice. The increased rate of lung metastasis observed in Shb deficient mice may have several explanations. One possibility is increased vascular permeability due to reduced pericyte coverage. However, gene expression profiling showed less expression of various markers for cytotoxic CD8+ lymphocytes in the Shb +/− vasculature and this may offer an alternative suggestion. Due to some yet unknown feature of the Shb deficient vasculature, passage of CD8+ cells over the vascular barrier into the tumor is specifically hampered thus causing less CD8+ cell infiltration into the tumor. This may contribute to increased metastasis since it is well established that CD8+ cytotoxic T cells combat melanoma growth and metastasis [37-39]. Indeed, treatment with an activator of CD8+ T cells, Ipilimumab (anti-CTLA-4), together with an inhibitor of VEGF signaling (Bevacizumab) causes perivascular CD8+ cell accumulation [40], thus confirming the relevance of the vasculature for tumor-infiltration of cytotoxic T cells. The bone marrow transplantation experiments further support this notion, since the metastatic phenotype followed the recipient genotype, i.e. more metastasis was seen in Shb +/− recipients with a Shb deficient vasculature despite these having a wild type bone marrow producing wild type blood cells. The decrease in tumor infiltration of CD8+ cells was indeed surprising, considering that vascular permeability was increased these conditions. Apparently, specific mechanisms operating in the vascular component control lymphocyte endothelial transmigration. Lymphocyte extravasation depends on numerous endothelial processes but selective mechanisms operating for lymphocytes and in particular for CD8+ lymphocytes are poorly defined.
The microarray analysis provides no obvious explanation for leaky vascular phenotype, although changes in the expression of numerous cytokines/chemokines could contribute to this. PlGF is one factor that has been suggested to increase vascular permeability [41-43] but more recent studies suggest a modest role, if any, of PlGF for vascular permeability [44,45]. Reduced PDGFRA expression could be explained as downregulation of its expression in a subset of pericytes, the B-pericyte [25]. Apart from the reduction in PDGFRA expression, our study shows decreased perivascular desmin staining, a feature shared by these B-pericytes. Alternatively, reduced PDGFRA expression might indicate a reduction in myofibrillar cells. PDGFRA-expressing cells can be found on tumor-associated fibroblasts infiltrating B16 melanomas [46] but their association with the vasculature has not been investigated. It is likely that in certain malignancies, fibroblasts might be found in the vicinity of the vasculature [47], but this occurs normally in conjunction with alpha-smooth muscle actin expression, conferring these cells a myofibroblast phenotype. In our study, as analyzed by microarray, we did not detect significant differences in alpha smooth muscle actin expression, indicating that the decrease in PDGFRA expression may indeed reflect down-regulation of the B-type subset of pericytes. It is tempting to speculate that the shift towards an increase in type-A pericytes could confer the microvascular environment with features that enhance malignant cell intravasation. Increased expression of Pten is, however, likely to have profound effects on endothelial function since this gene reduces phosphatidyl-3’-inositol levels and thus suppresses PI3K and Akt activities. These play major roles for angiogenesis and vascular permeability [48] and thus a reduction in these would be predicted to be deleterious for vascular integrity.
The compromised vasculature in Shb deficient mice increases the risk of intravasation of melanoma cells allowing them to disseminate in blood and infiltrate target tissues such as lung. Apparent is the fact that lung seeding of metastases after tail vein injections was not different between the genotypes, further implicating local vascular changes in the primary tumors as responsible for the effects. We were unable to detect increased metastasis to the liver of Shb +/− insulinomas [15] and the discrepancy between those findings and the current may lie in the difference in the local angiogenic milieu, which is probably dependent on a multitude of factors in melanomas whereas RIP-Tag2 insulinomas are highly dependent on VEGF and FGF-2 [49].
Conclusions
Absence of Shb promotes B16F10 tumor metastasis due to increased vascular permeability and reduced pericyte/myofibrillar cell coverage of endothelial cells, thus allowing intravasation and vascular dissemination of tumor cells. The data support a model in which tumor metastasis is affected in a context-dependent manner by the absence of Shb, contingent on the local angiogenic environment and how it affects vascular permeability and immune cell recruitment. Since Shb is a signaling protein participating in angiogenic responses, this finding has implications for choosing an appropriate strategy for inhibiting tumor expansion by anti-angiogenic treatment without simultaneously increasing tumor metastasis.
Acknowledgements
We are grateful to Xin Wang for help in performing real-time RT-PCR, Anna Bereza-Jarocinska for immune staining and Ross Smith for tail vein injections. The study was supported by grants from the Swedish Cancer Foundation (130618, 120831, 2013–0782), Swedish Research Council (54x-10822), Swedish Diabetes fund (DIA 2012) and Family Ernfors fund. The microarrays were performed by the Uppsala Array Platform, Department of Medical Science, Science for Life Laboratory, Uppsala University, Entrance 61, 3rd floor, Uppsala University Hospital 751 85 Uppsala.
Abbreviations
- VEGF
Vascular endothelial growth factor
- VEGFR
VEGF receptor
- SHB
Src homology-2 domain protein B
- FGF
Fibroblast growth factor
- PDGF
Platelet-derived growth factor
- VE-cadherin
Vascular endothelial-cadherin
- PlGF
Placental growth factor
- IL
Interleukin
- CCL
Chemokine C-C motif ligand
- CCR
CCL receptor
- CXCL
Chemokine C-X-C motif ligand
- CXCR
CXCL receptor
- GM-CSF
Granulocyte macrophage colony-stimulating factor
- NOS
Nitric oxide synthase
- FAK
Focal adhesion kinase
- ERK
Extracellular signal-regulated kinase
- RT2
Rat insulin promoter driven SV40 large T antigen-2
Additional files
B16F10 melanoma growth (A) and times of tumor resection and animal sacrifice (B). B16F10 melanoma cells (2X105) were injected subcutaneously on day zero on the backs of Shb +/+ and +/− mice. Tumor sizes on day 10, 12, 14 and at removal are shown. After tumor removal, mice were maintained for some time until sacrificed, at which numbers of metastases were determined. Times of tumor resection and animal sacrifice are given. Means ± SEM for n = 5 on day 10, n = 24 on day 12 and n = 23 (+/+) and n = 19 (+/−) on day 14. For time of resection and sacrifice, n = 16-21.
Lung seeding of tail vein-injected B16F10 melanoma cells in Shb +/+ and +/− mice. (A) Numbers of lung seeding metastases for each mouse at three weeks after tail vein injections of B16F10 cells (p > 0.77). (B) Numbers of metastases determined as categories (1 = 1-5, 2 = 5-10, 3 > 10). Means ± SEM are given (p > 0.61). Nine mice of each genotype were injected with 200000 cells.
Gene expression in Shb +/- vascular fragments relative wild type. Values are log2 expression wild type, log2 fold change in expression (+/-) and p values for five preparations of vascular fragments from each genotype.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors participated in experimental design, in performing the experiments and in analysis and interpretation of the data. The specific contributions were: tumor growth and metastasis (GZ), tumor morphology (GZ, MJ, MW), bone marrow transplantation (KG), isolation of vascular fragments or endothelial cells (KG, GG, JH), RNA isolation (GZ, KG, MJ, GG, JH), qPCR (GZ, KG, MJ, JH) and Ingenuity analysis (GG, MW). MW wrote the paper and all authors provided comments. All authors read and approved the final manuscript.
Contributor Information
Guangxiang Zang, Email: guangxiang.zang@medbio.umu.se.
Karin Gustafsson, Email: Karin.gustafsson@mcb.uu.se.
Maria Jamalpour, Email: Maria.jamalpour@mcb.uu.se.
JongWook Hong, Email: Jong.hong@ki.se.
Guillem Genové, Email: Guillem.genove@ki.se.
Michael Welsh, Email: Michael.welsh@mcb.uu.se.
References
- 1.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339(6219):58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 4.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anneren C, Lindholm CK, Kriz V, Welsh M. The FRK/RAK-SHB signaling cascade: a versatile signal-transduction pathway that regulates cell survival, differentiation and proliferation. Curr Mol Med. 2003;3(4):313–24. doi: 10.2174/1566524033479744. [DOI] [PubMed] [Google Scholar]
- 7.Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, et al. The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J Biol Chem. 2004;279(21):22267–75. doi: 10.1074/jbc.M312729200. [DOI] [PubMed] [Google Scholar]
- 8.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437(2):169–83. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 9.Calounova G, Livera G, Zhang XQ, Liu K, Gosden RG, Welsh M. The Src homology 2 domain-containing adapter protein B (SHB) regulates mouse oocyte maturation. PLoS One. 2010;5(6):e11155. doi: 10.1371/journal.pone.0011155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kriz V, Mares J, Wentzel P, Funa NS, Calounova G, Zhang XQ, et al. Shb null allele is inherited with a transmission ratio distortion and causes reduced viability in utero. Dev Dyn. 2007;236(9):2485–92. doi: 10.1002/dvdy.21257. [DOI] [PubMed] [Google Scholar]
- 11.Akerblom B, Barg S, Calounova G, Mokhtari D, Jansson L, Welsh M. Impaired glucose homeostasis in Shb−/− mice. J Endocrinol. 2009;203:271–9. doi: 10.1677/JOE-09-0198. [DOI] [PubMed] [Google Scholar]
- 12.Gustafsson K, Calounova G, Hjelm F, Kriz V, Heyman B, Gronvik KO, et al. Shb deficient mice display an augmented TH2 response in peripheral CD4+ T cells. BMC Immunol. 2011;12:3. doi: 10.1186/1471-2172-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gustafsson K, Willebrand E, Welsh M. Absence of the adaptor protein Shb potentiates the T 2 response in a mouse model of atopic dermatitis. Immunology. 2014;143:33–41. doi: 10.1111/imm.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funa NS, Kriz V, Zang G, Calounova G, Akerblom B, Mares J, et al. Dysfunctional microvasculature as a consequence of shb gene inactivation causes impaired tumor growth. Cancer Res. 2009;69(5):2141–8. doi: 10.1158/0008-5472.CAN-08-3797. [DOI] [PubMed] [Google Scholar]
- 15.Akerblom B, Zang G, Zhuang ZW, Calounova G, Simons M, Welsh M. Heterogeneity among RIP-Tag2 insulinomas allows vascular endothelial growth factor-A independent tumor expansion as revealed by studies in Shb mutant mice: Implications for tumor angiogenesis. Mol Oncol. 2012;6:333–46. doi: 10.1016/j.molonc.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christoffersson G, Zang G, Zhuang ZW, Vagesjo E, Simons M, Phillipson M, et al. Vascular adaptation to a dysfunctional endothelium as a consequence of Shb deficiency. Angiogenesis. 2012;15(3):469–80. doi: 10.1007/s10456-012-9275-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zang G, Christoffersson G, Tian G, Harun-Or-Rashid M, Vagesjo E, Phillipson M, et al. Aberrant association between vascular endothelial growth factor receptor-2 and VE-cadherin in response to vascular endothelial growth factor-a in Shb-deficient lung endothelial cells. Cell Signal. 2013;25(1):85–92. doi: 10.1016/j.cellsig.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Crowson AN, Magro C, Miller A, Mihm MC., Jr The molecular basis of melanomagenesis and the metastatic phenotype. Semin Oncol. 2007;34(6):476–90. doi: 10.1053/j.seminoncol.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355(1):51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 20.Wallgard E, Larsson E, He L, Hellstrom M, Armulik A, Nisancioglu MH, et al. Identification of a core set of 58 gene transcripts with broad and specific expression in the microvasculature. Arterioscler Thromb Vasc Biol. 2008;28(8):1469–76. doi: 10.1161/ATVBAHA.108.165738. [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson K, Heffner G, Wenzel PL, Curran M, Grawe J, McKinney-Freeman SL, et al. The Src homology 2 protein Shb promotes cell cycle progression in murine hematopoietic stem cells by regulation of focal adhesion kinase activity. Exp Cell Res. 2013;319(12):1852–64. doi: 10.1016/j.yexcr.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 22.Gustafsson K, Jamalpour M, Trinh C, Kharas MG, Welsh M. The Src homology-2 protein Shb modulates focal adhesion kinase signaling in a BCR-ABL myeloproliferative disorder causing accelerated progression of disease. J Hematol Oncol. 2014;7(1):45. doi: 10.1186/1756-8722-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stackpole CW. Distinct lung-colonizing and lung-metastasizing cell populations in B16 mouse melanoma. Nature. 1981;289(5800):798–800. doi: 10.1038/289798a0. [DOI] [PubMed] [Google Scholar]
- 24.Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29(5):630–8. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 25.Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science. 2011;333(6039):238–42. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- 26.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Straume O, Salvesen HB, Akslen LA. Angiogenesis is prognostically important in vertical growth phase melanomas. Int J Oncol. 1999;15(3):595–9. doi: 10.3892/ijo.15.3.595. [DOI] [PubMed] [Google Scholar]
- 28.Streit M, Detmar M. Angiogenesis, lymphangiogenesis, and melanoma metastasis. Oncogene. 2003;22(20):3172–9. doi: 10.1038/sj.onc.1206457. [DOI] [PubMed] [Google Scholar]
- 29.Mahabeleshwar GH, Byzova TV. Angiogenesis in melanoma. Semin Oncol. 2007;34(6):555–65. doi: 10.1053/j.seminoncol.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu L, Wu X, Cheng Z, Lee CV, LeCouter J, Campa C, et al. Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest Ophthalmol Vis Sci. 2008;49(2):522–7. doi: 10.1167/iovs.07-1175. [DOI] [PubMed] [Google Scholar]
- 31.Nisancioglu MH, Betsholtz C, Genove G. The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor-A blockade. Cancer Res. 2010;70(12):5109–15. doi: 10.1158/0008-5472.CAN-09-4245. [DOI] [PubMed] [Google Scholar]
- 32.Richmond A, Yang J, Su Y. The good and the bad of chemokines/chemokine receptors in melanoma. Pigment Cell Melanoma Res. 2009;22(2):175–86. doi: 10.1111/j.1755-148X.2009.00554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schramm SJ, Campain AE, Scolyer RA, Yang YH, Mann GJ. Review and cross-validation of gene expression signatures and melanoma prognosis. J Invest Dermatol. 2012;132(2):274–83. doi: 10.1038/jid.2011.305. [DOI] [PubMed] [Google Scholar]
- 34.Vianello F, Papeta N, Chen T, Kraft P, White N, Hart WK, et al. Murine B16 melanomas expressing high levels of the chemokine stromal-derived factor-1/CXCL12 induce tumor-specific T cell chemorepulsion and escape from immune control. J Immunol. 2006;176(5):2902–14. doi: 10.4049/jimmunol.176.5.2902. [DOI] [PubMed] [Google Scholar]
- 35.Girard E, Strathdee C, Trueblood E, Queva C. Macrophage migration inhibitory factor produced by the tumour stroma but not by tumour cells regulates angiogenesis in the B16-F10 melanoma model. Br J Cancer. 2012;107(9):1498–505. doi: 10.1038/bjc.2012.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoejberg L, Bastholt L, Schmidt H. Interleukin-6 and melanoma. Melanoma Res. 2012;22(5):327–33. doi: 10.1097/CMR.0b013e3283543d72. [DOI] [PubMed] [Google Scholar]
- 37.Gao K, Li X, Zhang L, Bai L, Dong W, Shi G, et al. Transgenic expression of IL-33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett. 2013;335(2):463–71. doi: 10.1016/j.canlet.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Lee CH, Kakinuma T, Wang J, Zhang H, Palmer DC, Restifo NP, et al. Sensitization of B16 tumor cells with a CXCR4 antagonist increases the efficacy of immunotherapy for established lung metastases. Mol Cancer Ther. 2006;5(10):2592–9. doi: 10.1158/1535-7163.MCT-06-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakasone Y, Fujimoto M, Matsushita T, Hamaguchi Y, Huu DL, Yanaba M, et al. Host-derived MCP-1 and MIP-1alpha regulate protective anti-tumor immunity to localized and metastatic B16 melanoma. Am J Pathol. 2012;180(1):365–74. doi: 10.1016/j.ajpath.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Immunol Res: Cancer; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao R, Xue Y, Hedlund EM, Zhong Z, Tritsaris K, Tondelli B, et al. VEGFR1-mediated pericyte ablation links VEGF and PlGF to cancer-associated retinopathy. Proc Natl Acad Sci U S A. 2010;107(2):856–61. doi: 10.1073/pnas.0911661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Odorisio T, Schietroma C, Zaccaria ML, Cianfarani F, Tiveron C, Tatangelo L, et al. Mice overexpressing placenta growth factor exhibit increased vascularization and vessel permeability. J Cell Sci. 2002;115(Pt 12):2559–67. doi: 10.1242/jcs.115.12.2559. [DOI] [PubMed] [Google Scholar]
- 43.Taylor AP, Rodriguez M, Adams K, Goldenberg DM, Blumenthal RD. Altered tumor vessel maturation and proliferation in placenta growth factor-producing tumors: potential relationship to post-therapy tumor angiogenesis and recurrence. Int J Cancer. 2003;105(2):158–64. doi: 10.1002/ijc.11059. [DOI] [PubMed] [Google Scholar]
- 44.Deissler HL, Deissler H, Lang GK, Lang GE. VEGF but not PlGF disturbs the barrier of retinal endothelial cells. Exp Eye Res. 2013;115:162–71. doi: 10.1016/j.exer.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 45.Gaal EI, Tammela T, Anisimov A, Marbacher S, Honkanen P, Zarkada G, et al. Comparison of vascular growth factors in the murine brain reveals placenta growth factor as prime candidate for CNS revascularization. Blood. 2013;122(5):658–65. doi: 10.1182/blood-2012-07-441527. [DOI] [PubMed] [Google Scholar]
- 46.Anderberg C, Li H, Fredriksson L, Andrae J, Betsholtz C, Li X, et al. Paracrine signaling by platelet-derived growth factor-CC promotes tumor growth by recruitment of cancer-associated fibroblasts. Cancer Res. 2009;69(1):369–78. doi: 10.1158/0008-5472.CAN-08-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruan GX, Kazlauskas A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012;31(7):1692–703. doi: 10.1038/emboj.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]