

Abstract
The WNT16 locus is a major determinant of cortical bone thickness and nonvertebral fracture risk in humans. The disability, mortality and costs caused by osteoporosis-induced nonvertebral fractures are enormous. We demonstrate here that Wnt16-deficient mice develop spontaneous fractures as a result of low cortical thickness and high cortical porosity. In contrast, trabecular bone volume is not altered in these mice. Mechanistic studies revealed that WNT16 is osteoblast derived and inhibits human and mouse osteoclastogenesis both directly by acting on osteoclast progenitors and indirectly by increasing expression of osteoprotegerin (Opg) in osteoblasts. The signaling pathway activated by WNT16 in osteoclast progenitors is noncanonical, whereas the pathway activated in osteoblasts is both canonical and noncanonical. Conditional Wnt16 inactivation revealed that osteoblast-lineage cells are the principal source of WNT16, and its targeted deletion in osteoblasts increases fracture susceptibility. Thus, osteoblast-derived WNT16 is a previously unreported key regulator of osteoclastogenesis and fracture susceptibility. These findings open new avenues for the specific prevention or treatment of nonvertebral fractures, a substantial unmet medical need.
Osteoporosis is a common skeletal disease, affecting millions of individuals worldwide. Although substantial progress has been made in the therapeutic reduction of vertebral fracture risk in individuals with osteoporosis, nonvertebral fracture risk has been improved only marginally by currently available treatments, defining an unmet medical need1. Cortical bone, comprising 80% of the skeleton, is a major determinant of bone strength and nonvertebral fracture susceptibility, whereas vertebral fracture risk is determined mainly by trabecular bone mass2–4. Currently used anti-resorptive drugs reduce the risk of vertebral fractures by up to 70%, whereas the risk for nonvertebral fractures is only reduced by 20% with these drugs1, suggesting that trabecular and cortical bone might respond differently to signals involved in the regulation of skeletal homeostasis. Therefore, new insights into the biology of these compartments could be of great clinical and therapeutic importance.
WNT proteins belong to a family of secreted cysteine-rich glycoproteins that signal through both the WNT–β-catenin pathway, also termed the canonical WNT pathway, and noncanonical WNT pathways5–8. Activation of canonical β-catenin signaling increases bone mass, and rare human genetic mutations affecting bone have been identified in components of the canonical WNT signaling machinery5,9–13. Notably, mouse genetic studies have confirmed that activation of the canonical WNT signaling pathway increases bone formation by promoting osteoblast differentiation and activity while inhibiting bone resorption indirectly by reducing osteoclastogenesis, mainly by regulating OPG14–22. A direct effect of canonical WNT signaling on osteoclastogenesis was also proposed recently, as mice lacking β-catenin in osteoclast precursors develop osteopetrosis because of reduced osteoclast numbers and activity23. Furthermore, activation of noncanonical WNT signaling by osteoblast-produced WNT5a has been shown to increase osteoclastogenesis through a receptor tyrosine kinase-like orphan receptor 2 (ROR2)–FZD receptor complex24. Thus, there is increasing evidence for the importance of noncanonical WNT signaling in bone and crosstalk between the pathways5.
We and others recently performed genome-wide association studies demonstrating that the WNT16 locus reproducibly associates with bone mineral density (BMD), cortical bone thickness and nonvertebral fractures4,25–29. A preliminary screening of multiple (n ≈ 4,500) gene-targeted mouse models within Lexicon Pharmaceuticals reported that a Wnt16−/− mouse strain displays reduced cortical bone thickness, but these studies did not explore the mechanisms of action for WNT16 in bone4,30.
To explore the mechanisms by which WNT16 regulates both general bone homeostasis and specifically the cortical bone compartment, we generated and analyzed several mouse strains with global as well as cell-specific Wnt16 inactivation. We demonstrate that Wnt16 deletion reduces cortical bone thickness and increases cortical bone porosity, leading to spontaneous fractures in these mice. Most notably, this effect is restricted to cortical bone, whereas trabecular bone is not affected, thereby demonstrating a dissociation between the regulation of cortical and trabecular bone homeostasis. Mechanistically, we found that WNT16 is derived from osteoblast-lineage cells and activates both canonical and noncanonical WNT signaling to inhibit human and mouse osteoclastogenesis through direct effects on osteoclast progenitors and indirect effects that increase Opg expression in osteoblasts.
RESULTS
Lower cortical but not trabecular bone mass in Wnt16−/− mice
We assessed Wnt16 mRNA levels in several tissues from mice using real-time PCR analysis and found the highest levels in cortical bone. We found that Wnt16, together with Wnt5a, is highly expressed in adult cortical bone, and its expression is markedly higher than that of the other WNT ligands examined (Fig. 1a and Supplementary Fig. 1a). Wnt16 mRNA levels were high in primary cultured osteoblasts, whereas we detected no expression in osteoclast cultures (Fig. 1a). Immunohistochemistry revealed abundant WNT16 staining in osteoblasts lining the surfaces of cortical bone (Fig. 1b).
Figure 1.

Wnt16−/− mice have reduced cortical but not trabecular bone mass. (a) Wnt16 mRNA levels in different tissues. Cort., cortical; gastroc., gastrocnemius; WAT, white adipose tissue; BAT, brown adipose tissue; Obl, primary cultured osteoblasts; Ocl, primary cultured osteoclasts; M, macrophage colony-stimulating factor (M-CSF) stimulation; M/RL, M-CSF and RANKL stimulation; ND, not detectable (n = 6). (b) Immunohistochemistry analysis of WNT16 immunostaining (red) and nuclear staining (blue, DAPI) in the cortical bone (C) of WT and Wnt16−/− mice. BM, bone marrow. The white outlines indicate the cortical bone surfaces. Scale bars, 50 μm. (c) Schematic of the Wnt16 gene in the generated Wnt16−/−, exon 1-4 mice. (d) Whole-mount Alizarin red and Alcian blue staining of Wnt16−/− and WT embryos (E18.5) with close ups of the paws (middle) and tibia and fibula (right). Scale bars, 2.5 mm. (e–g) Femur lengths, cortical bone mineral content (BMC) and trabecular BMD of the tibia as measured by peripheral quantitative CT (pQCT) in Wnt16−/− and WT mice at 5 (n = 9), 8 (n = 9) and 11 (female WT, n = 9; female Wnt16−/−, n = 8; male WT, n = 11; male Wnt16−/−, n = 12) weeks of age. (h,i) Cortical thickness (h) and distal metaphyseal trabecular bone volume per total volume (BV/TV; i) as measured by micro-CT in the femurs of 5- and 11-week-old female Wnt16−/− and WT mice (5 weeks old: WT, n = 9; Wnt16−/−, n = 8; 11 weeks old: n = 6). Left, representative micro-CT images of the experiment described to the right. Scale bars, 500 μm. (j) Bone volume per total volume as measured by histomorphometry of trabecular bone in the L4 vertebra of 11-week-old female Wnt16−/− (n = 6) and WT (n = 7) mice. All values are given as the mean ± s.e.m. **P < 0.01, Student’s t test compared to WT.
To determine the role of WNT16 in the skeleton, we evaluated two separate mouse models with global Wnt16 inactivation. The first model has all four exons of Wnt16 deleted (Wnt16−/−, exon 1-4; Fig. 1c), and the second mouse model has the first three exons of Wnt16 deleted (Wnt16−/−, exon 1-3)4. The phenotypes of the Wnt16−/−, exon 1-4 (Figs. 1 and 2 and Supplementary Tables 1–4) and Wnt16−/−, exon 1-3 mouse models (Supplementary Fig. 1b–h and Supplementary Tables 5 and 6) are identical, and we refer to both mouse models as Wnt16−/− mice, if not otherwise specified. Wnt16−/− embryos (embryonic day (E) 18.5) showed normal embryonic skeletal development and were born apparently healthy (Fig. 1d,e and Supplementary Table 1). The cortical bone thickness, cortical cross-sectional area and cortical mineral content of the long bones were substantially lower in young Wnt16−/− mice compared to age- and sex-matched wild-type (WT) mice (5–12 weeks old; Fig. 1f,h, Supplementary Figs. 1b–d and 2d and Supplementary Tables 1, 2, 5 and 6). In contrast, trabecular bone parameters were the same in Wnt16−/− and WT mice (Fig. 1g,i,j, Supplementary Fig. 1e and Supplementary Tables 1, 3, 5 and 6). Notably, 16-week-old Wnt16−/−, exon 1-3 mice displayed markedly lower cortical thickness compared to WT mice (Supplementary Fig. 1d). Similar to the results in younger mice, trabecular bone was not affected in the 16-week-old mice (Supplementary Fig. 1e). These findings suggest a role for WNT16 in the maintenance of cortical bone homeostasis during not only skeletal growth but also skeletal maturity. Although we observed the cortical bone phenotype in both male and female mice, it was generally more pronounced in females compared with males (8-week-old mice; tibia cortical bone thickness in females −58 ± 3% (mean ± s.e.m.; P < 0.01) and males (–22 ± 6%, P < 0.05) compared to WT mice).
Figure 2.
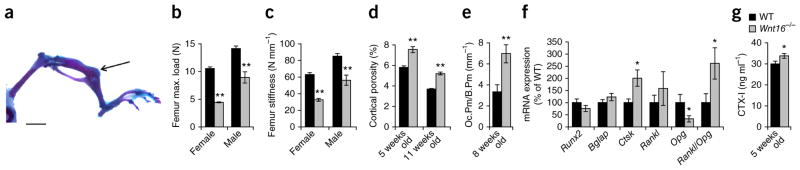
Spontaneous fractures as a result of several defects of cortical bone in Wnt16−/− mice. (a) Spontaneous fracture of the tibia (arrow) in a Wnt16−/− mouse. Scale bar, 500 μm. (b,c) Maximal (max.) load (b) and stiffness (c) at failure determined by three-point bending of the femur in 8-week-old Wnt16−/− mice compared to WT mice (n = 9). (d) Cortical porosity in the femur diaphysis as measured by micro-CT of female Wnt16−/− compared to WT mice (5 weeks old: WT, n = 9; Wnt16−/−, n = 8; 11 weeks old: n = 6). (e) Osteoclast surface/bone surface (Oc.Pm/B.Pm) in femur cortical bone of 8-week-old female Wnt16−/− (n = 9) compared to WT (n = 7) mice. (f) mRNA expression analyses of femur cortical bone in 8-week-old female Wnt16−/− (n = 8) and WT (n = 9) mice. Rankl/Opg ratio is the ratio of the two transcripts. (g) Serum levels of the bone resorption marker CTX-I in 5-week-old Wnt16−/− and WT mice (n = 9). *P < 0.05, **P < 0.01, Student’s t test compared to WT. All values are given as the mean ± s.e.m.
Cortical bone defects result in fractures in Wnt16−/− mice
About one-third of Wnt16−/− mice developed spontaneous tibia diaphyseal fractures (Fig. 2a and Supplementary Figs. 1f and 2a). We verified lower bone strength in the long bones of Wnt16−/− mice by three- point bending analyses of excised femurs compared to WT mice (Fig. 2b,c). The lower bone strength was caused by both a thinner cortical bone and a higher cortical porosity (Fig. 2d and Supplementary Fig. 1g).
Wnt16−/− mice displayed higher cortical bone resorption compared to WT mice, as demonstrated by more osteoclasts and larger osteoclast-covered surface in cortical bone at the endocortical surface and higher expression of the osteoclast-specific transcript Ctsk in cortical bone, as well as higher serum levels of the bone resorption marker CTX-I (type I collagen fragments; Fig. 2e–g and Supplementary Table 6), despite no changes in trabecular bone resorption parameters (Supplementary Tables 3 and 6). Notably, these effects were associated with lower Opg expression, resulting in a higher ratio of Rankl to Opg expression in cortical bone (Fig. 2f and Supplementary Fig. 1h).
Histomorphometric analyses of the endosteal surface of cortical bone in the tibia diaphysis demonstrated unchanged mineral apposition rate (MAR) and endosteal osteoid surface to bone surface in Wnt16−/− mice (Supplementary Tables 2 and 6). The periosteal MAR was not affected in the cortical bone of 8-, 11- or 12-week-old Wnt16−/− mice, but there was a nonsignificant trend of lower periosteal MAR in 6-week-old Wnt16−/− mice compared to WT mice (Supplementary Tables 2 and 6). Neither bone formation nor the number of osteoclasts per bone surface were affected in the trabecular bone of Wnt16−/− mice (Supplementary Tables 3 and 6), and the serum plasma levels of calcium homeostasis parameters were similar in Wnt16−/− and WT mice (Supplementary Table 4).
Micro computed tomography (micro-CT) analyses of the periosteal and endosteal circumferences in 11-week-old Wnt16−/− mice indicated that the lower cortical thickness in the tibia compared to WT mice was due to a slightly larger endosteal circumference, whereas in the femur, both the periosteal and endosteal circumferences were smaller in Wnt16−/− mice compared to WT mice (Supplementary Fig. 2b–d). Thus, the lower cortical thickness observed after global depletion of WNT16 appears to be mostly the result of higher endosteal resorption. In addition, it is probable that a slower periosteal apposition at early time points may also contribute to the thinning of the cortical bone observed at later time points.
Osteoblast-derived WNT16 inhibits osteoclastogenesis
We then further identified the source and target cells of WNT16 in the bone microenvironment. In vitro cultures of calvarial osteoblasts demonstrated that Wnt16 expression increased during osteoblastic differentiation (Fig. 3a) and that osteoblastic proliferation and differentiation were similar in cultures from WT and Wnt16−/− mice (Fig. 3b–d). FACS analysis showed that the number of osteoclast progenitors in bone marrow cells was not altered between WT and Wnt16−/− mice (Supplementary Fig. 3a). We observed no difference in the formation of tartrate-resistant acid phosphatase–positive multinucleated osteoclasts (TRAP+MuOCLs) or in the release of TRAP5b or CTX-I when we cultured RANKL-stimulated bone marrow cells from WT or Wnt16−/− mice on plastic or bone slices (Fig. 3e,f). In addition, we noted no difference in the formation of TRAP+MuOCLs in purified bone marrow macrophage (BMM) cultures from WT and Wnt16−/− mice in response to RANKL stimulation (Supplementary Fig. 3b). These studies demonstrate that there is no major cell-autonomous effect of WNT16 in osteoblast or osteoclast proliferation or functions.
Figure 3.

Osteoblast-derived WNT16 inhibits osteoclastogenesis. (a–d) Calvarial osteoblast cultures. (a) Wnt16 mRNA expression (bottom) and staining for alkaline phosphatase (top) during differentiation of osteoblasts. Scale bars, 500 μm. d, day. (b) Proliferation of Wnt16−/− and WT osteoblasts. (c) Alizarin red staining of mineralized nodules in Wnt16−/− and WT osteoblast cultures (left) and quantification of eluted Alizarin red (ALZ, right). Scale bars, 1 cm (top); 500 μm (bottom). (d) mRNA expression of Alpl and Runx2. (e) Number of TRAP+MuOCLs and release of TRAP5b into the medium in RANKL-stimulated bone marrow cultures from Wnt16−/− and WT mice on plastic (e, top images and left graph) or bone (e, bottom images and right graph). Scale bars, 400 μm. (f) Release of CTX-I from RANKL-stimulated bone marrow cells from Wnt16−/− mice cultured on bone. (g) Cocultures of BMMs and calvarial osteoblasts from Wnt16−/− and WT mice. Representative of two separate TRAP staining carried out in triplicate (top) and quantification (bottom) of TRAP+MuOCLs in cocultures. Scale bars, 200 μm. *P < 0.05, Student’s t test compared to WT Obl and WT BMMs or WT Obl and Wnt16−/− BMMs. All values are given as the mean ± s.e.m.
We next evaluated the possible paracrine role of osteoblast-derived WNT16 on osteoclastogenesis using mix-and-matched cocultures of calvarial osteoblasts and BMMs from WT or Wnt16−/− mice. More osteoclasts were formed in cultures with osteoblasts from Wnt16−/− than from WT mice (Fig. 3g), whereas there was no difference when the BMMs in culture were from WT or Wnt16−/− mice (Fig. 3g), which is in line with the lack of Wnt16 expression in BMMs (Fig. 1a). These findings indicate that osteoblast-derived WNT16 either directly or indirectly inhibits osteoclastogenesis.
WNT16 increases Opg expression in osteoblasts
Because Opg expression was lower in cortical bone of Wnt16−/− compared to WT mice (Fig. 2f and Supplementary Fig. 1h), we determined whether this difference could be the result of an effect of WNT16 on Opg expression in osteoblasts. Indeed, osteoblastic MC3T3-E1 cells responded to recombinant WNT16 with substantially enhanced Opg mRNA expression (Fig. 4a).
Figure 4.

WNT16 increases OPG expression and signals through both canonical and noncanonical pathways in osteoblasts. (a) mRNA expression analyses of MC3T3-E1 cells treated with WNT16 (n = 3). (b) mRNA expression analyses of cortical bone isolated from WT and Wnt16−/− mice (n = 4). (c) mRNA expression analyses of MC3T3-E1 cells treated with vehicle, WNT3a or WNT16 (n = 3). (d) TOPflash luciferase assay of MC3T3-E1 cells treated with vehicle, WNT3a or WNT16. Data are presented as a fold change normalized to Renilla compared to vehicle-treated cells. (e) Western blot of phosphorylated and total LRP6 in MC3T3-E1 cells treated with WNT16. Blots are representative of three separate experiments carried out in triplicate. (f) Nonphosphorylated levels of β-catenin (non–pβ-catenin) and β-catenin in MC3T3-E1 cells treated with vehicle, 100 ng ml−1 WNT3a or 100 ng ml−1 WNT16. (g) mRNA expression analyses of MC3T3-E1 cells treated with or without WNT16 in the presence or absence of XAV939 (n = 3). Data are presented as a fold change normalized to 18S compared to vehicle-treated cells. (h) Phosphorylated levels of JNK in calvariae isolated from 3-day-old WT and Wnt16−/− mice. (i) Western blot of phosphorylated JNK and cJUN in MC3T3-E1 cells treated with WNT16. (j) Luciferase assay of HEK293 cells stably expressing ROR2-hTRK-luciferase (luc) treated with vehicle, WNT5a or WNT16. Data are presented as a fold change normalized to vehicle-treated cells. All values are given as the mean ± s.e.m. *P < 0.05, **P < 0.01, Student’s t test compared to WT or vehicle. ††P < 0.01, Student’s t test compared to WNT16-treated cells.
The expression of the canonical WNT target genes Axin2 and Tcf1 (also called Hnf1a) was significantly lower in cortical bone of Wnt16−/− mice compared to that of WT mice (Fig. 4b), and the expression of these genes was significantly higher in MC3T3-E1 cells treated with WNT16 compared to vehicle-treated cells (Fig. 4c). Confirming these findings, WNT16 treatment led to a mild but significant activation of the TOPflash reporter and to increased levels of phosphorylated LRP6 and nonphosphorylated β-catenin in osteoblasts (Fig. 4d–f). To provide further evidence that WNT16 signals through canonical WNT signaling in osteoblasts, we treated MC3T3-E1 cells with XAV939, a tankyrase inhibitor that has been shown to block β-catenin–dependent signaling by stabilizing Axin2 and the destruction complex31. XAV939 significantly inhibited a WNT16-dependent increase in Axin2 expression, and its effect on WNT16-dependent induction of Opg followed the same trend but did not reach significance (Fig. 4g). Overall, these data show that WNT16 has the capacity to signal through the classical canonical pathway in osteoblasts, albeit less efficiently than classical canonical WNT ligands such as WNT3a (Fig. 4c–g).
WNT16 has also been proposed to signal through the noncanoni-cal Jun–N-terminal kinase cascade through phosphorylation of JUN and JNK32. The levels of phosphorylated JNK (pJNK) were lower in calvarial bone from Wnt16−/− mice compared to WT mice (Fig. 4h), and pJNK and phosphorylated cJUN (p-cJUN) levels were markedly increased by WNT16 treatment of MC3T3-E1 cells (Fig. 4i). Because JNK activation is a downstream effector of ROR2 receptor signaling, we then examined whether WNT16 functions through the ROR2-JNK cascade using the noncanonical WNT-specific ROR2-hTRK luciferase assay24,33. As expected, treatment with WNT5a, a classical activator of the WNT-ROR2-JNK pathway24,34, led to a significant increase in luciferase activity (Fig. 4j). In contrast, WNT16 did not affect this ROR2 reporter assay. Collectively, these data demonstrate that WNT16 signals through both the classical canonical WNT pathway and the noncanonical Jun–N-terminal kinase cascade in osteoblasts, implying the presence of an autocrine loop in these cells, resulting in increased Opg expression and decreased osteoclastogenesis.
WNT16 directly inhibits osteoclastogenesis
In addition to its OPG-mediated effects on osteoclastogenesis, we found that WNT16 directly affects the response of osteoclast precursors to RANKL. When added to purified mouse BMM cultures in the absence of osteoblasts, WNT16 abolished RANKL-stimulated formation of TRAP+MuOCLs (Fig. 5a). WNT16 also robustly inhibited RANKL-stimulated formation of TRAP+MuOCLs, CTX-I release and the number of resorption pits when BMMs were seeded on bone slices (Fig. 5b). Notably, we also observed a dose-dependent inhibitory effect of WNT16 on osteoclast formation in RANKL-stimulated human CD14+ monocyte cultures (Fig. 5c). Reduced osteoclast formation by WNT16 was associated with a time- and dose-dependent blunted mRNA expression of osteoclast functional genes (Fig. 5d and Supplementary Fig. 3c,d).
Figure 5.

WNT16 inhibits osteoclast differentiation through noncanonical WNT pathways. (a,b) RANKL-stimulated osteoclastogenesis of mouse BMMs on plastic (a, TRAP staining) and bone surface (b, TRAP staining) treated with recombinant WNT16. Scale bars, 100 μm. Numbers of TRAP+MuOCLs on plastic (a) and bone (b, left graph) and release of CTX-I from bone slices (b, right graph). (c) WNT16 dose-dependent inhibition of RANKL-stimulated osteoclastogenesis in human CD14+ monocyte cultures. Left, TRAP staining of RANKL-stimulated osteoclastogenesis with or without WNT16. Scale bars, 100 μm. Right, number of TRAP+MuOCLs per well. (d) Time-dependent (left) and dose-dependent (right) inhibition of Acp5 mRNA by WNT16 in RANKL-stimulated BMM cultures. (e) mRNA expression of Nfkb2 (left) and Relb (right) in RANKL-stimulated BMM cultures with or without WNT16. (f) NF-κB luciferase gene reporter assay in BMMs cultured in M, M/RL or M/RL plus WNT16. (g) mRNA expression of Fos (left) and Nfatc1 (right) in RANKL-stimulated BMM cultures with or without WNT16. (h) Lack of effect by WNT16 on β-catenin protein levels as measured by western blot analysis. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (i) Axin2 mRNA levels in BMMs treated with WNT16. (j) Levels of phosphorylated JNK (left) and phosphorylated cJUN (right) measured by western blot in BMMs cultured with or without WNT16. Blots are representative of three separate experiments carried out in triplicate. All values are shown as the mean ± s.e.m. (n = 4). *P < 0.05, **P < 0.01, ***P < 0.005, Student’s t test compared to no WNT16.
We also examined the mechanism by which WNT16 could inhibit osteoclastogenesis. RANKL-induced activation of nuclear factor-κB (NF-κB) was inhibited by WNT16, as shown by decreased mRNA expression of Nfkb2 and Relb (Fig. 5e) and decreased NF-κB transcription in a reporter assay (Fig. 5f). However, RANKL-induced increases in Fos mRNA levels and AP-1 transcriptional activity were unaffected (Fig. 5g and Supplementary Fig. 3e). Notably, RANKL-induced upregulation of Nfatc1 mRNA, encoding NFATc1, which is regarded as the master regulator of osteoclast differentiation, was substantially decreased by WNT16 (Fig. 5g). These data show that WNT16 inhibits osteoclast formation by interfering with osteoclast differentiation at the early steps of RANK signaling.
In contrast to the findings in osteoblasts, WNT16 did not significantly regulate β-catenin protein levels or Axin2 expression in mouse osteoclast precursors (Fig. 5h,i) but did activate the noncanonical Jun–N-terminal kinase cascade, increasing pJNK and p-cJUN levels in a time-dependent manner in mouse osteoclast progenitors (Fig. 5j).
Osteoblast-lineage Wnt16 inactivation mimics global deletion
To assess the role of osteoblasts and osteocytes in the function of WNT16 in vivo, we generated conditional Wnt16-inactivated mouse models targeting osteoblasts or osteocytes. We first used homologous recombination to develop a mouse model with exon 3 of Wnt16 flanked by loxP sites (Wnt16flox/flox; Fig. 6a and Supplementary Fig. 2e). The skeletal characteristics of these Wnt16flox/flox mice were indistinguishable from those of WT mice (Supplementary Table 7). To achieve early inactivation of Wnt16 in the osteoblast lineage, we generated Runx2-creWnt16flox/flox mice, which express Cre recombinase driven by the Runx2 promoter, a promoter that is expressed specifically in osteoblast-lineage cells but not in osteoclasts35. Runx2-creWnt16flox/flox mice displayed substantial and specific recombination of the Wnt16flox allele in cortical bone, resulting in as much as 98% lower Wnt16 mRNA levels in cortical bone compared to cortical bone from control mice (Fig. 6b and Supplementary Fig. 4a). Runx2-creWnt16flox/flox mice displayed no change in tibia length, lower cortical thickness, spontaneous fractures at cortical bone sites, lower mechanical strength of the cortical bone, higher cortical porosity and lower Opg levels in cortical bone compared to control mice, whereas the trabecular bone was unaffected (Fig. 6c–h, Supplementary Fig. 4b,d and Supplementary Tables 8 and 9), thereby phenocopying global WNT16 deletion.
Figure 6.

Osteoblasts are the principal source of WNT16, with an impact on cortical bone and fracture susceptibility. (a) Schematic presentation of the WT allele, the floxed allele and the Cre-mediated recombination of Wnt16. (b) Wnt16 mRNA expression in the cortical bone (left) and uterus (right) of 5-week-old Runx2-creWnt16flox/flox (n = 9) and Dmp1-creWnt16flox/flox (n = 8) mice. (c–e) Bone length (c), femur diaphyseal cortical thickness (d) and trabecular bone volume to total volume (e) in the distal femur metaphysis of Runx2-creWnt16flox/flox and Dmp1-creWnt16flox/flox mice compared to littermate controls (Wnt16flox/flox; n = 8; 5 weeks old). Cortical thickness and trabecular BV/TV were analyzed using micro-CT. (d,e, left) Representative micro-CT images of the experiment described to the right. Scale bars, 500 μm. (f) Three-point bending of femur diaphysis demonstrating maximal load (left) and stiffness at failure (right) in female Runx2-creWnt16flox/flox and Dmp1-creWnt16flox/flox mice compared to littermate controls (Wnt16flox/flox; n = 8; 5 weeks old). (g) Micro-CT analysis of cortical porosity in the femurs of female Runx2-creWnt16flox/flox and Dmp1-creWnt16flox/flox mice compared to littermate controls (Wnt16flox/flox; n = 8; 5 weeks old). (h) Opg mRNA expression in cortical bone of 5-week-old Runx2-creWnt16flox/flox (n = 9) and Dmp1-creWnt16flox/flox (n = 8) mice. (i) WNT16 treatment of PAM2-induced calvarial bone loss after 5 d as measured by micro-CT (PBS, n = 6; PAM2, n = 6; PAM2 + WNT16, n = 5). (j) Proposed role of WNT16 in cortical bone. WNT16 is osteoblast derived and inhibits human and mouse osteoclastogenesis both directly by acting on osteoclast progenitors and indirectly by increasing OPG expression in osteoblasts. *P < 0.05, **P < 0.01, Student’s t test compared to control. All values are given as the mean ± s.e.m.
Wnt16 inactivation in osteocytes affects older mice
We next inactivated Wnt16 specifically in late osteoblasts and osteocytes by mating Wnt16flox/flox mice with Dmp1-cre mice expressing Cre recombinase specifically in osteocytes (Dmp1-creWnt16flox/flox mice)36. Dmp1-creWnt16flox/flox mice displayed substantial recombination of the Wnt16flox allele in cortical bone (Supplementary Fig. 4a), but Wnt16 mRNA levels were not affected in cortical bone, and we observed no skeletal phenotype in young mice (Fig. 6b–h, Supplementary Fig. 4c,d and Supplementary Table 9). Although Dmp1-creWnt16flox/flox mice had normal cortical bone thickness at a young-adult age (5 weeks; female mice at 12 weeks of age had bone thickness +0.1 ± 2.2% over that of control mice (nonsignificant); Fig. 6d), they developed slightly but significantly (male mice, −10.0 ± 2.9%; P < 0.05) thinner cortical bone during aging (52 weeks of age) as a result of substantially lower Wnt16 mRNA expression in the cortical bone at 1 year of age (Supplementary Fig. 4e) compared to control mice. Taken together, these findings demonstrate that WNT16 is required for normal cortical bone homeostasis during aging. In addition, although osteoblasts and not late osteoblasts or osteocytes are the principal source of WNT16 in the cortical bone in young mice, late osteoblasts and osteocytes contribute to WNT16 expression in the cortical bone of elderly mice. These results strongly suggest that the role of WNT16 in cortical bone homeostasis is exerted by WNT16 derived from osteoblast-lineage cells.
Effects of local WNT16 treatment in vivo
To determine the possible beneficial effect of WNT16 treatment in vivo, we used a mouse model of inflammation-induced bone loss in calvarial bone37. Local treatment with the Toll-like receptor 2 (TLR2) agonist PAM2 (Pam2CSK4) resulted in thinner calvarial bone in the frontal region, whereas WNT16 delivered in liposomes prevented bone loss in PAM2-treated mice (Fig. 6i). These findings demonstrate that WNT16 has the capacity to improve bone status in vivo in a model of pathological bone loss.
To further evaluate the effect of WNT16 on the skeleton, we used a rat model in which we previously found that locally administered anti-resorptive bisphosphonates enhanced bone mass38. We administered local injection of WNT16 in liposomes directly into the proximal tibia of 10-week-old male rats (n = 6). We similarly injected empty liposomes in the contralateral tibia. Two weeks after injection of WNT16, bone mineral density at the injection site was significantly higher in the WNT16-treated tibia as compared with the contralateral tibia treated with empty liposomes (BMD; WNT16 liposomes: 300 ± 26 mg cm−3; empty liposomes: 206 ± 32 mg cm−3; P = 0.01, paired Student’s t test), further demonstrating that local WNT16 treatment in vivo has a positive effect on bone mass.
DISCUSSION
Cortical bone mass and composition are major determinants of bone strength and fracture risk in humans2. Only a limited number of studies have focused on the cellular and molecular mechanisms specifically regulating cortical bone homeostasis. Notably, WNT16 has been linked to cortical bone mass, thickness and fracture risk in several large human genome-wide association studies4,25–29, and in the present study, we found that Wnt16-deficient mice exhibited low cortical thickness, high cortical porosity and spontaneous fractures. Furthermore, we show that osteoblast-lineage cells are the major source of WNT16 in bone and that the mechanism by which WNT16 regulates cortical bone homeostasis is through its ability to inhibit osteoclastogenesis.
Human studies have demonstrated that genetic variants in the WNT16 locus associate with BMD, cortical bone thickness and fracture risk at nonvertebral bone sites but not with trabecular bone mass4,25–29. The specific role of WNT16 in cortical bone homeostasis is supported by our present finding that very high and specific Wnt16 expression is observed in cortical bone and that its deletion leads to decreased cortical strength and spontaneous fractures in mice, whereas trabecular bone is not affected. Notably, Wnt16−/− mice frequently developed spontaneous fractures at the tibia diaphysis, which is a bone site consisting mainly of cortical bone in mice. We propose that because of dimensional reasons, the tibia diaphyseal region might be especially sensitive to cortical osteopenia, resulting in early tibia diaphyseal fractures in Wnt16−/− mice.
Spontaneous fractures are rare events in mouse models but have been described for mice with osteoblast- or osteocyte-specific inactivation of Wntless (Wls, also known as Gpr177), which is required for the secretion of WNT ligands from cells39. However, mice with Wls inactivation in osteoblasts or osteocytes display a marked reduction of both trabecular and cortical bone mass39, whereas the Wnt16−/− mice here displayed substantial cortical but not trabecular bone loss. Thus, it is possible that the cortical bone phenotype in mice with osteoblast- or osteocyte-specific inactivation of Wls is caused, at least in part, by loss of WNT16 secretion, whereas the trabecular bone phenotype is mediated by loss of secretion of other WNTs. As WNT10b−/− mice develop an age-dependent loss of trabecular bone specifically, it is probable that WNT10B is at least one of the WNTs explaining the trabecular bone phenotype in mice with Wls inactivation in osteoblasts or osteocytes40,41.
Three-point bending analyses demonstrated substantial lower mechanical bone strength in Wnt16−/− compared to WT mice, explaining the high frequency of spontaneous fractures. Thinner cortical bone clearly contributes to impaired cortical bone strength in Wnt16−/− mice. The ~30% increase in cortical porosity may contribute to the substantially reduced cortical bone strength of Wnt16−/− mice2. The role of WNT16 in cortical porosity should be emphasized, as cortical porosity is a major determinant of bone strength and probably also of fracture risk in humans2. In contrast, in mice trabecular bone volume is not affected in the absence of WNT16. Clinical investigations have demonstrated that trabecular and cortical bone respond differently to treatment1. However, the mechanisms by which this occurs have not been elucidated previously. Our findings, therefore, provide one of the first insights into the mechanisms by which specific cortical bone homeostasis is ensured and show that WNT16 is a major component of this homeostatic machinery. WNT16 expression is higher in cortical than trabecular bone, suggesting that the cortical but not the trabecular bone microenvironment enhances osteoblastic WNT16 expression in cortical bone specifically. Furthermore, compensatory mechanisms might exist to preserve bone mass in the trabecular but not cortical bone compartment in the absence of WNT16. Thus, the data presented in this study corroborate the clinical findings and suggest that the cortical and trabecular compartments are indeed biologically different, their homeostases being independently regulated by different signaling molecules, all of which are involved in the regulation of bone mass.
The cortical bone defects in the Wnt16−/− mice seem to be caused primarily by higher cortical bone resorption as a result of more osteoclasts in cortical bone. Our study demonstrates that osteoblast- derived WNT16 inhibits osteoclastogenesis. We found that Opg expression was lower in the cortical bone of Wnt16−/− compared to WT mice, probably because of a direct autocrine effect on osteoblasts, as WNT16 robustly enhanced Opg mRNA levels in the osteoblastic cell line MC3T3-E1. In addition, WNT16 abolished osteoclast formation in RANKL-stimulated mouse and human osteoclast progenitor cultures, demonstrating that WNT16 inhibits osteoclastogenesis not only indirectly by increasing OPG expression in osteoblasts but also directly by acting on osteoclast progenitors (Fig. 6j). Our findings that WNT16 inhibited NF-κB activation and Nfatc1 expression suggest that WNT16 inhibits osteoclast formation by interfering with early steps in RANKL signaling and osteoclast differentiation.
Although some in vitro studies have suggested that WNT16 does not activate canonical WNT signaling29,42, other studies have shown that WNT16 has the capacity to signal through the canonical WNT cascade32,43–46. Our results clearly demonstrate that WNT16 activates both canonical WNT signaling and the noncanonical Jun–N-terminal kinase WNT signaling cascade in osteoblasts. However, it is noteworthy that the effect of WNT16 on canonical WNT signaling activation in osteoblasts is milder than the effect of the classical canonical WNT ligand, WNT3a, albeit with strong enough effects to efficiently increase Opg expression. Although activation of noncanonical WNT signaling in osteoblasts and osteoclast precursors might explain other aspects of the phenotype observed in Wnt16−/− mice, such as the potent impairment of RANKL-induced osteoclast differentiation, its effects on osteoblasts are not clear. It has been reported previously that β-catenin in osteoblasts and osteocytes is a positive regulator of OPG15. The findings that a WNT16-dependent increase in Axin2 expression is abolished by blocking canonical WNT signaling confirm that WNT16 functions at least in part through canonical WNT signaling. Notably, our findings that the WNT16-dependent increase in Opg expression is blunted, but not significantly, by XAV939 treatment suggest that other WNT16-activated signaling pathways, including noncanonical WNT signaling (which is activated by WNT16 in osteoblasts), may be responsible for the regulation of OPG. Nevertheless, the finding that recombinant WNT16 activates JNK in osteoblasts but does not affect the ROR2 luciferase reporter gene assay suggest that the observed activation of JNK is independent of the ROR2 receptor tyrosine kinase cascade. These results are in agreement with the findings that although WNT5a binds to the Ror2 receptor and activates the Ror2 receptor signaling pathway, other WNT ligands, including WNT3a, WNT7b, WNT10b and WNT16, do not bind to the ROR2 receptor or do not activate the ROR2 signal24,34. Further studies will be required to better understand the role of WNT16-induced noncanonical WNT signaling.
Although WNT16 did not activate the β-catenin pathway in mouse osteoclast progenitors, it strongly increased pJNK and p-cJUN levels, indicating that noncanonical pathways are involved in the inhibitory effect of WNT16 on osteoclast progenitors. The inhibitory effect of WNT16 is in contrast to observations showing that noncanonical signaling by either WNT5b, acting through the RYK receptor, or WNT5a, acting through ROR2, results in potentiation of RANKL-induced osteoclastogenesis24,47. Interestingly, WNT5a-induced potentiation is associated with increased JNK and JUN activation24, whereas WNT16-induced activation of JNK and JUN is associated with inhibition of osteoclastogenesis. These observations demonstrate that WNT signaling is complex and that the pathways are not as linear as initially thought48. Thus, WNT16 in cortical bone may signal by activating distinct WNT signaling complexes in osteoblasts and osteoclasts, which may independently contribute to skeletal homeostasis. Because of the many possible crosstalks between canonical and noncanonical cascades, as well as the fact that several WNTs have been shown to act on bone cells, different WNT signaling pathways might converge distally to activate the same downstream effectors and, thus, regulate bone homeostasis6.
To determine the in vivo role of WNT16 derived from cells in the osteoblast lineage, we developed conditional Wnt16-inactivated mouse models with inactivation of Wnt16 early in both osteoblasts and osteocytes (Runx2-cre) or in late osteoblasts and osteocytes (Dmp1-cre). In young-adult mice, inactivation of Wnt16 in both early osteoblasts and osteocytes resulted in a marked reduction of cortical thickness, as well as in spontaneous fractures, whereas inactivation of Wnt16 in late osteoblasts and osteocytes did not significantly affect cortical thickness, demonstrating that osteoblasts, but not osteocytes, are the principal source of WNT16 in cortical bone in young mice. Together with the findings that 16-week-old mice globally null for Wnt16 show persistence of the cortical bone phenotype, our data suggest that WNT16 has a role in cortical homeostasis until skeletal maturity. Notably, Wnt16 inactivation in late osteoblasts and osteocytes leads to decreased cortical bone mass only after 3 months of age, as indicated by lower Wnt16 mRNA expression and cortical bone thickness at 1 year but not at 3 months of age. This finding demonstrates that the cortical bone phenotype is not generated during development and growth only. A role of WNT16 in cortical bone homeostasis during not only development and growth but also aging is also supported by our previous human genetic finding that an amino acid–altering SNP in WNT16 associates robustly with cortical bone thickness in subjects over a broad range of ages (15.5–78.7 years of age), and no significant impact of age for this association was observed4. These in vivo studies establish WNT16 derived from osteoblast-lineage cells as a crucial regulator of cortical bone mass and fracture susceptibility.
To evaluate the possible beneficial effect of WNT16 treatment in vivo, we used a mouse model of inflammation-induced bone loss in calvarial bone. Our finding that WNT16 treatment prevented PAM2-induced calvarial bone loss demonstrates that WNT16 has the capacity to improve bone mass in vivo in a model of pathological bone loss. We also showed that local WNT16 treatment in rat tibiae increased bone mineral density, further demonstrating that local WNT16 treatment in vivo has a positive effect on bone mass.
In conclusion, we have shown that in mice, an absence of WNT16 results in lower cortical thickness and higher cortical porosity and, thus, higher fracture susceptibility. Mechanistic studies revealed that osteoblast-derived WNT16 inhibits human and mouse osteoclastogenesis both directly by acting on osteoclast progenitors and indirectly by increasing Opg expression in osteoblasts (Fig. 6j). WNT16 signaling in osteoblasts is both canonical and noncanonical, whereas it is only noncanonical in osteoclast progenitors. New treatment strategies targeting the regulation of WNT16 might be useful to reduce fracture risk at cortical bone sites.
ONLINE METHODS
Generation of global Wnt16−/−, exon 1-4 mice
The mouse strain used for the generation of global Wnt16−/−, exon 1-4 mice was created from a C57BL/6N embryonic stem (ES) cell clone (10802A-B4) obtained from the National Center for Research Resources, US National Institutes of Health (NIH)- supported KOMP Repository (https://www.komp.org/) and generated by Regeneron Pharmaceuticals, Inc. for the NIH-funded Knockout Mouse Project (KOMP). The ES cell clone was injected into blastocysts of C57BL/6N female mice. Methods used to create the Velocigene-targeted alleles have been described previously49. The Wnt16 gene includes four exons, and translation of the WNT16 protein starts from position 281. In the Wnt16−/−, exon 1-4 mice, nucleotides between position 266 and 10025 are replaced by a ZEN Ub1 cassette in C57BL/6N ES cells. This leads to a complete lack of Wnt16 mRNA transcription of the targeted locus (Fig. 1a–c and Supplementary Fig. 4a). The Wnt16−/− and WT control littermates were generated by breeding male Wnt16+/− mice with female Wnt16+/− mice using an inbred C57BL/6N mouse strain throughout the study. The phenotype of this Wnt16−/−, exon 1-4 mouse model has not been reported previously. The following primer pairs were used for genotyping the WT mice: forward primer (5′-GCAGCCTCTGTT CCACATACACTTCA-3′) and reverse primer (5′-CCTTCCTTCCTACCT TCCCTCTTTCC-3′). The following primers were used for genotyping the Wnt16−/− mice: forward primer (5′-CATGACCGAATGTTCCTGTG-3′) and reverse primer (5′-CCGCTTCGTTGTTGTGTAGA-3′). To analyze the degree of Wnt16 deletion at the DNA level in different tissues, TaqMan custom assays from Life Technologies were used. Wnt16 intron 2 was analyzed using forward primer (5′-GGATAGTAGATTTCCTTTCCAAATATTCACACA-3′), reverse primer (5′-CTTGTTGTCTTAGGAGGCAATCGA-3′) and probe (6Fam 5′-TTTTCAAGCGTTTACAGAGAAT-3′ MGBNFQ). As the ZEN Ub1 cassette is retained in the Wnt16−/−, exon 1-4 mice, we evaluated whether the expression of the two closest neighboring genes (Fam3c and Cped1, located downstream and upstream, respectively, of the Wnt16 gene) was affected. However, the expression of these two genes in cortical bone was normal in 8-week-old Wnt16−/−, exon 1-4 mice, suggesting that the ZEN Ub1 cassette did not affect neighboring genes (data not shown). The mice were studied at different time points, including prenatal mice (E18.5), prepubertal mice (5 weeks old), postpubertal mice (8 weeks old) and adult mice (11 weeks old). As the Wnt16−/− mice frequently developed spontaneous fractures directly after sexual maturation, we did not characterize mice older than 11 weeks of age. Animal care was in accordance with guidelines from the Animal Committee of the University of Gothenburg, and the study was approved by the local ethical committee in Gothenburg.
Generation of global Wnt16−/−, exon 1-3 mice
C57BL/6-129SvEv Wnt16−/−, exon 1-3 mice with deletion of exons 1–3 were obtained from Lexicon Pharmaceuticals Inc. (The Woodlands, TX)4. Mice were backcrossed for at least five generations with an inbred C57BL/6 mice. Because of this background, Wnt16−/−, exon 1-3 and their WT age- and gender-matched littermates were used in all studies. All animal protocols were approved by the Harvard Medical School Institutional Animal Care and Use Committee policies.
Generation of conditional Wnt16flox/flox mice
We used homologous recombination to develop a mouse model with exon 3 of Wnt16 flanked by loxP sites (Wnt16flox/flox; Fig. 6a and Supplementary Fig. 2e). The targeting vector for the Wnt16 gene (HTGRS6013_A_H08) was obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM), and the sequence was further confirmed by PCR, restriction enzyme mapping and sequencing. G4 ES cells derived from (129S6 × C57BL/6NCr)F1 were cultured on a neomycin- resistant primary embryonic fibroblast feeder layer, and 106 cells were electroporated with 30 μg of linearized targeting construct digested by AsiSI. After electroporation, the cells were exposed to G418 (300 μg ml−1; Sigma), and colonies (155) were picked 7–9 d later and expanded by culturing the individual colonies for an additional 2–3 d. Proper targeting was confirmed by long-range PCR reactions spanning both the 5′- and 3′-homologous arms. Using the primers WNT16Arm5UF1 (5′-GCTCTTCTGAAGGTTCTGAA-3′) and LAR3 (5′-CACAACGGGTTCTTCTGTTAGTCC-3′), a proper PCR product was generated corresponding to the 4,854-bp-long targeting arm at the 5′ region. Similarly, a PCR product with a proper size was generated with the primer pair RAF (5′-CACACCTCCCCCTGAACCTGAAAC-3′) and WNT16Arm3DR1 (5′-AATGCTCCTGTCTTTGATGA-3′) corresponding to the 6,264-bp-long 3′-homologous arm. Both of the PCR products were also partially sequenced to further confirm the right targeting.
In order to delete Neo and LacZ cassettes and generate a floxed allele (Wnt16+/flox), the correctly targeted ES cells were electroporated with a plasmid expressing the Flp recombinase (pCAGGS-Flpe, Gene Bridges GmbH, Heidelberg, Germany). After electroporation, the cells were plated on 100-mm cell culture dishes, and colonies were picked after 3–5 days and expanded further by culturing on 96-well plates. The proper excision of the cassettes was confirmed by performing a PCR analysis using the primers R1 (5′-AAGGCGCATAACGATACCAC-3′) and Wnt16Arm5DR2 (5′-AAATGTGTAACCTTCACGAG-3′), and the PCR products formed were confirmed by sequencing.
ES cells from two correctly targeted clones were injected into C57BL/6N blastocysts, and chimeric male mice with germline transmission were produced. Furthermore, heterozygous F1 males born from the chimeric mouse breeding were subsequently bred with C57BL/6N females for the colony expansion. Genotyping of the WT and targeted allele (Wnt16flox) was performed by PCR using the primers Wnt165ArmSe1 (5′-CATAAAGCCAGCTGCACTGC-3′) and Wnt16Arm5DR2 (5′-AAATGTGTAACCTTCACGAG-3′). A PCR product 370 bp in length was produced from the WT allele, and the Wnt16flox allele produced a 522-bp-long product.
The following primer pairs were used for genotyping of the presence or absence of the loxP sequence: 5′-CATAAAGCCAGCTGCACTGC-3′ and 5′-AAATGTGTAACCTTCACGAG-3′. The presence of the Runx2-cre gene was determined using the three-primer strategy. The common 5′ primer (5′-CCAGGAAGACTGCAAGAAGG-3′) binds to the Runx2 promoter of the endogenous gene and the transgene. The 3′ primer (5′-TGGCT TGCAGGTACAGGAG-3′) binds to the cre sequence, and another 3′ primer (5′-GGAGCTGCCGAGTCAATAAC-3′) binds to the endogenous Runx2. The presence of the Dmp1-cre gene was determined using the following primer pair: 5′-CCAGGCTAAGTGCCTTCTCTACA-3′ and 5′-AATGCT TCTGTCCGTTTGCCGGT-3′.
To generate specific knockout of Wnt16 in the early osteoblast lineage, floxed Wnt16 mice were crossed with the previously described Runx2-cre mice35. These Runx2cre-Wnt16flox/flox mice display early osteoblast-specific cre expression and have the capacity to recombine loxP-flanked DNA sequences in an early osteoblast-specific manner. We have previously demonstrated that efficient recombination occurs at all sites of endochondral and intramembranous bone formation, particularly in periosteal cells, osteoblasts and osteocytes but not osteoclasts, when Runx2-cre mice were crossed to a Rosa26 reporter strain35. To generate mice depleted of Wnt16 in osteoblasts, female mice with two floxed Wnt16 alleles (Wnt16flox/flox) were mated with male mice having one floxed Wnt16 allele and one allele of Runx2cre (Runx2-creWnt16flox/w). The littermate control mice were Wnt16flox/flox. Wnt16flox/flox and Runx2-cre mice have an unchanged skeletal phenotype compared to WT mice (Supplementary Tables 7 and 10).
To generate mice depleted of Wnt16 in late osteoblasts and osteocytes, Wnt16flox/flox female mice were crossed with heterozygous Wnt16flox/w male mice expressing Cre recombinase driven by the 10-kb Dmp1 promoter specifically expressed in late osteoblasts and osteocytes (Tg(Dmp1-cre)1Jqfe mice) (Dmp1-cre mice)36. The littermate control mice were Wnt16flox/flox. We have previously demonstrated that efficient recombination occurs in osteocytes but not osteoblasts or osteoclasts when Dmp1-cre mice were crossed to a Rosa26 reporter strain50.
Power calculation, blinding of investigators and randomization of mouse samples
The predesigned primary endpoint in the mouse studies was to record the effect of WNT16 inactivation on cortical bone thickness. Our power analysis suggested that when using eight WT and eight Wnt16−/− mice, we would have 80% power to detect a biological significant effect with a 1.51 s.d. change in cortical bone thickness, and therefore we aimed to use eight mice per group in the different mouse studies. All in vivo experiments and subsequent assessments of the outcomes from these experiments were done in total blinding of the investigators. No experiments requiring randomization of sample groups were performed. All animals of the required genotype were included in the analysis.
Whole-mount histological staining
Dissection of prenatal and adult mouse skeletons and detection of cartilaginous and bony elements by Alcian blue and Alizarin red staining was performed using standard histology methods51.
pQCT of Wnt16−/−, exon 1-4 mice
CT scans were performed with the pQCT XCT RESEARCH M (version 4.5B, Norland) operating at a resolution of 70 μm, as described previously52. The scan was positioned in the metaphysis of the femur at a distance proximal from the distal growth plate corresponding to 3.4% of the total length of the femur, and the trabecular bone region was defined as the inner 45% of the total cross-sectional area. Cortical bone parameters were analyzed in the mid-diaphyseal region of the tibia53.
High-resolution micro-CT of Wnt16−/−, exon 1-4 mice
High-resolution micro-CT analyses were performed on the distal femurs using a 1172 model micro-CT (Bruker micro-CT, Aartselaar, Belgium). The femurs were imaged with an X-ray tube voltage of 50 kV and a current of 201 μA, with a 0.5-mm aluminum filter. The scanning angular rotation was 180°, and the angular increment was 0.70°. The voxel size was 4.48 μm isotropically. NRecon (version 1.6.9) was employed to perform the reconstruction after the scans. In the femurs, the trabecular bone proximal to the distal growth plate was selected for analyses within a conforming volume of interest (cortical bone excluded) commencing at a distance of 538.5 μm from the growth plate and extending a further longitudinal distance of 134.5 μm in the proximal direction. Cortical measurements were performed in the diaphyseal region of the femur starting at a distance of 3.59 mm from the growth plate and extending a further longitudinal distance of 134.5 μm in the proximal direction. For BMD analysis, the equipment was calibrated with ceramic standard samples.
Micro-CT of Wnt16−/−, exon 1-4 mice
Micro-CT analyses were performed on the distal tibias using a model 1072 scanner (Skyscan) and imaged with an X-ray tube voltage of 100 kV and current of 98 μA, with a 1-mm aluminum filter54. The scanning angular rotation was 180°, and the angular increment was 0.90°. The voxel size was 6.51 μm isotropically. Data sets were reconstructed using a modified Feldkamp algorithm and segmented into binary images using adaptive local thresholding55. In the tibias, the trabecular bone distal to the proximal growth plate was selected for analyses within a conforming volume of interest (cortical bone excluded) commencing at a distance of 338.5 μm from the growth plate and extending a further longitudinal distance of 488 μm in the distal direction. Cortical measurements were performed in the diaphyseal region of the femur starting at a distance of 5.2 mm from the distal growth plate and extending a further longitudinal distance of 163 μm in the proximal direction. Trabecular thickness and separation were calculated by the sphere-fitting local thickness method56.
Micro-CT and bone histomorphometry of Wnt16−/−, exon 1-3 mice
High-resolution images of the femora were obtained using a micro-CT 35 (Scanco Medical) according to the recommended guidelines. A 7-μm isotropic voxel size, 50 kVp and 144 μA were applied to analyze trabecular bone microarchitecture in the distal femoral metaphysis and cortical bone at the femoral midshaft. For dynamic bone histomorphometry, mice were subcutaneously injected with calcein (20 mg per kg body weight) and demeclocycline (20 mg per kg body weight) (both from Sigma-Aldrich). Tibiae were removed, fixed in 70% alcohol and embedded in methyl methacrylate. 5-μm sagittal sections were used. Von Kossa staining was performed following standard protocols. Quantitative bone histomorphometric measurements were performed semiautomatically with the OsteoMeasure image analyzer (Osteometrics Inc) according to standardized protocols57.
Histomorphometric analyses of Wnt16−/−, exon 1-4 mice
Trabecular bone
L4 vertebrae were fixed in 4% paraformaldehyde, dehydrated in 70% EtOH and embedded in plastic (L R White Resin, Agar Scientific). The trabecular bone was analyzed by Pharmatest Services, Ltd. using longitudinal coronary sections of the vertebrae. Sections 4-μm thick were stained with Masson-Goldner tri-chrome58,59. The parameters were measured using the OsteoMeasure histomorphometry analysis system with software version 2.2 (OsteoMetrics) and following the guidelines of the American Society for Bone and Mineral Research57.
Cortical bone
Tibia were fixed in Bürkhardt’s fixative, dehydrated in 70% EtOH and embedded in plastic (L R White Resin, Agar Scientific). For the measurement of dynamic parameters, the mice were double labeled with calcein, which was injected (intraperitoneally) into the mice 1 and 8 d before termination. Histomorphometric analyses of cortical bone were done using 20-μm-thick transverse cross-sectional sections in the mid-diaphyseal region of the tibia.
Three-point bending analyses
Immediately after the dissection, the femurs were fixed in Bürkhardt’s fixative for 2 d and then stored in 70% ethanol. Before mechanical testing, the bones were rinsed in PBS. The three-point bending test (span length, 4.4 mm; loading speed 0.155 mm/s) at the mid femur was made using an Instron universal testing machine (Instron 3366, Instron Corp., Norwood, MA). Based on the recorded load deformation curves, the biomechanical parameters were acquired from raw files produced by Bluehill 2 software version 2.6 (Instron) with custom-made Excel macros.
Quantitative real-time PCR analysis
Total RNA was prepared using TRIzol reagent (Life Technologies) followed by the RNeasy Mini Kit (Qiagen). The RNA was reverse transcribed into cDNA, and real-time PCR analysis was performed using the iCyclerIQ (BioRad) with Fast Start Universal SYBR Master Mix (Roche) or using predesigned real-time PCR assays and the StepOnePlus Real-Time PCR system (Applied Biosystems). We used predesigned real-time PCR assays from Applied Biosystems for the analysis of Wnt16 (Mm00446420_m1), Runx2 (Mm00501580_m1), Alpl (Mm00475834_m1), Acp5 (Trap; Mm00475698_m1), Ctsk (Mm00484036_m1), Nfatc1 (Mm00479445_m1), Rankl (Tnfsf11; Mm00441908_m1), Opg (Tnfrsf11b; Mm0043545_m1), Calcr (Mm00432282_m1), Mmp9 (Mm00600163_m1), Oscar (Mm00558665_m1), Tcirg1 (Mm00469394_m1), Fos (Mm00487425_m1), Irf8 (Mm00492567_m1), Axin2 (Mm00443610_m1), Tcf1 (Hnf1a; Mm00493434_m1), Relb (Mm00485664_m1) and Nfkb2 (Mm00479810_g1). The mRNA abundance of each gene was adjusted for the expression of 18S or Actb.
Immunohistochemistry
Bones were fixed in 4% formaldehyde and decalcified in 10% EDTA, and 5-μm-thick paraffin-embedded sections were obtained. Antigen retrieval was performed in citrate buffer (pH 6.0) for 15 min at 94–96 °C. The sections were incubated with 10% normal serum for 1 h. Expression of WNT16 was investigated using a rabbit polyclonal primary antibody (H-96, sc-20964, 1:200, Santa Cruz Biotechnology, Santa Cruz, CA), followed by fluorescent donkey anti-rabbit Cy3 (711-165-152, 1:400, Jackson Laboratories, Bar Harbor, ME), before mounting with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA). Images were captured using a Nikon Eclipse E800 microscope equipped with an Olympus DP70 digital camera.
Analyses of osteoclasts in vivo
Femurs were fixed in Bürkhardt’s fixative and embedded and undecalcified in methyl-methacrylate resin (Medim-Medizinische Diagnostik, Giessen, Germany), and 4-μm longitudinal sections were prepared for analysis using OsteoMeasure software (v3.2.1.0, OsteoMetrics Inc., GA). For measurements of osteoclast surface and osteoclast numbers, sections were stained for TRAP activity as described previously60.
Analyses of osteoids in vivo
Tibiae were fixed in Bürkhardt’s fixative, dehydrated in 70% EtOH and embedded in plastic (L R White Resin, Agar Scientific). Longitudinal 4-μm sections were stained using a Masson-Goldner trichrome58,59 assay, and analysis of the osteoids was done using OsteoMeasure software.
Serum analyses
Analyses were performed according to the manufacturer’s instructions for serum concentrations of PTH (ELISA, Immutopics, San Clemente, CA), calcium (QuantiChrom Calcium Assay Kit (DICA-500), Hayward, CA), 1,25 dihydroxy vitamin D (1,25(OH)2D; RIA, Immunodiagnostic Systems, Herlev, Denmark) and phosphate by the phosphomolybdate- ascorbic acid method using the COBAS INTEGRA system (Roche Diagnostics GmbH, Mannheim, Germany). As a marker of bone resorption, serum levels of type I collagen fragments were assessed using an ELISA RatLaps kit (CTX-I) (Immunodiagnostic Systems).
Flow cytometry
Bone marrow cells from female mice were harvested by flushing PBS through the bone cavity of the humerus using a syringe. After centrifugation, cells were resuspended in Tris-buffered 0.83% NH4Cl solution (pH 7.29) for 5 min to lyse erythrocytes and then washed in PBS. Bone marrow cells were resuspended in PBS supplemented with 10% FBS and 5 mM EDTA. Cells were stained with phycoerythrin (PE) anti-mouse Ly-6G (Gr-1) (12-5931-83, 1:50, eBioscience), V450 anti-mouse CD11b (560455, 1:50, BD Bioscience) and allophycocyanin (APC) anti-mouse CSF-1R (135510, 1:10, BioLegend). The cells were then subjected to FACS on a FACSCalibur (BD Pharmingen, Franklin Lakes, NJ) and analyzed using FlowJo software. The monocytes were gated from the side scatter (SSC) versus forward scatter (FSC). The pre-osteoclasts were defined as CSF-1R–positive, CD11b-positive and Gr1-low/negative monocytes and are presented as a cell frequency (%) of bone marrow cells.
Cell culture media
Human and mouse cells were cultured in complete α-MEM medium (MEM alpha medium (Gibco, 22561-021) supplemented with 10% heat-inactivated FBS (Sigma, F7524), 2 mM GlutaMAX (Gibco, 35050-038), 50 μg ml−1 gentamicin (Gibco, 15750-037), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Gibco, 15140-148)). For osteoblast differentiation and mineralization assays, mouse calvarial osteoblast were cultured in complete osteogenic α-MEM supplemented as described above and with 10 mM β-glycerophosphate disodium salt hydrate (BGP; Sigma, G9422) and 0.2 mM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Asc-2P; Sigma, A8960).
Isolation and culture of primary mouse calvarial osteoblast cells
Primary calvarial osteoblasts were isolated from newborn mouse calvaria by sequential digestions as described previously61. Calvarial bones were incubated in buffer containing 137 mM NaCl, 2.7 mM KCl, 3 mM NaH2PO4 (pH 7.2) and 4 mM Na2EDTA. This solution was used for the first three digestions, and then EDTA was substituted by 180 U ml−1 of Worthington bacterial collagenase type 2. The bones were placed in a shaking water bath at 37 °C. Cells were released by 10 min of sequential digestions, and cells released from the first five digestions were discarded. Cells from subsequent digestions six through ten were used. Cells were cultured for 4–6 d before subculture in 48-well plates at 40,000 cells cm−2 and induction of osteogenic differentiation with BGP and Asc-2P.
Analyses of osteoblast proliferation, differentiation and mineralization
Cell growth was followed by detaching cells with trypsin solution and cell counting using Nucleocassettes and Nucleocounter (Chemometec). Differentiation of osteoblasts was determined by staining for ALP activity (ALP kit, Sigma) or by Alizarin red (ALZ) staining, as described previously62, at the indicated time points. The total area of ALZ-stained noduli was analyzed using OsteoMeasure software. ALZ amounts were determined by a quantitative de-staining procedure using 10% cetylpyridinium chloride and absorbance at 562 nm, as described previously62. Gene expression was analyzed by lysing cells at the indicated time points in RLT buffer (Qiagen) followed by RNA purification, cDNA synthesis and quantitative real-time PCR as described above using commercial TaqMan assays.
In vitro studies using MC3T3-E1 cells and HEK293 cells
MC3T3-E1 cells were cultured in α-MEM, 10% FBS and 1% penicillin-streptomycin. To evaluate the effect of WNT3a and WNT16b, cells were starved overnight in α-MEM containing 1% FBS and then treated with WNT16b (0.2 or 1 μg ml−1, R&D, 7790-WN-025/CF) or WNT3a (0.2 μg ml−1). For experiments with the tankyrase inhibitor, XAV939, cells were treated with the inhibitor (1 μM) for 16 h before the WNT3a and WNT16b treatments. HEK293 cells stably expressing ROR2-hTRK-luc were cultured in DMEM containing 10% FBS and 1% penicillin-streptomycin.
Luciferase reporter assay used in MC3T3-E1 and HEK293 cells
MC3T3-E1 cells were co-transfected with 400 ng TOPflash-luc reporter plasmid and 10 ng control pCMV-Renilla luciferase (Promega) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Six hours after transfection, the medium was changed, and cells were cultured in α-MEM containing 1% FBS overnight. Cells were then treated with WNT16b (0.2 μg ml−1) or WNT3a (0.2 μg ml−1) for 24 h, followed by luciferase assay using the Dual-Glo Assay system (Promega) according to the manufacturer’s instructions. Data were normalized by Renilla firefly activity and are presented as a fold change compare to vehicle-treated cells. HEK293 cells stably expressing ROR2-hTRK-luc were incubated with WNT16 (0.2 μg ml−1) or WNT5a (0.2 μg ml−1) for 24 h. Luciferase activities were determined as described above.
Osteoclast formation using crude bone marrow cells
Bone marrow cells were flushed from the femur and tibia. Osteoclast formation in crude bone marrow cells was studied by seeding 400,000 bone marrow cells in 40 μl complete α-MEM per 48-well plate (plastic) or in 96-well plates with bovine bone slices (IDS Nordic). Thereafter, complete α-MEM with 30 ng ml−1 mouse M-CSF (R&D, 416-ML-050) with or without 4 ng ml−1 mouse RANKL (R&D, 462-TEC-010) was added. The culture medium was changed every 3 d, and at indicated time points, the cells were stained for TRAP. As a measure of osteoclastogenesis, the total amount of TRAP5b was analyzed in the culture medium using commercial TRACP 5b ELISA (IDS Diagnostic). In addition, the release of C-terminal telopeptides of type I collagen (CTX-I) from the bone slices into the culture media was followed from day 3 to day 12 and measured using commercial ELISA (IDS Nordic).
Osteoclast formation using mouse BMMs
Mouse BMMs were obtained as described previously63,64. Briefly, bone marrow cells were centrifuged and resuspended in ice-cold water for 1 min to remove red blood cells. Cells were cultured in complete α-MEM with 30 ng ml−1 M-CSF in a suspension culture dish (Corning Costar Ins., NY), to which stromal cells and lymphoid cells cannot adhere, at 37 °C for 2–3 d. Cells were washed vigorously with PBS to remove any nonadherent cells and then washed with cold (4 °C) 0.02% EDTA in PBS to release the attached BMMs. Cells were spot seeded in either 96-well plates for counting of osteoclasts (5,000 cells in 5 μl complete α-MEM) or 24-well plates for gene expression analysis (40,000 cells in 40 μl complete α-MEM). For osteoclast generation, cells were cultured in 30 ng ml−1 M-CSF and 4 ng ml−1 RANKL with or without WNT16b protein (62–1,000 ng ml−1) (R&D, 7790-WN-025/CF). For gene expression analysis, cells were lysed in RLT buffer (Qiagen), followed by RNA purification, cDNA synthesis and real-time PCR as described above.
Cocultures of osteoblasts and osteoclasts
Mouse calvarial osteoblast cells from WT and Wnt16−/− mice were seeded in 24-well plates (2 × 104 cells ml−1) in complete osteogenic α-MEM. After 6 d, BMMs from WT and Wnt16−/− mice were added (2 × 105 cells ml−1), and osteoclastogenesis was induced by culturing cells in complete osteogenic α-MEM with 10 nM 1,25(OH)2-vitD3 and 1 μM PGE2. After TRAP staining, the number of osteoclasts with more than five nuclei was counted.
Osteoclast formation using human CD14+ monocyte cultures
Peripheral blood mononuclear cells were prepared by Ficoll-Paque PLUS separation, and CD14+ cells were labeled with CD14 MicroBeads and isolated using a MACS column (Miltenyi). Cells were then seeded in 96-well plates (3 × 105 cells cm−2) containing α-MEM supplemented with 10% FBS and 30 ng ml−1 human M-CSF (R&D, 216-MC-025). For osteoclast generation, 2 ng ml−1 recombinant mouse RANKL was added without or with recombinant WNT16 protein (31–500 ng ml−1). After 4 d, with a medium change at day 3, cells were stained for TRAP, and the total number of TRAP+ cells containing three or more nuclei was counted and recorded as TRAP+MuOCLs.
Analyses of osteoclast differentiation and bone resorption
The surface and number of TRAP-positive cells with three or more nuclei were analyzed using OsteoMeasure software. As an additional measure of osteoclastogenesis, TRAP5b was analyzed in the culture medium using commercial TRACP 5b ELISA (IDS Diagnostic). Gene expression was analyzed using commercial TaqMan assays. Bone resorption was analyzed by measuring the release of C-terminal telopeptides of type I collagen (CTX-I) in bone cell culture supernatants using commercial ELISA (IDS Nordic).
Western blot analysis
Cells were lysed in NP-40 lysis buffer (Novex/Life Technologies) and a cocktail of protease inhibitors (cOmplete ULTRA Tablets, Roche) and Pefabloc SC PLUS (Roche). The insoluble material was sedimented by centrifugation (12,000g, 10 min, 4 °C), and the supernatants were collected and frozen at −80 °C. Protein content was determined using a BCA Protein Assay kit (Pierce, Rockford, IL). Equivalent amounts of protein were electrophoresed on 4–12% Bis-Tris gels (Life Technologies) and transferred to polyvinylidene difluoride membranes (Life Technologies). Membranes were blocked and incubated with primary antibodies (1:1,000; β-catenin: #610154, BD Biosciences; GAPDH: #ab37168, Abcam; β-actin: #13E5; LRP6, #2560; pLRP6, #S1490; SAPK/JNK: #9252S; pSAPK/JNK (Thr183/Tyr185), #9255S; cJUN: #9165S; pcJUN (Ser73) #9164S, Cell Signaling Technology, Inc., Danvers, MA) overnight, followed by incubation with horseradish peroxidase (HRP)-linked secondary antibodies (#7074S and #7076S, 1:1,000, Cell Signaling Technology). The protein bands were visualized with ImmunoStar Western C (Bio-Rad, Hercules, CA) and the ChemiDoc XRS+ System (Bio-Rad).
Reporter gene assays used in BMM experiments
We used commercially available lentiviral signal transduction reporter assays (Qiagen Cignal Lenti Reporter Assays). The vectors express the luciferase reporter gene under the control of either the AP-1 (TPA) or NF-κB response elements. BMMs were transduced in M-CSF–containing medium with the reporter vectors at a multiplicity of infection of 10 for 18 h. Thereafter, the vector-containing media were replaced by vector-free media containing M-CSF for 24 h. Then, osteoclast differentiation medium without or with WNT16 was added. Cells were lysed at different time points after addition of the test substances, and luciferase was measured using a Luciferase Assay System (Promega) according to the manufacturer’s instructions and analyzed by a Mithras LB 940 luminometer (Berthold Technologies, Bad Wildbad, Germany).
Generation of WNT16 liposomes
As the WNT16 protein is hydrophobic, the protein was packed into liposomes65. To generate the liposomes, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC; 850345C, Avanti Polar Lipids, Inc., Alabaster, AL) dissolved in chloroform was dried to a thin film in a round-bottom flask, first using a gentle nitrogen gas stream and then in a vacuum for at least 2 h. The so-obtained film was then hydrated in 40 μg ml−1 WNT16 protein (in PBS) prewarmed to 32 °C. The final lipid concentration was 13.2 mg ml−1. Control liposomes were produced by hydration in PBS. This was followed by vortexing until the solution was cloudy and no lipids were visible on the bottom of the flask. After hydration, the liposomes were extruded by pressing the mixture 51 times through a 100-nm polycarbonate membrane (Whatman, UK) at 32 °C. After cooling to room temperature, the liposomes were stored at 4 °C until further use. Prior to use in the mouse study, the WNT16 liposomes were incubated with additional WNT16 protein (20 μg ml−1 in PBS) in a ratio of 1:2, giving a final WNT16 protein concentration of 27 μg ml−1. For the rat study, the WNT16 liposomes were incubated with additional WNT16 protein to a final concentration of 100 μg ml−1.
Mouse study of inflammation-induced bone loss in calvaria
Six-week-old female C57BL/6N mice from Charles River Laboratories, Inc. (Germany) were used. Animal care was in accordance with guidelines from the Animal Committee of the University of Gothenburg, and the study was approved by the local ethical committee in Gothenburg. Inflammation-induced bone loss in calvarial bone was initiated by subcutaneous injection of 75 μg synthetic diacylated lipopeptide, Pam2CSK4 (PAM2; InvivoGen, Toulouse, France)37. PAM2 is an agonist of Toll-like receptor 2. PBS was used as the vehicle. Mice received either 150 μl WNT16 liposomes (27 μg ml−1), empty liposomes or PBS by subcutaneous injections over the calvaria three times, i.e., 1 d before they were given PAM2, the day they were given PAM2 and the day after they were given PAM2. All injections were done under anesthesia with isoflurane (Baxter Medical AB, Kista, Sweden). Five days after the injection of PAM2, the mice were bled and euthanized by cervical dislocation under anesthesia. The calvaria was dissected, and the calvarial bone thickness in the frontal region was analyzed by micro-CT. The average of ten separate calvaria thickness measurements per calvaria was used (PBS, n = 6; PAM2 + PBS, n = 6; PAM2 + WNT16 liposomes, n = 5). Empty liposomes did not reverse the PAM2-induced bone loss (data not shown).
Local WNT16 injection into rat metaphyseal bone
Male Sprague-Dawley rats, 10 weeks old, received an injection of 20 μl of WNT16 (100 μg ml−1) in liposomes into their right proximal tibia with a 0.4-mm needle. Empty liposomes were similarly injected into the left proximal tibia. The procedure was performed aseptically and under anesthesia according to approval by the Regional Ethical Committee on Animal Research. The medial aspect of the proximal tibia was exposed as described previously66, and the needle was inserted perpendicular to the surface 3 mm below the growth plate. After 2 weeks, the animals were euthanized, and the tibia was collected for micro-CT. By micro-CT, we confirmed that the distance between the injection canal and the growth plate was as intended (3.1 ± 0.7 (s.d.) mm), with no statistically significant difference between the left and right legs (P = 0.29). Micro-CT analysis was done on a 1174 Skyscan system (1174, Bruker, MA), in 180°, with a 0.5-mm Al filter and at 50 kV. Corrections for ring artifacts and beam hardening were made. A pixel size of 13 μm, a rotation step of 0.4° and a frame averaging of 4 was used. The volume of interest (VOI) was defined as a 1.4-mm-diameter cylinder in the marrow compartment centered around the visible hole after injection. The cylinder length was 1.5 mm. It extended 1.5 mm into the marrow compartment, starting 0.2 mm away from the endosteum. The VOI was analyzed for BMD.
Statistical analyses
Numerical data and histograms are expressed as the mean ± s.e.m. Student’s t test was performed between two groups, and a difference was considered statistically significant with P < 0.05.
Supplementary Material
Acknowledgments
We are grateful to C. Uggla, M. Petersson, A. Hansevi, A. Lie, I. Lundgren, B. Aleksic and the staff at the Turku Center for Disease Modeling (TCDM) for technical assistance. We thank M. Johansson at Bergman Labora for help with photographing the embryos. We thank R. Brommage (Lexicon Pharmaceuticals Inc.) for providing the Wnt16−/−, exon 1-3 mice. This study was supported by the Swedish Research Council, the Swedish Foundation for Strategic Research, COMBINE, the Avtal om Läkarutbildning och Forskning/Läkarutbildningsavtalet (ALF/LUA) research grant in Gothenburg, Linköping University, Swedish National Graduate School in Odontological Sciences, the Lundberg Foundation, the Torsten and Ragnar Söderberg’s Foundation, the Swedish Rheumatism Association, the Royal 80 Year Fund of King Gustav V, the Novo Nordisk Foundation and the German Research Foundation (SPP 1468 Tu220/6, Immunobone). The work at TCDM was supported by funding provided by University of Turku and Biocenter Finland. This work was supported in part by a grant to R.B. from the US National Institutes of Health (R01AR064724). The HSDM micro-CT core facility performed the micro-CT analyses of the Wnt16−/−, exon 1-3 mice.
Footnotes
AUTHOR CONTRIBUTION
S.M.-S., P.H. and X.L. contributed equally to this work. S.M.-S. was responsible for the breeding, phenotyping and treatment of the Wnt16−/−, exon 1-4 and conditional Wnt16flox/flox mouse strains. P.H. conducted the in vitro culture experiments and analyses of primary osteoblasts, cocultures and osteoclasts. X.L. was responsible for the breeding and phenotyping of the Wnt16−/−, exon 1-3 mouse colony and conducted in vitro and ex vivo experiments. H.S. assisted with animal experiments and performed initial bone histomorphometry analyses. K.N. performed bone histomorphometry analyses. A.E.B. assisted with animal experiments and performed micro-CT analysis. K.S., S.H.W., H.F., H.I. and C.E. assisted with animal experiments. B.K. performed the FACS analysis. A. Koskela and J. Tuukkanen performed the three-point bending experiments. F.-P.Z. and M.P. generated the conditional Wnt16flox/flox mice. E.E.E., F.Z. and L.S. performed the immunohistochemistry. A.H. performed western blots. M.B. produced the WNT16 liposomes. A. Kassem and C.L. performed the inflammation-induced calvarial bone loss model. O.S. and P.A. performed the rat metaphyseal WNT16 injections. J.Q.F. provided the Dpm1-cre mice. J. Tuckermann provided the Runx2-cre mice. S.M.-S., P.H., R.B., U.H.L., F.G. and C.O. designed and supervised the project and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
References
- 1.Baron R, Hesse E. Update on bone anabolics in osteoporosis treatment: rationale, current status, and perspectives. J Clin Endocrinol Metab. 2012;97:311–325. doi: 10.1210/jc.2011-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zebaze RM, et al. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet. 2010;375:1729–1736. doi: 10.1016/S0140-6736(10)60320-0. [DOI] [PubMed] [Google Scholar]
- 3.Holzer G, von Skrbensky G, Holzer LA, Pichl W. Hip fractures and the contribution of cortical versus trabecular bone to femoral neck strength. J Bone Miner Res. 2009;24:468–474. doi: 10.1359/jbmr.081108. [DOI] [PubMed] [Google Scholar]
- 4.Zheng HF, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 2012;8:e1002745. doi: 10.1371/journal.pgen.1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 6.Cadigan KM, Peifer M. Wnt signaling from development to disease: insights from model systems. Cold Spring Harb Perspect Biol. 2009;1:a002881. doi: 10.1101/cshperspect.a002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenny A. Planar cell polarity signaling in the Drosophila eye. Curr Top Dev Biol. 2010;93:189–227. doi: 10.1016/B978-0-12-385044-7.00007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohn AD, Moon RT. Wnt and calcium signaling: β-catenin–independent pathways. Cell Calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 9.Gong Y, et al. LDL receptor–related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 10.Little RD, et al. A mutation in the LDL receptor–related protein 5 gene results in the autosomal dominant high–bone-mass trait. Am J Hum Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyden LM, et al. High bone density due to a mutation in LDL-receptor–related protein 5. N Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 12.Brunkow ME, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balemans W, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kramer I, et al. Osteocyte Wnt/β-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–3085. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glass DA, II, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Miclea RL, et al. Adenomatous polyposis coli–mediated control of β-catenin is essential for both chondrogenic and osteogenic differentiation of skeletal precursors. BMC Dev Biol. 2009;9:26. doi: 10.1186/1471-213X-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett CN, et al. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22:1924–1932. doi: 10.1359/jbmr.070810. [DOI] [PubMed] [Google Scholar]
- 18.Kato M, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606–615. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmen SL, et al. Essential role of β-catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–21168. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 21.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Wei W, et al. Biphasic and dosage-dependent regulation of osteoclastogenesis by β-catenin. Mol Cell Biol. 2011;31:4706–4719. doi: 10.1128/MCB.05980-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeda K, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18:405–412. doi: 10.1038/nm.2653. [DOI] [PubMed] [Google Scholar]
- 25.Estrada K, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44:491–501. doi: 10.1038/ng.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medina-Gomez C, et al. Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLoS Genet. 2012;8:e1002718. doi: 10.1371/journal.pgen.1002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koller DL, et al. Meta-analysis of genome-wide studies identifies WNT16 and ESR1 SNPs associated with bone mineral density in premenopausal women. J Bone Miner Res. 2013;28:547–558. doi: 10.1002/jbmr.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.García-Ibarbia C, et al. Missense polymorphisms of the WNT16 gene are associated with bone mass, hip geometry and fractures. Osteoporos Int. 2013;24:2449–2454. doi: 10.1007/s00198-013-2302-0. [DOI] [PubMed] [Google Scholar]
- 29.Hendrickx G, et al. Variation in the Kozak sequence of WNT16 results in an increased translation and is associated with osteoporosis related parameters. Bone. 2014;59:57–65. doi: 10.1016/j.bone.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 30.Brommage R, Liu J, Revelli J, Kirkpatrick L, Powwell D. Gene knockouts of Wnt10b and Wnt16 in mice result in low bone mass. Bone. 2007;40:S187. [Google Scholar]
- 31.Huang SM, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 32.Teh MT, et al. Role for WNT16B in human epidermal keratinocyte proliferation and differentiation. J Cell Sci. 2007;120:330–339. doi: 10.1242/jcs.03329. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Ross JF, Bodine PV, Billiard J. Homodimerization of Ror2 tyrosine kinase receptor induces 14–3-3β phosphorylation and promotes osteoblast differentiation and bone formation. Mol Endocrinol. 2007;21:3050–3061. doi: 10.1210/me.2007-0323. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Rubin B, Bodine PV, Billiard J. Wnt5a induces homodimerization and activation of Ror2 receptor tyrosine kinase. J Cell Biochem. 2008;105:497–502. doi: 10.1002/jcb.21848. [DOI] [PubMed] [Google Scholar]
- 35.Rauch A, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11:517–531. doi: 10.1016/j.cmet.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y, et al. DMP1-targeted Cre expression in odontoblasts and osteocytes. J Dent Res. 2007;86:320–325. doi: 10.1177/154405910708600404. [DOI] [PubMed] [Google Scholar]
- 37.Kim J, et al. Lipoproteins are an important bacterial component responsible for bone destruction through the induction of osteoclast differentiation and activation. J Bone Miner Res. 2013;28:2381–2391. doi: 10.1002/jbmr.1973. [DOI] [PubMed] [Google Scholar]
- 38.Wermelin K, Suska F, Tengvall P, Thomsen P, Aspenberg P. Stainless steel screws coated with bisphosphonates gave stronger fixation and more surrounding bone. Histomorphometry in rats. Bone. 2008;42:365–371. doi: 10.1016/j.bone.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Zhong Z, et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc Natl Acad Sci USA. 2012;109:E2197–E2204. doi: 10.1073/pnas.1120407109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett CN, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA. 2005;102:3324–3329. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevens JR, et al. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res. 2010;25:2138–2147. doi: 10.1002/jbmr.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green JL, et al. Use of a molecular genetic platform technology to produce human Wnt proteins reveals distinct local and distal signaling abilities. PLoS ONE. 2013;8:e58395. doi: 10.1371/journal.pone.0058395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo X, et al. Wnt/β-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–2417. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu D, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2004;101:3118–3123. doi: 10.1073/pnas.0308648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugimura R, et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. 2012;150:351–365. doi: 10.1016/j.cell.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Z, Von den Hoff JW, Torensma R, Meng L, Bian Z. Wnt16 is involved in intramembranous ossification and suppresses osteoblast differentiation through the Wnt/β-catenin pathway. J Cell Physiol. 2014;229:384–392. doi: 10.1002/jcp.24460. [DOI] [PubMed] [Google Scholar]
- 47.Santiago F, Oguma J, Brown AM, Laurence J. Noncanonical Wnt signaling promotes osteoclast differentiation and is facilitated by the human immunodeficiency virus protease inhibitor ritonavir. Biochem Biophys Res Commun. 2012;417:223–230. doi: 10.1016/j.bbrc.2011.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 49.Valenzuela DM, et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol. 2003;21:652–659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- 50.Windahl SH, et al. Estrogen receptor-α in osteocytes is important for trabecular bone formation in male mice. Proc Natl Acad Sci USA. 2013;110:2294–2299. doi: 10.1073/pnas.1220811110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maes C, et al. Increased skeletal VEGF enhances β-catenin activity and results in excessively ossified bones. EMBO J. 2010;29:424–441. doi: 10.1038/emboj.2009.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Windahl SH, Vidal O, Andersson G, Gustafsson JA, Ohlsson C. Increased cortical bone mineral content but unchanged trabecular bone mineral density in female ERβ−/− mice. J Clin Invest. 1999;104:895–901. doi: 10.1172/JCI6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vidal O, et al. Estrogen receptor specificity in the regulation of skeletal growth and maturation in male mice. Proc Natl Acad Sci USA. 2000;97:5474–5479. doi: 10.1073/pnas.97.10.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Movérare S, et al. Differential effects on bone of estrogen receptor α and androgen receptor activation in orchidectomized adult male mice. Proc Natl Acad Sci USA. 2003;100:13573–13578. doi: 10.1073/pnas.2233084100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waarsing JH, Day JS, Weinans H. An improved segmentation method for in vivo microCT imaging. J Bone Miner Res. 2004;19:1640–1650. doi: 10.1359/JBMR.040705. [DOI] [PubMed] [Google Scholar]
- 56.Hildebrand T, Ruegsegger P. Quantification of bone microarchitecture with the structure model index. Comput Methods Biomech Biomed Engin. 1997;1:15–23. doi: 10.1080/01495739708936692. [DOI] [PubMed] [Google Scholar]
- 57.Parfitt AM, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 58.Baron R, Vignery A, Neff L, Silverglate A, Santa Maria A. Processing of undecalcified bone specimens for bone histomorphometry. In: Recker R, editor. Bone Histomorphometry: Techniques and Interpretation. CRC Press Inc; Boca Raton: 1983. pp. 13–35. [Google Scholar]
- 59.Eriksen E, Axelrod D, Melsen F. Bone Histomorphometry. Raven; New York: 1994. [Google Scholar]
- 60.Hayman AR, Macary P, Lehner PJ, Cox TM. Tartrate-resistant acid phosphatase (Acp 5): identification in diverse human tissues and dendritic cells. J Histochem Cytochem. 2001;49:675–684. doi: 10.1177/002215540104900601. [DOI] [PubMed] [Google Scholar]
- 61.Granholm S, Henning P, Lindholm C, Lerner UH. Osteoclast progenitor cells present in significant amounts in mouse calvarial osteoblast isolations and osteoclastogenesis increased by BMP-2. Bone. 2013;52:83–92. doi: 10.1016/j.bone.2012.09.019. [DOI] [PubMed] [Google Scholar]
- 62.Stanford CM, Jacobson PA, Eanes ED, Lembke LA, Midura RJ. Rapidly forming apatitic mineral in an osteoblastic cell line (UMR 106–01 BSP) J Biol Chem. 1995;270:9420–9428. doi: 10.1074/jbc.270.16.9420. [DOI] [PubMed] [Google Scholar]
- 63.Granholm S, Lundberg P, Lerner UH. Calcitonin inhibits osteoclast formation in mouse haematopoetic cells independently of transcriptional regulation by receptor activator of NF-κB and c-Fms. J Endocrinol. 2007;195:415–427. doi: 10.1677/JOE-07-0338. [DOI] [PubMed] [Google Scholar]
- 64.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- 65.Morrell NT, et al. Liposomal packaging generates Wnt protein with in vivo biological activity. PLoS ONE. 2008;3:e2930. doi: 10.1371/journal.pone.0002930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Agholme F, Li X, Isaksson H, Ke HZ, Aspenberg P. Sclerostin antibody treatment enhances metaphyseal bone healing in rats. J Bone Miner Res. 2010;25:2412–2418. doi: 10.1002/jbmr.135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.