

Abstract
Capillary zone electrophoresis-tandem mass spectrometry (CZE-MS/MS) has recently attracted attention as a tool for shotgun proteomics. However, its performance for this analysis has fallen far below that of reversed phase liquid chromatography (RPLC)-MS/MS. Here, we report the use of a CZE method with a wide separation window (up to 90 min) and high peak capacity (~300). This method is coupled to an Orbitrap Fusion mass spectrometer via an electro-kinetically pumped sheath flow interface for analysis of complex proteome digests. Single-shot CZE-MS/MS identified over 10 000 peptides and 2 100 proteins from a HeLa cell proteome digest in ~100 min. This performance is nearly an order of magnitude superior to earlier CZE studies and is within a factor of 2 to 4 of state-of-the-art nano ultrahigh pressure LC system.
Keywords: CZE-MS/MS, improved electro-kinetically pumped sheath flow interface, shot-gun proteomics, Orbitrap Fusion
In shotgun proteomics, samples are first digested with enzymes to generate peptides, followed by reversed phase liquid chromatography (RPLC) separation and tandem mass spectrometry (MS/MS) detection. The resulting MS/MS spectra are then analyzed using correlation against a database.[1-3]
Recently, capillary zone electrophoresis (CZE)-electrospray ionization (ESI)-MS/MS has been suggested as an alternative to RPLC in shotgun proteomics.[4-6] CZE-MS and RPLC-MS generate complementary peptide identifications, and the combination of these two techniques can improve protein sequence coverage.[7-10] CZE-MS/MS can generate superior performance to RPLC-MS/MS for trace peptide analysis.[9,10] However, the performance of CZE-MS/MS for shotgun proteomics has been disappointing. To date, single shot CZE-MS/MS proteomic analyses have identified ~1,000 peptides and ~300 proteins,[10,11] which is at least 10-times less than the state-of-art RPLC-MS/MS system.[12-15]
Two obstacles limit CZE-MS performance. First, the sample loading capacity of CZE is typically 10-times less than RPLC; this issue hinders the detection of low abundance proteins. Second, the separation window in CZE-MS is usually <20 min. In contrast, RPLC routinely generates separation windows in excess of 300 min, which provides ample opportunity for peptide identification by MS/MS.
To improve the number of identifications using single-shot CZE-MS/MS for complex proteome analysis, we report two innovations. First, we employ electrophoretic conditions that generate separation windows up to 90 min and stacking injection conditions that increase sample-loading amount. Second, we couple a state-of-the-art mass spectrometer with an efficient and robust CZE electrospray interface. The MS system combines a mass filter, collision cell, high-field Orbitrap analyzer, and a dual cell linear ion trap analyzer geometry (Q-OT-qIT, Orbitrap Fusion) to provide up to 20 Hz MS/MS acquisition speed.[15] The combination of a long separation window with high MS/MS sampling rates generates ~50,000 tandem mass spectra in a single CZE separation, producing remarkable improvements in proteomic coverage over earlier reports.
Figure 1 (top trace) presents the CZE-MS/MS analysis of a HeLa cell proteome digest, generating a ~90 min wide separation window. This separation window is the widest, to our knowledge, for CZE-MS/MS analysis of peptides. We used an unsupervised nonlinear least squares routine to fit a Gaussian function to the extracted ion electropherograms of the 100 most intense peptide peaks in Figure 1 (top trace). The median peak width, measured as the standard deviation of the Gaussian function, was 8.5 s; the median number of theoretical plates was 100,000; and the peak capacity was 300.[16]
Figure 1.
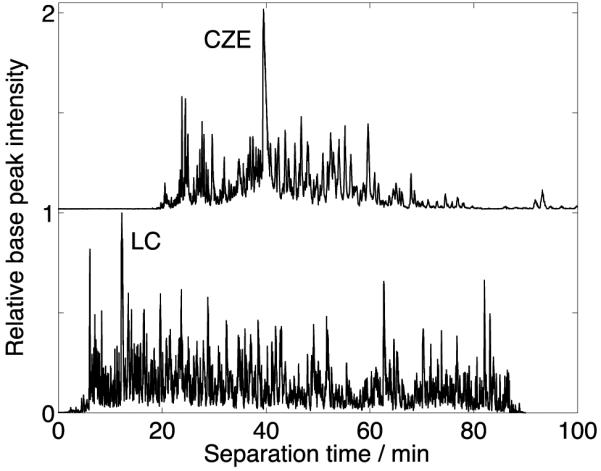
HeLa cell lysate digest data after single-shot CZE-MS/MS (top trace) and RPLC-MS/MS (bottom trace) analysis. ~400 ng and 300 ng of sample were loaded for CZE and RPLC analysis, respectively.
The wide separation window results from the use of a linear polyacrylamide coated capillary and a high concentration of acetic acid (5% (v/v)) in the separation buffer to produce very low electroosmotic flow. Separation efficiency improves from dissolution of samples in 0.03-0.04% (v/v) formic acid containing 30-40% (v/v) acetonitrile. The conductivity of the sample buffer is much lower than the separation buffer, producing sample stacking and peak sharpening. We used a second-generation electro-kinetically pumped sheath flow interface[17] to couple CZE to the mass spectrometer, which reduced sample diffusion in the spray emitter, compared with our first-generation interface.[18]
We used this system to analyze E. coli, yeast, and HeLa cell proteomes, Table 1. The single-shot CZE-MS/MS system identified about 1 000 proteins and 4 700 peptides from an E. coli tryptic digest in 1.5 h. The number of peptide and protein identifications (IDs) is three times larger than an earlier report of the use of CZE-MS/MS for the study of an unfractionated E. coli proteome.[10] The improved performance is a result of the generation of six-times more MS/MS spectra over a ~two-times longer separation period.
Table 1.
Summary of peptide and protein group identifications (IDs) from single-shot CZE-MS/MS for different samples.
| Peptide IDs[a] | Protein group IDs |
Samples | Analysis duration (min) |
|---|---|---|---|
| 4741 | 956 | Escherichia coli | 90 |
| 5961 | 1529 | Yeast | 105 |
| 10274 | 2149 | HeLa | 105 |
Proteome Discoverer 1.4 software with Mascot (version 2.2.4) search engine was employed for data analysis. False discovery rates of peptide identifications are < 1%.
We also performed the first application of CZE-MS/MS for analysis of the yeast proteome. The system identified > 1 500 proteins and ~ 6 000 peptides in ~100 min, Table 1. Roughly 4 500 proteins are expressed during log-phase yeast growth;[19] our single-shot CZE-MS/MS data covered 1/3 of that proteome. We further separated the yeast proteome digest to three fractions using RPLC. CZE-MS/MS analysis of the fractions identified 2 512 yeast proteins in 5 h, which covers >50% of the yeast proteome. This proteome dataset is the largest generated using CZE-MS/MS to date.
Application of CZE-MS/MS to the HeLa cell proteome produced > 2 100 proteins and > 10 000 peptides in 105 min, Table 1. These results are a ~10-times improvement in the number of peptide and protein IDs over the previous state-of-the-art single-shot CZE-MS/MS analysis. The results significantly reduce the gap between single-shot CZE-MS/MS and the state-of-art single-shot RPLC-MS/MS for analysis of a mammalian proteome.
Next, we evaluated the dynamic range of the identified yeast proteome with the CZE-MS/MS system. We performed a database search using MaxQuant (version 1.3); [20] 1 529 and 2 213 protein group IDs were identified from unfractionated and fractionated yeast samples, respectively, with both peptide and protein level false discovery rates < 1%, which are consistent with the data from Proteome Discoverer in Table 1. Based on the protein intensity information from MaxQuant,[14] we estimated the dynamic range of identified yeast proteome to be roughly 5 orders of magnitude for the unfractionated sample, which is similar to a published comprehensive yeast proteome study based on state-of-art single-shot RPLC-MS/MS.[14] Protein identifications for these experiments are provided in Supporting Material II, and replicate CZE runs for E. coli and yeast are provided as S-Figures 1-2 (Supporting Material I).
A recent multiple reaction monitoring study examined the detectability of 127 proteins that represent the full range of yeast protein expression.[21] Our single-shot CZE-MS/MS data identified 11 out of 12 proteins classified as “less than 50 copies/cell” and “western-blot band not quantifiable” in that work, although this classification may need to be revisited.[22]
We generated high-quality RPLC-MS/MS data using both the HeLa and yeast samples, Figure 1 (bottom trace) and S-Figures 3 (Supporting Material I). RPLC generated > 5 000 protein and 40 000 peptide IDs in 90 min from the HeLa sample; these values are ~2.5 and ~4 times higher than CZE. While 70% of the CZE peptides are detected by RPLC, CZE tends to identify larger peptides than RPLC, S-Figure 4 (Supporting Material I), consistent with our previous work.[10] The poorer performance of CZE is due to a ~2.5 times larger average peak width than RPLC, which is due to the large injection volume (>5% of the capillary volume) used in the CZE experiment. Duplicate runs for the yeast sample generated reproducible separation profiles and base peak intensities (S-Figures 2-3).
There is a small literature on the use of CZE for bottom-up analysis of complex proteomes; we summarize the data in Figure 2 and S-Table 1 (Supporting Material I). This learning curve demonstrates improved performance that results from a number of advancements. There is a steady increase in the complexity of the proteome, which increases the number of protein targets. There also is an improvement in the speed and mass accuracy of mass spectrometers, which leads to a concomitant increase in the number of IDs. Finally, improved electrophoresis conditions yield longer runs and increase number of IDs.
Figure 2.
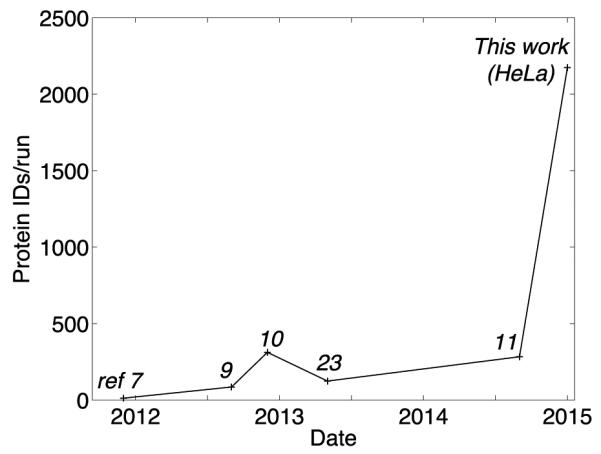
Number of protein IDs from a single CZE-MS/MS analysis of complex proteomes. Reference number for each point is listed in the figure. The average number of protein IDs per run from the analysis of multiple fractions was used for ref 7, 9, and 23.
It is interesting to speculate on further improvements. Improved mass spectrometer speed and resolution will allow identification of more peptides per run. Also, the peaks generated in this separation are ~10-times broader than expected for a diffusion-limited separation. Narrower peaks will come from optimization of injection conditions, and should produce a two- to four-fold improvement in peptide and protein IDs. In this case, the performance of CZE-MS/MS should equal that of the very best RPLC-MS/MS system for bottom-up proteomics. Such technology will have at least three important advantages. First, CZE instrumentation is inherently simpler and less expensive than ultrahigh pressure RPLC systems. Second, CZE samples a different peptide pool than RPLC; combination of the results from the two technologies will allow deeper sequencing and higher protein coverage. Third, as we will show elsewhere, CZE migration time can be easily predicted from a peptide’s sequence, which improves confidence in peptide IDs.
Experimental Section
The experimental details are provided in supporting material I.
Supplementary Material
Acknowledgements
We thank Dr. William Boggess in the Notre Dame Mass Spectrometry and Proteomics Facility for his help with this project. This work was funded by the National Institutes of Health (R01GM096767 – NJD) and (R01GM080148 – JJC) and the National Science Foundation (0701846 – JJC).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Wenger CD, Coon JJ. J. Proteome Res. 2013;12:1377–1386. doi: 10.1021/pr301024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Elias JE, Gygi SP. Nature Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- [3].Yates JR, Speicher S, Griffin PR, Hunkapiller T. Anal. Biochemistry. 1993;214:397–408. doi: 10.1006/abio.1993.1514. [DOI] [PubMed] [Google Scholar]
- [4].Heemskerk AA, Deelder AM, Mayboroda OA. Mass Spectrom. Rev. 2014 doi: 10.1002/mas.21432. doi: 10.1002/mas.21432. [DOI] [PubMed] [Google Scholar]
- [5].Sun L, Zhu G, Yan X, Dovichi NJ. Curr. Opin. Chem. Biol. 2013;17:795–800. doi: 10.1016/j.cbpa.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sun L, Zhu G, Yan X, Champion MM, Dovichi NJ. Proteomics. 2014;14:622–628. doi: 10.1002/pmic.201300295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li Y, Champion MM, Sun L, Champion PA, Wojcik R, Dovichi NJ. Anal. Chem. 2012;84:1617–1622. doi: 10.1021/ac202899p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Faserl K, Sarg B, Kremser L, Lindner H. Anal. Chem. 2011;83:7297–7305. doi: 10.1021/ac2010372. [DOI] [PubMed] [Google Scholar]
- [9].Wang Y, Fonslow BR, Wong CC, Nakorchevsky A, Yates JR., III Anal. Chem. 2012;84:8505–8513. doi: 10.1021/ac301091m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhu G, Sun L, Yan X, Dovichi NJ. Anal. Chem. 2013;85:2569–2573. doi: 10.1021/ac303750g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sun L, Zhu G, Mou S, Zhao Y, Champion MM, Dovichi NJ. J. Chromatogr. A. 2014;1359:303–308. doi: 10.1016/j.chroma.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kelstrup CD, Young C, Lavallee R, Nielsen ML, Olsen JV. J. Proteome Res. 2012;11:3487–3497. doi: 10.1021/pr3000249. [DOI] [PubMed] [Google Scholar]
- [13].Pirmoradian M, Budamgunta H, Chingin K, Zhang B, Astorga-Wells J, Zubarev RA. Mol. Cell. Proteomics. 2013;12:3330–3338. doi: 10.1074/mcp.O113.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nagaraj N, Kulak NA, Cox J, Neuhauser N, Mayr K, Hoerning O, Vorm O, Mann M. Mol. Cell. Proteomics. 2012;11:M111.013722. doi: 10.1074/mcp.M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. Mol. Cell. Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Busnel JM, Schoenmaker B, Ramautar R, Carrasco-Pancorbo A, Ratnayake C, Feitelson JS, Chapman JD, Deelder AM, Mayboroda OA. Anal. Chem. 2010;82:9476–9483. doi: 10.1021/ac102159d. [DOI] [PubMed] [Google Scholar]
- [17].Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ. Angew. Chem. Int. Ed. Engl. 2013;52:13661–13664. doi: 10.1002/anie.201308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wojcik R, Dada OO, Sadilek M, Dovichi NJ. Rapid Commun. Mass Spectrom. 2010;24:2554–2560. doi: 10.1002/rcm.4672. [DOI] [PubMed] [Google Scholar]
- [19].Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- [20].Cox J, Mann M. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- [21].Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Cell. 2009;138:795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Thakur SS, Geiger T, Chatterjee B, Bandilla P, Fröhlich F, Cox J, Mann M. Mol. Cell. Proteomics. 2011;10:M110.003699. doi: 10.1074/mcp.M110.003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yan X, Essaka DC, Sun L, Zhu G, Dovichi NJ. Proteomics. 2013;13:2546–2551. doi: 10.1002/pmic.201300062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.