

Abstract
Adults with Down syndrome (DS) develop Alzheimer’s disease (AD) neuropathology by 40 years of age. Synaptophysin (SYN) consistently declines with age and is further reduced with sporadic AD. Thus, we hypothesized that SYN would be reduced in DS with AD. The gene for synaptojanin-1 (SYNJ1), involved in synaptic vesicle recycling, is on chromosome 21. We measured SYN and SYNJ1 in an autopsy series of 39 cases with DS and 28 without DS, along with 7 sporadic AD cases were examined. SYN was significantly lower in DSAD compared with DS alone and similar to sporadic AD. Reduced SYN is associated with AD neuropathology and with Aβ levels in DS, as is seen in sporadic AD. SYNJ1 was significantly higher in DS and correlated with several measures of Aβ. SYNJ1 was higher in DSAD and significantly higher than SYNJ1 in sporadic AD. Although significantly higher in DS, SYNJ1 is further increased with AD neuropathology suggesting interesting differences in a synapse-associated protein that is overexpressed in trisomy 21.
Keywords: beta-amyloid, neuroinflammation, oligomers, synapses, synaptophysin, synaptojanin, trisomy 21
Introduction
The most common known genetic cause of intellectual disability is Down syndrome (DS), also called trisomy 21. The primary cause of DS is triplication of chromosome 21 [1], resulting in a phenotype that is accompanied by altered brain development and other neurologic features [2,3]. The lifespan of adults with DS has been improving, leading to a higher prevalence of individuals with DS at middle and older ages. [4]. However, a key challenge for adults with DS as they age is the increasing risk for developing Alzheimer’s disease (AD) [5]. Sporadic AD is a progressive neurodegenerative disease associated with cognitive decline and dementia, the pathological hallmarks of which are beta-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs) in the brain. AD neuropathology (including Aβ plaques/deposition and NFTs) appears in virtually all adults with trisomy 21 after 40 years of age [6-8]. Aβ is cleaved from the larger beta-amyloid precursor protein which is encoded on chromosome 21, and likely accounts for increased amount of Aβ deposition seen in DS. [9].
A consistent observation in the brains of individuals with sporadic AD in comparison to brain specimens from nondemented elderly control brains is synapse loss, a finding which reflects dementia severity [10-12]. In addition, several studies show a reduction in proteins associated with synapses, with one of the most robust observations being a reduction in synaptophysin (SYN)[13]. SYN is a 38 kDa integral membrane glycoprotein located in presynaptic vesicles [14]. Previous studies in individuals with AD report SYN protein losses (e.g. [15-18]. There are a few papers describing SYN loss in autopsy samples from older adults with DS [19,20], but the sample sizes are small and none were characterized as having AD. There is a need for a more systematic evaluation of the age of onset of SYN protein losses relative to AD neuropathology in DS.
Another synapse associated protein that is of key interest in DS is synaptojanin 1 (SYNJ1). The gene for SYNJ1 is on chromosome 21 and present in triplicate in DS [21]. SYNJ1 is a brain enriched phosphoinositide phosphatase [22] that is involved with endocytosis and synaptic vesicle cycling [23,24]. Several studies report increased protein levels of SYNJ1 in DS brain [25], particularly within the frontal cortex [26] using immunocytochemical or western blotting approaches. Interestingly, increasing SYNJ1 can ameliorate synaptic and behavioral impairments in a mouse model of AD, Tg2576 mice [27]. SYNJ1 expression declines in the brain of individuals with the sporadic form of AD [28] but this finding has not been tested in DS. However, there are no reports of the effects of aging or the presence of AD in DS with respect to SYNJ1 levels.
This study examined the hypothesis that SYNJ1 would be higher overall in DS due to overexpression but may also decline in DS with AD (DSAD). We also hypothesized that SYN would be reduced in DSAD as reported in sporadic AD. Further, we predicted that SYN and SYNJ1 protein levels would be decreased with increasing AD neuropathology as characterized in part by, Aβ and oligomeric Aβ accumulation. To test these hypotheses, we conducted western blotting studies of SYN and SYNJ1 protein in frontal cortex samples from DS and DSAD autopsy cases in comparison to age-matched control cases and sporadic AD cases. Previously published Aβ measures combined with the western blot measures of SYN and SYNJ1 from the current study allowed us to look at changes in synaptic protein levels with Aβ accumulation.
Materials and Methods
Subjects
Autopsy brain tissue was obtained from several sources including the University of California at Irvine Alzheimer’s Disease Research Center and the Maryland Developmental Disorders Brain Bank. Human tissue collection and handling conformed to University of Kentucky/UC Irvine Institutional Review Board guidelines. All cases selected ranged from 1 to 88 years based, in part, on the availability of frozen frontal cortex. Control cases were subsequently selected to match for age and PMI as closely as possible to the DS cases. AD cases were selected based on PMI only to match DSAD cases (DS cases that had sufficient neuropathology for a post-mortem diagnosis of AD)[29]. Since individuals with DSAD typically come to autopsy at younger ages than sporadic AD cases, it was not possible to match for age at death. As a result, tissues from 5 autopsy groups were available: young controls (YC, age matched to young DS; N=13), old controls (OC, age matched to DSAD; N=15), DS (N=8), DSAD (N=31), and AD alone (N=7). Both males and females were included in the study, but given the challenges of matching cases, we did not match for gender. The level of premorbid ID was not available in these cases and thus it was not possible to use this variable as a co-variate in the analysis.
As shown in Table 1, DS cases were divided into two groups: with findings for AD (DSAD) or without (DS). In the former group, we required sufficient pathology for a diagnosis of AD [30]. All DSAD cases were over the age of 40 years. Thus for the current study, control cases were split into two groups, less than or equal to 40 years (YC) or older than 40 years (OC) at death. The post mortem interval (PMI) was different across groups, with the DSAD group overall having a lower PMI (F(4,73)=7.3 p<.0005). Group comparisons of SYN and SYNJ1 were thus adjusted for PMI when necessary.
Table 1.
Characteristics of Autopsy Cases
| Characteristic | Control <40 years |
Control >40 years |
DS | DSAD | AD |
|---|---|---|---|---|---|
| n=13 | n=15 | n=8 | n=31 | n=7 | |
| Mean (Range) | |||||
| Age (yrs) | 17.8 (1-39) | 51.5 (41-67) | 15.8 (1-39) | 51.4 (40-66) | 80.9 (67-88) |
| PMI (hrs) | 16.2 (6-28) | 14.9 (3-28) | 17.0 (12-28) | 7.6 (2-25) | 8.4 (6-10) |
| Number (%) | |||||
| Female (%) | 5 (38) | 8 (53) | 2 (25) | 18 (58) | 2 (29) |
Western blotting
Frozen samples from the mid-frontal cortex (BA46) were homogenized in 1% SDS extraction buffer (150 mg/ml) with protease inhibitors (Roche, Indianapolis, IN). Protein concentration was determined by BCA as described previously [31]. A 10-20% SDS page criterion gel (Bio-Rad Laboratories, Hercules, CA) was used to separate proteins and transferred to a nitrocellulose membrane (Bio-Rad Laboratories). Membranes were probed with anti-synaptophysin (Millipore Corp., Temcula, CA, 1:800,000, Monoclonal) or anti-synaptojanin 1 (Sigma Prestige Antibodies, St. Louis, MO, 1:1000, Polyclonal) and then incubated with either anti-rabbit or anti-mouse secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Detection was done using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL). To ensure that densitometric measurements of protein levels of SYN and SYNJ1 were not saturated, a standard curve was derived for each antibody as published in a previous study [31]. To establish equal protein loading, membranes were stripped reprobed with anti-α-tubulin (Abcam, Cambridge, MA, 1:80,000, Monoclonal)
Aβ ELISAs
Aβ was extracted from tissue measured as previously described [32]. Briefly, frozen cortical samples were extracted sequentially in ice cold phosphate buffered saline (PBS, pH 7.4) with a complete protease inhibitor cocktail (PIC; Amresco, Solon, OH) and centrifuged at 20,800 × g for 30 min. at 4°C. Following centrifugation, the supernatant was collected and the pellets were sonicated (10 × 0.5 sec pulses at 100W, Sonic Dismembrator, Fisher Scientific, Pittsburgh, PA) in 2% SDS with PIC and centrifuged at 20,800 × g for 30 min. at 14°C. The supernatant was again collected and the remaining pellets were sonicated in 70% formic acid (FA), followed by centrifugation at 20,800 × g for 1 hour at 4°C. Samples were stored at °80°C until time of assay.
FA-extracted material was initially neutralized by a 1:20 dilution in TP buffer (1 M Tris base, 0.5 M Na2HPO4), followed by a further dilution as needed (1:5 to 1:20, for a final dilution of 1:100 to 1:400) in Antigen Capture buffer (AC) (20mM Na3PO4, 0.4% Block Ace (AbD Serotec, Raleigh, NC), 0.05% NaN3, 2mM EDTA, 0.4M NaCl, 0.2% BSA, 0.05% CHAPS, pH 7) . SDS soluble fractions were diluted as needed (1:20 to 1:50) in AC buffer alone. PBS fractions were diluted 1:4 in AC buffer alone.
Aβ was measured using a standard, well-characterized two-site sandwich ELISA as described previously [47]. Briefly, an Immulon 4HBX plate was coated with 0.5 µg antibody per well, incubated overnight at 4°C, and blocked with a solution of Synblock (AbD Serotec, as per the manufacturer’s instructions). Antigen capture was performed using monoclonal antibody Ab9 (against Human Aβ 1-16). Antigen detection was performed using biotinylated antibodies 13.1.1 (end-specific for Aβ 1-40) and 12F4 (end-specific for Aβ 1-42; Covance, Princeton, NJ), followed by NeutraVidin-HRP (Pierce Biotechnologies, Rockford, IL)
A synthetic Aβ peptide standard was run on the same plate for comparison, and standards and samples were run at least in duplicate; Aβ values were determined by interpolation relative to the standard curve. Plates were washed between steps with standard PBS containing 0.05% Tween-20 (2-4x) followed by PBS (2-4x). Plates were developed with TMB reagent (KPL, Inc., Gaitherburg, MD), stopped with 6% o-phosphoric acid, and read at 450 nm using a multiwell plate reader (BioTek, Winooski, VT).
Oligomeric Aβ from the SDS-soluble fraction was measured using a single-site sandwich ELISA similar to the one described above, except the same antibody (4G8; Covance, Princeton, NJ) was used for capture and detection. SDS samples were diluted 1:50 in AC buffer. Synthetic Aβ42 oligomers were used to prepare a standard curve; oligomeric Aβ values were determined by interpolation relative to the standard curve.
Data Analysis
For synapse protein measures, multiple western blots were required. In order to standardize across gels, one DS with AD case was loaded on each gel with increasing concentrations (1,5,10 µg) to serve as an internal control [31] . Optical density measures for each gel were then calculated as total protein (µg) of this internal control case. We examined the relationship between synapse markers and post mortem interval to determine if this needed to be included as a covariate in subsequent analyses. In each group analysis, cases were classified as YC (<40 yrs), DS (<40), OC (>40 years), DSAD (>40) and AD. The AD, DS and DSAD groups were compared to the appropriate control group (YC, OC). Using this method, we were able to determine whether protein expression differed from the control group A two-way univariate analysis of variance was used to compare group differences (Young <40 & >40 Old; AD present vs. not present) with gender serving as a co-variate. Post-hoc pairwise comparisons of the means for all analyses were made using the Bonferroni test. Pearson correlation coefficients were used to test for the linear association between synapse proteins and age within the DS and control groups, separately. To determine the correlation between SYN and SYNJ1 with measures of Aβ, we first log transformed the raw Aβ data (log10+10) as most associations were nonlinear and calculated partial correlations with age as a co-variate. We further confirmed correlations between SYN and SYNJ1 with Aβ using a stepwise linear regression that included age at death, gender and PMI.
Results
Figure 1 shows a representative western blot experiment, a single band representing SYN was observed at ~38 kDa and SYNJ1 showed 1 or 2 bands at 140 and 180 kDa (Fig 1A). Given differences in PMI across our groups, we calculated the correlation between PMI and our outcome measures. Longer PMI was associated with higher protein levels of SYN (r=0.40 p=<.0005) but not with SYNJ1. Therefore, we decided to adjust for PMI in analyses involving SYN.
Figure 1.

SYN and SYNJ1 in DS. Representative western blots with SYN and SYNJ1 in YC, OC, DS, DSAD, and AD cases showing a ~38kDa and ~150 kDa bands, respectively. α-tubulin was used as a loading control and did not show systematic differences predicting SYNJ1 or SYN protein levels (A). SYN was significantly lower in DSAD and sporadic AD cases relative to control or DS cases alone. DSAD was not different from sporadic AD in SYN level. *p<.05 DSAD < DS, YC, OC, **p<.05 AD < DS, YC, OC. (B). SYNJ1 was significantly higher in DS overall and further increased in DSAD. DSAD cases had significantly higher levels of SYNJ1 than sporadic AD cases. *p<.05 DSAD>AD, OC. Bars represent group means and error bars are standard error of the mean.
As a first level of analysis, a comparison of the five groups (YC, OC, DS, DSAD and AD) was made using an analysis of variance and the Bonferonni correction was used for post hoc comparisons. Significant group differences were found for SYN (F(4,73)=2.75 p=0.035) and for SYNJ1 (F(4,73)=3.2 p=0.017). Figure 1B shows that SYN was significantly lower in DSAD and AD cases compared to DS or both YC and OC. SYNJ1 levels were lowest in the AD cases relative to younger controls and to DS cases and significantly lower than DS with AD cases Fig. 1C).
Next, we used a two factor univariate analysis that compared genotype (DS vs. CTL) and group (<40 or >40 years for controls vs. DS or DSAD) to determine the contribution of each factor independently to SYN and SYNJ1 protein levels (Table 2). Sporadic AD cases were not included in this analysis. For SYN, after co-varying for PMI, there was a significant main effect of genotype (i.e. the presence of DS) (F(1,67)=3.36 p=0.07). If PMI is removed from this analysis to increase the power, the presence of DS is a significant contributor to SYN protein levels (F(1,67)=3.91 p=0.05) as is the age group (<40 years vs > 40 years) (F(1,67)=6.4 p=0.014). The interaction was not significant suggesting that with age in controls and DSAD, there is a parallel decrease in SYN protein level. Overall, in DSAD SYN protein levels were ~50% of SYN levels in DS alone.
Table 2.
SYN and SYNJ1 as a function of genotype and age group.
| Synaptophysin* | ||||
| Age Group | <40 years | >40 years | Total | |
| Genotype | CTL | 13.8 (7.4) (n=13) | 10.2 (6.9) (n=15) | 11.9 (7.3) (n=28) |
| DS | 10.6 (7.4) (n=8) | 5.6 (5.1) (n=31) | 6.6 (5.9) (n=39) | |
| Total | 12.6 (7.4) (n=21) | 7.1 (6.1) (n=46) | 8.8 (7.0) (n=67) | |
| Synaptojanin 1* | ||||
| Age Group | <40 years | >40 years | Total | |
| Genotype | CTL | 5.7 (2.6) (n=13) | 4.3 (3.0) (n=15) | 5.0 (2.9) (n=28) |
| DS | 7.8 (7.5) (n=8) | 11.0 (10.7) (n=31) | 10.3 (10.1) (n=39) | |
| Total | 6.5 (5.0) (n=21) | 8.8 (9.5) (n=46) | 8.1 (8.3) (n=67) | |
Mean (Std Dev)
SYNJ1 was significantly different overall in DS when compared to control cases (F(1,67)=5.1 p=0.03)(Table 2). SYNJ1 also differed by age group (Young vs. Old) in controls and DSAD cases (F(1,67)=6.4 p=0.014). The interaction between age group and presence of DS was not significant (F(1,67)=0.16 p=0.69). The lack of interaction effect may be due to opposite effects of age or the presence of AD neuropathology in controls and DS cases, respectively. In controls, there was a lowering of SYNJ1 levels in cases over 40 years (young mean=5.70 (±2.57); old mean=4.33(±3.03); there was an opposite effect with increasing levels of SYNJ1 observed in DSAD (DS mean=7.77 (±7.51); DSAD mean=10.97 (±10.73). In DS overall, SYNJ1 levels were ~25% higher than age-matched controls. SYNJ1 levels were increased ~90% in DSAD cases as compared to OCs, and ~40% increased as compared to DS without AD.
We next determined whether synapse protein levels declined as a function of age in controls or DS groups, separately. Within the control group only (no sporadic AD cases), there were no significant age-associated decreases in SYN or SYNJ1. If sporadic AD cases were added into the analysis – there was a significant correlation between age at death and SYN (r=−0.45 p=0.007) and SYNJ1 (r=−0.35 p=0.039)(Fig 2A and C). In DS cases, SYN (r=−0.48 p=0.002) decreased as a function of age (Fig.2B) but no significant age effects were observed for SYNJ1 (Fig. 2D).
Figure 2.
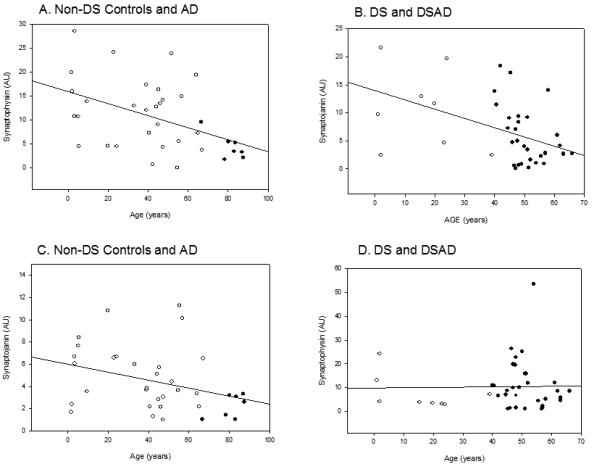
Changes in SYN and SYNJ1 with age. (A). Cases without DS showed a progressive decrease in SYN with age (open circles), with AD cases (closed circles) who were the oldest of the autopsy cases showed the lowest SYN overall. (B). In DS (open circles) and DSAD (closed circles), a similar significant decrease in SYN with age is observed. (C). SYNJ1 showed a progressive but not significant decrease with age in control cases (open circles) with AD cases (closed circles) being lower overall. (D). No age related changes were observed in SYNJ1 in DS (open circles) and DSAD (closed circles). Open circles are individual YC, OC, DS and DSAD cases. Closed circles are sporadic AD cases. Lines represent the linear regression.
Increasing Aβ and Aβ oligomer accumulation is associated with the disruption of synaptic function (reviewed in [33-35]). Thus, we compared Aβ and Aβ oligomer levels to the amount of SYN and SYNJ1 in DS only using partial correlations that corrected for age at death given that this comparison includes both age as well as AD neuropathology and it is challenging to distinguish these two variables. Higher amounts of oligomeric Aβ, SDS-soluble Aβ 1-40 and SDS-soluble Aβ 1-42 were associated with lower SYN protein levels (Table 3). Exploratory stepwise regressions predicting SYN and SYNJ1 from all of the Aβ measures (including PMI, genotype and age at death) confirmed the association between SYN level and SDS-soluble Aβ 1-42 (R2=0.16, p=0.001). For SYNJ1, the best predictor was FA extracted Aβ 1-42 (R2=0.074 p=0.03) but this was much weaker than that seen for SYN with SDS Aβ 1-42).
Table 3.
Correlations between Aβ and synapse proteins in DS cases (corrected for age).
| Aβ | Synaptophysin | Synaptojanin 1 |
|---|---|---|
| Oligomers | r=−0.008 p=0.95 | r=0.13 p=0.3 |
| PBS Aβ 40 | r=−0.23 p=0.07 | r=0.24 p=0.06 |
| SDS Aβ 40 | r=-−0.20 p=0.13 | r=0.24 p=0.05* |
| FA Aβ 40 | r=−0.23 p=0.07 | r=0.27 p=0.03* |
| PBS Aβ 42 | r=−0.16 p=0.21 | r=0.08 p=0.6 |
| SDS Aβ 42 | r=−0.25 p=0.05* | r=0.28 p=0.03* |
| FA Aβ 42 | r=−0.22 p=0.09 | r=0.27 p=0.03* |
Discussion
SYN protein levels in the frontal cortex of DSAD were significantly lower than DS alone and comparable to AD in persons without DS. Further, lower SYN was associated with higher soluble (SDS) Aβ and oligomeric Aβ levels in DS cases. SYN was also correlated with the amount of oligomeric and SDS extracted Aβ in cases with DS. These results suggest that as with sporadic AD, SYN is significantly reduced in DSAD and is associated with Aβ neuropathology. In contrast, higher overall levels of SYNJ1 were observed in DS, which is consistent with overexpression. However, novel results in the current study suggest that in contrast to our original hypothesis that SYNJ1 would decrease with DSAD, we found a further increase in SYJN1 in DSAD.
Previous studies of AD robustly report synaptic protein losses and particularly in SYN (e.g. [15-18]). These previous findings formed the rationale for selecting SYN as a protein marker to determine if there was a similar synaptic protein loss in DSAD compared to sporadic AD. The presence of AD neuropathology in DS was also associated with a similar level of SYN as in sporadic AD, which in turn was significantly lower than in DS cases without AD, suggesting this protein is affected by AD overall. In sporadic AD, SYN is associated with cognition and lower SYN is associated with poorer cognition [17]. Interestingly, higher SYN is observed in autopsy cases with normal cognition but with significant AD neuropathology at autopsy when compared to typical AD cases suggesting a possible protective effect or that SYN is critical to intact cognition [31,36]. Similar studies have not been described in DS primarily due to the challenge of obtaining cognitively characterized autopsy cases, but it is likely that a similar link is present.
SYNJ1 is a brain-enriched phosphoinositide phosphatase [22] that is engaged during endocytosis and synaptic vesicle cycling [23]. There is also evidence that SYNJ1 is involved with astrogliosis [37] and overexpression of SYNJ1 by 50% in Ts65Dn mice is associated with a 36% increase in glial fibrillary acidic protein (GFAP) but not S100β (a gene also on Chromosome 21) levels. SYNJ1 protein levels are increased in the DS brain [25], , particularly within the frontal cortex [26]. SYNJ1 is overexpressed in the Ts65Dn mouse model of DS and may play a role in the neuronal and behavioral deficits observed in these animals [38]. Indeed, behavioral deficits can be rescued in Ts65Dn mice by restoring SYNJ1 to disomy [39]. Further, also in DS mouse models overexpressing SYNJ1, several studies show cognitive dysfunction, early endosome enlargement and altered synaptic function [24,39,40]. These features closely mimic observations in DS autopsy studies showing endosomal enlargement that appears to be associated with the earliest Aβ deposition with age [41-43]. In contrast, knocking out SYNJ1 in mice leads to premature death, along with neurological dysfunction and abnormal synaptic function [23], thus lower levels of SYNJ1 in sporadic AD may be partially involved with cognitive dysfunction.
Chromosome 21 also contains the gene for DRYK1a, which can phosphorylate SYNJ1 at multiple sites [44], but the resulting link between these two proteins has yet to be established in DS brain. Oligomeric forms of Aβ, typically observed in AD brain, can destabilize SYNJ1 and leave individuals with DS with excessive levels of SYNJ1 vulnerable to Aβ toxicity [45]. However, increased expression of SYNJ1 in older adults with DS and AD relative to DS alone may be beneficial or predict the development of AD. Despite the presence of significant AD neuropathology in DS by the age of 40 years, the presence of dementia is typically delayed until after 50 years [5].
The results of the current study provide several novel insights. SYN decreases in DSAD as it does in sporadic AD and to approximately the same extent. Further, decreased SYN in DS is associated with increased Aβ neuropathology. SYN protein losses in the DS brain with AD may be linked to reports of neuronal loss [46-51] observed with age and with the presence of AD in these individuals. However, these reports are in brain regions including the entorhinal cortex and locus coereuleus with less information available on the frontal cortex. In contrast, SYNJ1 is consistently overexpressed in DS and may be an important component of synaptic function that leads to developmental cognitive deficits. DSAD is associated with higher SYNJ1 levels than DS alone and further associated with higher levels of SDS and FA extracted Aβ 1-40 and Aβ 1-42. The positive correlation between SYNJ1 and Aβ independently of age at death suggests one possibility; higher Aβ leads to reduced turnover of SYNJ1 in DSAD. The functional consequences of increased SYNJ1 in DSAD compared with DS may be twofold. First, given there is a 10 year delay between age of onset of AD neuropathology and signs of dementia [5], higher SYNJ1 may be beneficial as hypothesized for other proteins that are overexpressed due to trisomy 21 [52]. However, it is important to consider the multiple genes that are present in triplicate in DS and the many proteins that are overexpressed in DS can have multiple and interacting detrimental and beneficial effects on neuronal function. Further, sporadic AD is also associated with a > 10 year prodromal phase that as of yet, has not been linked to SYNJ1 [53]. Higher SYNJ1 in DSAD compared to DS may also be detrimental, and as discussed previously, transgenic mice overexpressing SYNJ1 show abnormal neurological function [24,39,40].
The current study of aging and AD neuropathology in DS shows that SYN protein losses occur in DSAD as it does in sporadic AD. Interestingly, SYNJ1, which is overexpressed in DS is higher with the presence of AD neuropathology in DS. Given the significant number of genes on chromosome 21 that involve multiple synapse associated pathways, this should be a very interesting area to explore that may lead to novel interventions.
Acknowledgments
Research reported in this manuscript was supported by Eunice Kennedy Shriver National Institute of Child Health and Development of the National Institutes of Health under award number R01HD064993 to EH and FAS, R01HD065160 and R01AG16573 to ITL. Autopsy tissue was obtained from the UCI-ADRC (P50AG16573), from the UK ADC (P30AG28383) and from the NICHD Brain and Tissue Bank for Developmental Disorders of the University of Maryland, Baltimore, MD, contract HHSN275200900011C (N01HD90011). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- [1].Lejeune J, Gautier M, Turpin R. Etude des chromosomes somatiques de neuf enfants mongoliens. Comptes Rendus Hebdomadaires des Seances de L'Academie des Sciences. 1959;248:1721–2. [PubMed] [Google Scholar]
- [2].Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–21. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Roizen NJ, Patterson D. Down's syndrome. Lancet. 2003;361(9365):1281–9. doi: 10.1016/S0140-6736(03)12987-X. [DOI] [PubMed] [Google Scholar]
- [4].Strauss D, Eyman RK. Mortality of people with mental retardation in California with and without Down syndrome, 1986-1991. Am J Ment Retard. 1996;100(6):643–53. [PubMed] [Google Scholar]
- [5].Schupf N, Sergievsky GH. Genetic and host factors for dementia in Down's syndrome. British journal of psychiatry. 2002;180:405–10. doi: 10.1192/bjp.180.5.405. [DOI] [PubMed] [Google Scholar]
- [6].Mann DMA, Esiri MM. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down's syndrome. J Neurol Sci. 1989;89:169–79. doi: 10.1016/0022-510x(89)90019-1. [DOI] [PubMed] [Google Scholar]
- [7].Wisniewski K, Howe J, Williams G, Wisniewski HM. Precocious aging and dementia in patients with Down's syndrome. Biological Psychiatry. 1978;13(5):619–27. [PubMed] [Google Scholar]
- [8].Wisniewski K, Wisniewski H, Wen G. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol. 1985;17:278–82. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- [9].Tanzi RE, McClatchey AI, Lamperti ED, Villa-komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer's diase. Nature. 1988;333:528–30. doi: 10.1038/331528a0. [DOI] [PubMed] [Google Scholar]
- [10].DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- [11].Scheff SW, Price DA. Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24(8):1029–46. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- [12].Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27(10):1372–84. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- [13].Ozcelik T, Lafreniere RG, Archer BT, 3rd, Johnston PA, Willard HF, Francke U, Sudhof TC. Synaptophysin: structure of the human gene and assignment to the X chromosome in man and mouse. Am J Hum Genet. 1990;47(3):551–61. [PMC free article] [PubMed] [Google Scholar]
- [14].Wiedenmann B, Franke WW. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell. 1985;41(3):1017–28. doi: 10.1016/s0092-8674(85)80082-9. [DOI] [PubMed] [Google Scholar]
- [15].Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol. 1991;138(1):235–46. [PMC free article] [PubMed] [Google Scholar]
- [16].Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett. 1989;103(2):234–9. doi: 10.1016/0304-3940(89)90582-x. [DOI] [PubMed] [Google Scholar]
- [17].Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Annals of Neurology. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- [18].Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr., Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7(2):103–17. doi: 10.3233/jad-2005-7203. discussion 73-80. [DOI] [PubMed] [Google Scholar]
- [19].Downes EC, Robson J, Grailly E, Abdel-All Z, Xuereb J, Brayne C, Holland A, Honer WG, Mukaetova-Ladinska EB. Loss of synaptophysin and synaptosomal-associated protein 25-kDa (SNAP-25) in elderly Down syndrome individuals. Neuropathol Appl Neurobiol. 2008;34(1):12–22. doi: 10.1111/j.1365-2990.2007.00899.x. [DOI] [PubMed] [Google Scholar]
- [20].Bugiani O, Giaccone G, Verga L, Pollo B, Ghetti B, Frangione B, Tagliavini F. Alzheimer patients and Down patients: abnormal presynaptic terminals are related to cerebral preamyloid deposits. Neurosci Lett. 1990;119(1):56–9. doi: 10.1016/0304-3940(90)90754-w. [DOI] [PubMed] [Google Scholar]
- [21].Cremona O, Nimmakayalu M, Haffner C, Bray-Ward P, Ward DC, De Camilli P. Assignment of SYNJ1 to human chromosome 21q22.2 and Synj12 to the murine homologous region on chromosome 16C3-4 by in situ hybridization. Cytogenetics and cell genetics. 2000;88(1-2):89–90. doi: 10.1159/000015493. [DOI] [PubMed] [Google Scholar]
- [22].McPherson PS, Takei K, Schmid SL, De Camilli P. p145, a major Grb2-binding protein in brain, is co-localized with dynamin in nerve terminals where it undergoes activity-dependent dephosphorylation. J Biol Chem. 1994;269(48):30132–9. [PubMed] [Google Scholar]
- [23].Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99(2):179–88. doi: 10.1016/s0092-8674(00)81649-9. [DOI] [PubMed] [Google Scholar]
- [24].Cossec JC, Lavaur J, Berman DE, Rivals I, Hoischen A, Stora S, Ripoll C, Mircher C, Grattau Y, Olivomarin JC, de Chaumont F, Lecourtois M, Antonarakis SE, Veltman JA, Delabar JM, Duyckaerts C, Di Paolo G, Potier MC. Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum Mol Genet. 2012;21(14):3156–72. doi: 10.1093/hmg/dds142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cheon MS, Kim SH, Ovod V, Kopitar Jerala N, Morgan JI, Hatefi Y, Ijuin T, Takenawa T, Lubec G. Protein levels of genes encoded on chromosome 21 in fetal Down syndrome brain: challenging the gene dosage effect hypothesis (Part III) Amino Acids. 2003;24(1-2):127–34. doi: 10.1007/s00726-002-0340-6. [DOI] [PubMed] [Google Scholar]
- [26].Arai Y, Ijuin T, Takenawa T, Becker LE, Takashima S. Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev. 2002;24(2):67–72. doi: 10.1016/s0387-7604(01)00405-3. [DOI] [PubMed] [Google Scholar]
- [27].McIntire LB, Berman DE, Myaeng J, Staniszewski A, Arancio O, Di Paolo G, Kim TW. Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer's disease. J Neurosci. 2012;32(44):15271–6. doi: 10.1523/JNEUROSCI.2034-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci. 2008;28(6):1410–20. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer's disease. Neurobiol Aging. 1997;18(4 Suppl):S85–8. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- [31].Head E, Corrada MM, Kahle-Wrobleski K, Kim RC, Sarsoza F, Goodus M, Kawas CH. Synaptic proteins, neuropathology and cognitive status in the oldest-old. Neurobiol Aging. 2009;30(7):1125–34. doi: 10.1016/j.neurobiolaging.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Beckett TL, Niedowicz DM, Studzinski CM, Weidner AM, Webb RL, Holler CJ, Ahmed RR, LeVine H, 3rd, Murphy MP. Effects of nonsteroidal anti-inflammatory drugs on amyloid-beta pathology in mouse skeletal muscle. Neurobiology of disease. 2010;39(3):449–56. doi: 10.1016/j.nbd.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nimmrich V, Ebert U. Is Alzheimer's disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev Neurosci. 2009;20(1):1–12. doi: 10.1515/revneuro.2009.20.1.1. [DOI] [PubMed] [Google Scholar]
- [34].Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid {beta} associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24(45):10191–200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Arnold SE, Louneva N, Cao K, Wang LS, Han LY, Wolk DA, Negash S, Leurgans SE, Schneider JA, Buchman AS, Wilson RS, Bennett DA. Cellular, synaptic, and biochemical features of resilient cognition in Alzheimer's disease. Neurobiol Aging. 2013;34(1):157–68. doi: 10.1016/j.neurobiolaging.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Herrera F, Chen Q, Fischer WH, Maher P, Schubert DR. Synaptojanin-1 plays a key role in astrogliogenesis: possible relevance for Down's syndrome. Cell Death Differ. 2009;16(6):910–20. doi: 10.1038/cdd.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gardiner K. Predicting pathway perturbations in Down syndrome. J Neural Transm Suppl. 2003;(67):21–37. doi: 10.1007/978-3-7091-6721-2_2. [DOI] [PubMed] [Google Scholar]
- [39].Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, Arancio O, Davisson MT, Antonarakis SE, Gardiner K, De Camilli P, Di Paolo G. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down's syndrome. Proc Natl Acad Sci U S A. 2008;105(27):9415–20. doi: 10.1073/pnas.0803756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chang KT, Min KT. Upregulation of three Drosophila homologs of human chromosome 21 genes alters synaptic function: implications for Down syndrome. Proc Natl Acad Sci U S A. 2009;106(40):17117–22. doi: 10.1073/pnas.0904397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–86. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, Carlson EJ, Staufenbiel M, Nixon RA. APP gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of Down syndrome. J Neurosci. 2003;23(17):6788–92. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25(10):1263–72. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- [44].Adayev T, Chen-Hwang MC, Murakami N, Wang R, Hwang YW. MNB/DYRK1A phosphorylation regulates the interactions of synaptojanin 1 with endocytic accessory proteins. Biochem Biophys Res Commun. 2006;351(4):1060–5. doi: 10.1016/j.bbrc.2006.10.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Berman DE, Dall'armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G. Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci. 2008;11(5):547–54. doi: 10.1038/nn.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sadowski M, Wisniewski HM, Tarnawski M, Kozlowski PB, Lach B, Wegiel J. Entorhinal cortex of aged subjects with Down's syndrome shows severe neuronal loss caused by neurofibrillary pathology. Acta Neuropathol (Berl) 1999;97(2):156–64. doi: 10.1007/s004010050968. [DOI] [PubMed] [Google Scholar]
- [47].Mann DM, Royston MC, Ravindra CR. Some morphometric observations on the brains of patients with Down's syndrome: their relationship to age and dementia. J Neurol Sci. 1990;99(2-3):153–64. doi: 10.1016/0022-510x(90)90152-d. [DOI] [PubMed] [Google Scholar]
- [48].Marcyniuk B, Mann DM, Yates PO, Ravindra CR. Topography of nerve cell loss from the locus caeruleus in middle aged persons with Down's syndrome. J Neurol Sci. 1988;83(1):15–24. doi: 10.1016/0022-510x(88)90016-0. [DOI] [PubMed] [Google Scholar]
- [49].Casanova MF, Walker LC, Whitehouse PJ, Price DL. Abnormalities of the nucleus basalis in Down's syndrome. Ann Neurol. 1985;18(3):310–3. doi: 10.1002/ana.410180306. [DOI] [PubMed] [Google Scholar]
- [50].Ball MJ, Nuttall K. Neurofibrillary tangles, granulovacuolar degeneration, and neuron loss in Down Syndrome: quantitative comparison with Alzheimer dementia. Ann Neurol. 1980;7(5):462–5. doi: 10.1002/ana.410070512. [DOI] [PubMed] [Google Scholar]
- [51].Karlsen AS, Pakkenberg B. Total Numbers of Neurons and Glial Cells in Cortex and Basal Ganglia of Aged Brains with Down Syndrome--A Stereological Study. Cerebral cortex. 2011 doi: 10.1093/cercor/bhr033. [DOI] [PubMed] [Google Scholar]
- [52].Head E, Lott IT, Patterson D, Doran E, Haier RJ. Possible compensatory events in adult Down syndrome brain prior to the development of Alzheimer disease neuropathology: targets for nonpharmacological intervention. J Alzheimers Dis. 2007;11(1):61–76. doi: 10.3233/jad-2007-11110. [DOI] [PubMed] [Google Scholar]
- [53].Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68(1):1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]